

Abstract
Background
Hedgehog (Hh) signaling pathway-related genes have important roles in several physiological and disease processes that involve cell proliferation. Long non-coding region RNAs (lncRNAs) have a regulatory role on gene expression. Keloid is characterized by excessive proliferation of scar tissue following trauma. The aims of this study were to evaluate the Hh signaling pathway in keloid skin tissues and its downstream gene expression and lncRNAs, compared with normal skin.
Material/Methods
Four pairs of keloids and adjacent normal skin epidermis underwent total RNA extraction. Gene chip high-throughput real-time quantitative polymerase chain reaction (qPCR) was used to examine the differential expression profiles of the Hh signaling pathway-related lncRNAs and mRNAs in the human keloid and normal skin. The differentially expressed mRNAs were analyzed by Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) to identify their biological roles.
Results
In keloid tissue, differential expression of 33 mRNAs and 30 lncRNAs relating to the Hh pathway, were verified by gene chip qPCR. The results of GO and KEGG analysis showed that the upregulated mRNAs were involved in cell proliferation, cell growth, and tissue repair, and down-regulated mRNAs were involved in apoptosis. The lncRNA, AC073257.2, affected cell keloid growth and proliferation by its upstream target the GLI2 gene at the transcriptional level. The lncRNA, HNF1A-AS1, affected cell keloid growth and proliferation by its neighboring target gene, HNF1A.
Conclusions
Differential expression occurred in Hh signaling pathway-related lncRNAs and mRNAs, which may provide further insight into the development of keloid.
MeSH Keywords: Hedgehog Proteins; Keloid; RNA, Long Noncoding
Background
Keloid is a proliferative reaction or hyperplasia of skin scar tissue following trauma and consists of the excessive proliferation of fibroblasts and the deposition of disordered collagen fibers and extracellular matrix (ECM) [1]. The clinical appearance of keloid includes areas of elevated patches or papules, which exceed the boundaries of the primary wound. Keloid does not regress spontaneously and is prone to recurrence after excision. The chest, shoulders, upper arms, earlobes, and cheeks are sites of predilection for keloid scars [2]. Currently, the pathogenesis of keloid remains unclear, but skin type, trauma, and local inflammation are generally considered as the most common causes. Because the specific mechanisms of keloid are unclear, and because of the effects of keloid formation on cosmesis, there are both clinical and social reasons to determine the underlying pathogenesis to prevent or treat keloid formation.
Long non-coding RNA (lncRNA) is a protein non-coding sequence with a length of more than 200 nucleotides (nt), which is widely conserved in mammalian cells and is formed by RNA polymerase transcription. Due to the lack of an effective open reading frame, lncRNA seldom encodes but has diverse functions and interactions that include RNA-RNA base pairing, RNA-protein interactions, and RNA-DNA interactions. Studies have shown that lncRNAs are involved in the regulation of several processes, including genomic imprinting, chromatin epigenetic modification, and the expression of homologous genes [3], with variable frequency and stages of disease development, and gene regulation [4,5]. Previously published studies have shown that lncRNAs are involved in cell growth and proliferation in wound healing and in the development of keloid [6,7]. However, the mechanism of the role of lncRNAs in keloid formation remains unclear.
The Hedgehog (Hh) signaling pathway-related genes, involving many secretory signaling proteins, have recently been studied in human physiological and disease processes, but was first found in Drosophila (fruit flies), with the spikes appearing on the body surface of Drosophila larvae following mutation, which explains the name, ‘hedgehog’ [8].
The main target genes of the Hh signaling pathway are PTCH1, PTCH2, and GLI1. The Hh signaling pathway has been described as the Hh-Ptch-Smo-Gli signaling axis, and although Hh is nonactivated, PTCH and SMO genes constitute the receptor complex, which inhibits the activity of SMO. Once Hh combines with PTCH, the inhibiting effect of SMO is removed and the SMO gene is activated to pass signaling down to the transcription factor Gli, which then enters into the cell nucleus. Downstream target genes are then activated and expressed to regulate cell differentiation, proliferation, epithelial-mesenchymal transition (EMT), invasion, migration, drug resistance, and apoptosis [9]. Also, the Hh pathway plays a key role in cell differentiation and proliferation during embryonic development and wound healing of many organs, and is also associated with recurrence, invasion, and metastasis of multiple tumors [10]. The Hh pathway has previously been shown to be involved in cutaneous fibrosing disorders [11]. A previously published study has shown that Hh pathway related lncRNAs could modulate the development of gastric cancer [12]. However, the expression, effects, and mechanisms of lncRNAs remain unclear in the keloid epidermis.
Therefore, the aims of this study were to examine samples of human keloid epidermis obtained at surgery, with adjacent normal epidermis, using the LncPath Human Hedgehog Pathway Array (8 × 15K) hybridization technique to screen differential expression profiles of Hh pathway-related lncRNAs and mRNAs. Gene Ontology (GO) analysis was used to investigate the mRNAs involved in the biological process of keloid formation, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to explore the signaling pathway-related mRNAs, which were involved in the occurrence and development of keloid. The study also aimed to analyze differentially expressed lncRNAs and mRNAs to investigate their roles on the occurrence and development of keloid using the Arraystar lncRNA database to explore potential targets for the treatment of keloid.
Material and Methods
Patients
The study was approved by the Ethics Committee of First Affiliated Hospital of Nanchang University and the Ethics Committee of Jiangxi Maternal and Child Health Hospital. Patients consented to provide skin tissue samples that included four cases of excised keloids and adjacent normal skin. The patient ages ranged from 18 to 50 years, with an average age of 26 years. There were two cases of keloid following burn injury, and two cases of earlobe keloid. The keloids had a clinical appearance of intumescent plaques or papules with an area exceeding the primary wound boundaries and none of them spontaneously regressed. None of the cases of keloid had received any drug therapy or radiotherapy surgery, and the patients did not suffer from systemic diseases or malignancy. Two tissue samples, keloid and normal skin, were taken from each patient, and then the dermis was removed so that the epidermal tissue remained. RNA storage fluid was added to the epidermal tissues, which were stored in a freezer at −80°C until RNA extraction.
Reagents and instruments
The LncPath™ Human Hedgehog (Hh) Pathway Array (ArrayStar, Rockville, MD, USA), RNA storage fluid (ComWin Biotech, Beijing, China), TRIzol, diethyl pyrocarbonate (DEPC) (Invitrogen, Carlsbad, CA, USA), gentamycin for injection (North China Pharmaceutical, Shijiazhuang, China), mRNA-ONLY™ mRNA isolation kit, sodium acetate, formaldehyde (Shanghai Chemical Reagent, Shanghai, China), ethylenediaminetetraacetic acid (EDTA), ethidium bromide (Cusabio Technology, Wuhan, China), agarose (Sangon Biotech, Shanghai, China), primers (ThermoFisher Scientific, Waltham, MA, USA), polymerase chain reaction (PCR) master mix, (ArrayStar, Rockville, MD, USA), Image-Pro Plus version 6.0 Image Analysis Software (Media Cybernetics, Rockville, MD, USA), chip scanner G2565CA, Feature Extraction v11.0.1.1, American GeneSpring (GX version 12) software (Agilent, Santa Clara CA, USA).
Extraction and separation of total RNA from epidermal tissue
Fresh tissue samples (100 mg) of human keloid epidermis and normal skin epidermis were macerated in a mortar at high temperature to sterilize the tissue, and then 1 ml of RNA extracting solution, TRIzol was added. The mixture was transferred to 1.5 ml Eppendorf tubes by pipette and mixed by repeated inversion (ten times). The mixtures were left to stand at room temperature for 10 min to separate the nucleic acids and protein. The mixtures were centrifuged at 11,180×g at 4°C, the supernatant was removed and mixed with 0.2 ml trichloromethane for 15 s and then centrifuged again at 11,180×g at 4°C. The upper phase solutions (about 500 uL) containing RNA were transferred to new tubes, leaving the middle and bottom layer, which contained DNA and protein.
RNA precipitation
Using new tubes, 0.5 ml of ice-cold isopropanol was added to the RNA solution, and then stored at −20°C for 30 min after mixing, then centrifuged at 11,180×g at 4°C for 10 min. The RNA precipitates were often invisible before centrifugation, and the colloidal precipitates of RNA were found at the tube sides or bottom after centrifugation. Then RNA precipitates were collected and dissolved in 1ml of 75% ethyl alcohol (prepared with DEPC), centrifuged at 6,288×g at 4°C for 5 min after vortex oscillation. The supernatant was discarded, and 10 min later, the RNA precipitates were dried. Finally, the precipitate was dissolved and the mixed solution was divided into several samples and stored at −70°C or reversed transcription immediately.
Detection of total RNA concentration, purity, and integrity
Firstly, 0.1% of DEPC water was added dropwise and 1 ul of RNA was diluted with a ratio of 1: 50 with 0.1% DEPC water and the concentration was measured using a Nanodrop ND-1000 spectrophotometer. The absorption peak of RNA was 260 nm while that of protein was 280 nm. The ideal purity of RNA had a ratio of A260/A280 between 1.8 and 2.1. If the ratio was more than 2.1 or less than 1.8, this meant that the RNA samples were contaminated by protein or enzyme. The RNA was diluted with DEPC water and mixed with ethidium bromide (EtBr) (1.0 mg/mL) before the RNA integrity was tested by agarose gel electrophoresis. RNA integrity was demonstrated by RNA bands that were clear and bright, with the ration of bands of 28S: 18S rRNA more than or approximated to 2: 1.
Gene microarray, image acquisition, and data analysis
The total RNAs of the two groups were given to KangChen Biotech Inc. (Shanghai, China) for chip hybridization after the quality assessment was satisfactory. The chip used was the lncPath™ Human Hedgehog Pathway Array (8×15K) (ArrayStar, Rockville, MD, USA). The lncRNAs were selected from authoritative public transcriptome databases with a high impact factor (including RefSeq, UCSC Known Genes, and GENCODE). The rRNAs were removed from the total RNAs, according to the manufacturer’s protocol for the mRNA-ONLY™ Eukaryotic mRNA Isolation Kit (Epicentre Biotechnology, San Diego, CA, USA). Random priming was used to amplify each sample and transcribe it into cRNAs with fluorescence and purified and labeled using the RNeasy Mini Kit (Qiagen, Hilden, Germany). The concentration and activity of the cRNAs were tested by NanoDrop ND-1000 (Thermo Fisher, Waltham, MA, USA). Then, cRNAs were hybridized with the LncPath™ Human Hedgehog Pathway Array, and the obtained hybrid chips were washed and fixed by Agilent DNA Microarray Scanner (G2565CA). A spectrophotometer was used to test the efficiency of fluorescence labeling.
Standardization and primary analysis of original data
After hybridization, the chips were scanned using the Agilent G2565BA scanner and washed. The original data was processed by Agilent Feature Extraction version 11.0.1.1 software and the Agilent Gene Spring GX version 12 software was used for data analysis. Screening of the differentially expressed genes was undertaken to include the fold-change ≥1.5, and p<0.05. DAVID Bioinformatics Resources version 6.7 and FunRich software were used to conduct Gene Ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, clustering analysis of function annotation and functional enrichment analysis.
Based on the Gene Ontology database, the obtained differential protein-coding gene mRNA was performed for enrichment analysis of gene function using GO analysis (http://www.geneontology.org), which is an international standard classification system of gene function, which mainly includes the biological process (BP), molecular function (MF) and cellular component (CC). This study focused on the biological process (BP), and the p-value, which correlated with the degree of enrichment in GO terms using the GO database default statistics, with the lower the p-value, the more important the GO term. A p-value of <0.05 was assumed to be statistically significant. The p-value was converted to log base 10 (the common logarithm), the enrichment score was obtained for differentially expressed mRNA. The higher the enrichment score was, the more significant it was.
The differentially expressed genes by chip imaging were conducted for metabolic pathway analysis after being input into the signaling pathway of the KEGG database, for pathway analysis, which was the database for systematic analysis of gene functions and relevant information of genome. Using the network of molecular interaction in a cell, this process linked a series of acquired genes in the genome into a pathway or a compound, to show the biological functions at a higher level. In this way, all the pathways involved in the differentially expressed genes would be obtained. As with GO analysis, a p-value of <0.05 was assumed to be statistically significant.
Quality assessment of the gene chip data
Box plot and scatter plot were adopted to evaluate the quality of the screened differentially expressed lncRNAs and mRNAs, which aimed to preliminarily judge the expression levels and differentially expressed lncRNAs and mRNAs. On the basis of obtained standardized data, cluster analysis was used to analyze and classify the expression patterns of lncRNAs and mRNAs in the two groups, and the differentially expressed genes among the samples were displayed by heat maps.
Real-time quantitative polymerase chain reaction (qPCR) for verification of the reliability of the lncRNA chip testing result
On the basis of obtained standardized data, cluster analysis was used to analyze the randomly selected differentially expressed two lncRNAs and four mRNAs (more than 1.5 times). The epidermis of normal skin was used as control and GAPDH as an internal reference to carry out the verification of the qPCR results for the Hh pathway-related lncRNAs and mRNAs in the epidermis of the keloid tissue. The serial numbers of primers are shown in Table 1. The reaction conditions of the PCR were as follows: 95°C, 10 min; 40 PCR cycles at 95°C, 10 s; 60°C, 60 s. When the cycle was finished, a curve was used to detect the product specificity, and the product was slowly heated from 60°C to 90°C. Fluorescence signals were gathered five times at each temperature. The quantitative analysis used the 2−Δ ΔCt method and the fold-change of differentially-expressed genes in the skin epidermal keloid.
Table 1.
Hedgehog (Hh) pathway-related long non-coding region RNA (lncRNA) and mRNA gene primers for real-time quantitative polymerase chain reaction (qPCR).
| LncRNA and mRNA gene | Primer sequence (5′-3′) | Size (bp) | |
|---|---|---|---|
| Forward | Reverse | ||
| LOC100271722 | CCACAGAACTGAGCAGAACTAAT | GCAGCAGGTTAAGGACACTT | 199 |
| HNF1A-AS1 | CGCCAGAATCTAAGCCTTGAC | CTCTAGCACCTCGGAACCTT | 148 |
| SFRP2 | GGAGACCAAGAGCAAGACCAT | TGAGCCACAGCACCGATT | 77 |
| WIF1 | CAGCACACGCCTTCACTT | AGATGTCGGAGTTCACCAGAT | 81 |
| WNT7B | ATCCCTCATCTCATCCCTCATC | AGTGTCTCGGTGGCATCA | 170 |
| HNF1A | GCCTTGTTCTGTCACCAATGTA | TGCCTGCCTTCCCTGTTA | 196 |
Statistical analysis
Statistical analysis was performed using SPSS Version 19 software. The t-test (two-tailed) was used for the evaluation of the differentially expressed lncRNAs or mRNAs in the microarray data and qPCR data to determine whether there were differentially expressed lncRNAs or mRNAs between the keloid group and control group. A p-value of <0.05 was considered to represent statistical significance.
Results
RNA quality testing
The total RNAs of keloid epidermis and normal epidermis were extracted and the optical density (OD) ratios were tested by the NanoDrop ND-1000 spectrophotometer (Table 2). The A260/A280 ratio of RNAs extracted from the keloid and normal groups were 1.98 and 1.88, which were between 1.8 and 2.1, indicating that there was no RNA degradation, and no protein and DNA contamination. The A260/A230 ratios were 2.21 and 2.27 (<1.8), which indicated they were not affected by the organic solvent. The imaging of the agarose gel electrophoresis showed the total RNAs of the 18 s and 28 s bands were very clear, which demonstrated that the sample quality was well preserved.
Table 2.
RNA quantification and quality assurance of two samples.
| Sample ID | OD260/280 Ratio | OD260/230 Ratio | Conc. (ng/μl) | Volume (μl) | Quantity (ng) | QC result Pass or Fail |
|---|---|---|---|---|---|---|
| Keloid | 1.98 | 2.21 | 724.41 | 60 | 43464.6 | Pass |
| Normal skin | 1.88 | 2.27 | 404.10 | 30 | 12123.0 | Pass |
Chip hybridization
The clustering method was used for preliminary analysis of the expression pattern of the Hedgehog (Hh) signaling pathway-related long non-coding region RNAs (lncRNAs) and mRNAs in keloid epidermis. The heat map (Figure 1) showed that the Hh pathway-related lncRNAs and mRNAs showed differential expression in the keloid epidermis, which indicated that the differentially expressed lncRNAs might be involved in the occurrence and development of keloid.
Figure 1.

Expression of Hedgehog (Hh) signaling pathway-related long non-coding region RNAs (lncRNAs). (A) Thirty long non-coding region RNAs (lncRNAs) were differentially expressed relating to the Hedgehog (Hh) pathway. (B) Thirty-three lncRNAs were differentially expressed relating to the Hh pathway. There are 30 color bars in A and 33 color bars in B. Each color bar in the heat maps represent a lncRNA or mRNA, and the name of lncRNA or mRNA is on the right of the Figure. The red bar indicates upregulation of lncRNA or mRNA and the green bar indicates the down-regulation. [N] represents normal skin epidermis, and [K] represents the keloid epidermis. A is the lncRNA heat map and B is the mRNA heat map.
There were 214 mRNAs and 314 lncRNAs evaluated in the study. The expression profile of the Hh pathway-related lncRNAs and mRNAs by gene chip was acquired in keloid epidermis. Compared with normal epidermis, there were 30 lncRNAs and 33 mRNAs with differential expression in keloid epidermis. There were 16 lncRNAs that were upregulated, which included AK055628, MIAT, MIR31HG, RP11-264F23.3, and AC073257.2, and 14 lncRNAs were downregulated, which included RP11-12M9.3, XLOC_007437, XLOC_009485, RP5-1042I8.7, and HNF1A-AS1 (Table 3). There were 13 mRNAs that were upregulated, which included SFRP2, ANGPT2, APC2, IVL, and GLI2, and 20 mRNAs that were downregulated, which included WIF1, KRT1, CCND1, BCL2, and NOTCH2 (Table 4).
Table 3.
Hedgehog (Hh) pathway-related long non-coding region RNA (lncRNA) upregulation and down-regulation in keloid.
| Probe ID | Fold change | Regulation | Seqname |
|---|---|---|---|
| ASPWP0002024 | 2.34 | Up | ENST00000539135 |
| ASPWP0002926 | 1.70 | Up | ENST00000561261 |
| ASPWP0006522 | 2.00 | Up | ENST00000424191 |
| ASPWP0008593 | 35.88 | Up | AK055628 |
| ASPWP0008612 | 1.67 | Up | AF080092 |
| ASPWP0008613 | 1.55 | Up | FJ209302 |
| ASPWP0008625 | 9.14 | Up | NR_033321 |
| ASPWP0008670 | 3.99 | Up | NR_027054 |
| ASPWP0093960 | 1.62 | Up | TCONS_00002016 |
| ASPWP0115436 | 1.63 | Up | ENST00000573934 |
| ASPWP0123565 | 1.79 | Up | uc002lfu.1 |
| ASPWP0130865 | 1.54 | Up | ENST00000413991 |
| ASPWP0134856 | 1.81 | Up | ENST00000455309 |
| ASPWP0135363 | 1.55 | Up | ENST00000413525 |
| ASPWP0142205 | 1.70 | Up | NR_027036 |
| ASPWP0142208 | 1.70 | Up | uc003bgs.3 |
| ASPWP0002025 | −1.54 | Down | ENST00000537370 |
| ASPWP0002195 | −1.53 | Down | ENST00000537361 |
| ASPWP0002461 | −1.77 | Down | ENST00000566949 |
| ASPWP0008623 | −1.58 | Down | NR_002733 |
| ASPWP0008640 | −1.65 | Down | NR_051960 |
| ASPWP0046898 | −1.69 | Down | ENST00000473840 |
| ASPWP0113335 | −1.86 | Down | TCONS_00016040 |
| ASPWP0132397 | −1.77 | Down | TCONS_00020094 |
| ASPWP0142037 | −1.51 | Down | ENST00000423293 |
| ASPWP0142198 | −1.59 | Down | ENST00000451118 |
| ASPWP0143423 | −1.95 | Down | ENST00000399188 |
| ASPWP0155090 | −1.57 | Down | ENST00000511127 |
| ASPWP0184407 | −1.56 | Down | ENST00000411473 |
| ASPWP0231943 | −1.54 | Down | uc001aam.4 |
Table 4.
Hedgehog pathway-related mRNA upregulation and down-regulation in keloid.
| Probe ID | Fold change | Regulation | Seqname |
|---|---|---|---|
| ASPWP0000156 | 1.88 | Up | NM_198968 |
| ASPWP0001546 | 1.85 | Up | NM_000435 |
| ASPWP0001595 | 1.65 | Up | NM_001147 |
| ASPWP0001679 | 6.82 | Up | NM_003013 |
| ASPWP0003294 | 2.17 | Up | NM_005250 |
| ASPWP0003585 | 1.94 | Up | NM_001466 |
| ASPWP0003794 | 2.47 | Up | NM_005883 |
| ASPWP0010789 | 1.93 | Up | NM_004625 |
| ASPWP0010937 | 10.09 | Up | NM_003013 |
| ASPWP0011247 | 6.32 | Up | NM_001147 |
| ASPWP0011533 | 2.08 | Up | NM_012193 |
| ASPWP0011760 | 1.62 | Up | NM_005270 |
| ASPWP0012305 | 2.28 | Up | NM_005547 |
| ASPWP0000050 | −1.52 | Down | ENST00000269576 |
| ASPWP0001079 | −1.68 | Down | NM_013409 |
| ASPWP0001348 | −2.11 | Down | NM_007191 |
| ASPWP0001372 | −3.36 | Down | NM_006121 |
| ASPWP0001716 | −1.66 | Down | NM_005524 |
| ASPWP0004262 | −2.17 | Down | NM_003507 |
| ASPWP0005425 | −2.07 | Down | NM_000427 |
| ASPWP0006917 | −2.02 | Down | NM_005438 |
| ASPWP0009248 | −1.53 | Down | NM_001077628 |
| ASPWP0009873 | −2.38 | Down | NM_053056 |
| ASPWP0010027 | −1.65 | Down | NM_024408 |
| ASPWP0010382 | −2.19 | Down | NM_000633 |
| ASPWP0011103 | −1.70 | Down | NM_005618 |
| ASPWP0011307 | −1.99 | Down | NM_002467 |
| ASPWP0011396 | −1.64 | Down | NM_004089 |
| ASPWP0011429 | −1.92 | Down | NM_031866 |
| ASPWP0011572 | −1.54 | Down | NM_000610 |
| ASPWP0011800 | −2.06 | Down | NM_000214 |
| ASPWP0012306 | −1.51 | Down | NM_000427 |
| ASPWP0012416 | −6.64 | Down | NM_007191 |
The LncPath™ array profile lncRNAs and their target gene mRNAs were pathway-focused or disease-specific. By quantifying lncRNAs (at the transcription level) and mRNAs (at both gene and transcription levels) in parallel, the LncPath™ pathway focused on lncRNA microarrays that could provide comprehensive insights into the expressional relationship between lncRNAs and mRNAs. The connections between the lncRNA regulatory mechanisms and their biological functions are shown in Table 5.
Table 5.
Hedgehog (Hh) pathway-related long non-coding region RNA (lncRNA) predicted the regulation of mRNA of genes in keloid.
| Genesymbol | Fold change | Regulation | Genomic relationship | mRNA symbol |
|---|---|---|---|---|
| RP11-264F23.3 | 2.34 | Up | Overlapping | CCND2 |
| RP11-12M9.3 | −1.95 | Down | Upstream | RBX1 |
| XLOC_007437 | −1.86 | Down | Downstream | GAS1 |
| XLOC_009485 | −1.77 | Down | Upstream | CCND1 |
| RP5-1042I8.7 | −1.77 | Down | Upstream | NOTCH2 |
| LOC100271722 | 1.70 | Up | Downstream | WNT7B |
| LINC00594 | 1.70 | Up | Upstream | DISP2 |
| LINC00594 | 1.70 | Up | Upstream | DISP2 |
| XLOC_000166 | 1.62 | Up | Downstream | PTCH2 |
| WI2-85898F10.1 | −1.59 | Down | Upstream | WNT7B |
| AC010090.1 | 1.55 | Up | Upstream | ZEB2 |
| RP11-264F23.4 | −1.54 | Down | Overlapping | CCND2 |
| AC073257.2 | 1.54 | Up | Upstream | GLI2 |
| HNF1A-AS1 | −1.53 | Down | Overlapping | HNF1A |
| RP11-12M9.4 | −1.51 | Down | Downstream | RBX1 |
Gene Ontology (GO) database analysis of target genes
The DAVID Bioinformatics Resources version 6.7 that was adopted to conduct GO analysis for mRNA identified 87 enriched GO terms that were screened according to statistical significance (p<0.05). The results of the first 20 enrichments are shown in Table 6. The differentially expressed mRNAs in GO database were involved in cellular biological processes. The upregulated mRNAs in the keloid epidermis mainly participated in biological processes including cell proliferation, cell growth, as well as tissue repair. While the significantly downregulated genes took part in cell necrosis, apoptosis, programmed cell death, and cell metabolism. FunRich software was used to analyze GO data for mRNAs. Figure 2 shows seven of the enriched metabolic pathways, which were mainly involved in embryonic development, gene expression, epigenetic inheritance, cell growth, cell communication, maintenance of cell morphology, and signal transduction, which might be associated with the activations of cell pathways and cell proliferation.
Table 6.
Gene Ontology (GO) analysis of differentially expressed mRNA in biological processes.
| Term | Genes | P-Value |
|---|---|---|
| GO: 0017147 Wnt-protein binding |
FZD8, SFRP2, WIF1, FZD2, FZD4, FZD7 | <0.001 |
| GO: 0060070 canonical Wnt signaling pathway |
FZD8, CCND1, FZD2, WNT7A, FZD4, MYC, FZD7 | <0.001 |
| GO: 0007219 Notch signaling pathway |
NOTCH3, HES1, NOTCH2, APH1A, DLL1, JAG1, MYC | <0.001 |
| GO: 0042813 Wnt-activated receptor activity |
FZD8, SFRP2, FZD2, FZD4, FZD7 | <0.001 |
| GO: 0007275 multicellular organism development |
NOTCH2, FOXL1, SFRP2, DZIP1, DLL1, WIF1, JAG1, FZD2, FZD4 | <0.001 |
| GO: 0007220 Notch receptor processing |
NOTCH3, APH1A, DLL1, JAG1 | <0.001 |
| GO: 0035567 non-canonical Wnt signaling pathway |
FZD8, SFRP2, FZD2, WNT7A | <0.001 |
| GO: 0045944 positive regulation of transcription from RNA polymerase II promoter |
NOTCH3, HES1, FZD8, SFRP2, DLL1, JAG1, GLI2, WNT7A, FOSL1, MYC | <0.001 |
| GO: 0030178 negative regulation of Wnt signaling pathway |
CCND1, APC2, SFRP2, WIF1 | <0.001 |
| GO: 0000122 negative regulation of transcription from RNA polymerase II promoter |
NOTCH3, HES1, FZD8, TSC22D3, CCND1, FST, GLI2, MYC | <0.001 |
| GO: 0005515 protein binding |
LOR, FZD8, APC2, APH1A, DZIP1, FST, DLL1, JAG1, FZD2, GLI2, FZD4, FZD7, NOTCH3, HES1, NOTCH2, CCND1, CD44, BCL2, KRT1, WIF1, FOSL1, IVL, MYC, ANGPT2, WNT7A | <0.001 |
| GO: 0010812 negative regulation of cell-substrate adhesion |
ANGPT2, FZD4, FZD7 | <0.001 |
| GO: 0016055 Wnt signaling pathway |
APC2, SFRP2, WIF1, WNT7A, FZD4 | <0.001 |
| GO: 0030216 keratinocyte differentiation |
LOR, KRT10, JAG1, IVL | <0.001 |
| GO: 0030165 PDZ domain binding |
FZD8, FZD2, FZD4, FZD7 | <0.001 |
| GO: 0001709 cell fate determination |
NOTCH2, DLL1, JAG1 | <0.001 |
| GO: 0030182 neuron differentiation |
FZD8, FZD2, FZD4, FZD7 | <0.001 |
| GO: 0097150 neuronal stem cell population maintenance |
HES1, DLL1, JAG1 | <0.001 |
| GO: 0043066 negative regulation of apoptotic process |
NOTCH2, CD44, BCL2, GLI2, WNT7A, MYC | <0.001 |
| GO: 0033077 T cell differentiation in thymus |
FZD8, BCL2, FZD7 | <0.001 |
Figure 2.
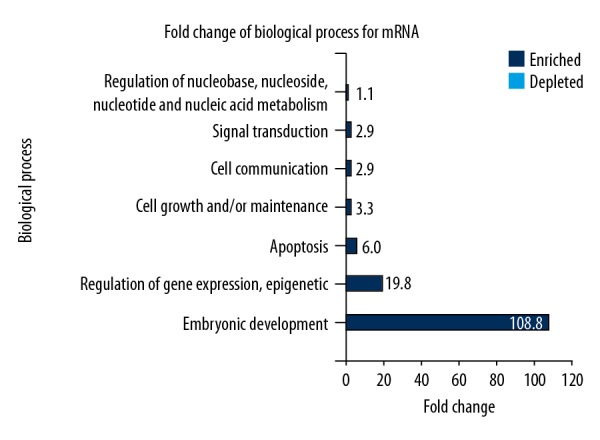
The biological effects of mRNA determined by enrichment of fold-change by Gene Ontology (GO) with FunRich software analysis. Seven enriched metabolic processes were identified, which mainly participated in embryonic development, gene expression, epigenetic inheritance, cell growth or communication, maintenance of cell morphology, and signal transduction.
Enrichment analysis of the Kyoto Encyclopedia of Genes and Genomes (KEGG) biological pathways of target genes
On the basis of the latest database of the Kyoto Encyclopedia of Genes and Genomes (KEGG), the Hh pathway-related differentially expressed mRNAs were analyzed in keloid epidermis. Biological pathways showed the main enrichment of differentially expressed mRNAs. The DAVID Bioinformatics Resources version 6.7 was used to conduct KEGG analysis for mRNA, and 14 significantly enriched KEGG pathways were screened according to statistical significance (p<0.05). The results are shown in Table 7. The pathways of significant enrichment were analyzed, among which the pathways associated with cell proliferation and apoptosis included the following: the Wnt signaling pathway (which promotes cell proliferation or survival and prevents cells from differentiation after activation); the Hippo signaling pathway (which promotes cell apoptosis, prevents limits cell size, and the size of organs after activation); the Notch signaling pathway (which affects the morphogenesis of normal cell growth, including cell proliferation, formation of cell boundaries, differentiation of multipotent progenitors, and cell apoptosis); the thyroid hormone signaling pathway (which controls hormone secretion and impacts on cell proliferation at the molecular level); the signaling pathways regulating pluripotency of stem cells; the proteoglycans and pathways in cancer.
Table 7.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of differentially expressed mRNA.
| Term | Genes | P-Value |
|---|---|---|
| Wnt signaling pathway | FZD8, CCND1, APC2, SFRP2, WIF1, FZD2, WNT7A, FZD4, FOSL1, MYC, FZD7 | <0.05 |
| Hippo signaling pathway | FZD8, CCND1, APC2, FZD2, GLI2, WNT7A, FZD4, MYC, FZD7 | <0.05 |
| Basal cell carcinoma | FZD8, APC2, FZD2, GLI2, WNT7A, FZD4, FZD7 | <0.05 |
| Notch signaling pathway | NOTCH3, HES1, NOTCH2, APH1A, DLL1, JAG1 | <0.05 |
| HTLV-I infection | FZD8, CCND1, APC2, FZD2, WNT7A, FZD4, FOSL1, MYC, FZD7 | <0.05 |
| Pathways in cancer | FZD8, CCND1, APC2, BCL2, FZD2, GLI2, WNT7A, FZD4, MYC, FZD7 | <0.05 |
| Proteoglycans in cancer | FZD8, CCND1, CD44, FZD2, WNT7A, FZD4, MYC, FZD7 | <0.05 |
| Signaling pathways regulating pluripotency of stem cells | FZD8, APC2, FZD2, WNT7A, FZD4, MYC, FZD7 | <0.05 |
| MicroRNAs in cancer | NOTCH3, NOTCH2, CCND1, CD44, APC2, BCL2, MYC | <0.05 |
| Melanogenesis | FZD8, FZD2, WNT7A, FZD4, FZD7 | <0.05 |
| Colorectal cancer | CCND1, APC2, BCL2, MYC | <0.05 |
| Thyroid hormone signaling pathway | NOTCH3, NOTCH2, CCND1, MYC | <0.05 |
| Endometrial cancer | CCND1, APC2, MYC | <0.05 |
| Small cell lung cancer | CCND1, BCL2, MYC | <0.05 |
FunRich software was adopted to perform the KEGG analysis for mRNAs. Figure 3 shows 10 significantly enriched metabolic pathways, which mainly involved the activation of the Notch signaling pathway and cellular signaling, promoting cell proliferation and inhibiting cell differentiation and apoptosis. These findings might explain the basis for the growth characteristics of a keloid.
Figure 3.
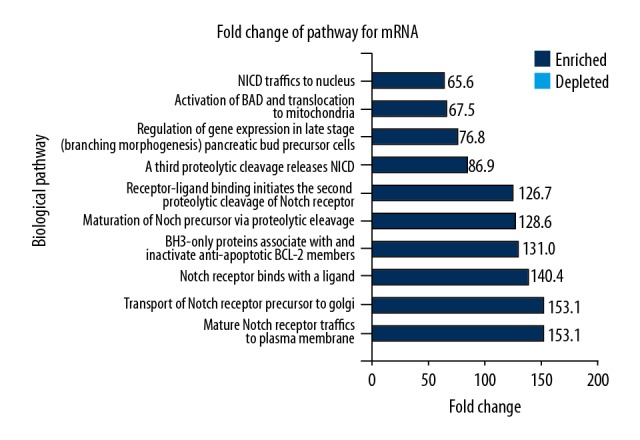
The biological effects of mRNA determined by enrichment of fold-change by the Kyoto Encyclopedia of Genes and Genomes (KEGG) with FunRich software analysis. Ten enriched metabolic pathways were identified, which mainly involved the activation of the Notch signaling pathway, control of apoptosis, and regulation of gene expression.
Real-time quantitative polymerase chain reaction (qPCR) verification
Real-time quantitative polymerase chain reaction (qPCR) was used to verify the reliability of gene chip results. The lncRNAs, HNF1A-AS1 and LOC100271722, and the mRNAs SFRP2, WIF1, WNT7B, and HNF1A were selected at random for qPCR verification. The expression of Hh pathway-related lncRNA LOC100271722 was upregulated and HNF1A-AS1 was downregulated in the keloid epidermis. The mRNAs, SFRP2, WNT7B, and HNF1A were upregulated and WIF1 was downregulated, which supported the findings of the microarray data, indicated that the data acquired by the gene chips were highly reliable. Figure 4 shows the comparison of the gene chip results and the qPCR results.
Figure 4.
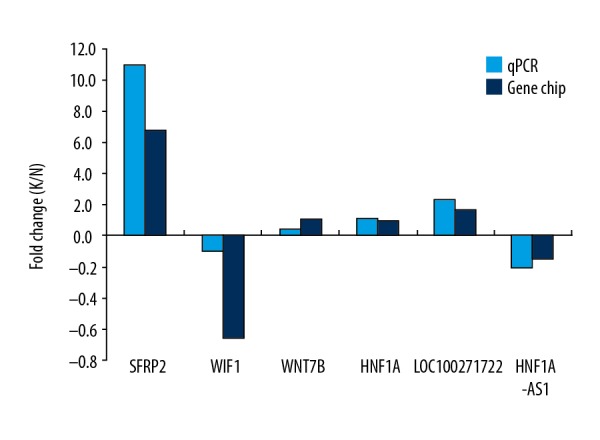
Comparison of gene chip analysis and high-throughput real-time quantitative polymerase chain reaction (qPCR).
Discussion
When the skin is injured from trauma or burns, fibroblasts are activated and undergo proliferation, with excessive but disorganized deposition of collagen and expression of growth factors that are associated with wound healing. Scar tissue eventually forms in normal wound healing but is found in excess in hypertrophic scar and keloid, which are similar skin lesions [13]. Hypertrophic scar does not involve the skin beyond the boundary of the initial injury or wound, has self-limited growth and does not relapse after excision. However, keloid scar tissue exceeds the boundaries of the initial wound, continues to grow, has infiltrative growth, and often recurs after surgical excision [14]. Currently, research on keloid has shown that the formation of keloid is mainly due to the excessive proliferation of fibroblasts, excessive deposition of collagen and extracellular matrix, changes in the expression of growth factors and apoptosis factors, and changes in signaling pathways for transforming growth factor-beta (TGF-beta)/Smad, MAPK, Wnt/beta-catenin, RhoA/ROCK and TNF-alpha/nuclear factor κB (NFκB) [15]. These previous findings have not yet resulted in more effective treatments for keloid.
Published studies have shown that Hedgehog (Hh) signaling pathway-related genes have an important role in processes including embryonic development and tissue repair, especially in the development of malignant tumors, with abnormal activation of the Hh signaling pathway being present in pancreatic cancer, liver cancer, and lung cancer [16–18]. Continuous activation of the Hh signaling pathway has been shown to result in excessive hyperplasia of the damaged liver and to promote hepatic fibrosis [19,20]. Because of these previously published findings, and the similarities with the biological processes found in keloid, the aims of this study were to evaluate the Hh signaling pathway in keloid skin tissues and its downstream mRNA expression and long non-coding region RNAs (lncRNAs), compared with normal skin.
The results of the present study are summarized in Tables 1–7, which show the differentially expressed lncRNAs and mRNAs. The function of many lncRNAs was unclear, but the function of their target gene mRNAs was known. Therefore we conducted Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis for target gene mRNAs to analyze their function in keloid. By GO analysis, the results showed that the function of the mRNAs was in maintaining the stability of the cellular and extracellular environment, cell transcription, translation and expression of genes, biochemical metabolic processes, cell proliferation and differentiation, signal transduction and apoptosis. By KEGG analysis, the mRNAs were shown to be involved in the Notch pathway, the Hippo pathway, the Wnt pathway, and cell cycle pathways. Therefore, it is possible that lncRNAs are involved in these processes in the pathogenesis of keloid.
To further explore the relationship between lncRNAs and mRNAs, the predicted regulation for lncRNAs and mRNAs was performed (Table 5). In particular, the expression of mRNA of Gli and HNF1α are of importance. Gli is the zinc finger transcription factor in the Hh signal transduction pathway and was first shown to be abnormally expressed in malignant glioma [21]. Gli not only plays an important role in the regulation of differentiation of embryonic tissue but also the occurrence and development of tumors. Gli2 has been shown to enhance the expression of Gli1, and both are important transcription factors in Hh signaling pathways [22]. The increased expression of Gli1 and Gli2 could directly lead to changes in the transcription level of target genes downstream of the Hh signaling pathway [22]. Gli2 was found to be highly expressed in hepatocellular carcinoma, and after knockout of the gene, the growth, proliferation, differentiation of the tumor cells and tumor invasion were significantly inhibited [23]. The findings of the present study showed that lncRNA AC073257.2 and its target gene Gli2 were both upregulated in keloid. By predicted regulation using the Arraystar lncRNA database, it was indicated that lncRNA AC073257.2 might have the role of a gene enhancer and could positively regulate the expression of its upstream target gene Gli2 at the transcription level to affect cell growth and proliferation in keloid.
Hepatocyte nuclear factor 1α (HNF1α) is one of the transcription factors that regulate the expression of liver genes, and mainly participates in liver detoxification, the metabolism of sugar, fat, steroid and amino acids, and maintains homeostasis [24]. The results of the present study showed that lncRNA HNF1α-AS1 was significantly downregulated in keloid, while expression of its target gene, HNF1α, was upregulated. HNF1α-AS1 was first described in gastric cancer and was shown by next-generation sequencing in pancreatic cancer, but with low expression [25,26]. In gastric cancer low expression of HNF1α-AS1 was associated with tumor size [25]. The findings of the present study showed that HNF1α-AS1 was downregulated and its target gene, HNF1α, was upregulated in keloid. By predicted regulation using the Arraystar lncRNA database, it was indicated that lncRNA HNF1α-AS1 could negatively regulate the expression of its neighboring target gene HNF1α at the transcription or post-transcription level to affect the growth of keloid.
Finally, the results of the present study were based on existing bioinformatics data in the databases used. However, to better understand the role of lncRNAs in the occurrence and development of keloid, studied should be undertaken to investigate the regulation of the identified lncRNAs in the keloid. Future studies are recommended, based on the findings of this study, to investigate the relationship between the lncRNAs and functional mechanisms in keloid. The aim of future studies will be to investigate potential new targets for the treatment of keloid.
Conclusions
The results of this study showed that differential expression of Hedgehog (Hh) signaling pathway-related long non-coding region RNAs (lncRNAs) were identified in keloid. In particular, the lncRNA AC073257.2 may participate in cell growth and proliferation in keloid by regulating the expression of its target gene Gli2, and the lncRNA HNF1α-AS1 may affect the growth of keloid by regulating the expression of its neighboring target gene, HNF1α. Therefore, the differentially expressed lncRNAs may provide novel insight into the occurrence and development of keloid, which may provide a basis for future studies to develop treatments for keloid.
Footnotes
Source of support: Departmental sources
References
- 1.Profyris C, Tziotzios C, Do Vale I. Cutaneous scarring: Pathophysiology, molecular mechanisms, and scar reduction therapeutics Part I. The molecular basis of scar formation. J Am Acad Dermatol. 2012;66:1–12. doi: 10.1016/j.jaad.2011.05.055. [DOI] [PubMed] [Google Scholar]
- 2.Butler PD, Longaker MT, Yang GP. Current progress in keloid research and treatment. J Am Coll Surg. 2008;206:731–41. doi: 10.1016/j.jamcollsurg.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–76. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao L, Kong H, Sun H, et al. LncRNA-PVT1 promotes pancreatic cancer cells proliferation and migration through acting as a molecular sponge to regulate miR-448. J Cell Physiol. 2017;10:1002. doi: 10.1002/jcp.26072. [DOI] [PubMed] [Google Scholar]
- 5.Chen X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci Rep. 2015;5:13186. doi: 10.1038/srep13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Si Y, Bai J, Wu J, et al. LncRNA PlncRNA1 regulates proliferation and differentiation of hair follicle stem cells through TGFbeta1mediated Wnt/betacatenin signal pathway. Mol Med Rep. 2018;17:1191–97. doi: 10.3892/mmr.2017.7944. [DOI] [PubMed] [Google Scholar]
- 7.Nong Q, Li S, Wu Y, Liu D. LncRNA COL1A2-AS1 inhibits the scar fibroblasts proliferation via regulating miR-21/Smad7 pathway. Biochem Biophys Res Commun. 2018;495:319–24. doi: 10.1016/j.bbrc.2017.11.027. [DOI] [PubMed] [Google Scholar]
- 8.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 9.Agyeman A, Jha BK, Mazumdar T, Houghton JA. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget. 2014;5:4492–503. doi: 10.18632/oncotarget.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skoda AM, Simovic D, Karin V, et al. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn J Basic Med Sci. 2018;18(1):8–20. doi: 10.17305/bjbms.2018.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goyal A, Linskey KR, Kay J, et al. Differential expression of hedgehog and snail in cutaneous fibrosing disorders: Implications for targeted inhibition. Am J Clin Pathol. 2016;146:709–17. doi: 10.1093/ajcp/aqw192. [DOI] [PubMed] [Google Scholar]
- 12.Peng W, Wu J, Fan H, et al. LncRNA EGOT promotes tumorigenesis via hedgehog pathway in gastric cancer. Pathol Oncol Res. :2017. doi: 10.1007/s12253-017-0367-3. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 13.Gao FL, Jin R, Zhang L, Zhang YG. The contribution of melanocytes to pathological scar formation during wound healing. Int J Clin Exp Med. 2013;6:609–13. [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart SA, Dougall GM, Tafuro EM. The use of silgel STC-SE, a topical silicone gel for the treatment and reduction of hypertrophic and keloid scars. Plast Reconstr Surg Glob Open. 2016;4:e1183. doi: 10.1097/GOX.0000000000001183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C, Ogawa R. Fibroproliferative disorders and their mechanobiology. Connect Tissue Res. 2012;53:187–96. doi: 10.3109/03008207.2011.642035. [DOI] [PubMed] [Google Scholar]
- 16.Leprieur EG, Tolani B, Li H, et al. Membrane-bound full-length Sonic Hedgehog identifies cancer stem cells in human non-small cell lung cancer. Oncotarget. 2017;8:103744–57. doi: 10.18632/oncotarget.21781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y, Yu W, Shrivastava A, et al. Sanguinarine inhibits pancreatic cancer stem cell characteristics by inducing oxidative stress and suppressing sonic hedgehog-Gli-Nanog pathway. Carcinogenesis. 2017;38:1047–56. doi: 10.1093/carcin/bgx070. [DOI] [PubMed] [Google Scholar]
- 18.Sheng X, Sun X, Sun K, et al. Inhibitory effect of bufalin combined with Hedgehog signaling pathway inhibitors on proliferation and invasion and metastasis of liver cancer cells. Int J Oncol. 2016;49:1513–24. doi: 10.3892/ijo.2016.3667. [DOI] [PubMed] [Google Scholar]
- 19.Hyun J, Choi SS, Diehl AM, Jung Y. Potential role of Hedgehog signaling and microRNA-29 in liver fibrosis of IKKbeta-deficient mouse. J Mol Histol. 2014;45:103–12. doi: 10.1007/s10735-013-9532-5. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Choi SS, Michelotti GA, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–29. doi: 10.1053/j.gastro.2012.07.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinzler KW, Bigner SH, Bigner DD, et al. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 22.Faiao-Flores F, Alves-Fernandes DK, Pennacchi PC, et al. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene. 2017;36:1849–61. doi: 10.1038/onc.2016.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Javelaud D, Alexaki VI, Dennler S, et al. TGF-beta/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011;71:5606–10. doi: 10.1158/0008-5472.CAN-11-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shih DQ, Bussen M, Sehayek E, et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat Genet. 2001;27:375–82. doi: 10.1038/86871. [DOI] [PubMed] [Google Scholar]
- 25.Dang Y, Lan F, Ouyang X, et al. Expression and clinical significance of long non-coding RNA HNF1A-AS1 in human gastric cancer. World J Surg Oncol. 2015;13:302. doi: 10.1186/s12957-015-0706-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller S, Raulefs S, Bruns P, et al. Next-generation sequencing reveals novel differentially regulated mRNAs, lncRNAs, miRNAs, sdRNAs and a piRNA in pancreatic cancer. Mol Cancer. 2015;14:94. doi: 10.1186/s12943-015-0358-5. [DOI] [PMC free article] [PubMed] [Google Scholar]