

Abstract
We report the case of a previously well 80-year-old man who presented with subacute bilateral painful optic neuropathy with initial response to corticosteroids but ultimately progressed to a fatal skull base syndrome. Initial presentation of steroid-responsive painful bilateral posterior optic neuropathy, preliminary normal enhanced MRI, normal cerebrospinal fluid and inflammatory markers indicated atypical optic neuritis. However, this progressed to a bilateral orbital apex syndrome with ophthalmoplegia and evidence of abnormal skull base enhancement on subsequent MRI. Biopsy of radiologically abnormal dura was non-diagnostic and negative for fungal stains. He deteriorated and died 8 months after initial presentation. At postmortem, fungal skull base infection was diagnosed. This case demonstrates that chronic skull base fungal infection can: (1) present in elderly immunocompetent patients, (2) show initial improvement with corticosteroids and (3) evade diagnosis on biopsy. We encourage a high index of suspicion for fungal skull base infection in similar cases.
Keywords: infection (neurology), neuroimaging, cranial nerves, visual pathway
Background
Intracranial aspergillosis is not uncommon in immunosuppressed patients, but it is rarely reported in the immunocompetent.1–4 Fungal infections in the central nervous system (CNS) represent a notorious diagnostic challenge.1
We describe a previously well 80-year-old Caucasian man who presented with a subacute bilateral painful optic neuropathy with initial response to immunosuppression but subsequent progression resulted in his death. Despite extensive investigation, including tissue biopsy, a definitive diagnosis could not be reached. At postmortem, skull base Aspergillus fumigatus was diagnosed.
Case presentation
The patient initially presented with facial and periorbital discomfort, a right relative afferent pupillary defect and visual acuity of 6/18 in the right eye and 6/4 in the left eye. He had a history of mild asthma, for which he used a salbutamol inhaler but did not require steroids. He had no other drug history and no relevant family history. He was a retired export salesman. He was a non-smoker and drank a glass of wine in the evenings. He had no history of recent vaccinations, immunosuppression or travel history. He lived with his wife and was independent with all activities of daily living. He played golf twice per week and enjoyed woodwork.
Investigations
At presentation, cerebrospinal fluid (CSF) examination, standard blood tests including white blood cell differential and inflammatory markers including C reactive protein (CRP) and erythrocyte sedimentation rate (ESR) were normal. Antineutrophil antibodies (ANA), Antineutrophil cytoplasmic antibodies (ANCA), immunoglobulins (including IgG4, which is associated with an immune-mediated fibro-inflammatory process that is steroid-responsive), protein electrophoresis, serum ACE, aquaporin-4 and myelin-oligodendrocyte-glycoprotein antibodies (MOG; which are commonly associated with bilateral or sequential optic neuritis) were negative, as were Lyme, HIV and syphilis serology. Additionally, a temporal artery biopsy, obtained 10 days after starting steroids on initial suspicion of giant cell arteritis, was unremarkable.
CT imaging of the body revealed a left renal mass. He underwent left nephrectomy, and histological examination of the renal mass unexpectedly revealed an oncocytoma, a rare benign tumour made up of oncocytes, epithelial cells characterised by an excess of mitochondria. Pre-exchange paraneoplastic antibody testing, including anti-Hu, Ri, Yo, Ma1, Ma2, CV2/CRMP-5, amphiphysin, SOX-1, Zic-4 and anti-Tr antibodies, was negative.
MRI orbits with gadolinium contrast at presentation was normal. A second MRI orbits with gadolinium, 2 months following initial presentation and shortly after development of a right sixth nerve palsy post nephrectomy, showed minor enhancement of the right posterior optic nerve (figure 1) but no other abnormality.
Figure 1.
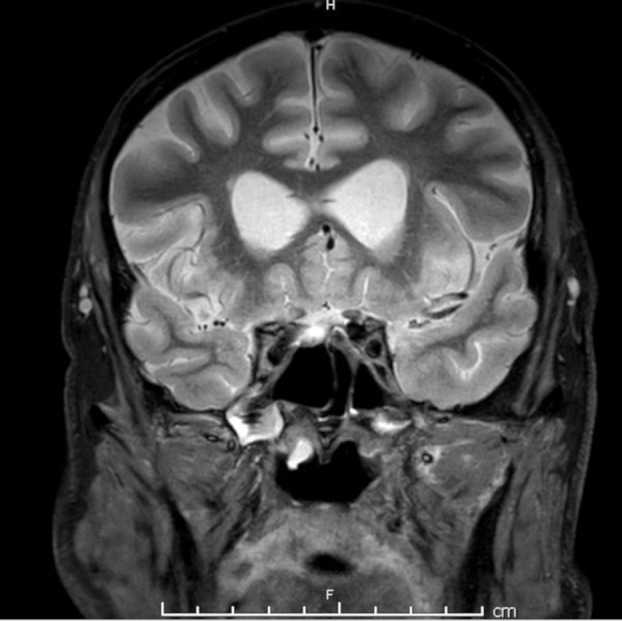
Coronal Short TI Inversion Recovery (STIR) MRI brain at 2 months following presentation showing enhancement of right optic nerve.
Four months after presentation, his vision and ophthalmoplegia deteriorated further and a third MRI orbits with gadolinium revealed enhancement of the skull base (figure 2). Repeat autoimmune screen (as iterated above) and routine CSF constituents were normal, but his oligoclonal bands in the CSF, previously negative, were now positive and unmatched, suggesting CNS inflammation.
Figure 2.
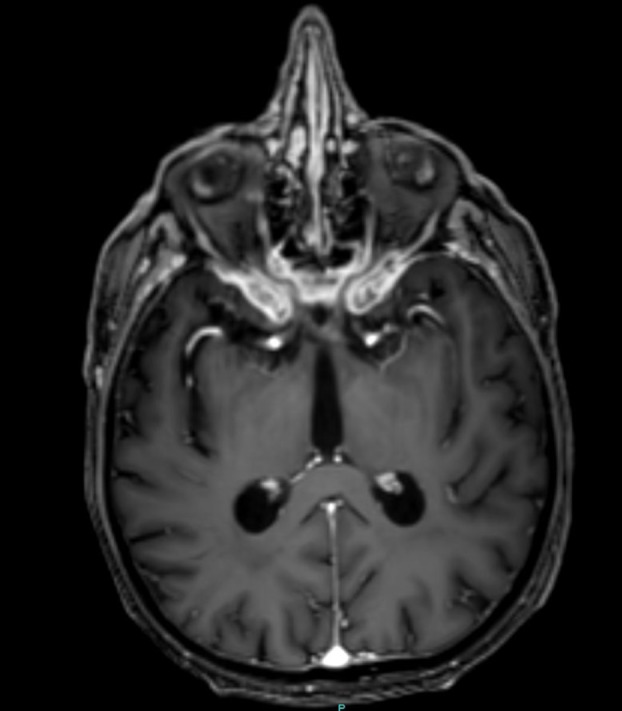
Axial T1 Gadolinium (GAD) at 4 months following presentation showing abnormal enhancement of skull base, dural thickening, narrowing of optic foramina and distortion of optic nerves.
Right subtemporal craniotomy revealed abnormally thickened dura overlying the skull base. Multiple biopsies were taken, but extensive histological examination, including fungal stains, was non-diagnostic and revealed mild non-specific inflammation.
Differential diagnosis
The differential diagnosis of an initial presentation of bilateral, sequential painful optic neuropathy with early steroid response, in an otherwise healthy 80-year-old man, includes giant cell arteritis, optic neuritis, neoplastic and compressive causes. Painless causes of optic neuropathy including Leber’s hereditary optic neuropathy, nutritional and toxic aetiology can probably be discounted in this case.
The initially normal MRI orbital imaging with gadolinium, normal inflammatory markers and negative temporal artery biopsy help to exclude giant cell arteritis, neoplastic and compressive causes leading to a diagnosis of optic neuritis. Typical optic neuritis presents as a painful subacute unilateral optic neuropathy in the younger patient and is associated with a significant risk of multiple sclerosis. Therefore, in our case, the clinical picture was that of atypical optic neuritis.
The differential of atypical optic neuritis includes antibody mediated (aquaporin-4, myelin-oligodendrocyte-glycoprotein antibodies (MOG) and collapsin response mediator protein 5 (CRMP 5) antibodies, chronic relapsing inflammatory optic neuropathy, acute disseminated encephalomyelitis, sarcoidosis, systemic lupus erythematosus (SLE), vasculitides and infective causes (typically syphilis, Lyme or viral causes). Blood tests revealed the absence of relevant antibodies, or markers for sarcoidosis, SLE and vasculitis. The absence of neuroretinitis on fundoscopic examination, negative blood tests and normal CSF help to exclude infective causes. The discovery of the renal oncocytoma prompted consideration of a paraneoplastic optic neuritis; however, this is very rare, there was no improvement with tumour removal, and extensive screening of paraneoplastic antibodies were negative.
The evolution of an ophthalmoplegia and skull base enhancement on MRI prompts reconsideration of sarcoidosis and also IgG4 disease, neoplastic and infective causes that may have been missed during the initial investigation. Biopsy of the abnormal skull base enhancement was not diagnostic and revealed no evidence of infection. The differential diagnosis was not advanced further in the patient’s lifetime.
Treatment
Initial concerns about giant cell arteritis prompted urgent treatment with oral prednisolone, which was associated with early but unsustained improvement in pain and vision. Steroid withdrawal, when further investigation made giant cell arteritis unlikely, was associated with relapse such that within 3 weeks after presentation, his visual acuity had reduced to hand movements on the right and 6/18 on the left.
Reinstitution of steroids, initially with pulsed methylprednisolone followed by oral prednisolone, provided temporary improvement. Further visual deterioration prompted five sessions of plasma exchange with minor improvement that relapsed when treatment was reduced to steroids alone and he progressed to complete blindness.
He continued to deteriorate, and by 4 months after presentation, he had developed a comprehensive ophthalmoplegia, ataxic gait and facial paraesthesia. With a non-diagnostic skull base biopsy and initial unsustained steroid response, refractory inflammatory disease was suspected. He was given one cycle of intravenous pulsed cyclophosphamide, with no symptomatic improvement.
Outcome and follow-up
Before his second cycle of cyclophosphamide, he clinically worsened with urinary incontinence, facial weakness, cognitive symptoms, dysarthria and drowsiness. At this stage, the patient was palliated, and he died 8 months after initial presentation.
Autopsy revealed nodular tissue covering the skull base (figure 3). Histological examination revealed the abundant presence of Aspergillus fumigatus (figure 4).
Figure 3.
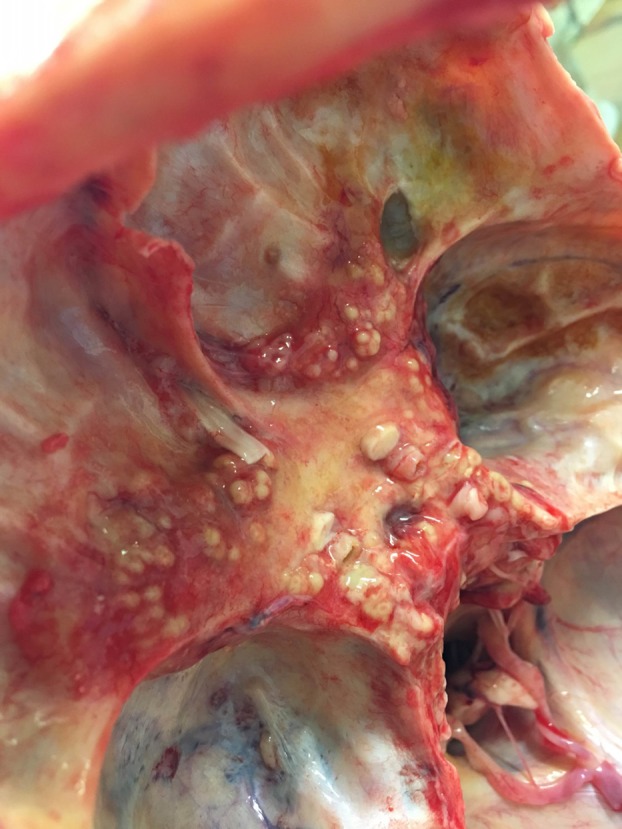
Postmortem skull base showing nodular dural thickening.
Figure 4.
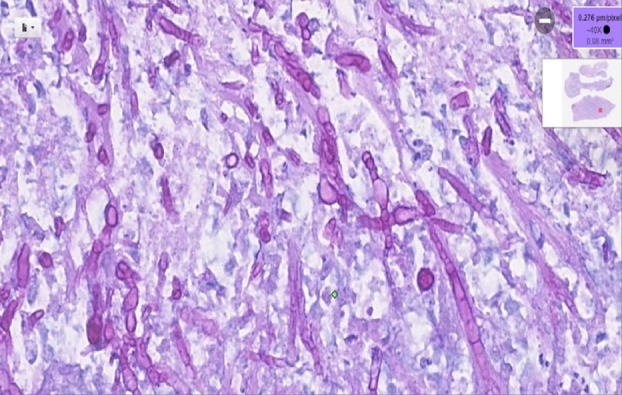
Diastase PAS staining highlighting Aspergillus fumigatus in connective tissue from the region of the optic chiasm.
Discussion
CNS aspergillosis
Opportunistic fungi are ubiquitous, and the second most frequent extrapulmonary disease from Aspergillus is in the CNS.5 Aspergillus is angiotropic; thus, it can cause necrotising angiitis, secondary thrombosis and haemorrhage.1 The primary port of entry is the respiratory tract, and intracranial infection results directly from nasal sinuses or via spread of infection in the systemic circulation.1 As stated in a review article by Nadkarni et al,1 fungal infections in the CNS can be diagnostically challenging. Intracranial aspergillosis is a rare and often fatal condition, generally affecting immunocompromised patients.1 2 Risk factors for intracranial aspergillosis include allergic rhinosinusitis, nasal polyps and recurrent sinus infections, as well as immunosuppression.2 6 7 The disease can be slowly progressive, and symptoms may persist for months.1 The prognosis is poor, and early diagnosis and aggressive management, with surgical debridement and adjuvant anti-fungal chemotherapy, can help to reduce mortality.1 6 8
Similar published cases
A study of 21 patients with CNS Aspergillus in South India revealed that 76% of patients have intracranial invasion by contiguous spread from paranasal sinuses, with over half the patients presenting with skull base syndromes.8 One Ukrainian case presented with left retro-orbital pain and ipsilateral monocular visual loss, with rapid progression to the contralateral temporal visual field, ptosis and reduced eye movements, secondary to an acute chiasmal abscess caused by Aspergillus fumigatus infection, highlighting invasive sino-orbital aspergillosis in an immunocompetent patient.6 Further case reports have also demonstrated intracranial complications of Aspergillus in immunocompetent patients.2–4 9 In many of these case reports, patients presented with headache and retro-orbital pain and were misdiagnosed as temporal arteritis and given steroid treatment similar to our patient.6
Case discussion
This case demonstrates that dural-based fungal infection can be refractory to extensive investigation, and a partial response to immunosuppression can be misleading. On review of history with relatives, it was found that he had a history of recurrent sinusitis and last complained of a ‘head cold’ a week before his initial presentation, which was not explored when the patient had first presented. Though recurrent sinusitis is common and may be unrelated, it is possible that this may have been the initial source of fungal infection. No persuasive premorbid immunosuppressive state was identified, although his age and the presence of oncocytoma may have contributed. It is clear that his initial atypical optic neuritis was not caused by iatrogenic immunosuppression, although subsequent progression may have been augmented by steroids, plasma exchange and cyclophosphamide. The discovery of the oncocytoma briefly raised the suspicion of a novel paraneoplastic optic neuritis, although no known relevant anti-neuronal antibody was found.
Steroid-responsive primary aspergillosis of the orbital apex has been reported previously, as well as CNS aspergillosis in immunocompetent patients.1 6 8 10 However, the initial presentation of apparent steroid-responsive atypical optic neuritis, with normal initial enhanced MRI, normal CSF and inflammatory markers, is unreported.
We hypothesise that:
Steroid treatment reduced the reactive skull base inflammation leading to a brief improvement in pain and vision but ultimately impairing the immune response and augmenting overall progression.
The skull base dura reactive inflammation was attempting to loculate and contain the fungal infection and that the biopsy was of reactive dura but missed the locules of infection. This is supported by the finding of nodular tissue at postmortem from which pus containing fungal elements could be expressed.
Repeat biopsy may have been helpful, but at the time, further biopsy was declined on discussion with the patient and the neurosurgical team. Even with cessation of immunosuppressants, consideration of a fungal cause, and institution of antifungal medications, the prognosis for this patient remained poor.1 8 We suspect that age-related decline in immune function perhaps allowed atypical fungal pathogenicity here.11
In conclusion, we would advise vigilance for fungal infection in elderly patients presenting with steroid-responsive polycranial neuropathy, and we highlight the importance of obtaining a tissue diagnosis prior to escalation of immunosuppression.
Learning points.
Old age is a cause of immunosuppression.
Fungal skull base infection may show an early improvement with steroid treatment.
Always check for a history of sinus infection in the context of atypical optic neuritis.
Where possible, always obtain a tissue diagnosis before treating otherwise undiagnosed skull base disease.
Acknowledgments
We would like to acknowledge Dr Paul Smith, Neuroradiologist, Southmead Hospital, and Dr Fred Mayall, Pathologist, Musgrove Park Hospital, for helping us with selection of images. We would like to thank the next of kin of the case discussed who kindly provided permission to produce this report.
Footnotes
Contributors: PS and LB discussed the topic and idea of report. PS produced the manuscript and performed necessary editing. CP and MF provided critical feedback and editing of manuscript. LB finalised the draft and supervised the manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Next of kin consent obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Nadkarni T, Goel A. Aspergilloma of the brain: an overview. J Postgrad Med 2005;51 Suppl 1:S37–41. [PubMed] [Google Scholar]
- 2. Curone M, D’Amico D, Maccagnano E, et al. Fatal Aspergillus brain abscess in immunocompetent patient. Neurol Sci 2009;30:233–5. 10.1007/s10072-009-0049-3 [DOI] [PubMed] [Google Scholar]
- 3. Kawakami N, Nishizaki T, Sugiyama S, et al. Aspergillus brain abscess in a patient with normal immunity–case report. Neurol Med Chir 1994;34:237–40. 10.2176/nmc.34.237 [DOI] [PubMed] [Google Scholar]
- 4. Kim DG, Hong SC, Kim HJ, et al. Cerebral aspergillosis in immunologically competent patients. Surg Neurol 1993;40:326–31. 10.1016/0090-3019(93)90145-Q [DOI] [PubMed] [Google Scholar]
- 5. Segundo JB, da Silva MA, Filho WE, et al. Cerebral aspergillosis in a patient with leprosy and diabetes: a case report. BMC Res Notes 2014;7:689 10.1186/1756-0500-7-689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leyngold I, Olivi A, Ishii M, et al. Acute chiasmal abscess resulting from perineural extension of invasive sino-orbital aspergillosis in an immunocompetent patient. World Neurosurg 2014;81:203.e1–203.e6. 10.1016/j.wneu.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wiggins RE. Invasive aspergillosis. A complication of treatment of temporal arteritis. J Neuroophthalmol 1995;15:36–8. [PubMed] [Google Scholar]
- 8. Murthy JM, Sundaram C, Prasad VS, et al. Aspergillosis of central nervous system: a study of 21 patients seen in a university hospital in south India. J Assoc Physicians India 2000;48:677–81. [PubMed] [Google Scholar]
- 9. Jayashree P, Puranik R, Kulkarni MH. Cerebral aspergilloma presenting as atypical meningioma in an immunologically competent patient: a case report. Indian J Pathol Microbiol 2007;50:367–9. [PubMed] [Google Scholar]
- 10. Slavin ML. Primary aspergillosis of the orbital apex. Arch Ophthalmol 1991;109:1502–3. 10.1001/archopht.1991.01080110036024 [DOI] [PubMed] [Google Scholar]
- 11. Grubeck-Loebenstein B, Della Bella S, Iorio AM, et al. Immunosenescence and vaccine failure in the elderly. Aging Clin Exp Res 2009;21:201–9. 10.1007/BF03324904 [DOI] [PubMed] [Google Scholar]