

Abstract
We describe two cases of agenesis of the corpus callosum (ACC) with interhemispheric cyst (IHC). Case 1: a male infant was born at 36 weeks gestation with a history of second trimester fetal ultrasound (US) scan and MRI showing ACC with IHC. His head circumference at birth and 5 months was at 90th centile. He developed infantile spasm and electroencephalogram showed hypsarrhythmia at 5 months of age. Seizures were controlled. He is under evaluation for surgical treatment. Case 2: ACC with a midline cyst was reported in the midtrimester US scan of a male infant. Subsequent fetal scans showed increasing size of the cyst. At birth, he had macrocephaly with a head circumference above 97th centile. MRI of the brain confirmed ACC with IHC. The parents refused a cystoperitoneal shunt offered. The child displayed gross neurodevelopmental delay with progressive hydrocephalus on follow-up and succumbed to aspiration pneumonia at 22 months of age.
Keywords: neuroimaging, hydrocephalus, epilepsy and seizures
Background
The corpus callosum is the most important supratentorial cerebral commissure bridging the cerebral hemispheres, facilitating the transfer of sensor, motor and cognitive information between them. Partial or complete agenesis of the corpus callosum (ACC) occurs either in isolation, or together with other cerebral malformations. Developmental anomalies of corpus callosum associated with interhemispheric cyst (IHC) are infrequent. The detection of ACC with IHC in antenatal scan has significant clinical implications and outcome. Fetal MRI can provide more detailed information on ACC and IHC, aiding in accurate diagnosis and explicit parental counselling. A knowledge of the natural history of IHCs, their evolution, subtypes of cysts, associated complications and possible neurological outcome can help in planning antenatal management, as well as postnatal care.
Case presentation
Case 1
A male infant was born at 36 weeks of gestation to a gravida 2 mother with no history of consanguinity. The antenatal ultrasound (US) scan at 21 weeks of gestation showed ACC, IHC and the absence of cavum septum pellucidum. Fetal MRI performed at 22 weeks confirmed the anomalies detected in US scan but with mass effect. Maternal serologies were negative and the couple decided to continue with the pregnancy.
The mother had a late preterm birthing and parturition was uneventful. The infant’s birth weight was 3201 g and head circumference was 36 cm (90th centile). He had hypertelorism, prominent forehead and polysyndactyly of the big toes and both thumbs (figure 1). The rest of the clinical examination was unremarkable. He had normal tone and elicitable primitive reflexes. He passed the newborn hearing screen and his eye examination was normal. At 4 months of age, the infant developed afebrile generalised seizures and was controlled with lorazepam and maintained on carbamazepine based on electroencephalogram (EEG) report. At 5 months, he could turn over and was able to reach and grasp objects. He was readmitted at 5 months of age with infantile spasm and the seizure was controlled with vigabatrin. On follow-up at 9 months of age, his neurological examination revealed low truncal tone and swaying to the left on pulling to sit up. Speech is limited to babbling and parents feel that he is disinterested in the surroundings. He has no difficulty in feeding and no excessive drooling. His head circumference is currently at 97th centile and the anterior fontanelle is widely open.
Figure 1.
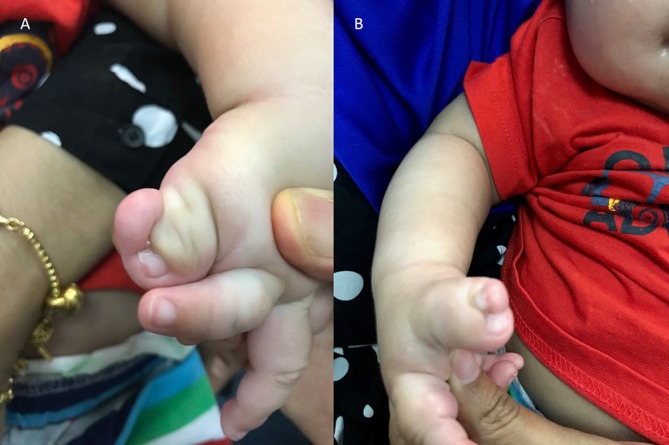
(A) Case 1 with polysyndactyly of left thumb and (B) right thumb as part of Acrocallosal syndrome.
Case 2
A male term infant was born to a primigravida mother from a non-consanguineous marriage. The antenatal period was uneventful. Fetal US at 20 weeks of gestation revealed a single large midline cyst with ACC. Subsequent US scans revealed an increase in cyst size with progressive dilatation of the ipsilateral ventricle. The parents were counselled and they decided to continue with the pregnancy. The right and left lateral ventricles measured 14 and 11 mm, respectively, across the atria of the posterior horn of lateral ventricles at 24 weeks of gestation. The infant was delivered at term gestation and weighed 2800 g. The occipitofrontal circumference was at 97th centile; and the rest of the systemic examination was unremarkable. The infant passed the neonatal hearing screen and a formal eye examination at 2 months was unremarkable.
On follow-up, there was a rapid increase in the infant’s head circumference, which measured 44.5 cm at 6 months of age (>97th centile) and he weighed 8200 g. His neck control was poor and was unable to turn over. He had occasional social smile. Truncal tone was variable. He had decreased tone in the extremities and tendon reflexes were diminished. He passed the high-risk hearing screen and the vision was normal. There was no history of seizures or abnormal movements. He was lost to follow-up for a year. He was last reviewed at 18 months of age, with a head circumference measuring 53 cm (>97th centile) and weight of 14.5 kg (>97th centile).
Investigations
Case 1
Fetal MRI scan at 22 weeks gestation showed an irregular contour of right cerebral hemisphere and disorganised signals involving the frontal lobe. A midline cyst was noted and the frontal horn was not visualised. An interhemispheric fissure was observed extending down to the roof of the third ventricle and nodular heterotopia was noted on the right cerebrum. The third ventricle was dilated and the cisterna magna was prominent (figure 2A,B). US scan of the brain on day 1 of life showed a midline multiloculated cystic structure. The largest cystic component measured 6.1×6.0×3.2 cm and extended into the right frontal region exerting a mass effect on to the right cerebral hemisphere, with the resulting midline shift to the left compressing the left lateral ventricle. The left occipital horn and third ventricle were dilated. MRI of the brain on day 2 of life showed ACC with left colpocephaly, multiloculated IHC, dilated third ventricle and mega cisterna magna. The anterior commissure was visible. The sulci and grey matter in the right frontal and temporal lobes were dysplastic (figure 3A,B). An EEG done during the readmission at 5 months of age for afebrile seizures showed hypsarrhythmia consistent with a diagnosis of infantile spasm. His US kidneys showed an exophytic cyst measuring 0.3 cm in the right kidney and a lower pole cyst in the left kidney. Echocardiography was unremarkable. His karyotyping was normal but parent’s deferred further genetic studies due to financial constraints.
Figure 2.
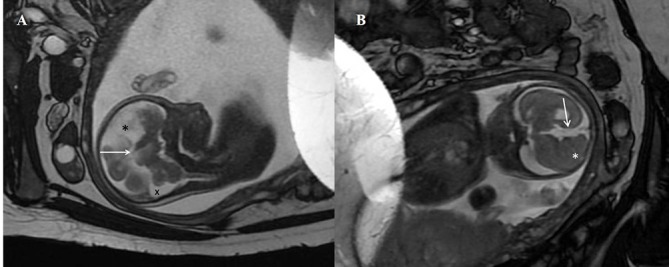
(A) Antenatal MRI sagittal T2-weighted image in the midline shows absence of the corpus callosum in the expected position (arrow). A midline cyst is noted anteriorly (*). The cisterna magna is slightly prominent (x). (B) Antenatal MRI coronal T2-weighted image shows the interhemispheric fissure extending down to the roof of the third ventricle (arrow). Nodular heterotopia is noted on the right (*).
Figure 3.
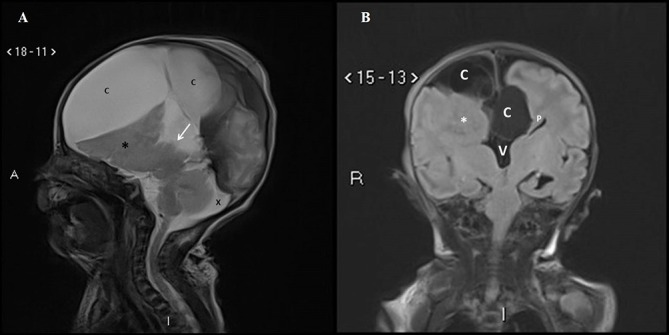
(A) Postnatal MRI sagittal T2-weighted image in the midline confirms the absence of the corpus callosum in the expected position (arrow). The midline cyst (C) is much larger in size compared to the antenna scan and is compressing on the underlying brain. Nodular heterotopia (*) is noted. The prominent cisterna magna is still seen (X). (B) Postnatal MRI coronal T2-weighted image shows the interhemispheric cysts (C), left Probst bundle (P), high-riding third ventricle (V) and right nodular heterotopia (*).
Case 2
Fetal US scan at 20 weeks of gestation showed ACC and a midline cystic lesion within the brain (figure 4). A postnatal cranial US scan showed complete ACC, a large right IHC with septations and a dilated right lateral ventricle. MRI confirmed the presence of a right paramidline cyst, extending from anterior to posterior and right colpocephaly (figure 5A). The anterior commissure was not seen. The midline sagittal image showed IHC with complete callosal agenesis (figure 5B). His karyotype was 46 XY. The echocardiogram, renal and abdominal US scans were unremarkable.
Figure 4.
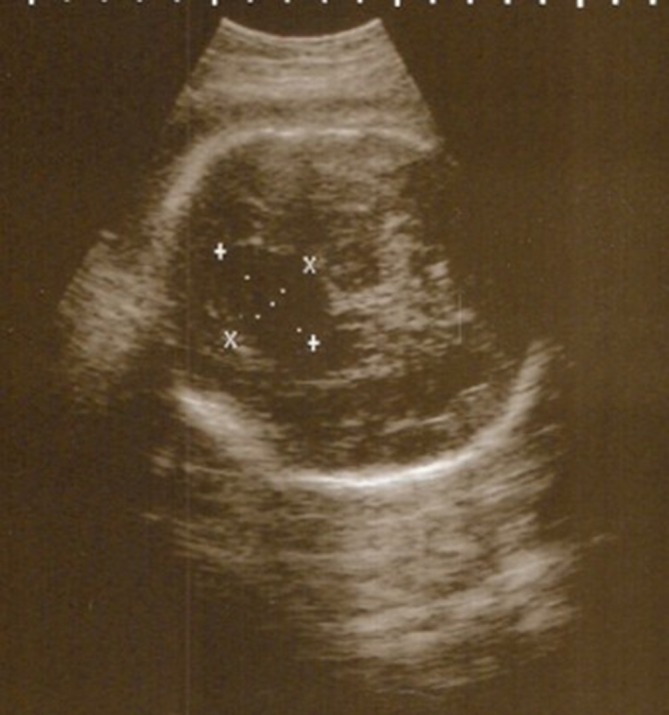
Fetal scan. A transabdominal sonogram of the fetus showing a midline cystic lesion within the brain.
Figure 5.

(A) T1-weighted MRI: axial images of the brain show the presence of a right paramidline cyst, extending from anterior to posterior. The atrium of the right lateral ventricle is dilated, consistent with colpocephaly. (B) T1-weighted MRI: midline sagittal image shows the right paramidline cyst with absent corpus callosum.
Treatment
Case 1
His seizures are under control for the last 3 months. The neurosurgical team is observing his progress. A detailed evaluation is planned after a seizure-free interval to decide on the need for surgical intervention.
Case 2
The neurosurgical team offered the insertion of a cystoperitoneal shunt but the parents refused any form of surgical intervention and opted for treatment with complementary medicine.
Outcome and follow-up
Case 1
Paediatric, neurology and neurosurgical teams are following up the infant. His motor and mental milestones have delayed since the onset of infantile spasm. His growth and neurodevelopmental assessments are carried out periodically.
Case 2
There was profound delay in attaining age-appropriate milestones. The infant was admitted at 22 months of age following milk aspiration and succumbed to severe aspiration pneumonia.
Discussion
The corpus callosum is the most complex and largest white matter tract in the human brain, which normally begins to develop at the 8–10th week of fetal life from genu, and continues posteriorly along the body to the splenium and completes at the rostrum. A mature corpus callosum is seen at 20 weeks of gestation. Axons of corpus callosum and anterior commissure originate from the small pyramidal neurons of the mature cortex. The corpus callosum is believed to play an important role in cognition: helping the integration of information and the mediation of complex behaviours.1 ACC is a clinically and genetically heterogeneous congenital anomaly with an estimated incidence of 1:4000.2 Approximately 10% of all ACC have been known to be associated with chromosomal anomalies, and 20%–35% of cases have recognisable genetic syndromes with male dominance. Male predominance was also noted in our reported cases. ACC can be inherited as either an autosomal recessive trait or an X-linked dominant trait. It can also result from an infection or injury during the 12th to 22nd week of gestation, leading to developmental aberrations of the fetal brain. Fetal alcohol syndrome due to intrauterine exposure to alcohol can result in ACC.2–4
ACC, which can be partial or complete, results from the disruption of several developmental steps beginning from early midline telencephalic formation. One proposed mechanism of embryogenesis of ACC is the failure of directional migration of commissural fibres in the developing brain due to the development of a ventricular diverticulum/cyst. Imaging of the brain may show the presence of arachnoid cyst or lipoma with partial or complete ACC. These cysts or lipoma actually do not originate in the corpus callosum but from the primitive meninx. Beginning at about 5 weeks of embryonic stage, mesencephalic neural crest migration forms the entire cerebral leptomeninges that may terminally differentiate as arachnoid cysts or as lipomata. Since these neural crest migrations occurs from 5 weeks gestation in humans and the first pioneer callosal axons traverse the midline at 10 weeks gestation, any IHC formation prevents the formation of the corpus callosum.5
ACC more often occurs as an isolated anomaly and its concurrence with IHC has been well recognised. In complete ACC, the third ventricle tends to be widened and extends superiorly into the interhemispheric fissure resulting in a cerebrospinal fluid collection referred to as IHC.6 The IHC associated with ACC may be arachnoid, neurenteric or ependymal in origin. The location of cysts, morphology and the imaging characteristics often provide important clues about the origin of the cysts.7 8
In 2001, Barkovich et al presented a new system for classification of IHC associated with ACC based on morphological appearance on postnatal imaging.9 Type 1 cysts communicate with the ventricles. Type 1a IHC has no other cerebral malformations whereas type 1b has hydrocephalus secondary to malformations hindering the egress of the cerebrospinal fluid from the third ventricle into the aqueduct of Sylvius. Type 1c IHC has cerebral hemispheric hypoplasia and small head. These cysts are diverticula of the lateral or third ventricles. Type 2 cysts have no communication with the ventricles. Type 2 is further subdivided into type 2a when there are no abnormalities other than ACC, and types 2b and 2c that are associated with Aicardi syndrome and subcortical heterotopia, respectively.
Arachnoid cysts are mostly extra-axial and believed to develop following a minor aberration in the genesis of arachnoid leading to either splitting or duplication of the membrane. Most of the arachnoid cysts are found in the middle cranial fossa; but 25% of supratentorial arachnoid cysts are IHC.10 Ependymal cysts are thought to develop from the sequestration of developing neuroectoderm during embryogenesis. Though usually located deep in the brain parenchyma, subarachnoid and intraventricular ependymal cysts have been described.11 12 Neurenteric cysts are rare and are believed to be of an endodermal origin, though their pathogenesis within neuroectodermal tissue of the central nervous system is still unclear. These cysts are mostly found in the posterior cranial fossa, but rarely reported in the supratentorial region.13 14
The functional outcome in an individual with isolated primary ACC is generally favourable, with little impact on general cognitive capacity; but the consistently observed disability seen with isolated cases include impairment in abstract reasoning, problem solving, generalisation and category fluency. The clinical manifestations and the natural history in ACC with IHC vary from being asymptomatic, with spontaneous regression of cysts, to significant neurological disabilities (macrocrania secondary to hydrocephalus, signs of raised intracranial pressure (ICP), seizures, hemiparesis and significant psychomotor delays).15–18 The presence of polymicrogyria, pachygyria and heterotopia, detected using MRI studies, correlates with severe neurological impairment with moderate to severe developmental delay.19
The infant reported in case 1 has clinical features of a polymalformative syndrome described by Albert Schinzel, acrocallosal syndrome (ACS), characterised by ACC, macrocephaly, minor facial anomalies like prominent forehead and hypertelorism, hallux and thumb duplication and mental retardation. ACS may be associated with arachnoid cyst, reported in one-third of cases. ACS is an autosomal recessive disease. Mutations of the kinesin KIF7(15Q26.1) and the transcriptional activator GL13(7p14.1) genes are responsible for ACS.20 In the reported case 1, he has ACC, IHC, macrocephaly, facial features of prominent forehead and hypertelorism, polysyndactyly in fingers and toes (figure 1) and neurodevelopmental delay consistent with a diagnosis of ACS. A genetic testing to confirm ACS was suggested but parents deferred.
The second trimester US scan and fetal MRI usually establishes the prenatal diagnosis of ACC and IHC. In IHC, serial imaging is recommended for the precise location of cysts and pressure effects on surrounding structures.19 21 ACC is considered isolated when antenatal MRI or genetic tests do not indicate additional anomalies. ACC can be syndromic or part of a genetic disease if there is a positive family history of epilepsy or mental retardation and/or consanguinity. Volpe et al reported that the outcomes of both isolated partial and complete ACC were similar. The most practical approach when encountering fetuses with large IHC and ACC in US and/or MRI would be to keep a close watch with serial US scans on the progress of the cyst size, number, dilatation of ventricles and pressure effect on nearby structures.21 On birth, the baby would require chromosomal studies and screening to rule out other associated anomalies, and a thorough neuroimaging, which greatly aids in neurosurgical intervention. The differentials to consider with postnatal neuroimaging are the alobar form of holoprosencephaly and porencephalic cysts. In alobar holoprosencephaly, there is midline fusion of cerebral parenchyma with lateral ventricles resulting in a monoventricle. Porencephalic cysts are intra-axial cysts communicating with lateral ventricles, often with no mass effect.19 21 22 As the anterior commissure forms at 7 weeks of embryonic life, which is 3 weeks earlier than corpus callosum, presence or absence of anterior commissure predicts the timing of pathogenesis.5 In case 1, MRI showed the anterior commissure whereas it was not noted in case 2. The presence of Probst bundles (aberrant bundles of axons that run in an anteroposterior direction rather than connecting the two hemispheres right to left) in brain MRI is a characteristic feature of ACC.23 Nodular heterotopia (figure 2B) is important in diagnosis and prognosis. Nodular heterotopia is indicative of cortical malformations due to impaired neuronal migration.24 The infant in case 1 who has nodular heterotopia has shown neurodevelopmental delay since the onset of infantile spasm.
There has been no consensus on the optimal surgical treatment for symptomatic and large asymptomatic IHC in children, with three surgical interventions (craniotomy, shunt and neuroendoscopic surgery) currently being practised.25–27 Caldarelli et al reported good results without any mortality using open craniotomy and cyst membrane excision with fenestration of the cyst into the ventricle or subarachnoid space,25 while Lena et al considered the use of cystoperitoneal shunts to be the optimal surgical treatment.27 In ACC with IHC, even after shunt insertion, a favourable neurodevelopmental outcome is expected in 75% of infants.21 22 25 Recently, neuroendoscopic surgery has replaced craniotomy as the treatment of choice as these minimally invasive surgeries avoid shunts and reported a success rate of 71%–81%.28 29
Learning points.
Agenesis of the corpus callosum with interhemispheric cyst is a complex congenital anomaly, with a wide spectrum of manifestations and implications on neurological outcomes.
Antenatal ultrasound scan will aid in identifying these anomalies; while serial imaging will aid in recognising changing pattern of cysts and significant neurological complications like progressive hydrocephalus.
Knowing the type of cysts, associated complications and natural history of the evolution of the cysts will help greatly in planning antenatal and postnatal intervention.
Neuroendoscopic surgical management remains the treatment of choice, but anatomical variations of the cyst may require cystoperitoneal shunt and/or craniotomy instead.
Acknowledgments
We would like to thank Assistant Professor Harvey Teo Eu Leong, Department of Diagnostic Imaging, Kandang Kerbau Women’s and Children’s Hospital for reporting the US and MRI.
Footnotes
Contributors: KGR: manuscript preparation and literature review. SC and VSR: manuscript preparation, literature review and finalisation of the manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent: Parental/guardian consent obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Hinkley LB, Marco EJ, Findlay AM, et al. The role of corpus callosum development in functional connectivity and cognitive processing. PLoS One 2012;7:e39804–16. 10.1371/journal.pone.0039804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bedeschi MF, Bonaglia MC, Grasso R, et al. Agenesis of the corpus callosum: clinical and genetic study in 63 young patients. Pediatr Neurol 2006;34:186–93. 10.1016/j.pediatrneurol.2005.08.008 [DOI] [PubMed] [Google Scholar]
- 3. Oh KY, Kennedy AM, Selden NR, et al. Asymmetric ventriculomegaly, interhemispheric cyst, and dysgenesis of the corpus callosum (AVID): an imaging triad. J Ultrasound Med 2012;31:1811–20. [DOI] [PubMed] [Google Scholar]
- 4. Edwards TJ, Sherr EH, Barkovich AJ, et al. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 2014;137:1579–613. 10.1093/brain/awt358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarnat HB. Embryology and malformations of the forebrain commissures : Sarnat HB, Curatolo P, Handbook of clinical neurology. 3rd series: Elsevier, 2008;87:68–87. [DOI] [PubMed] [Google Scholar]
- 6. Barkovich AJ. Congenital malformations of the brain and skull Barkovich AJ, Pediatric neuroimaging. Philadelphia, Pennsylvania: Lippincott – Raven Publishers, 1996:177–275. [Google Scholar]
- 7. Pavone P, Barone R, Baieli S, et al. Callosal anomalies with interhemispheric cyst: expanding the phenotype. Acta Paediatr 2005;94:1066–72. 10.1080/08035250510027372 [DOI] [PubMed] [Google Scholar]
- 8. Stroustrup Smith A, Levine D. Appearance of an interhemispheric cyst associated with agenesis of the corpus callosum. AJNR Am J Neuroradiol 2004;25:1037–40. [PMC free article] [PubMed] [Google Scholar]
- 9. Barkovich AJ, Simon EM, Walsh CA. Callosal agenesis with cyst: a better understanding and new classification. Neurology 2001;56:220–7. 10.1212/WNL.56.2.220 [DOI] [PubMed] [Google Scholar]
- 10. Gosalakkal JA. Intracranial arachnoid cysts in children: a review of pathogenesis, clinical features, and management. Pediatr Neurol 2002;26:93–8. 10.1016/S0887-8994(01)00329-0 [DOI] [PubMed] [Google Scholar]
- 11. Pawar SJ, Sharma RR, Mahapatra AK, et al. Giant ependymal cyst of the temporal horn – an unusual presentation. Pediatr Neurosurg 2001;34:306–10. 10.1159/000056043 [DOI] [PubMed] [Google Scholar]
- 12. Moriyama E, Nishida A, Sonobe H. Interhemispheric multiloculated ependymal cyst with dysgenesis of the corpus callosum: a case in a preterm fetus. Childs Nerv Syst 2007;23:807–13. 10.1007/s00381-006-0275-4 [DOI] [PubMed] [Google Scholar]
- 13. Turel MK, Chacko G, Chacko AG. Interhemispheric neurenteric cyst. Neurol India 2013;61:563–5. 10.4103/0028-3886.121959 [DOI] [PubMed] [Google Scholar]
- 14. Mittal S, Petrecca K, Sabbagh AJ, et al. Supratentorial neurenteric cysts-A fascinating entity of uncertain embryopathogenesis. Clin Neurol Neurosurg 2010;112:89–97. 10.1016/j.clineuro.2009.11.001 [DOI] [PubMed] [Google Scholar]
- 15. Sotiriadis A, Makrydimas G. Neurodevelopment after prenatal diagnosis of isolated agenesis of the corpus callosum: an integrative review. Am J Obstet Gynecol 2012;206:337.e1–5. 10.1016/j.ajog.2011.12.024 [DOI] [PubMed] [Google Scholar]
- 16. Fischer M, Ryan SB, Dobyns WB. Mechanisms of interhemispheric transfer and patterns of cognitive function in acallosal patients of normal intelligence. Arch Neurol 1992;49:271–7. 10.1001/archneur.1992.00530270085023 [DOI] [PubMed] [Google Scholar]
- 17. Imamura T, Yamadori A, Shiga Y, et al. Is disturbed transfer of learning in callosal agenesis due to a disconnection syndrome? Behav Neurol 1994;7:43–8. 10.1155/1994/146072 [DOI] [PubMed] [Google Scholar]
- 18. David AS, Wacharasindhu A, Lishman WA. Severe psychiatric disturbance and abnormalities of the corpus callosum: review and case series. J Neurol Neurosurg Psychiatry 1993;56:85–93. 10.1136/jnnp.56.1.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shevell MI. Clinical and diagnostic profile of agenesis of the corpus callosum. J Child Neurol 2002;17:895–9. 10.1177/08830738020170122601 [DOI] [PubMed] [Google Scholar]
- 20. Koenig R, Bach A, Woelki U, et al. Spectrum of the acrocallosal syndrome. Am J Med Genet 2002;108:7–11. 10.1002/ajmg.10236 [DOI] [PubMed] [Google Scholar]
- 21. Pierre-Kahn A, Hanlo P, Sonigo P, et al. The contribution of prenatal diagnosis to the understanding of malformative intracranial cysts: state of the art. Childs Nerv Syst 2000;16:618–26. 10.1007/s003810000316 [DOI] [PubMed] [Google Scholar]
- 22. Garel C, Moutard ML. Main congenital cerebral anomalies: how prenatal imaging aids counseling. Fetal Diagn Ther 2014;35:229–39. 10.1159/000358519 [DOI] [PubMed] [Google Scholar]
- 23. Probst M. Über den Bau des vollständig balkenlosen Großhirns. Arch Psychiatr 1901;34:709–86. [Google Scholar]
- 24. González G, Vedolin L, Barry B, et al. Location of periventricular nodular heterotopia is related to the malformation phenotype on MRI. AJNR Am J Neuroradiol 2013;34:877–83. 10.3174/ajnr.A3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Caldarelli M, Di Rocco C. Surgical options in the treatment of interhemispheric arachnoid cysts. Surg Neurol 1996;46:212–21. 10.1016/0090-3019(96)00155-3 [DOI] [PubMed] [Google Scholar]
- 26. Ulu MO, Kafadar AM, Dashti R, et al. Treatment of symptomatic interhemispheric arachnoid cysts by cystoperitoneal shunting. J Clin Neurosci 2010;17:700–5. 10.1016/j.jocn.2009.09.029 [DOI] [PubMed] [Google Scholar]
- 27. Lena G, van Calenberg F, Genitori L, et al. Supratentorial interhemispheric cysts associated with callosal agenesis: surgical treatment and outcome in 16 children. Childs Nerv Syst 1995;11:568–73. 10.1007/BF00300994 [DOI] [PubMed] [Google Scholar]
- 28. Talamonti G, D’Aliberti G, Picano M, et al. Intracranial cysts containing cerebrospinal fluid-like fluid: results of endoscopic neurosurgery in a series of 64 consecutive cases. Neurosurgery 2011;68:788–803. 10.1227/NEU.0b013e318207ac91 [DOI] [PubMed] [Google Scholar]
- 29. Cinalli G, Peretta P, Spennato P, et al. Neuroendoscopic management of interhemispheric cysts in children. J Neurosurg 2006;105:194–202. 10.3171/ped.2006.105.3.194 [DOI] [PubMed] [Google Scholar]