

ABSTRACT
Sorafenib has been demonstrated to be a beneficial treatment for advanced hepatocellular carcinoma (HCC). Emerging evidence indicates that caspase-1 activation plays a crucial role in HCC progression. However, the relationship between caspase-1 and sorafenib has rarely been reported. In this study, we showed that caspase-1 was essential for lipopolysaccharide (LPS)-induced epithelial-mesenchymal transition (EMT). Moreover, sorafenib treatment could inhibit LPS-stimulated caspase-1 overexpression through restricting the nuclear transport of p65, which contributed to inactivation of NF-κB. Co-immunoprecipitation (Co-IP) experiments and immunoblot analysis indicated that sorafenib treatment decreased the SUMOylation of p65 via inhibiting TLR4/stat3/SUMO1 signaling cascades. In conclusion, the results of this study suggest that sorafenib inhibits caspase-1 expression through suppressing the nuclear translocation of p65 and provide new insights into the mechanisms of sorafenib treatment in HCC.
Keywords: HCC, sorafenib, caspase-1, LPS
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignancy and leading cause of cancer related death worldwide.1 Due to its high rate of postsurgical metastasis and recurrence, the prognosis of patients diagnosed with HCC remains extremely poor.1,2 Despite the advances in diagnosis and improvements in surgical skills during the past several decades, the 5-year survival rate is less than 40%.3 Therefore, it is imperative to find novel strategies to treat HCC.
Caspase-1 is one of cysteine-protease involved in inflammation and apoptosis and responsible for maturation of interleukin 1β (IL1b) and interleukin 18 (IL18).4,5 Once stimulated by LPS or cellular stress, activated caspase-1 cleaves pro-IL1b and pro-IL18 in inflammasomes.5,6 Accumulating data indicate that caspase-1 plays a critical role in tumor progression, including HCC.4,6-10 Caspase-1 was demonstrated to be activated by HMGB1 released from hypoxic HCC cells and contributed to the invasion and metastasis.7 NF-κB has been reported to be necessary for aspase-1 activation in various types of cells.4,5 NF-κB pathway is related to the progression of HCC and nuclear p65 is an active form of NF-κB pathway.11 In addition, SUMOylation of p65 mediated by SUMO1 has been demonstrated to be essential for nuclear transport of p65.12
Sorafenib, a multi-kinase inhibitor, is designed to target inhibition of Raf/MEK/ERK signaling pathway and suppressing several receptor tyrosine kinases, including vascular endothelial growth factor receptor 2 (VEGFR-2), VEGFR-3, Flt-3, c-KIT and platelet-derived growth factor receptor (PDGFR).13-15 Sorafenib has been demonstrated to be the first-line systematic therapy for advanced HCC.16,17 Evidences indicate that sorafenib treatment could improve 3 months survival of patients with advanced HCC.16,17 Accumulating data demonstrate that sorafenib inhibit the proliferation, invasion and metastasis of HCC cells both in vitro and vivo.13-15,18 However, the relationship between sorafenib and caspase-1 has not been studied.
Here, we reported that caspase-1 inhibition by caspase-1 siRNA as well as sorafenib treatment, respectively, counteracted the effects induced LPS administration on HCC cells. Soarfenib could decrease caspase-1 expression and retard LPS-induced caspase-1 overexpression. Furthermore, we demonstrated that p65 translocation into nuclear and SUMO1 mediated SUMOylation of p65 was inhibited by sorafenib. We also verified that decrease of SUMOylation of p65 was resulted from suppression of TLR4/stat3/SUMO1 cascades caused by sorafenib. These data suggest a novel role of sorafenib treatment in regulating caspase-1 expression.
Results
LPS promotes the invasion of HCC via upregulating caspase-1
LPS has been determined to promote EMT process in HCC, thus contributing to invasion and metastasis.19,20 EMT is a critical step in tumor metastasis.21,22 Interestingly, except the induction of EMT, we observed that caspase-1 expression was upregulated both in two HCC cell lines, HepG2 and HCCLM3, treated by LPS for 24h (Figure 1A,B). Furthermore, LPS modulated caspase-1 expression in a dose-dependent manner (Figure 1A,B). To investigate the relationship between caspase-1 and EMT in LPS-treated cells, caspase-1 siRNA was used to silence the expression of capase-1. Western blot and qPCR experiments were performed to check the silencing efficiency of caspase-1 siRNA on caspase-1 expression (Figure S1A,B). With the down-regulation of caspase-1, LPS-induced EMT process in HepG2 and HCCLM3 was both inhibited, which suggested that caspase-1 was essential for EMT process mediated by LPS (Figure 1C,D). Consistently, data from transwell experiments confirmed that caspase-1 inhibition could repress invasive ability in LPS-treated cells (Figure 1E).
Figure 1.
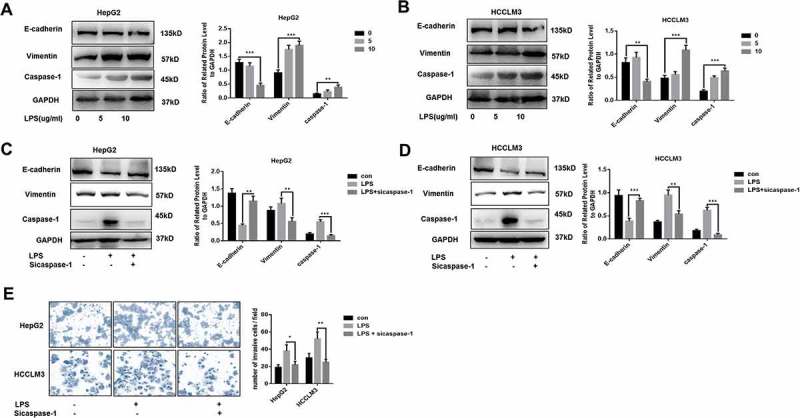
LPS promotes the invasion of HCC via upregulating caspase-1(A,B) Western blot analysis indicates that LPS induces EMT process and caspase-1 expression. HepG2 and HCCLM3 cells were incubated with 0, 5, 10 µg/ml LPS for 24 h. Protein levels of E-cadherin, Vimentin and caspae-1 were analyzed by western blot and normalized to GAPDH. (C-D) Western blot analysis indicates that caspase-1 is essential for LPS induced EMT process. HepG2 and HCCLM3 cells were incubated with 10ug/ml LPS for 24 h. Protein levels of E-cadherin, Vimentin and caspae-1 were analyzed by western blot and normalized to GAPDH. (E) Invasion experiments were determined by transwell assays and number of invaded cells was counted. Data are means ± SEM from 3 independent experiments, * means p < 0.05, ** means p < 0.01, *** means p < 0.001 by unpaired student T test.
LPS is a critical inflammatory mediator in tumor microenvironment.20,23,24 We further explored the relationship of caspase-1, related inflammatory cytokines and EMT in HCC tissues (Figure S2A,B). We observed that caspase-1 expression was tightly associated with EMT phenotype and inflammatory microenvironment. Mounting evidence has demonstrated that caspase-1 is responsible for interleukin-1β maturation and release.5 We found that human IL1b treatment could partly rescue the impaired EMT phenotype in caspase-1 deficient cells, which indicated that IL1b is important for caspase-1 mediated EMT (Fig.SC-D). Collectively, these results suggest caspase-1 could be upregulated by LPS treatment and is necessary for EMT induction in HCC cells.
Sorafenib decreases caspase-1 expression and represses LPS-stimulated caspase-1 expression through restricting p65 nuclear translocation
Sorafenib is demonstrated to be effective in suppressing the development of HCC, including invasion and metastasis.16 To explore the role of sorafenib in caspase-1 expression, HCCLM3 cells were cultured in different concentration of sorafenib for 24h. We found that sorafenib decreased the expression of caspase-1 in a dose-dependent way (Figure 2A). Sorafenib (10 µm) treatment inhibited caspase-1 expression both in HepG2 and HCCLM3 cells (Figure 2B). Moreover, we observed that nuclear transport of p65, the active form of NF-κB, was repressed in cells treated by sorafenib and sorafenib treatment made no difference on the expression of total p65 (Figure 2C). Given activation of NF-κB pathway is necessary for caspsae-1 expression, we thought sorafenib inhibited caspase-1 expression via restricting p65 nuclear translocation.
Figure 2.
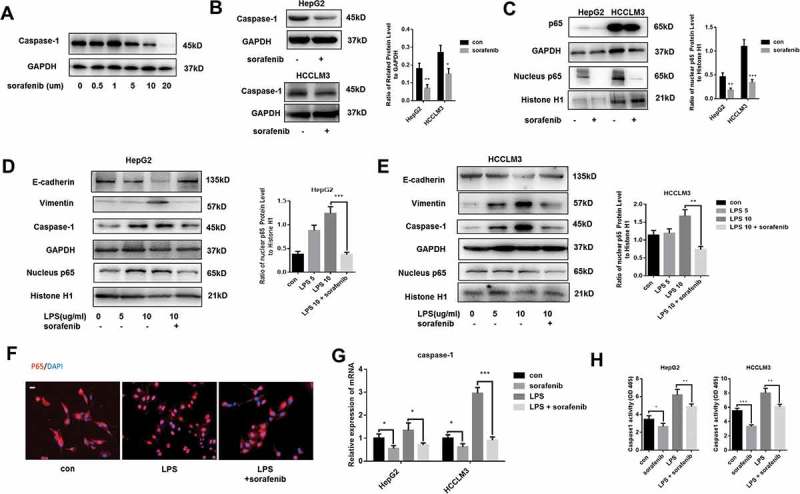
Sorafenib decreases caspase-1 expression and represses LPS-stimulated caspase-1 expression through restricting p65 nuclear translocation (A) Western blot analysis indicates that sorafenib decreases caspase-1 in a dose-dependent way. HCCLM3 cells were incubated with 0, 0.5, 1, 5, 10, 20 µm sorafeniib for 24 h. (B) Western blot analysis indicates that sorafenib decreases caspase-1. HepG2 and HCCLM3 cells were incubated with 10 um sorafeniib for 24 h. Protein levels of caspase-1 were analyzed by western blot and normalized to GAPDH. (C) Western blot analysis indicates that sorafenib restricts the nuclear transport of p65. Nuclear protein levels of p65 were analyzed by western blot and normalized to Histone H1. (D,E) Western blot analysis indicates that sorafenib suppresses the nuclear transport of p65 caused by LPS. Nuclear protein levels of p65 were analyzed by western blot and normalized to Histone H1. (F) Representative images show the distribution of p65 was visualized by performing immunofluorescence experiments. Scale bars = 20 µm. (G) q-PCR analysis indicates that sorafenib decreases caspase-1 expression and represses LPS-induced caspase-1 overexpression. (H) Caspase-1 enzymatic activity was measured by caspase-1 activity assays. Data are means ± SEM from 3 independent experiments, * means p < 0.05, ** means p < 0.01, *** means p < 0.001 by unpaired student T test.
Mounting evidence showed that caspase-1 expression in varied types of cells could be upregulated through activating NF-κB signaling.4,5,11 We already verified that caspase-1 was responsible for LPS-induced EMT. In Figure 2D,E, we showed that nuclear transport of p65 was enhanced by LPS treatment and sorafenib could obviously down-regulate the nuclear translocation of p65, as a result of which, LPS-triggered caspase-1 expression and EMT process was suppressed in both HepG2 and HCCLM3 cells. To visualize the distribution of p65 in HCCLM3 cells, immunofluorescence experiments were conducted, which showed that nuclear transport of p65 was enhanced by LPS and sorafenib inhibited the LPS-induced translocation (Figure 2F). Moreover, qPCR experiments were carried out to measure the transcriptional level of caspase-1 (Figure 2G). Results from qPCR experiments further confirmed that sorafenib inhibited caspase-1 expression and counteracted the effects of LPS treatment on caspase-1 expression. Using caspase-1 activity assays, we demonstrated that sorafenib both inhibited caspase-1 enzymatic activity of cells treated with or without LPS, which indirectly suggested caspase-1 expression was modulated by sorafenib. Taken together, our data indicate that sorafenib could repress caspase-1 expression through restricting p65 nuclear translocation.
Sorafenib inhibits SUMO1 mediated sumoylation of p65
SUMOylation of p65 plays a critical role in p65 translocation from cytoplasm into nucleus.12,25 Recent studies demonstrated that SUMOylation mediated by the interaction of SUMO1 and p65 promoted the nuclear transport of p65 and was associated with the development of HCC.12 Firstly, we found that SUMO1 expression was down-regulated by sorafenib in HepG2 and HCCLM3 (Figure 3A). Co-immunoprecipitation experiments were performed to investigate the interaction of p65 and SUMO1. We found that cells treated with sorafenib, compared to untreated cells, were characterized with decreased interaction of SUMO1 and p65 (Figure 3B). Furthermore, immunofluorescence staining p65 and SUMO1 were performed, which indicated that LPS enhanced the co-localization of p65 and SUMO1 and also sorafenib treatment inhibited LPS-induced co-localization by decreasing SUMO1 expression (Figure 3C). However, Collectively, these results suggest that sorafenib restricts p65 nuclear translocation through down-regulating SUMO1-meadiated SUMOylation of p65.
Figure 3.
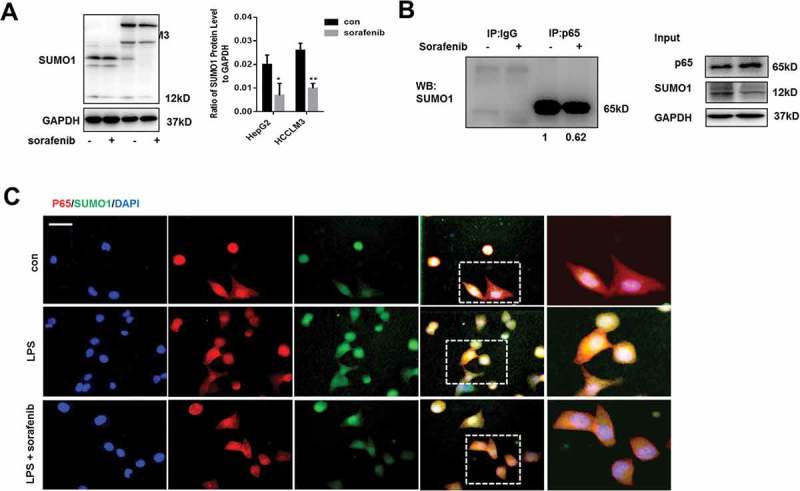
Sorafenib inhibits SUMO1 mediated SUMOylation of p65(A) Western blot analysis indicates that sorafenib decreases SUMO1. HepG2 and HCCLM3 cells were incubated with 10 µm sorafenib for 24h. Protein levels of SUMO1 were analyzed by western blot and normalized to GAPDH. (B) Co-immunoprecipitation experiments indicate sorafenib inhibits the interaction of p65 and SUMO1. (C) Immunofluorescences staining p65 and SUMO1 were performed. Data are means ± SEM from 3 independent experiments, * means p < 0.05, ** means p < 0.01, *** means p < 0.001 by unpaired student T test.
LPS-induced caspase-1 overexpression was dependent on TLR4/stat3/SUMO1 signaling pathway
In Figure 4A, we observed that LPS treatment could upregulate the expression of TLR4 and SUMO1 and enhance the phosphorylation of stat3. To explore the relationship between TLR4/stat3 signaling and SUMO1, LPS-treated cells were cultured with stattic, an inhibitor of stat3 phosphorylation. With the decreased level of p-stat3, SUMO1 and nuclear transport of p65 were inhibited, thus resulting in the downregulation of caspase-1, which indicated that activation of TLR4/stat3 was essential for LPS-induced caspase-1 overexpression through enhancing SUMO1-mediated SUMOylation of p65. Additionally, we found that the decrease of nuclear p65 caused by stattic treatment did not affect the expression of TLR4 (Figure 4A). NF-κB inhibitor BAY 11-7082 also failed to repress LPS-induced TLR4 expression, which indicated that NF-κB activation was not responsible for LPS-induced TLR4 expression (Figure S3A). Through immunofluorescence analysis, compared to cells treated by LPS, stattic treatment contributed to a decrease of p65 nuclear translocation in LPS treated cells and repressed the invasive ability induced by LPS (Figure 4B). Moreover, LPS-mediated EMT process was obviously repressed by static treatment (Figure 4C). We also performed qPCR experiments to measure the transcription level of caspase-1 and found that stattic treatment repressed LPS-mediated caspase-1 overexpression (Figure 4D). We also demonstrated that stattic could abolish the effects of LPS on caspase-1 enzymatic activity (Figure 4E). These data indicated that LPS stimulated caspase-1 expression through TLR4/stat3/SUMO1 signaling cascades.
Figure 4.
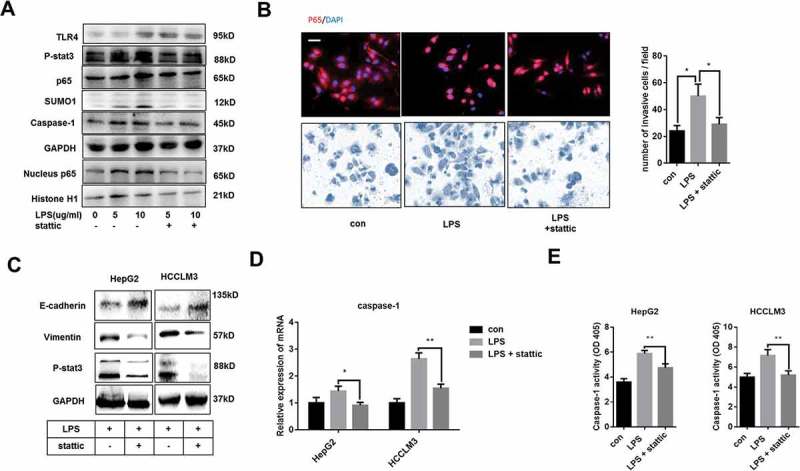
LPS-induced caspase-1 overexpression was by activating TLR4/stat3/SUMO1 signaling pathway(A) Western blot analysis indicates that TLR4/stat3 signaling is essential for LPS-induced caspase-1 expression. LPS-treated HCCLM3 cells were cultured with or without 10 µm stattic for 24 h. (B) Immunofluorescence staining p65 and transwell experiments were performed. LPS-treated HepG2 and HCCLM3 cells were cultured with or without 10 µm stattic for 24 h. (C) Western blot analysis indicates that stat3 signaling is essential for LPS-induced EMT phenotype. (D) q-PCR analysis reveals that Stat3 activation is essential for LPS-induced caspase-1 overexpression. (E) Caspase-1 enzymatic activity was measured by caspase-1 activity assays. Data are means ± SEM from 3 independent experiments, * means p < 0.05, ** means p < 0.01, *** means p < 0.001 by unpaired student T test.
Sorafenib inhibits caspase-1 expression by downregulating tlr4/stat3/sumo1 signaling pathway
We already verified that sorafenib inhibited caspase-1 expression and repressed LPS-induced caspase-1 overexpression via modulating nuclear transport of p65. To investigate the effects of sorafenib on TLR4/stat3 signaling, HepG2 and HCCLM3 cells were cultured with sorafenib for 24h and data showed that TLR4 expression and p-stat3 level were inhibited by sorafenib (Figure 5A). At transcriptional level, sorafenib decreased the expression of TLR4 and repressed LPS-induced TLR4 overexpression (Figure 5B). Furthermore, sorafenib suppressed the activation of TLR4/stat3/SUMO1 caused by LPS (Figure 5C). In conclusion, as shown in Figure 5D, we demonstrated that TLR4/stat3/SUMO1 played a crucial role in LPS-induced caspase-1 expression and sorafenib downregulated caspase-1 expression through inactivating TLR4/stat3/SUMO1 pathway.
Figure 5.
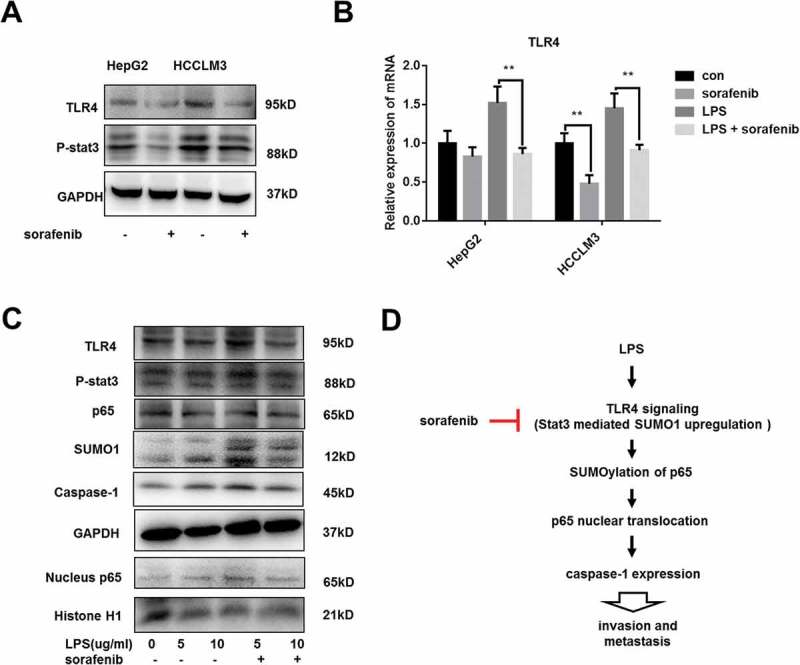
sorafenib inhibits LPS-mediated activation of TLR4/stat3/SUMO1 pathway (A) Western blot analysis indicates that TLR4/stat3 signaling was repressed by sorafenib. HepG2 and HCCLM3 cells were incubated with 10 µm sorafeniib for 24 h. (B) q-PCR analysis reveals that sorafenib decreases TLR4 expression and represses LPS-induced TLR4 overexpression. (C) Western blot analysis indicates that LPS-induced activation of TLR4/stat3 signaling is repressed by sorafenib. LPS-treated HCCLM3 cells were cultured with or without 10 µm sofanib for 24 h. (D) Schematic diagram. Sorafenib decreases caspase-1 expression and suppresses LPS-induced caspase-1 overexpression through inhibiting TLR4/stat3/SUMO1 signaling cascades. Data are means ± SEM from 3 independent experiments, * means p < 0.05, ** means p < 0.01, *** means p < 0.001 by unpaired student T test.
Discussion
HCC is the leading cause of cancer associated death worldwide.1 Most of HCC are developed from chronic inflammatory liver diseases, such as hepatic B or C virus infection and Non-alcoholic fatty liver disease (NAFLD).26 Tumor inflammatory microenvironment, including inflammatory cytokines and cells, is considered as one hallmark of HCC and indicates poor prognosis of patients with HCC.23,24
As one of inflammatory caspases, caspase-1 activation has been demonstrated to be associated with the development of HCC, especially in metastasis.7,9,10 Recent studies have shown that hypoxia condition could stimulate caspase-1 activation in HCC via promoting the release of endogenous HMGB1, thus resulting in invasion and metastasis.7
Mounting evidence has shown that gut-derived LPS plays a critical role in tumorigenic response of the liver and contributes to HCC development.19 Recent studies indicated that LPS promoted the invasion and metastasis of HCC through facilitating the formation of cancer stem cells (CSCs) and leading to induction of epithelial-mesenchymal transition (EMT).19,20,27 LPS and other cellular stresses are verified to stimulate caspase-1 activation. In our investigation, LPS-induced caspase-1 activation is responsible for LPS-mediated EMT. Silencing caspase-1 by using siRNA could suppressing EMT caused by LPS in HCC, thus inhibited the invasion and metastasis.
As the first-line systematic therapy for advanced HCC, sorafenib has been known to repress the progression of HCC.16 In our study, we demonstrated that sorafenib could decrease caspase-1 expression at the transcriptional level and also suppress LPS-mediated caspase-1 upregulation, which contributed to the repression of invasion. Recent studies have suggested NF-κB is essential for caspase-1 activation.5,11 Here, our study observed that sorafenib restricted the nuclear translocation of p65, an active form of NF-κB, and repressed LPS-induced p65 translocation into nucleus. Moreover, we verified the repression of p65 translocation caused by sorafenib was determined by the down-regulation of SUMOylation of p65 mediated by SUMO1. Through Co-IP and immunoblot analysis experiments, we found sorafenib could decease SUMO1 expression and suppress the SUMOylation of p65, contributing to restriction of nuclear transport of p65 and thus decreasing caspase-1 expression.
As the major receptor of LPS, TLR4 has been demonstrated to be related the development of HCC and commonly expressed in HCC cells.28-31 Accumulating data indicated that TLR4 was indispensable for LPS-induced EMT and CSCs formation.19,20,27 IL-6/stat3 signaling was stimulated by LPS and responsible for CSCs formation.20,27 Furthermore, studies indicated that IL-6/stat3 signaling played a part in EMT.21 We observed that stat3 inhibitor could repress LPS-induced caspase-1 expression and nuclear transport of p65. Our study confirmed that stat3 inhibitor suppressed LPS-induced expression of SUMO1. Moreover, we found that sorafenib could inhibit stat3 activation and repress LPS-induced stat3 activation via down-regulating TLR4.
In summary, we show that caspase-1 activation is essential for LPS-induced EMT. Moreover, sorafenib could decrease caspase-1 expression and repress LPS-induced caspase-1 activation through restricting p65 nuclear translocation, which was caused by the down-regulation of TLR4/stat3/SUMO1 signaling cascades. Our results generate novel insights on the relationship between sorafenib and caspase-1.
Materials and methods
Patients and specimens
Thirty HCC patients were collected from Medical School of Nanjing University Affiliated Drum Tower Hospital. Informed consent was obtained from each recruited patient, and the protocol was approved by the Institutional Research Ethics Committee. The clinical signatures of all patients are summarized in Supplementary Table 1.
Chemical reagents and antibodies
LPS were purchased from Sigma-Aldrich (No. L2630-25MG, USA). Recombinant Human IL-1β (rhIL1b) was purchased from PeproTech (No.200-01B, USA). Sorafenib was purchased from Selleck Chemicals (No.S7397, USA). Stattic was purchased from Selleck Chemicals (No.S7024, USA). BAY 11-7082 was purchased from Selleck Chemicals (No. S2913, USA). All chemical reagents were diluted according to manufactural instructions. Primary antibodies were purchased from Cell Signaling Technology (Boston, MA, USA).
Cell culture
The human HCC cell line HCCLM3 and HepG2 were achieved from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Two HCC cells were al cultured in 4.5g/L glucose DMEM containing 10% FBS, penicillin (100U/mL) and streptomycin (100μg/mL). All cells were incubated at 37°C in humidified air with 5% CO2.
Western blot analysis
Total protein was extracted by lysing cells in RIPA buffer containing protease inhibitor cocktail. Protein samples boiled with 1x loading buffer were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with 5% BSA in TBS-T, membranes were incubated with the primary antibody at 4°C overnight. Goat-anti-rabbit or mouse IgG conjugated to horseradish peroxidase (HRP) was used as the secondary antibody. Protein was imaged by a camera (Tanon, China).
Q-PCR analysis
Total RNA of HCC cells was isolated using Trizol reagent (Life Technology). 2 μg of total RNA were reverse-transcribed using the PrimeScript RT reagent Kit (Takara, Kyoto, Japan). The PCR was carried out in triplicate using SYBR Green real-time PCR master mix (Takara, Kyoto, Japan). All results are normalized to 18S rRNA expression. The specific primers used to amplify relevant genes are shown in Supplementary Table 2.
SiRNA transfection
Caspase-1 siRNA and control siRNA (Riobio, China) were transfected into cells using lipofectin 2000 according to the manufacturer’s instructions. At the end of the siRNA treatment (48–72 h), the cells were collected for western blot analysis and q-PCR. The specific primers used to amplify relevant genes are shown in Supplementary Table 1.
Immunofluorescence
Immunofluorescence analysis was processed according to protocols. Cells were implanted in 24-well dishes and fixed by 4% paraformaldehyde for 24h. Fixed cells were stained with p65 (1:200) (Cell Signaling Technology) followed by Cy3–conjugated anti–rabbit IgG (1:200) (abcam, USA). Representative images were detected by fluorescent microscopy (Leica, Germany) and data were processed via ImagePro Plus.
Immunohistochemistry staining
Immunohistochemistry of HCC samples were performed. Briefly, after incubation with caspase-1 (Abcam, USA), HMGB1 (Abcam, USA), IL1b (Cell Signaling Technology, USA), E-cadherin (Cell Signaling Technology, USA), Vimentin (Cell Signaling Technology, USA), the sections were stained in an Envision System (Dako Cytomation, USA).
Caspase-1 enzymatic activity assay
Caspase-1 enzymatic activity assays were purchased from Beyotime (No. C1101, China) and were performed according to manufacturer’s instructions.
Transwell experiments
The invasive ability of HCC cells was measured by 24-well transwell chambers separated by polycarbonate membranes with 8-µm pores and precoated with Matrigel. The lower chamber was filled with complete DMEM as a chemoattractant. Treated cells were seeded at 5 × 104 in the upper chamber and incubated at 37°C in a humidified incubator containing 5% CO2. Cells that migrated to the underside of the membrane were fixed and stained with Giemsa (Sigma-Aldrich, USA), detected, and calculated with a microscope (Leica, Wetzlar, Germany).
Co-immunoprecipitation experiments
HCCLM3 cells treated with or without 10 µm sorafenib for 24 hours were harvested and lysed in ice-cold IP buffer for 30 minutes. Total protein extracts were centrifuged at 12,000g for 10 min at 4°C. 500ul of the total lysate was incubated at 4°C with 2 μg of corresponding antibodies or IgG as control and 50 μl protein A/G beads (Santa Cruz, USA) to immunoprecipitate p65. The interacted complexes were then washed three times with lysis buffer. After centrifuged at 12,000g for 10 min at 4°C, pellets were suspended in 100ul lysis buffer and boiled with 1x SDS loading buffer and then processed by western blot analysis.
Statistical analysis
All experiments were performed at least three times. Quantitative data were expressed as mean ± SD for each experiment. Significance between groups was performed using Student’s t-test. Statistical significances are indicated by *: P < 0.05, **: P < 0.01, and ***: P < 0.001. GraphPad Prism 6 was used for all statistical analyses. P < 0.05 was considered statistically significant.
Funding Statement
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81670566), Jiangsu Province’s Key Provincial Talents Program (Grant No. ZDRCA2016066).
Disclosure of interest
None.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Han ZG. 2012; Functional genomic studies: insights into the pathogenesis of liver cancer. Annu Rev Genomics Hum Genet. 13:171–205. doi: 10.1146/annurev-genom-090711-163752. [DOI] [PubMed] [Google Scholar]
- 2.Portolani N, Coniglio A, Ghidoni S, Giovanelli M, Benetti A, Tiberio GAM, Giulini SM. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Ann Surg. 2006;243(2):229–235. doi: 10.1097/01.sla.0000197706.21803.a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rampone B, Schiavone B, Confuorto G. Current management of hepatocellular cancer. Curr Oncol Rep. 2010;12(3):186–192. doi: 10.1007/s11912-010-0094-3. [DOI] [PubMed] [Google Scholar]
- 4.Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57(3):642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 5.Davis BK, Wen H, Ting JP. 2011; The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, Dinarello CA, Fujita M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem. 2010;285(9):6477–6488. doi: 10.1074/jbc.M109.064907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, Monga SPS, Geller DA, Lotze MT, Tsung A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55(6):1863–1875. doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celardo I, Grespi F, Antonov A, Bernassola F, Garabadgiu AV, Melino G, Amelio I. 2013; Caspase-1 is a novel target of p63 in tumor suppression. Cell Death Dis. 4:e645. doi: 10.1038/cddis.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu Q, Jiang Y, Zhang W, Xu C, Du W, Tuguzbaeva G, Qin Y, Li A, Zhang L, Sun G, et al. Pyroptosis is involved in the pathogenesis of human hepatocellular carcinoma. Oncotarget. 2016;7(51):84658–84665. doi: 10.18632/oncotarget.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A. 2010;107(50):21635–21640. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Tao X, Zhang J, Wang P, Sha M, Ma Y, Geng X, Feng L, Shen Y, Yu Y, et al. Small ubiquitin-related modifier 1 is involved in hepatocellular carcinoma progression via mediating p65 nuclear translocation. Oncotarget. 2016;7(16):22206–22218. doi: 10.18632/oncotarget.8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu LP, Ho RLK, Chen GG, Lai PBS. Sorafenib inhibits hypoxia-inducible factor-1alpha synthesis: implications for antiangiogenic activity in hepatocellular carcinoma. Clin Cancer Res. 2012;18(20):5662–5671. doi: 10.1158/1078-0432.CCR-12-0552. [DOI] [PubMed] [Google Scholar]
- 14.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66(24):11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 15.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 16.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, De Oliveira AC, Santoro A, Raoul J-L, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 17.Cheng A-L, Kang Y-K, Chen Z, Tsao C-J, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang T-S, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 18.Kuo YC, Lin W-C, Chiang I-T, Chang Y-F, Chen C-W, Su S-H, Chen C-L, Hwang -J-J. Sorafenib sensitizes human colorectal carcinoma to radiation via suppression of NF-kappaB expression in vitro and in vivo. Biomed Pharmacother. 2012;66(1):12–20. doi: 10.1016/j.biopha.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 19.Jing YY, Han Z-P, Sun K, Zhang -S-S, Hou J, Liu Y, Li R, Gao L, Zhao X, Zhao Q-D, et al. 2012; Toll-like receptor 4 signaling promotes epithelial-mesenchymal transition in human hepatocellular carcinoma induced by lipopolysaccharide. BMC Med. 10:98. doi: 10.1186/1741-7015-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai FB, Liu W-T, Jing -Y-Y, Yu G-F, Han Z-P, Yang X, Zeng J-X, Zhang H-J, Shi R-Y, Li X-Y, et al. Lipopolysaccharide supports maintaining the stemness of CD133(+) hepatoma cells through activation of the NF-kappaB/HIF-1alpha pathway. Cancer Lett. 2016;378(2):131–141. doi: 10.1016/j.canlet.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 21.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28(1–2):15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 22.Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC, Stiles B, Rountree CB. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology. 2010;52(3):945–953. doi: 10.1002/hep.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010;29(2):285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine. 2008;43(3):374–379. doi: 10.1016/j.cyto.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Hildebrandt T, Knetzger S-M, Schrauder MG, Jud SM, Hein A, Rauh C, Fasching PA, Beckmann MW, Thiel FC. 2015; Small ubiquitin-related modifier 2/3 interacts with p65 and stabilizes it in the cytoplasm in HBV-associated hepatocellular carcinoma. BMC Cancer. 15:675. doi: 10.1186/s12885-015-1584-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. [DOI] [PubMed] [Google Scholar]
- 27.Liu W-T, Jing -Y-Y, Yu G-F, Han Z-P, Yu -D-D, Fan Q-M, Ye F, Li R, Gao L, Zhao Q-D, et al. Toll like receptor 4 facilitates invasion and migration as a cancer stem cell marker in hepatocellular carcinoma. Cancer Lett. 2015;358(2):136–143. doi: 10.1016/j.canlet.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 28.Sepehri Z, Kiani Z, Kohan F, Alavian SM, Ghavami S. 2017; Toll like receptor 4 and hepatocellular carcinoma; A systematic review. Life Sci. 179:80–87. doi: 10.1016/j.lfs.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 29.Lin A, Wang G, Zhao H, Zhang Y, Han Q, Zhang C, Tian Z, Zhang J. TLR4 signaling promotes a COX-2/PGE2/STAT3 positive feedback loop in hepatocellular carcinoma (HCC) cells. Oncoimmunology. 2016;5(2):e1074376. doi: 10.1080/2162402X.2015.1074376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Li M, Zheng QC. 2015; Emerging role of Toll-like receptor 4 in hepatocellular carcinoma. J Hepatocell Carcinoma. 2:11–17. doi: 10.2147/JHC.S44515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Yan J, Lin H, Hua F, Wang X, Liu H, Lv X, Yu J, Mi S, Wang J, et al. Toll-like receptor 4 activity protects against hepatocellular tumorigenesis and progression by regulating expression of DNA repair protein Ku70 in mice. Hepatology. 2013;57(5):1869–1881. doi: 10.1002/hep.26234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.