

Abstract
Acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF) is a devastating condition that frequently occurs in the advanced stage of IPF. However, the clinical features in AE of connective tissue disease-associated interstitial pneumonia (AE-CTD-IP) have not been well-established. The aim of this study was to clarify the clinical features of AE-CTD-IP and to compare them with those of AE-IPF. Fifteen AE-CTD-IP patients and 48 AE-IPF patients who were diagnosed and treated at our hospital were retrospectively studied. Compared with AE-IPF patients, AE-CTD-IP patients had a significantly higher %FVC (median, 94.8 vs. 56.3%; p < 0.001) and a lower extent of honeycombing on HRCT (p = 0.020) within 1 year before AE. At AE, AE-CTD-IP patients showed higher white blood cell counts (12.0 vs. 9.9 × 103/μL; p = 0.023), higher CRP (10.2 vs. 6.7 mg/dL; p = 0.027), and longer period from admission to the beginning of AE treatment (4 vs. 1 days; p = 0.003) than AE-IPF patients. In addition, patients with AE-CTD-IP had poor prognosis as in those with AE-IPF (log-rank; p = 0.171). In conclusion, AE-CTD-IP occurred even in the early stage of IP and had more inflammatory status than in AE-IPF.
Keywords: Connective tissue disease, interstitial pneumonia, acute exacerbation, idiopathic pulmonary fibrosis, rheumatoid arthritis
Introduction
Acute exacerbation of idiopathic pulmonary fibrosis (AE-IPF) is a devastating condition that occurs in the clinical course of IPF.1–4 During the heterogeneous clinical course of IPF, patients with more severe disease seem to more frequently progress to AE-IPF.5 In addition, one of the most important causes of disease progression and death is AE-IPF,6,7 and AE-IPF accounts for 30–40% of the deaths in patients with IPF.8,9 The 2007 criteria for diagnosis of AE-IPF were extremely strict, requiring careful exclusion of pulmonary infection, left heart failure, pulmonary embolism, and identifiable causes of acute lung injury.3 However, some of these causes, including infection and drug toxicity, are allowed as triggers of AE in the 2016 updated criteria for AE-IPF.10 Because these triggers sometimes coexist with AE-IPF in clinical practice, these amendments to the criteria are now easier to apply.
AE occurs not only in IPF but also in other idiopathic interstitial pneumonia (IIP) or connective tissue disease-associated interstitial pneumonia (CTD-IP).11–14 Although AE of CTD-IP (AE-CTD-IP) occurs less frequently than AE-IPF, this condition also has a great impact on patient prognosis.12 Therefore, AE-CTD-IP is also the most important event in the clinical course of CTD. In patients with CTD, especially those with rheumatoid arthritis (RA), immunosuppressive agents such as steroids, immunosuppressants, and novel biological products are frequently administered to treat CTD. Thus, infectious pneumonia and drug-induced pneumonitis are important differential diagnoses, especially in this setting. However, the specific features of AE-CTD-IP in clinical practice, such as pulmonary function tests before AE and inflammatory findings or high-resolution computed tomography (HRCT) findings at AE, have not been well-established compared with those of AE-IPF.
In this study, we evaluated important clinical factors for differential diagnosis and prognosis of AE-CTD-IP. Furthermore, these factors were compared with those of AE-IPF. To the best of our knowledge, this is the first study to show detailed clinical differences between AE-CTD-IP and AE-IPF.
Patients and methods
Study design and subjects
Fifteen patients who were diagnosed and treated for AE-CTD-IP at our hospital between 1997 and 2015 were retrospectively reviewed. Thirty-seven patients (48 episodes in total) with AE-IPF, who were treated at our hospital in the same period, were also recorded and data were compared with those of AE-CTD-IP. All patients with IPF met the 2011 international consensus criteria for IPF.4
AE of IP was diagnosed according to the modified diagnostic criteria described by Collard et al.3 Briefly, the diagnostic criteria for diagnosis of AE-IP were as follows: (1) previous or concurrent diagnosis of IP; (2) unexplained worsening or development of dyspnea within 30 days; (3) HRCT with new bilateral ground-glass abnormality and/or consolidation superimposed on a background reticular or honeycomb pattern; (4) no evidence of pulmonary infection; and (5) exclusion of alternative causes including left heart failure, pulmonary embolism, or an identifiable cause of acute lung injury. Patients enrolled in this study also met the newly proposed criteria in 2016.10 The study protocol was approved by the Ethics Committee of Hamamatsu University School of Medicine (approval number E16-065). All procedures in this study were performed in accordance with the 1964 Helsinki declaration and its later amendments.
Data collection
Clinical data were obtained from medical records. Disease severity of IPF within 12 months before the AE event was assessed using pulmonary function tests and findings on HRCT. Blood samples were collected at AE-IP.
Evaluation of HRCT findings
The extent of lung opacities was measured on three HRCT slices: at the bifurcation of the trachea, at the bases of the lower lobes, and at the midpoint between the other two slices. The extent of fibrosis in each lobe was scored using the following system: 0, none; 1, 1–10%; 2, 11–25%; 3, 26–50%; 4, 51–75%; and 5, 76–100%. The sum of the scores from five lobes (0–25) was used to express the extent of reticular opacity, honeycombing, consolidation, and ground-glass opacity in each patient. The pattern of acute exacerbation of IP on HRCT was classified as (1) peripheral, (2) multifocal, or (3) diffuse as reported by Akira.15 The HRCT findings were reviewed by two observers.
Statistical analysis
Statistical analyses were performed using StatView J-4.5 (SAS Institute Inc., Cary, North Carolina, USA). Categorical data were compared using the χ 2 test or Fisher’s exact probability test for independence, and continuous data were compared using the unpaired t-test. Continuous data at different time points in the same patient were compared using the paired t-test. Overall survival of patient groups was estimated using Kaplan–Meier curves and was compared between groups using the log-rank test. The relationships between variables and mortality were evaluated using the Cox proportional hazards regression analysis. All tests were two-sided and statistical significance was set at p < 0.05.
Results
Clinical characteristics, physical examination findings, and treatments in all patients with AE-CTD-IP
Clinical characteristics of all patients with AE-CTD-IP, including physical examination findings and preceding treatments, are shown in Table 1. Patients had a median age of 71 years at AE-CTD-IP and 11 patients were male. The median observation period was 56 months. A breakdown of CTD-diagnosis is shown in Figure 1. Patients with AE-CTD-IP comprised nine with rheumatoid arthritis, three with microscopic polyarthritis, two with systemic sclerosis, and one with microscopic polyarteritis and Sjogren syndrome. CTD activity before AE was not high in all patients. IP preceding CTD diagnosis was found in 6 of 15 patients (Table 1). Surgical lung biopsy was performed in four patients. Two showed a usual interstitial pneumonia pattern and the other two showed a nonspecific interstitial pneumonia pattern.
Table 1.
Clinical characteristics, physiological data, and treatments in patients with AE-CTD-IP.
| n = 15, median (range) | |
|---|---|
| Age, year | 71 (57, 85) |
| Sex, male/female | 11/4 |
| Smoking, never/ex/current | 2/13/0 |
| Pack-year of smoking | 27.5 (0, 90) |
| Surgical lung biopsy, ± | 4/11 |
| Period from CTD-diagnosis to AE, months | 64 (0, 365) |
| IP preceding CTD-diagnosis, ± | 6/9 |
| Dyspnea on effort, mMRC 0/1/2/3/4/unknown | 1/1/3/2/6/2 |
| Clubbed finger, ± | 3/12 |
| Preceding treatments with steroids, ± | 9/6 |
| Administration dose of steroids, mg/day | 5 (0, 30) |
| Preceding treatments with immunosuppresant, ± | 8/7 |
| Preceding treatments with methotrexate, ± | 5/10 |
| Preceding oxygen therapy, ± | 4/11 |
| Observation period, months | 56 (0, 228) |
AE: acute exacerbation, CTD: connective tissue disease, IP: interstitial pneumonia, mMRC: modified Medical Research Council.
Figure 1.
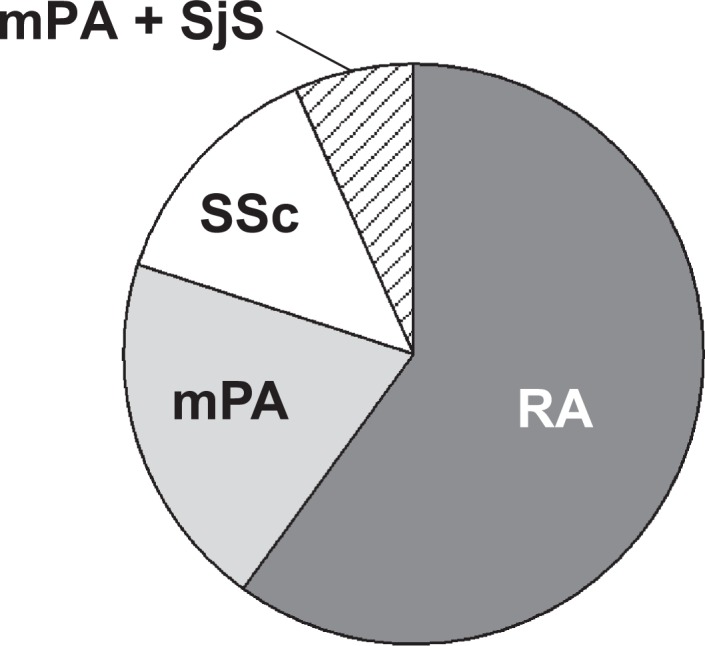
A breakdown of connective tissue disease (CTD)-diagnosis in 15 patients with acute exacerbation of CTD-associated interstitial pneumonia (AE-CTD-IP). Patients with AE-CTD-IP comprised nine with rheumatoid arthritis, three with microscopic polyarthritis, two with systemic sclerosis, and one with microscopic polyarteritis and Sjogren syndrome. RA: rheumatoid arthritis, mPA: microscopic polyarthritis, SSc: systemic sclerosis.
Treatment with steroids was preceded in nine patients. Treatment with immunosuppressants was preceded in eight, five of whom with methotrexate. In total, 11 patients with CTD-IP were treated with steroids and/or immunosuppressants. Regarding anti-fibrotic medicine, pirfenidone was administered in one patient. Four patients received long-term oxygen therapy before AE-CTD-IP. During the observation period, all patients presented with their first AE episode and no patient repeated AE-CTD-IP.
Comparison of data between patients with AE-CTD-IP and those with AE-IPF
Clinical data, including physiological data, laboratory findings, HRCT findings, and treatments of patients with AE-CTD-IP (shown in Table 2) were compared with clinical data from AE-IPF patients. While all patients with AE-CTD-IP presented with their first AE episode, several patients with AE-IPF repeated AE (p = 0.098). Percent-predicted forced vital capacity (%FVC) within 1 year before AE in patients with AE-CTD-IP was significantly higher than in those with AE-IPF (median 94.8 and 56.3%, respectively, p < 0.001, Figure 2(a)). Furthermore, the extent of honeycombing on HRCT within 1 year before AE in patients with AE-CTD-IP was significantly less severe than in those with AE-IPF (p = 0.020, Figure 2(b)). The extent of honeycombing at AE was also less severe (p = 0.007; Online Supplemental Table S1). Frequency of preceding treatments, including long-term oxygen therapy, was not different between groups. Administration of methotrexate was discontinued in all cases. At AE, the PaO2/FiO2 (P/F) ratio, extent score of all findings on HRCT, and HRCT pattern were not different between groups. However, the white blood cell count (12.0 vs. 9.9 × 103/μL; p = 0.023) and neutrophils (11.3 vs. 7.4 × 103/μL; p = 0.010) in peripheral blood and serum C-reactive protein (CRP, 10.2 vs. 6.7; p = 0.027) were significantly higher in patients with AE-CTD-IP than in those with AE-IPF (Table 2). In addition, the incidence of fever tended to be higher in patients with AE-CTD-IP than in those with AE-IPF (53.3 vs. 29.1%, p = 0.086). All patients with AE-CTD-IP or AE-IPF were treated with steroid pulse therapy after hospital admission (methylprednisolone 1000 mg/day for 3 days) followed by a tapering dose of prednisolone. Treatments with an immunosuppressant (cyclophosphamide or cyclosporine) and/or direct hemoperfusion with a polymyxin B-immobilized fiber column (PMX-DHP) (PMX; Toray Medical Co., Ltd, Tokyo, Japan)16,17 were added to steroid therapy in a large proportion of patients. These treatments were commenced as soon as possible after admission concomitantly with the administration of antibiotics. These AE treatments were not different between groups. However, the period from admission to the beginning of AE treatments (median, 4 vs. 1 day; p = 0.003) and that to the beginning of PMX-DHP (median, 10 vs. 1 day; p < 0.001) were significantly longer in patients with AE-CTD-IP than in those with AE-IPF (Table 2).
Table 2.
Comparison of data between patients with AE-CTD-IP and those with AE-IPF.
| AE-CTD-IP (n = 15, median (range)) | AE-IPF (n = 48, median (range)) | p Value | |
|---|---|---|---|
| Age, year | 71 (57, 85) | 69 (50, 84) | 0.273 |
| Sex, male/female | 11/4 | 45/3 | 0.049a |
| Smoking, never/ex/current | 2/13/0 | 4/39/5 | 0.386 |
| Pack-year of smoking | 28 (0, 90) | 37 (0, 81) | 0.574 |
| Observation period, months | 56 (0, 228) | 53 (2, 205) | 0.829 |
| The number of AE, 1/2/3 | 15/0/0 | 36/11/1 | 0.098 |
| Preceding treatments, ± | 11 / 4 | 31/17 | 0.755a |
| Preceding treatment with steroids, ± | 9 / 6 | 23 / 25 | 0.414 |
| Preceding oxygen therapy, ± | 4 / 11 | 16 / 32 | 0.756a |
| Fever at AE, ± | 8 / 7 | 14 / 34 | 0.086 |
| Peripheral blood WBC at AE, ×103/μL | 12.0 (7.4, 22.8) | 9.9 (1.9, 20.0) | 0.023 |
| Peripheral blood neutrophils at AE, ×103/μL | 11.3 (6.3, 19.4) | 7.4 (1.5, 18.2) | 0.010 |
| Peripheral blood platelets at AE, ×103/μL | 25.7 (8.1, 84.0) | 21.0 (11.3, 46.2) | 0.001 |
| Serum CRP at AE, mg/dL | 10.2 (1.5, 31.8) | 6.7 (0.9, 23.7) | 0.027 |
| Serum LDH at AE, IU/L | 405 (202, 815) | 347 (183, 693) | 0.089 |
| Serum KL-6 at AE, U/mL | 1483 (445, 2779) | 1550 (481, 6404) | 0.243 |
| Serum SP-D at AE, ng/mL | 285 (74, 2950) | 366 (23, 1330) | 0.284 |
| P/F ratio at AE | 181 (57, 290) | 168.5 (38, 386) | 0.814 |
| Extent scores on HRCT at AE (full score: 25) | 19 (10, 25) | 21 (13, 25) | 0.314 |
| HRCT pattern at AE, peripheral/multifocal/diffuse/unknown |
1/3/10/1 |
3/5/37/3 |
0.998 |
| Period from admission to the beginning of AE-treatment, day | 4 (0, 13) | 1 (0, 17) | 0.003 |
| Period from admission to the beginning of PMX-DHP, day | 10 (7, 13) | 1 (0, 17) | <0.001 |
| Administration of steroid pulse therapy at AE, ± | 15/0 | 48/0 | NS |
| Administration of immunosuppressant at AE, ± | 9/6 | 36/12 | 0.272a |
| Treatment with PMX-DHP, ± | 5/10 | 26/22 | 0.237a |
| Intubation at AE, ± | 5/10 | 12/36 | 0.523a |
AE: acute exacerbation, CTD: connective tissue disease, IP: interstitial pneumonia, HRCT: high-resolution computed tomography, CRP: C-reactive protein, LDH: lactate dehydrogenase, KL-6: Krebs von den Lungen-6, SP-D: surfactant protein D, P/F: PaO2/FiO2, PMX-DHP: direct hemoperfusion with a polymyxin B-immobilized fiber column. NS: not significant.
a Fisher’s exact probability test.
Figure 2.

Comparison of pulmonary function test or HRCT findings between AE-CTD-IP and AE-IPF. (a) %FVC within one year before AE was evaluated. %FVC of patients with AE-CTD-IP was significantly higher than in those with AE-IPF (median 94.8 and 56.3%, respectively, p < 0.001). (b) The extent of honeycombing on HRCT within 1 year before AE was graded. Honeycombing was significantly less severe in patients with AE-CTD-IP than in those with AE-IPF (p = 0.020). HRCT: high-resolution computed tomography; AE-CTD-IP: acute exacerbation of connective tissue disease-associated interstitial pneumonia; IPF: idiopathic pulmonary fibrosis; %FVC: percent-predicted forced vital capacity.
Mortality rate and prognostic factors
Of 15 patients with AE-CTD-IP, 5 died within 1 month of onset (mortality rate, 33.3%), seven died within 3 months (mortality rate, 46.7%) and 11 died during the study period (mortality rate, 73.3%). Within 3 months of AE-IPF onset, six patients died of respiratory failure caused by AE-CTD-IP and one died of gastrointestinal bleeding after beginning of steroid treatment. Kaplan–Meier survival curves from the first AE onset are shown in Figure 3. The 3-month survival rate was not significantly different between the two groups (log-rank test, p = 0.171). However, the survival rate of AE-CTD-IP tended to be worse than that of AE-IPF. The results of univariate Cox proportional hazards models of survival in both groups to identify prognostic factors within 3 months of AE onset are shown in Table 3. The period from IP diagnosis to AE was a significant prognostic factor in patients with AE-CTD-IP, while it was not in those with AE-IPF (hazard ratio (HR): 1.011, p = 0.023 and HR 0.999, p = 0.820, respectively). Intubation at AE was also a worse prognostic factor in patients with AE-CTD-IP (HR 8.080, p = 0.015). Higher neutrophils in peripheral blood at AE tended to be a worse prognostic factor in patients with AE-CTD-IP (HR: 1.000, p = 0.076), while it was a significant factor in those with AE-IPF (HR: 1.000, p = 0.002). The extent score on HRCT at AE tended to be a significant factor in both groups (HR: 1.211, p = 0.097 and HR: 1.210, p = 0.055, respectively). Neither treatment with immunosuppressant nor PMX-DHP was a prognostic factor in patients with AE-CTD-IP (HR: 1.999, p = 0.409 and HR 0.851, p = 0.815, respectively). Multivariate Cox proportional hazards analyses for prognostic factors could not be conducted because of low statistical power.
Figure 3.

Kaplan–Meier survival curves from the first AE onset. Of 15 patients with AE-CTD-IP, 5 died within 1 month of onset (mortality rate, 33.3%), seven died within 3 months (mortality rate, 46.7%) and 11 died during the study period (mortality rate, 73.3%). The 3-month survival rate was not significantly different between patients with AE-CTD-IP and those with AE of IPF (log-rank test, p = 0.171). The survival rate of AE-CTD-IP tended to be worse than that of AE-IPF. AE: acute exacerbation; AE-CTD-IP: acute exacerbation of connective tissue disease-associated interstitial pneumonia; IPF: idiopathic pulmonary fibrosis.
Table 3.
Univariate cox proportional hazards models of survival.
| Variables | AE-CTD-IP | AE-IPF | ||
|---|---|---|---|---|
| HR | p Value | HR | p Value | |
| Age, year | 1.030 | 0.497 | 1.023 | 0.616 |
| Sex, male | 3.178 | 0.286 | 0.811 | 0.843 |
| Period from IP-diagnosis to AE, months | 1.011 | 0.023 | 0.999 | 0.820 |
| Extent score on HRCT before AEa | 0.997 | 0.980 | 1.181 | 0.107 |
| FVC before AEa, % pred | 1.066 | 0.354 | 0.974 | 0.307 |
| Preceding oxygen therapy, + | 3.382 | 0.114 | 3.416 | 0.053 |
| Fever at AE, + | 1.263 | 0.760 | 0.505 | 0.388 |
| Peripheral blood WBC at AE, /μL | 1.000 | 0.208 | 1.000 | 0.025 |
| Peripheral blood neutrophils at AE, /μL | 1.000 | 0.076 | 1.000 | 0.002 |
| Serum CRP at AE, mg/dL | 1.035 | 0.354 | 0.999 | 0.961 |
| Serum LDH at AE, IU/L | 1.001 | 0.631 | 1.005 | 0.071 |
| Serum KL-6 at AE, U/mL | 1.000 | 0.579 | 1.000 | 0.970 |
| Serum SP-D at AE, ng/mL | 1.001 | 0.163 | 0.999 | 0.673 |
| P/F ratio at AE | 0.993 | 0.179 | 0.995 | 0.215 |
| Extent score on HRCT at AE | 1.211 | 0.097 | 1.210 | 0.055 |
| Period from admission to the beginning of AE-treatment, day | 1.020 | 0.821 | 1.072 | 0.369 |
| Intubation at AE, + | 8.080 | 0.015 | 3.045 | 0.079 |
AE: acute exacerbation, CTD: connective tissue disease, IP: interstitial pneumonia, HRCT: high-resolution computed tomography, FVC: forced vital capacity, CRP: C-reactive protein, LDH: lactate dehydrogenase, KL-6: Krebs von den Lungen-6, SP-D: surfactant protein D, P/F: PaO2/FiO2.
a Pulmonary function tests, severity scores, HRCT, and serum markers were evaluated within 12 months before AE-IPF.
Discussion
In this study, we retrospectively reviewed 15 patients with AE-CTD-IP and compared their clinical features with those of AE-IPF. Patients with AE-CTD-IP had significantly higher %FVC and lower extent of honeycombing on HRCT within 1 year before AE. At AE, patients with AE-CTD-IP showed a higher white blood cell count and CRP level, and the period from admission to the beginning of AE treatment, including PMX-DHP, was significantly longer than those of AE-IPF. Finally, patients with AE-CTD-IP had a poor prognosis as that of AE-IPF. To the best of our knowledge, this is the first report to show the detailed clinical features of AE-CTD-IP compared with those of AE-IPF.
AE-CTD-IP less frequently occurs than AE-IPF. The 1-year incidence of AE-CTD-IP was 1.25–3.3%,11,12 while that of AE-IPF was 4.8–14.2%,2,18 Along with AE-IPF, early diagnosis of AE is quite important to save patients with AE-CTD-IP. However, early diagnosis of AE-CTD-IP is sometimes difficult for an attending doctor in clinical practice because many patients with CTD-IP are receiving steroids, immunosuppressants, and biological products before AE. This is the contrasting treatment of IPF, in which immunosuppressive medication is ineffective or sometimes harmful in their clinical course.4,19 Under these treatments, the most important differential diagnosis is pulmonary infection, such as pneumocystis pneumonia and cytomegalovirus infection. In this study, patients with AE-CTD-IP showed more inflammatory findings and increased levels of white blood cells and CRP compared with patients with AE-IPF, and clinicians were reluctant to start treatments for AE. Although these infections were thoroughly excluded before the treatments for AE-CTD-IP at our hospital, this process took time and was related to the delay in commencing of treatments for AE. Furthermore, administration of methotrexate for many patients with RA (five of nine patients) increased the possibility of drug-induced pneumonitis in clinical practice. As just described, these two differential diagnoses were related to the treatment delay in patients with AE-CTD-IP.
AE-IPF occurs more often in a patient’s advanced stage than in early stage.5 Surprisingly, in AE-CTD-IP, %FVC was far higher and the extent of honeycombing was lower before AE than those of AE-IPF. These data indicate that AE suddenly occurs, even in the early stage of IP in patients with CTD-IP, which is not true for patients with IPF. Whereas the exact mechanism of this frequent incidence in the early stage has been unclear, many patients (11 of 15) with CTD-IP were treated with steroids and/or immunosuppressants before AE. In these settings, some viral infection or some drug toxicity may have been triggers for AE-CTD-IP. Therefore, clinicians should pay more attention to the appearance of AE under immunosuppressive therapies in patients with CTD-IP.
Regarding the prognosis of AE-CTD-IP, our previous study12 and Park et al.11 reported poor survival in patients with AE-CTD-IP. The mortality rates in these small studies were 83.3%12 and 100%,11 respectively. However, two other studies showed better survival and the mortality rates were 33% (90 days)13 and 30%,14 respectively. Thus, the prognosis of AE-CTD-IP remains controversial. In the present study, patients with AE-CTD-IP tended to have relatively worse prognosis than those with AE-IPF. This may result from the delay in commencing AE-treatment, including PMX-DHP, because of taking time to exclude infection and drug-induced pneumonitis. Although the period from admission to the onset of AE-treatments was not a prognostic factor, this may be because of the small number of patients in this study and delayed commencement of AE treatment in many of these patients. In these two decades, treatments for AE-IP have significantly evolved and novel treatments have emerged.10 Prompt diagnosis of AE-CTD-IP and early initiation of immunosuppressive therapies in combination with recombinant thrombomodulin20 or PMX-DHP,16,17 which showed promising treatment effects on AE-IPF, may improve this poor survival in the near future.
This study has a number of limitations. First, only a small number of patients with AE-CTD-IP were included. This matter limited the statistical power to find differences between patients with AE-CTD-IP and those with AE-IPF. Furthermore, these patients had several different CTDs; therefore, the patient cohorts were quite heterogenous. Second, the data were retrospectively collected. Finally, the treatment for AE-CTD-IP was not uniform. A larger and prospective study is needed to further evaluate the clinical features and treatments for AE-CTD-IP.
In conclusion, we evaluated 15 patients with AE-CTD-IP and compared their clinical features with those of AE-IPF. AE-CTD-IP suddenly occurred, even in the early stage of CTD-IP, and showed higher inflammatory status at AE compared with AE-IPF. The period from admission to the beginning of AE treatment, including PMX-DHP, was significantly longer than that of AE-IPF, and patients with AE-CTD-IP showed poor prognosis, similar to AE-IPF patients. These findings should encourage doctors in clinical settings to promptly diagnose and treat patients with AE-CTD-IP. Further validation is needed to improve the poor survival in patients with AE-CTD-IP.
Supplemental material
Supplementary_Table_AE-CTD-IP_662018 for Differences in clinical features of acute exacerbation between connective tissue disease-associated interstitial pneumonia and idiopathic pulmonary fibrosis by Noriyuki Enomoto, Yoshiyuki Oyama, Yasunori Enomoto, Hideki Yasui, Masato Karayama, Masato Kono, Hironao Hozumi, Yuzo Suzuki, Kazuki Furuhashi, Tomoyuki Fujisawa, Naoki Inui, Yutaro Nakamura, and Takafumi Suda in Chronic Respiratory Disease
Footnotes
Authors’ note: This study was assisted by the Study Group on Diffuse Lung Disease and the Scientific Research/Research on Intractable Diseases in the Ministry of Health, Labour and Welfare of Japan.
Author contributions: Conception and design: NE; administrative support: TS. Provision of patients: NE, YO, YE, HY, MK, MK, HH, YS, KF, TF, NI, YN, TS; collection and assembly of data: NE, YO, YE, HY, MK, MK, HH, YS, KF; data analysis and interpretation: NE, YO, YE, HY, MK, MK, HH, YS, KF, TF, NI, YN, TS; manuscript writing: NE, YO, YE, HY, MK, MK, HH, YS, KF, TF, NI, YN, TS; and final approval of manuscript: NE, YO, YE, HY, MK, MK, HH, YS, KF, TF, NI, YN, TS.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Noriyuki Enomoto  https://orcid.org/0000-0003-3187-4264
https://orcid.org/0000-0003-3187-4264
Supplemental material: Supplemental material for this article is available online.
References
- 1. Kim DS, Park JH, Park BK, et al. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J 2006; 27: 143–150. [DOI] [PubMed] [Google Scholar]
- 2. Song JW, Hong SB, Lim CM, et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37: 356–363. [DOI] [PubMed] [Google Scholar]
- 3. Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007; 176: 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Homma S, Sugino K, Sakamoto S. Usefulness of a disease severity staging classification system for IPF in Japan: 20 years of experience from empirical evidence to randomized control trial enrollment. Respir Investig 2015; 53: 7–12. [DOI] [PubMed] [Google Scholar]
- 6. Kondoh Y, Taniguchi H, Kawabata Y, et al. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest 1993; 103: 1808–1812. [DOI] [PubMed] [Google Scholar]
- 7. Kondoh Y, Taniguchi H, Katsuta T, et al. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2010; 27: 103–110. [PubMed] [Google Scholar]
- 8. Ley B, Collard HR, King TE., Jr Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 431–440. [DOI] [PubMed] [Google Scholar]
- 9. Natsuizaka M, Chiba H, Kuronuma K, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med 2014; 190: 773–779. [DOI] [PubMed] [Google Scholar]
- 10. Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med 2016; 194: 265–275. [DOI] [PubMed] [Google Scholar]
- 11. Park IN, Kim DS, Shim TS, et al. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest 2007; 132: 214–220. [DOI] [PubMed] [Google Scholar]
- 12. Suda T, Kaida Y, Nakamura Y, et al. Acute exacerbation of interstitial pneumonia associated with collagen vascular diseases. Respir Med 2009; 103: 846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tachikawa R, Tomii K, Ueda H, et al. Clinical features and outcome of acute exacerbation of interstitial pneumonia: collagen vascular diseases-related versus idiopathic. Respiration 2012; 83: 20–27. [DOI] [PubMed] [Google Scholar]
- 14. Toyoda Y, Hanibuchi M, Kishi J, et al. Clinical features and outcome of acute exacerbation of interstitial pneumonia associated with connective tissue disease. J Med Invest 2016; 63: 294–299. [DOI] [PubMed] [Google Scholar]
- 15. Akira M, Kozuka T, Yamamoto S, et al. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008; 178: 372–378. [DOI] [PubMed] [Google Scholar]
- 16. Enomoto N, Mikamo M, Oyama Y, et al. Treatment of acute exacerbation of idiopathic pulmonary fibrosis with direct hemoperfusion using a polymyxin B-immobilized fiber column improves survival. BMC Pulm Med 2015; 15: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Enomoto N, Suda T, Uto T, et al. Possible therapeutic effect of direct haemoperfusion with a polymyxin B immobilized fibre column (PMX-DHP) on pulmonary oxygenation in acute exacerbations of interstitial pneumonia. Respirology 2008; 13: 452–460. [DOI] [PubMed] [Google Scholar]
- 18. Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2010; 35: 821–829. [DOI] [PubMed] [Google Scholar]
- 19. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015; 192: e3–e19. [DOI] [PubMed] [Google Scholar]
- 20. Kataoka K, Taniguchi H, Kondoh Y, et al. Recombinant human thrombomodulin in acute exacerbation of idiopathic pulmonary fibrosis. Chest 2015; 148: 436–443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary_Table_AE-CTD-IP_662018 for Differences in clinical features of acute exacerbation between connective tissue disease-associated interstitial pneumonia and idiopathic pulmonary fibrosis by Noriyuki Enomoto, Yoshiyuki Oyama, Yasunori Enomoto, Hideki Yasui, Masato Karayama, Masato Kono, Hironao Hozumi, Yuzo Suzuki, Kazuki Furuhashi, Tomoyuki Fujisawa, Naoki Inui, Yutaro Nakamura, and Takafumi Suda in Chronic Respiratory Disease