

Summary
Podocytes, highly specialized epithelial cells, build the outer part of the kidney filtration barrier and withstand high mechanical forces through a complex network of cellular protrusions. Here, we show that Arp2/3-dependent actin polymerization controls actomyosin contractility and focal adhesion maturation of podocyte protrusions and thereby regulates formation, maintenance, and capacity to adapt to mechanical requirements of the filtration barrier. We find that N-WASP-Arp2/3 define the development of complex arborized podocyte protrusions in vitro and in vivo. Loss of dendritic actin networks results in a pronounced activation of the actomyosin cytoskeleton and the generation of over-maturated but less efficient adhesion, leading to detachment of podocytes. Our data provide a model to explain podocyte protrusion morphology and their mechanical stability based on a tripartite relationship between actin polymerization, contractility, and adhesion.
Keywords: N-WASP, Arp2/3, actin networks, actomyosin, focal adhesion, cell process generation, kidney filtration barrier, glomerular epithelial cells, podocytes
Graphical Abstract
Highlights
-
•
ARP3-dependent actin assembly is required for podocyte process formation
-
•
Arp2/3 thereby links process formation, podocyte adhesion and mechano-adaptation
-
•
Arp2/3 function is regulated by a reciprocal interplay with actomyosin
Glomerular podocytes maintain the kidney filtration barrier. Schell et al. show that the Arp2/3 machinery is required for podocyte process formation and adaptation to filtration barrier mechanical requirements via reciprocal interplay with the actomyosin network. Dendritic actin network loss results in actomyosin cytoskeleton activation and decreased adhesion, leading to podocyte detachment.
Introduction
Cellular protrusion formation is involved in many important biological functions ranging from tumor metastasis to embryogenesis (Ladoux et al., 2016, Bravo-Cordero et al., 2012, Hunter and Fernandez-Gonzalez, 2017, Krause and Gautreau, 2014). Among the best-studied cellular protrusions in 2D is the “classical” lamellipodium, which is generated in a well-coordinated interplay of polymerization, crosslinking and severing of actin filaments (Krause and Gautreau, 2014). Various protrusion types have been identified and differ not only in their molecular composition, but also in their context-dependent properties (Inagaki and Katsuno, 2017, Di Martino et al., 2016, Leijnse et al., 2015). However, not much is known about how extremely fine protrusions can produce outstanding mechanical durability while maintaining adaptive capacity in integrated tissues and organs.
Here, we focused on the podocyte, which is essentially involved in the formation of the kidney filtration barrier by enwrapping glomerular capillaries with its specialized cellular protrusions (namely, foot processes [FPs]; Pavenstädt et al., 2003). As podocyte FPs are required to maintain morphological stability and cell-matrix adherence against physical filtration forces, they serve as an ideal model to study the contribution of protrusion phenotypes and reciprocal cell-matrix interactions. Retraction and simplification of the complex podocyte FP network is often linked to detachment of podocytes from the glomerular basement membrane and presents a key feature of glomerular disease (Grahammer et al., 2013, Kriz et al., 2013). During glomerular development, podocytes undergo dramatic morphogenetic alterations, transforming from cuboidal epithelial cells into arborized podocytes with a network of highly branched FPs (Schell et al., 2014, Welsh and Saleem, 2011). Based on the identification of disease-causing mutations affecting core components of the actin cytoskeleton (e.g., ACTN4 or INF2), the concept of a tightly controlled cellular morphology for podocyte function has evolved (Brown et al., 2010, Kaplan et al., 2000). However, it is still incompletely understood how the podocyte process networks form, which molecular machineries are involved, and if different cytoskeletal pools exert specific functions.
Here, we show that the Arp2/3 complex controls the integrity of the kidney filtration barrier by influencing two distinct, yet interlinked processes. First, Arp2/3 complex-dependent actin polymerization critically determines the arborization and complexity level of podocyte FPs. Second, Arp2/3-controlled focal adhesion (FA) and traction forces influence podocyte adhesion. The Arp2/3 complex provides actin nucleation and filament-branching properties to generate highly branched actin networks, which are essential for the formation and propulsion of lamellipodia (Rotty et al., 2013). It was only recently demonstrated that Arp2/3 also influences the alignment of cell-matrix adhesions and thereby affects cell-matrix-dependent haptotaxis (Wu et al., 2012). In the classical model, Arp2/3-complex-mediated filament assembly is activated by nucleation-promoting factors (NPFs such as SCAR/WAVE and N-WASP) downstream of CDC42- and RAC1-dependent signaling axes (Campellone and Welch, 2010). Advanced models challenged this view, as they identified competition between the Arp2/3 complex and other nucleation machineries (such as the formin protein family) defining critical cellular responses (Lomakin et al., 2015, Wu et al., 2017). Despite many years of research on Arp2/3, little is known about its role in vivo beyond the importance for classical lamellipodia and adhesion structures.
Results
The Arp2/3 Complex Presents a Central Node in the Network of Cytoskeletal Proteins in Podocytes
Given the extensive characterization of the Arp2/3 complex in vitro, we reasoned that Arp2/3-dependent branched actin networks might be involved in process formation as well as FA initiation in glomerular epithelial cells. Podocytes derive from the metanephric mesenchyme (Schell et al., 2014) and exhibit a rather cuboidal morphology in early glomerular development. With ongoing maturation, cells shift their polarization pattern and extend numerous cellular protrusions (podocyte processes) toward the glomerular basement membrane (Figures 1A and 1B). These thin processes are interconnected with extensions from adjacent podocytes and provide anchorage to the glomerular basement membrane (protrusions directly evolving out of the cell body are termed primary processes, whereas further arborized and terminally branched protrusions are termed secondary, or FPs; Figures 1C and 1D). Podocytes reside on the outer surface of glomerular capillaries and are exposed to high physical filtration forces. Specialized adhesion machineries are required to prevent detachment of those cells into the urinary space. We hypothesized that the structure and composition of the actin cytoskeleton network might interconnect FP development with efficient adhesion regulation. To describe the composition of the podocyte cytoskeleton, we made use of our recently developed genetic-reporter-based protocol for highly efficient single-cell isolation and downstream mass spectrometry (MS) applications (Figure 1E). Based on our initial hypothesis, we aimed to identify proteins or complexes that are known to act at the interface of cellular protrusion formation and FA regulation. Here, GFP-labeled podocytes were isolated from genetic reporter strains and after fluorescence-activated cell sorting (FACS)-based enrichment, MS analysis was performed. Filtering this dataset for podocyte-enriched cytoskeletal components involved in actin binding, protrusion formation, and FAs resulted in a list of 114 proteins, where the N-WASP/Arp2/3 complex axis appeared as a central node (Figures 1F and S1; Table S1; comparing podocytes versus non-podocytes, i.e., mesangial and endothelial cells). Employing proteomics of enriched adhesion complexes (focal adhesome), cell fractionation protocols, as well as immunofluorescence, we could furthermore demonstrate that ARP3 is enriched in the adhesion complex of podocytes (Figures 1G–1J and S2; Table S2). These observations once more corroborated our initial hypothesis that the Arp2/3 complex might function at the interface of propulsive actin network generation and the FA complex regulation in podocytes. Using confocal microscopy on human kidney tissue, N-WASP and ARP3 were detected in the podocyte compartment (enriched in podocytes as indicated by co-labeling with the podocyte specific protein SYNPO; Figures 1K, 1L, and S2). Formation and generation of podocyte FPs occurs during glomerular maturation at the so-called capillary loop stage (Figure 1M). Immunofluorescence studies on different maturational stages of glomeruli revealed that ARP3 and N-WASP accumulated at the basal compartment of podocytes where FP specification and arborization occurs (Figures 1N and 1O). This localization pattern was also detected in completely maturated glomeruli of adult mice, where N-WASP except for glomeruli also showed minor accumulations in distal nephron parts (Figure S2). Together, these findings indicate a substantial requirement of N-WASP and ARP3 in the podocyte FP compartment not only during development but also in completely maturated glomeruli.
Figure 1.

The Arp2/3 Complex Presents a Central Node in the Network of Cytoskeletal Proteins in Podocytes
(A) Schematic depicting how podocytes reside on the outer surface of glomerular capillaries.
(B) During glomerular maturation podocytes undergo profound cell shape changes (GBM, glomerular basement membrane; FP, podocyte foot process).
(C) Colorized scanning electron microscopy (SEM) image of podocyte protrusions illustrating the complex interdigitating network of podocyte FPs covering glomerular capillaries.
(D) Transmission electron microscopy (TEM) demonstrates FPs attaching to the glomerular basement membrane (GBM; cap - glomerular capillary). Slits between individual FPs are bridged by specialized cell-cell contacts known as slit diaphragms.
(E) Schematic describing the strategy for isolation of genetically labeled primary podocytes and subsequent mass spectrometry (MS) analysis. Following single-cell isolation and further FACS-based purification, respective cell populations are analyzed.
(F) Proteome network analysis of podocyte-enriched proteins revealed high abundance of proteins contributing to distinct functional subsets such as actin binding or focal adhesion-associated proteins. Filtering of the enriched proteins for the indicated functional subsets confirmed components of the Arp2/3 complex as a central node (color coded: enrichment scores and functional groups are indicated in Table S1; proteins were defined as podocyte enriched by a detection ratio >2 for podocytes to non-podocyte glomerular cells).
(G) Focal adhesome proteomics generated out of immortalized human podocytes detected multiple members of the Arp2/3 complex as enriched components of the focal adhesome (FA) (enrichment scores are indicated in Table S2; proteins were hierarchically ranked according to their detection scores).
(H and I) Immunofluorescence of spreading primary wild-type podocytes revealed co-localization of ARP3 and the focal adhesion protein PAXILLIN during early spreading phases (white line indicates region for intensity line scan analysis; RFI, relative fluorescence intensity; FA, focal adhesion).
(J) Western blot analysis of focal adhesion (FA)-enriched cell fractions or total cell lysate (TCL) confirmed ARP3 as a FA-enriched protein in podocytes. PXN (Paxillin) and PDH (Pyruvate dehydrogenase E1) were used to demonstrate successful separation of the focal adhesion fraction from the whole cell compartment (see also Figure S2).
(K) Confocal laser scanning microscopy of human glomeruli confirmed distinct localization of the nucleation-promoting factor N-WASP in the podocyte compartment (dashed line boxes indicate areas of higher magnification inserts).
(L) A similar enrichment was also observed for ARP3 employing confocal laser scanning microscopy (inserts in (L) show different levels of z stack series 0.43 micrometers apart; dashed outlines indicate the podocyte compartment; synpatopodin (SYNPO) was used as a marker for the podocyte compartment).
(M) Schematic depicting stages of glomerular development (podocyte progenitors and podocytes are shown in green; endothelial cells and capillary loops are depicted in orange).
(N and O) At late capillary loop stage, ARP3 (N) and N-WASP (O) accumulated at the basal compartment of maturating podocytes, where podocyte FP formation takes place (in earlier developmental stages, this prominent accumulation was not apparent; dashed line boxes indicate inserts; arrows indicating the basal podocyte compartment; pod, podocyte; cap, capillary; NIDOGEN was employed as a marker of the glomerular basement membrane; mice glomeruli were analyzed at p0).
N-WASP and ARP3 Are a Prerequisite for Ordered Podocyte Development In Vivo
To test the relevance of N-WASP/Arp2/3-generated actin networks for the formation of podocyte FPs, we employed a set of conditional knockout mouse models. We previously observed that deletion of N-WASP using the podocyte-specific Nphs2Cre line resulted in a delayed onset of proteinuria, starting at 3 weeks after birth (Schell et al., 2013). It is known from earlier studies that the hNPHS2 promotor exerts activity starting at embryonic day E14.5 onward and specifically targets maturating podocytes at the late capillary loop stage (Moeller et al., 2002). Hence, complete and efficient deletion in early podocyte progenitors cannot be predicted. To circumvent potential compensatory actions of other actin NPFs, we employed the Six2Cre deleter strain (E11.5; Figure S3), which targets the whole nephron including podocyte progenitors from early nephron and glomerular maturation onward (Kobayashi et al., 2008). Here, we observed that loss of N-WASP resulted in conspicuous glomerular capillary aneurysms (Figures 2B–2G), a phenotype associated with disturbed podocyte process formation (Hartleben et al., 2013). The impact on the integrity of the kidney filtration barrier was marked as respective knockout animals exhibited proteinuria early after birth (Figure 2H). To assess the morphology of podocyte FPs, we employed electron microscopy and detected marked simplification of FP morphology in N-WASP∗Six2Cre knockout animals (Figures 2I–2K), indicating the prerequisite role for N-WASP in this morphogenetic process. Of note, primary processes appeared not to be affected. Aside from the impact of N-WASP deletion on the glomerular compartment, we observed significant reduction in kidney and body weight of respective knockout mice (Figure S3). This effect might be attributed to the deletion of N-WASP throughout the whole nephron (Figure S3, as previously shown [Reginensi et al., 2013]). To abolish Arp2/3 complex-mediated actin nucleation, the nucleation core component Arp3 was deleted by the use of the well-established Nphs2Cre line, which initiates recombination at the late capillary loop stage during glomerular development (Figures 2L and 2M). Loss of ARP3 in podocytes resulted in high levels of proteinuria already at birth, accompanied by decreased birth weight gain (Figures 2N–2P). This phenotype drastically progressed to chronic kidney disease characterized by glomerular sclerosis as well as overall reduced survival (Figures 2Q and S4). Remarkably, loss of ARP3 resulted in global simplification of podocyte FPs in a similar manner as loss of N-WASP, which we demonstrated by transmission electron microscopy (TEM) (Figures 2R and S4). Of note, primary processes were not obviously affected in terms of morphology and size (in line with our observations in the NWASP∗SixCre model). In addition, we also employed a recently established super resolution microscopy technique (Figures 2S–2U and S4) to visualize and quantitate FPs of wild-type and respective knockout animals (Siegerist et al., 2017). These studies corroborated our initial observation by TEM and overall support our initial hypothesis that propulsive actin networks, as provided by the N-WASP/Arp2/3 complex axis, are involved in the complex generation of podocyte FPs and accurate formation of the kidney filtration barrier. Of note, Arp3 knockout podocytes did not exhibit major differences in the expression of podocyte-specific proteins (Figure S4).
Figure 2.

N-WASP and ARP3 Are a Prerequisite for Ordered Podocyte Development In Vivo
(A) Schematic illustrating the crossing strategy for the generation of N-WASPfl/fl∗Six2Cre knockout mice: Cre recombination targets all tissues deriving from the metanephric mesenchyme, i.e., the whole nephron including podocytes.
(B–G) Histological evaluation revealed dilated and aneurysmal transformed glomerular capillaries indicating defective enclosing of podocyte foot processes ([B and D] glomeruli from control animals; [C and E] aneurysmatic capillaries in N-WASP∗Six2Cre knockout animals; red dotted lines highlight areas of dilated glomerular capillaries). Immunofluorescence for the podocyte marker NEPHRIN also demonstrated the defective invagination of podocytes toward the capillary compartment ([F] shows an example of a respective control animal, while in G impaired invagination in N-WASP∗SixCre knockout animals is shown; indicated by white arrows).
(H) Evaluation of albumin to creatinine ratio (mg/mg) detected proteinuria in respective knockout animals at p3 and p5 (at least 3 animals at each time point were analyzed; ∗∗∗∗p < 0.0001).
(I) Quantification of foot process (FP) width by TEM showed pronounced effacement and simplification in respective N-WASP KO animals (n = 3–4 animals were analyzed; ∗∗p < 0.01).
(J and K) Transmission electron microscopy of wild type (J) and of N-WASP KO (K) mice detected misconfigured podocyte FPs, without proper slit diaphragm junctions (yellow asterisks indicate misconfigured FPs; yellow arrows highlight SD junctions).
(L) Schematic depicting the crossing scheme for the generation of podocyte-specific Arp3 deletion using the Nphs2Cre line.
(M) Western blot of FACS-sorted primary podocyte confirmed loss of ARP3 in respective knockout cells (TUBA: alpha-tubulin was used as a loading control).
(N) Arp3 knockout animals showed shrunk and pale kidneys at p21.
(O) Body weight gain was decreased in Arp3 KO animals beginning with 3 weeks of age; respective heterozygous animals were unaffected (∗∗∗∗p < 0.0001).
(P) Measurement of albumin to creatinine ratio (mg/mg) detected increased levels of proteinuria in Arp3 knockout animals already at p1. Proteinuria persistently showed high levels for the whole observational period of 4 weeks (at least 6 animals per time point and genotype were analyzed; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
(Q) Kaplan-Meier analysis revealed that 50% of respective Arp3 knockout animals died within 30 days after birth.
(R) Quantification of FP width in Arp3 knockout animals revealed marked simplification and effacement (n = 3 animals per genotype were analyzed; ∗p < 0.05).
(S–U) Overview of 3D SIM microscopy in respective wild-type animals (S); single plane zoom-ins (T-1 for wild type animals, and T-2 for respective Arp3 KO animals) illustrate misconfigured filtration barrier morphology indicative of aberrant FP architecture. (U) Quantification of SIM data (measurements were accumulated over 2 individual animals per genotype; ∗∗∗p < 0.001). All data are represented as mean ± SEM.
ARP3-Mediated Actin Polymerization Is Required for Efficient Protrusion Formation
Podocyte FPs are specialized cellular protrusions, which interconnect adjacent podocytes to each other and enclose glomerular capillaries (Figures 1A–1D). We devised a simplified in vitro model to assess protrusion formation of primary podocytes (Figures 3A and 3B) and observed that finely arborized protrusions are mainly F-actin based as their in vivo counterparts (Suleiman et al., 2017). Blockage of actin polymerization using latrunculin resulted in a marked reduction of generated protrusions (Figures 3C and 3D). In order to establish a primary cell culture system of high purity, we made use of a genetically encoded reporter system (Figure 3E). Loss of N-WASP or ARP3 resulted in a marked decrease of protrusion formation under 3D-culture conditions (Figures 3F–3H and S4). When we classified the morphology of respective protrusions (Figure 3I), we found that wild-type podocytes produced a higher level of complexity in terms of terminally branched protrusions compared to either N-WASP or Arp3 primary knockout podocytes (Figure 3J). This inability for efficient and complex protrusion formation was also underlined by an overall decrease in length of those cellular protrusions (Figure 3K). Altogether, loss of ARP3 and N-WASP in primary podocytes resulted in overall shorter and less arborized protrusions. These findings were coupled by alteration in the structure of the F-actin cytoskeleton characterized by decreased levels of cortical actin and diffuse accumulation of coarse cytoplasmic actin speckles in Arp3 knockout podocytes (Figures 3L–3O). Similar findings of defective lamellipodia protrusion formation were also observed in 2D-culture environments in respective Arp3 knockout podocytes (Figure S4). Morphological assessment of FP structure and complex arborization levels in Arp3 knockout animals additionally revealed an overall simplification of FPs in vivo, paralleling our in vitro observations using respective primary podocytes in 3D-culture conditions (Figures 3P, 3Q, and 2R–2U). Together, the N-WASP/Arp2/3 axis appears to act as a prerequisite for efficient and complex arborized cellular protrusion formation of podocytes in vitro and FP formation in vivo (in the following the term protrusion will be used for 3D cellular extensions in vitro, and the term FPs for the in vivo situation).
Figure 3.

ARP3-Mediated Actin Polymerization Is Required for Efficient Protrusion Formation
(A and B) Primary podocytes in 3D culture conditions form protrusions with increasing levels of branching; immunofluorescence staining revealed that terminally branched protrusions consisted mainly of filamentous actin, whereas major protrusions also showed positivity for tubulin (B). (C) Acute treatment with the actin polymerization inhibitor Latrunculin A markedly suppressed protrusion formation, as shown by a quantifcation for main protrusions (D) (n = 38–41 analyzed cells; ∗∗∗∗p < 0.0001).
(E) Schematic depicting the generation of a fluorescence-based reporter line: after podocyte-specific Cre-recombination mGFP is selectively expressed in only the glomerular epithelial cell compartment. Single-cell isolation and further FACS-based purification results in a primary culture system with proven genetic origin.
(F and G) 3D reconstruction of primary podocytes in 3D culture systems (wild typ podocytes [F]) unmasked the deficiency of Arp3 knockout podocytes (G) to form terminally branched protrusions.
(H) Color code describing segmentation of individual protrusion classes into main, primary, and secondary order protrusions.
(I) Loss of ARP3 and N-WASP resulted in significantly less cells with branching protrusion phenotypes (at least 150 cells per condition were analyzed, and data were accumulated over at least 3 experiments; n.s., non significant, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
(J and K) Reconstructed images of individual z stacks were used as a basis for the segmentation analysis (J) and length measurements of protrusions (K): due to ARP3 and N-WASP deficiency, higher order branched protrusions were diminished and apparent protrusions were significantly shorter compared to control conditions (17 wild-type, 32 Arp3 KO, and 20 N-WASP KO primary podocytes were reconstructed and analyzed; n.s., non significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
(L–O) Analysis of the cortical actin cytoskeleton of wild-type (L) and respective KO (M) cells under 3D culture conditions. Representative line scans (N) of filamentous actin intensities in single planes demonstrated altered levels of cortical actin due to loss of ARP3 (19 wild-type and 16 Arp3 KO cells were reconstructed, and ratios between cortical and cytoplasmic actin intensities were measured at multiple localizations (O); orange dashed lines indicate areas of line scan analysis, black arrows mark individual representative cell borders; ∗∗∗∗p < 0.0001; MFI, mean fluorescence intensity).
(P) Scanning electron microscopy (SEM) detected pronounced simplification, reduced branching and misconfiguration of podocyte processes in Arp3 KO ((P-1) shows a representative image of wild type controls, whereas (P-2) and (P-3) demonstrate representative images from Arp3 KO animals; colorized SEM image (P-3) depicting segmentation of podocyte processes, each colorized dashed line indicates one individual major process; white arrows and red asterisks mark secondary processes). (Q) Podocyte process branching was quantified from semi-planar SEM areas (n = 5 wild-type and 4 Arp3 KO mice were analyzed at p8; ∗∗∗∗p < 0.0001). All data are represented as mean ± SEM.
Loss of ARP3 Results in Altered Focal Adhesion Morphology and Increased Actomyosin Activity
As efficient protrusion formation also involves interaction of cells with their surrounding extracellular matrix (ECM), we aimed to analyze how the N-WASP/Arp2/3 axis might affect FA function and respective morphology. Here, we observed that loss of ARP3 but not of N-WASP in primary podocytes results in the formation of FAs with distorted morphology and a plaque-like appearance. These FAs were linked to each other via dense F-actin webs (Figures 4A–4F and S4). The overall number of FA sites as well as their composition appeared not to be altered in Arp3 knockout cells, but the morphology was shifted toward plaque-like FA sites at the expense of nascent adhesions (Figures 4G, 4H, S4, and S5). These plaque-like FAs displayed an increased density and altered localization pattern of FA proteins such as PAXILLIN (Figures 4I and 4J). Remarkably, primary N-WASP knockout cells exhibited an unaltered FA morphology and distribution pattern, accompanied by prominent lamellipodial protrusions (Figures 4E, 4F, and S5). This difference between protrusion deficiency in 3D-culture conditions (N-WASP and ARP3) and FA morphology (only ARP3) might be explained by compensatory upstream NPFs in 2D conditions. Indeed, the NPF WAVE2 was detected at the lamellipodial leading edge of WT and N-WASP KO primary podocytes. Inhibition of the RAC1/WAVE2/Arp2/3 signaling axis in N-WASP KO podocytes resulted in a phenocopy of the distorted FA morphology of ARP3 knockout podocytes, supporting a compensatory concept of NPFs in a contextual manner (Figure S5). As FA function is directly linked to tensional control of the actomyosin cytoskeleton, we assessed the distribution of non-muscle myosin II (NMHC-IIA) and actinin-4 (ACTN4) in Arp3 wild-type versus knockout cells (Figures 4K, 4L, and S5). Here, we observed sarcomere-like distribution patterns for both proteins in Arp3 knockout cells, suggestive for increased inherent tension levels. We correlated phosphorylated levels of myosin-light-chain (MLC) with respective genotypes and observed an increased activity and circular distribution of activated actomyosin cytoskeleton in Arp3 knockout podocytes (Figures 4M–4O), whereas a more longitudinal pattern for pp-MLC was detected in wild-type cells. The individual levels of pp-MLC also appeared to be modulated by the substratum stiffness, as knockout cells exhibited even higher pp-MLC levels on softer surfaces when compared to control cells (Figure 4P). Similar observations were also made for mechanical tension markers pp-MLC and YAP on glomerular sections in vivo (Figures 4Q–4T and S5). These finding together indicate that a loss of ARP3-mediated actin branching results in increased intracellular tension linked to elevated levels of pp-MLC and nuclear translocation of YAP in podocytes in vivo. Our in vitro and in vivo results imply an interdependent crosstalk between the podocyte actin cytoskeleton composition and inherent intracellular tension levels, as provided by the actomyosin cytoskeleton.
Figure 4.

Loss of ARP3 Results in Altered Focal Adhesion Morphology and Increased Actomyosin Activity
(A and B) Staining for the focal adhesion (FA) component PAXILLIN (red) revealed confluent FA clusters in primary Arp3 KO podocytes (B-1), which were connected via filamentous actin webs (wild type (A) and KO (B)). F-actin (blue) was stained by Phalloidin.
(C and D) Quantification of average focal adhesion area (C) and major axis (D) showed increased values for both parameters in respective KO cells (25 wild-type and 28 Arp3 KO cells were analyzed; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
(E and F) Average focal adhesion area (E) and lamellipodium formation (F) was not obviously impaired in primary N-WASP KO podocytes in 2D culture conditions (n = 36 cells per genotype were analyzed, n.s., non significant; FA were stained by PAXILLIN and F-actin by phalloidin; white arrows indicate lamellipodia).
(G) Analysis of focal adhesion clusters due to their individual size indicated a shift toward enlarged adhesion clusters in Arp3 KO podocytes (25 WT and 26 Arp3 KO cells were analyzed; ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
(H) This shift in FA size was paralleled by a change in the distribution of individual FA classes: loss of Arp3 resulted in the accumulation of FA plaques (n = 18 cells per genotype were analyzed; n.s., non significant, ∗∗p < 0.01).
(I and J) Analysis of PAXILLIN fluorescence intensities (FI) in wild type podocytes (I-1) and in respective Arp3 KO cells (I-2) revealed altered intensity patterns and increased mean PAXILLIN intensity (J) per focal adhesion (n = 100 mature FA per genotype were analyzed; ∗∗∗∗p < 0.0001).
(K and L) Immunofluorescence for NMHC-IIA (K) demonstrated an increased sarcomere-like pattern of actin fibers (L) in respective Arp3 KO podocytes (yellow lines indicate positions for line scan measurements, and black arrows indicate myosin-2 peaks).
(M) Immunofluorescence staining for pp-MLC and the focal adhesion marker PAXILLIN illustrated prominent, fiber-like accumulations of pp-MLC linked to FAs in Arp3 KO podocytes.
(N) Staining for pp-MLC revealed increased MLC activation levels in Arp3 KO podocytes, condensed in arc regions between pseudopodial protrusions. Also a ring-like distribution was observed in the majority of Arp3 KO podocytes. (Yellow lines indicate representative positions for line scan measurements).
(O) Line scan profiles for pp-MLC showed highest activation levels close to the cell edge in Arp3 KO cells (values represent mean intensities of 6 cells per genotype; gray error bars represent SEM).
(P) Quantification of fluorescence intensity for pp-MLC on either glass or soft gels revealed higher MLC activation levels for Arp3 KO podocytes (37 wild-type and 41 Arp3 KO cells on glass; 31 wild-type and 32 Arp3 KO cells on gels were analyzed; ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
(Q and R) Staining for pp-MLC in glomeruli from either wild-type (Q-1) or Arp3 KO (Q-2) animals showed high levels of phosphorylated MLC in the podocyte compartment, as visualized with the podocyte-specific marker NEPHRIN (mean podocyte pp-MLC intensity of n = 4 WT and 4 KO animals were statistically analyzed; ∗p < 0.05; the mean pp-MLC intensity per animal was calculated from mean pp-MLC intensity of the segmented podocyte compartment of at least 20 glomeruli per animal).
(S and T) Immunofluorescence staining for the mechanical tension marker YAP in glomeruli from either wild-type or Arp3 KO (S) animals showed increased percentages of YAP-positive podocyte nuclei in KO animals (T). Podocyte nuclei were visualized by the podocyte-specific marker WT1 (the mean percentage of YAP-positive podocyte nuclei per glomerulus of n = 5 WT and 5 KO animals were analyzed; white arrow bars indicate podocyte nuclei with co-occurring positivity for YAP, ∗∗p < 0.01; the percentage of YAP positive podocyte nuclei per glomerulus per animal was calculated from at least 20 glomeruli per animal). All data are represented as mean ± SEM.
Competition between Different F-Actin Networks Modulates Protrusion Phenotypes and FA Morphology of Podocytes
Next, we addressed the question of whether and how Arp2/3-dependent actin networks compete with actomyosin-mediated contractility and how this interplay might translate into modulation of essential parameters such as protrusion formation and cell-matrix interaction. First, Arp3 as well as N-WASP knockout cells were seeded in 3D matrices and pretreated with blebbistatin to assess protrusion formation and their branching complexity. In both conditions, inhibition of myosin-2 resulted in a mitigation of the protrusion defect (Figures 5A, 5B, and S5). Similar results were also observed upon blockage of the Arp2/3 complex using specific inhibitory compounds in primary cells and to some extent in tissue culture approach (Figure S5). The Arp2/3 complex differs in its mode of actin polymerization from other actin nucleators (such as formins) by providing a branched actin network instead of only linearly polymerized actin filaments (Campellone and Welch, 2010). To elucidate the minimal requirement for actin-dependent protrusion formation in our system, we inhibited formin function and observed a phenocopy in terms of defective protrusion formation. In contrast, co-treatment with the myosin-2 inhibitor blebbistatin could not revert or modify this phenotype as observed for Arp3 or N-WASP knockout cells (Figure 5B). Interestingly, the branching level of protrusions (ratio of major to minor protrusions) in terms of arborization correlated to the presence or absence of ARP3, indicating that formin-mediated actin polymerization appears as a prerequisite for protrusion formation, whereas Arp2/3 predominantly determines the level of terminally branched protrusions and influences myosin-2 tension (Figures 5B and 5C).
Figure 5.

Competition between Different F-Actin Networks Modulates Protrusion Phenotypes and FA Morphology of Podocytes
(A and B) Decrease of intracellular tension in Arp3 KO primary podocytes with the myosin-2 inhibitor blebbistatin (Bleb) resulted in the formation of simplified cellular protrusions compared to blebbistatin-treated WT cells (A). Cell morphology and thereby protrusion generation was quantified by circularity scores (B). Quantification showed that co-treatment with blebbistatin results in a decrease of the circularity score in Arp3 as well as N-WASP KO podocytes, reflecting a more protrusive phenotype. Suppression of protrusion formation by SMIFH2-mediated formin inhibition is resistant to co-treatment with blebbistatin. (44 DMSO and 43 blebbistatin treated wild-type cells; 73 DMSO and 70 blebbistatin treated Arp3 KO cells; 50 DMSO and 53 blebbistatin treated N-WASP KO cells; 70 SMIFH2+DMSO and 100 SMIFH2+blebbistatin treated wild-type cells were analyzed; n.s., non significant, ∗∗∗∗p < 0.0001).
(C) Definition of a branching index (major protrusion per end tips (minor protrusions) allowed a more detailed analysis of protrusion complexity and quality. Quantification for this branching index revealed simplified cellular protrusions with reduced branching levels for blebbistatin treated Arp3 KO cell podocytes compared to blebbistatin treated WT cells. (35 WT and 36 Arp3 KO cells were analyzed; ∗∗∗∗p < 0.0001).
(D–J) Inhibition of myosin-2 via blebbistatin resulted in a dose-dependent decrease in focal adhesion (FA) size in Arp3 knockout podocytes (WT - (D-F) and Arp3 KO - (G-I)), indicating that FA plaques represent myosin-2-dependent structures in these cells. Quantification of FA area was performed ([J]; white arrows indicating FA sites; 20 cells per condition were analyzed; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
(K) Correlation of average focal adhesion (FA) size to the level of lamellipodium formation shows an inverse correlation between these cellular processes, indicating also an inverse correlation between Arp2/3 and (acto-) myosin-2 activity as an underlying mechanism. Calyculin-A and blebbistatin were used, respectively, to either activate or inhibit myosin-2 activity. Rac1 inhibition by NSC-23766 or CK-869 was used to inhibit Arp2/3-mediated actin nucleation; Nocodazol was used to stabilize focal adhesions (FA) in a myosin-independent fashion. (24 to 44 immortalized human podocytes were analyzed per condition; ∗p < 0.05, ∗∗p < 0.01; ∗∗∗∗p > 0.0001).
(L–P) Treatment with the formin inhibitor SMIFH2 resulted in a dissolution of mature FAs, which was accompanied by an increase in the number of nascent adhesions in wild-type cells (M) and a loss of focal adhesion formation in Arp3 KO podocytes (N) (compared to untreated Arp3 KO cells - (L); n.s., non significant; ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). (O) Focal adhesion area and (P) FA number were quantified.
(Q and R) Washout experiments employing cytochalasin-D and co-treatments with inhibitors for myosin-2 activity (blebbistatin), formin nucleation (SMIFH2), or Arp2/3 nucleation (CK-869) revealed specific actin network activities as a prerequisite for the inverse correlation between dynamic focal adhesion maturation (Q) and cell size spreading (R) of podocytes (31–104 cells per condition were analyzed for FA maturation and 40–215 cells were analyzed for cell size; ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p > 0.0001). All data are represented as mean ± SEM.
As a loss of ARP3 resulted in the formation of FA plaques (Figures 4A–4D), we reasoned that increased actomyosin contractility might modulate this phenotype. In fact, inhibition of myosin-2 with blebbistatin significantly reversed the formation of FA plaques in a dose-dependent manner and also mitigated migratory deficits of Arp3 knockout cells (Figures 5D–5J and S6). To exclude at this point that the FA-plaque phenotype is rather a result from generally inhibited actin polymerization, we tested a series of different Arp2/3 inhibitors as well as other chemical compounds affecting different modes of actin polymerization. Interestingly, we detected predominant formation of FA plaques only in Arp2/3-inhibited cells, underlining the specificity of branched actin networks involved in the generation of the FA-plaque phenotype (Figure S6). These observations were corroborated by complementary experiments where actomyosin contractility was titrated either by treatment with the phosphatase-inhibitor calyculin-A or inhibited with blebbistatin in wild-type cells (Figures 5K and S6). Moreover, employing inhibitors for RAC1 (as an established upstream regulator) or the Arp2/3 complex itself revealed that there is an inverse correlation between Arp2/3-dependent lamellipodia formation and FA size (Figure 5K). At this point, we asked whether and how a competitive interplay between the Arp2/3 complex and formin type actin nucleation might influence FA morphology. Inhibition of formins resulted in a significant dissolution of FA plaques in Arp3 knockout cells. Interestingly, a similar response was observed in wild-type cells but was accompanied by a pronounced increase in numbers of nascent FAs (Figures 5L–5P). These findings indicate that formin type nucleation machinery and the Arp2/3 complex feed into different FA types via generation of distinct actin networks. To further extend these observations, we performed washout and co-treatment experiments after dissolution of the actin cytoskeleton with cytochalasin D treatment. Here, control cells showed a time-dependent expansion in cell size accompanied by increasing numbers of FAs (Figures 5Q and 5R). In contrast, blockage of the Arp2/3 complex, myosin-2, or formins revealed again an inverse correlation of cell size expansion and assembly of FAs. Altogether, these observations suggest that an interplay between the formin type nucleation machinery, the Arp2/3 complex, and myosin-2 activity critically defines cellular protrusion formation and at the same time influences the assembly rate as well as maturational level of cell-matrix adhesions.
Loss of ARP3 Leads to Decreased Adhesive Properties of Podocytes under Mechanical Stress Conditions
Up to this point, we established a central role for Arp2/3 in podocytes linking protrusion formation, actomyosin tension, and FA maturation. However, the in vitro experiments were performed under static conditions. Podocytes reside on the outer surface of glomerular capillaries and are therefore exposed to high levels of physical filtration forces (Endlich and Endlich, 2006). To simulate this continuous physical stress, we cultured primary knockout podocytes under cyclic-isometric stress conditions (Figure 6A). Interestingly, this intervention led to a rearrangement in the F-actin cytoskeleton of wild-type cells characterized by an increased density of ventral stress fibers, whereas Arp3 knockout cells did not show an adaptive response (Figures 6B–6F and S6). Of note, wild-type cells still showed a lower stress fiber density compared to Arp3 knockout cells. In addition, FA morphology showed an adaptive change in wild-type cells under stress conditions while in ARP3-deficient cells, FA plaques remained stable and appeared unaffected (Figures 6G–6L). Together with our previous observations of increased actomyosin cytoskeleton activity, we wanted to directly test exerted traction forces of Arp3 knockout cells. Here, we observed that primary podocytes deficient in ARP3 exerted significantly higher traction forces on the underlying substratum paralleling the prior results of increased actomyosin activity in vitro and in vivo (Figures 6M–6O and 4N–4T). Surprisingly, we detected an increased tendency of detachment of Arp2/3-inhibited podocytes under mechanical stress conditions (Figures 6P, 6Q, and S6). Altogether, those observations suggested that a loss of Arp2/3 function results in a hyper-contractile status essentially impairing adaptive capacity in situations of continous mechano-physical stress conditions.
Figure 6.

Loss of ARP3 Leads to Decreased Adhesive Properties of Podocytes under Mechanical Stress Conditions
(A) Schematic illustrating the application of cyclic stretch on primary podocytes by culture on a flexible membrane.
(B–E) Immunofluorescence staining of the actin cytoskeleton in wild-type (B and C) and knockout podocytes (D and E) analyzed under non-stressed and stressed conditions, respectively.
(F) Quantification of stress fibers under stressed conditions revealed an adaptive deficit of actin filaments in Arp3 knockout podocytes when compared to wild-type controls (at least 50 cells per condition were analyzed; n.s., non significant, ∗∗p < 0.01;∗∗∗∗p < 0.0001).
(G–L) Analysis of focal adhesion morphology ((G and H) depict wild type podocyte FAs; (I and J) depict Arp3 KO podocytes) under stressed conditions indicated an increase in focal adhesion size in wild-type cells ((K) - FA area quantification and (I) - FA major axis quantification for each condition), whereas no changes were observed in respective knockout podocytes (at least 11 cells per condition and genotype, n.s., non significant, ∗p < 0.05; ∗∗p < 0.01).
(M and N) Quantification of traction force microscopy (M) on collagen-coated soft gel matrices showed increased traction forces in Arp3 KO podocyte and increased strain energy (N) (22 WT and 32 KO cells, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
(O) Pseudo-colored force maps of wild-type and Arp3 KO podocytes on soft matrices (note difference in individual traction scale between wild-type and KO cells).
(P) Immunofluorescence staining of Arp2/3 inhibited podocytes under stressed conditions. (Q) Quantification of adherent cells under stressed conditions (n = 3 individual experiments; ∗∗∗p < 0.001). All data are represented as mean ± SEM.
ARP3 Is Required for Efficient Podocyte Adhesion and Maintenance of Foot Process Morphology In Vivo
To transfer these observations to the in vivo setting, we analyzed our in vivo models for signs of podocyte detachment and could detect loss of WT1-positive cells from the glomerulus into the urine of respective Arp3 knockout animals (Figures 7A–7D). To address the question, whether these mechanisms are not only required during glomerular maturation but might also be relevant to maintenance conditions in fully developed podocytes, we generated an inducible knockout model for Arp3 specifically in podocytes (Figure 7E). Here, we observed a pronounced reduction of podocytes per glomerulus indicative of podocyte detachment (Figures 7F and 7G), paralleled by increasing levels of proteinuria and glomerular sclerosis (Figures 7H and 7I). Assessment of podocyte FP morphology employing scanning electron microscopy as well as super-resolution microscopy revealed marked simplification of podocyte FPs (Figures 7J–7L and S6). Together, these experiments in vitro and in vivo support the concept that ARP3-dependent actin networks not only determine the complexity level of podocyte protrusions (in vitro) or FPs (in vivo) but also are required for the establishment of efficient and mechano-adaptive podocyte adhesion properties under conditions of physical stress (Figures 6 and 7M–7O).
Figure 7.

ARP3 Is Required for Efficient Podocyte Adhesion and Maintenance of Foot Process Morphology
(A and B) Immunfluorescence for WT1-positive podocytes in wild type and Arp3 knockout animals.
(C) Quantification of podocyte number per glomerulus a pronounced decrease in conditional Arp3 knockout animals, indicating significant detachment of podocytes (at least 106 glomeruli of 3 mice per genotype and age; n.s., non significant, ∗∗∗∗p < 0.0001).
(D) Western blot for WT-1 positive cells in urine detected only detached podocytes in respective knockout animals.
(E) Schematic illustrating the crossing strategy for the generation of tetracycline inducible Arp3fl/fl∗hNphs2∗rtTA∗TetOCre knockout animals.
(F) Immunofluorescence for WT1-positive podocytes in Arp3 inducible knockout animals.
(G) Quantification of podocyte numbers in inducible Arp3 knockout animals.
(H) Albumin to creatinine ratio (mg/mg) measurements detected increased levels of proteinuria already in the second week of doxycycline induction: levels increased furthermore over a period of 4 weeks (3–11 animals per time point and genotype were analyzed; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
(I) PAS staining demonstrated focal segmental glomerulosclerosis in inducible Arp3 KO animals.
(J) Scanning electron microscopy demonstrates simplified FP morphology in Arp3 inducible knockout animals.
(K and L) SIM revealed aberrant kidney filtration morphology correlating to simplified FPs in Arp3 inducible knockout animals (measurements were accumulated over 2 individual animals per genotype; ∗∗∗p < 0.001).
(M–O) Graphical summary of proposed Arp2/3-mediated functions in podocytes (M): nucleation of dendritic actin networks by the Arp2/3 complex promotes FA assembly at cell protrusion sites and controls FA maturation by limiting non-muscle myosin II (NM-II) complex activity (N). (O) Arp2/3 allows the development of podocyte protrusions by modulating NM-II activity and directly promotes complex arborization of podocyte protrusions in vitro and in vivo. All data are represented as mean ± SEM.
Discussion
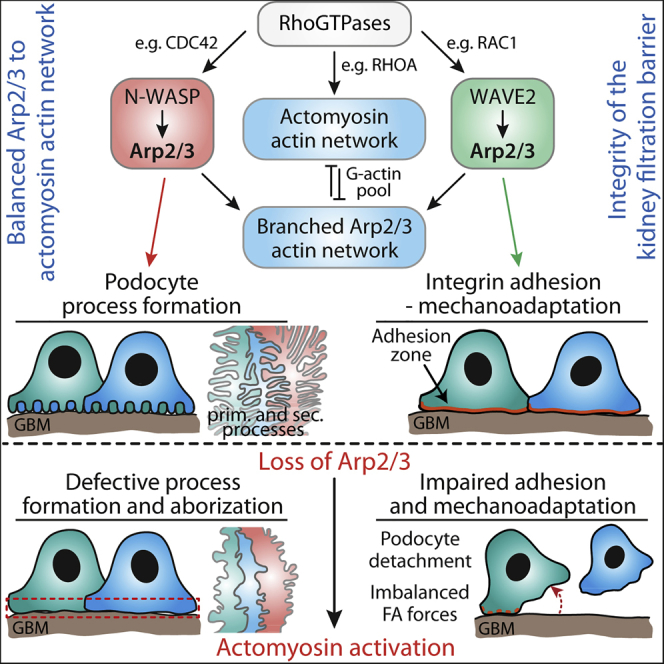
The complex cellular architecture of glomerular epithelial cells (namely podocytes), consisting of primary and secondary processes, is intimately linked with their function in providing the integrity of the kidney filtration barrier. Despite significant progress in podocyte biology research, it is still incompletely understood how those cellular protrusions are developed and maintained during later life (Schell et al., 2014, Pavenstädt et al., 2003, Liapis et al., 2013). Here, we employed in vitro and in vivo models to understand the contribution of Arp2/3-generated actin networks to control podocyte process networks. The Arp2/3 complex is an extensively in vitro characterized molecular machinery generating propulsive actin polymerization and branching, which is involved in diverse cellular processes such as migration or endocytosis (Swaney and Li, 2016, Soderling, 2009). However, it is still poorly understood which specific role this complex exerts in different biological contexts and cell types in vivo. Remarkably, we observed that the Arp2/3 complex influences not only the complexity level of cellular protrusions of podocytes (in vitro and in vivo) but also modulates their mechano-physical adaptive capacity under dynamic stress conditions.
Based on the identification of causative mutations for genetic podocyte diseases, previous work showed the relevance of certain actin-cytoskeleton related proteins (Perico et al., 2016). Based on 2D studies with cultured cells, it is well established that lamellipodial protrusions depend on propulsive dendritic actin meshworks generated by the Arp2/3 complex (Bisi et al., 2013, Dang et al., 2013). This mode of actin nucleation also seems to be required for the formation of more complex cellular protrusions in vivo, like it was recently shown for neuronal dendrites and oligodendrocytes (Zuchero et al., 2015, Kim et al., 2015, Kim et al., 2013). Given their highly arborized cellular protrusions, podocytes have several morphological features with these cells in common. However, biological function and exposure to physical forces differ strongly between these cell types (e.g., exposure to filtration forces in the case of podocytes), suggesting cell-specific mechanisms for protrusion formation and maintenance. Employing in vitro as well as in vivo models, we could delineate how N-WASP and the Arp2/3 complex influence the morphology and architecture of podocyte FPs (Figures 1, 2, and 3). Based on our in vivo observations, the onset and manifestation of phenotypes (comparing Arp3 and N-WASP podocyte-specific deletion) might be explained by the upstream-regulatory role of N-WASP and potential compensatory NPFs (e.g., WAVE2; as shown in Figure S5). Remarkably, we observed an indispensable role of formin-mediated actin polymerization in phases of initial 3D protrusion formation, whereas Arp2/3-dependent processes are mainly driving the complexity level of generated protrusions (Figures 2, 3, and 5). Besides established models for F-actin network modulation (such as Rho-GTPase signaling; Figures 7 and S7), recent reports proposed an additional model where different actin polymerization machineries are in competition for limited G-actin pools (Burke et al., 2014, Suarez et al., 2015, Rotty et al., 2015, Suarez and Kovar, 2016). In line with this, we identified formin-mediated actin polymerization as a competing factor in this network, modulating podocyte protrusion as well as FA formation (Figure 4). To date, the atypical formin INF2 is the best characterized formin family member in the context of podocytes based on the identification of INF2 disease-causing mutations (Brown et al., 2010). Previously, INF2 mutations were characterized to affect the interaction with mDIA, subsequently leading to altered RhoA signaling (Sun et al., 2011). Remarkably, knockin mice bearing disease-causing INF2 mutations do not show any obvious phenotype under basal conditions, in particular normal podocyte FP morphology was reported (Subramanian et al., 2016). However, stressing this system resulted in the collapse of podocyte FP architecture, highlighting the concept of balanced actin cytoskeleton pools to maintain podocyte FP morphology. Based on our results, it may be hypothesized that deletion of formins in vivo might result in a predominant defect of primary process formation (although, to our knowledge, there are no systematic studies published focusing on formin deletion in vivo). Consistently, the correlation of our in vitro and in vivo findings indicate that Arp2/3-controlled process formation predominantly affects most terminal processes instead of primary processes or protrusions (Figure 2). Together, these findings suggest that Arp2/3-mediated actin polymerization is essentially involved in critical morphogenetic programs as observed during the complex process of glomerular maturation, leading from only cuboidal epithelial cells to highly arborized podocytes (Figures 1 and 7).
Aside from the formation of complex, arborized networks of interdigitating FPs, podocytes are required to withstand high levels of physical forces given their extreme anatomical position on the surface of glomerular capillaries (Endlich and Endlich, 2006). The relevance of podocyte adhesion is exemplified by the identification of disease-causing mutations affecting certain integrin subunits as well as a series of genetic models targeting different components of the cell-matrix adhesion machinery (Has et al., 2012, Sachs et al., 2006, Pozzi et al., 2008). However, the complex and reciprocal interplay between cell-matrix adhesion and inherent contractility control is still unclear. Here, we demonstrated a high abundance of Arp2/3 complex components in the focal adhesome of podocytes in vivo and in vitro (Figure 1), highlighting a close link between this cytoskeletal machinery and the cell-adhesion nexus. Only very recently, it was shown that the Arp2/3 complex is influencing the alignment of FA complexes in a migration mode termed as haptotaxis (Wu et al., 2012), and it was demonstrated that ARP3 is interacting with FA components such as VINCULIN and FAK (Chorev et al., 2014, Serrels et al., 2007). Furthermore, we observed the formation of large FA plaques at the expense of smaller nascent FAs in Arp3 knockout cells (Figure 4). Based on biophysical models, retrograde actin flux due to Arp2/3-generated propulsive actin polymerization presents a critical factor in the maturation cycle of FAs by initiating nascent adhesions (Choi et al., 2008, Wu et al., 2017, Wu et al., 2012). In this context, it is of interest that Arp3 knockout cells were able to initialize nascent FAs but presented with hyper-maturated, plaque-like FAs (Figure 4). These observations indicate that the Arp2/3 complex might not be solely required for the initialization of nascent FAs but also for later maturational processes and, thereby, might influence the turnover of FAs (Wu et al., 2012). Remarkably, ARP3-deficient cells exhibited concomitantly an activated actomyosin cytoskeleton and exerted high traction forces (Figures 3, 4, and 6). We reasoned that loss of branched actin networks provokes increased inherent tension levels, and indeed lowering the activity of myosin-2 resulted in a reversal or at least mitigation of observed phenotypes such as FA plaques or impaired 3D protrusion formation (Figure 4). Besides established models for F-actin network modulation (such as Rho-GTPase signaling), recent reports proposed an additional model where different actin polymerization machineries are in competition for limited G-actin pools (Figures 7 and S7) (Leijnse et al., 2015, Liapis et al., 2013, Lomakin et al., 2015). In line with this, we identified formin-mediated actin polymerization as a competing factor in this network, modulating podocyte protrusion as well as FA formation (Figure 4).
By combining our in vitro and in vivo observations we demonstrated that ARP3-deficient cells show only impaired adhesive properties under dynamic stress conditions, despite large FA plaques and increased actomyosin activity (Figures 4 and 6). These findings indicate that a loss of branched actin networks not only results in a compensatory prevalence of formin-mediated actin polymerization but also might impede the adaptive capacity of podocytes in situations of continuous mechano-physical stress. A similar model was recently proposed for disease-causing mutations in the ACTN4 gene, and we could also establish a reciprocal interplay between podocyte FA proteins and actomyosin activity (Schell et al., 2017, Feng et al., 2018).
The concept of a tripartite relationship between FA maturation status, traction force, and ECM linkage/sensing suggests a model for FP effacement and consecutive podocyte detachment. Altering one component (e.g., impairing branched actin networks) will result in a compensatory response (increased tension, altered FA morphology), which might transiently tighten adherence toward the glomerular basement membrane, but ultimately impedes required mechanical adaptive plasticity in conditions of physical stress.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit ACTN4 | Abcam | Cat# ab108198; RRID: AB_10858236 |
| Rabbit ARP3 | Abcam | ab151729 |
| Rabbit CORTACTIN | Cell Signaling | Cat# 3503; RRID: AB_2115160 |
| Rabbit ITGB1 | Santa Cruz | Cat# M-106; RRID: AB_2130101 |
| Guinea pig NEPHRIN | Progene | Cat# Gp-N2 |
| Rat Nidogen (ELM1) | Nordic-MUbio | Cat# X1778M |
| Rabbit NMHC-IIA | Covance | Cat# PRB-440P; RRID: AB_291638 |
| Rabbit NWASP | Cell Signaling | Cat# 4848S; RRID: AB_10694415 |
| Mouse PAXILLIN | BD Transduction Lab. | Cat# 610051 |
| Mouse PDH E2/E3 | Abcam | Cat# ab110333; RRID: AB_10862029 |
| Rabbit P-MLC | Cell Signaling | Cat# 3671/3674; RRID: AB_2147464 |
| Rabbit PODOCIN | Santa Cruz | Cat# P0372; RRID: AB_2267439 |
| Mouse SYNPO | Progene | Cat# 65194 |
| Mouse Tubulin | Sigma | N/A |
| Mouse Vinculin | Abcam | Cat# SPM227; RRID: AB_883461 |
| Rabbit WAVE2 (WASF2) | Atlas Antibodies | Cat# HPA045288; RRID: AB_2679287 |
| Mouse WT-1 | Millipore | Cat# 05-753-clone 6F-H2; RRID: AB_309966 |
| Rabbit YAP | Cell Signaling | Cat# 14074S; RRID: AB_2218911 |
| Rabbit ZYXIN | Atlas Antibodies | Cat# HPA004835; RRID: AB_1080771 |
| Cy™3 AffiniPure Donkey AntiGuinea Pig IgG (H+L) | Jackson Immuno | Cat# 706-165-148 |
| Alexa fluor (488,555,633 – resp. species) | Thermo Fisher Scientifc | N/A |
| DAPI | Thermo Fisher Scientifc | Cat# D21490 |
| LTG | Vector Laboratories | Cat# FL-1321; RRID: AB_2336559 |
| DBA | Vector Laboratories | Cat# RL-1032; RRID: AB_2336396 |
| Alexa Fluor Phalloidin (546, 488) | Thermo Fisher Scientifc | Cat# A22283, A12379; RRID: AB_2315147 |
| Biological Samples | ||
| All biological samples were generated in this study and described in detail in the Results | University Medical Center Freiburg | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Blebbistatin | Sigma-Aldrich | B0560 |
| Calyculin A | Sigma-Aldrich | C5552 |
| CK312 | Calbiochem | 182518 |
| CK548 | Sigma-Aldrich | C7499 |
| CK636 | Sigma-Aldrich | C7374 |
| CK666 | Sigma-Aldrich | SML0006 |
| CK689 | Calbiochem | 182517 |
| CK869 | Calbiochem | 182516 |
| Cytochalasin D | Sigma-Aldrich | C8273 |
| Latrunculin A | Sigma-Aldrich | L5163 |
| Nocodazole | Sigma-Aldrich | M1404 |
| NSC23766 | Sigma-Aldrich | SML0952 |
| SMIFH2 | Sigma-Aldrich | S4826 |
| Y27632 | Calbiochem | 688000 |
| Doxycycline hydrochloride | Fagron, Germany | 803873 |
| Prolong Gold Antifade | Thermo Fisher Scientifc | P36930 |
| Ibidi Mounting Medium | Ibidi | 50001 |
| μ-Slide 8 Well, Collagen IV: #1.5 polymer | Ibidi | 80822 |
| μ-Dish 35 mm, high, ibiTreat | Ibidi | 81156 |
| μ-Dish 35 mm, high Glass Bottom | Ibidi | 81158 |
| Nunc Lab-Tek Chamber Slide system 8-well | Sigma-Aldrich | C7182 |
| BioFlex Culture Plate Collagen IV | Flexcell | BF-3001C-IV |
| Fibronectin | Corning | 354008 |
| Collagen IV from human placenta | Sigma-Aldrich | C5533 |
| Matrigel | Corning | 356231 |
| Bovine Type I Atelo-Collagen Solution | AdvancedBioMatrix | 5005 |
| 40% Acrylamide | Bio-Rad | 161-0140 |
| 2% Bis-acrylamide | Bio-Rad | 161-0142 |
| DSP | Sigma-Aldrich | 803200 |
| DPPB | Sigma-Aldrich | 16646 |
| Sulfo-SANPAH | ThermoFisher Scientific | 22589 |
| FluoSpheres | ThermoFisher Scientific | F8794 |
| Collagenase, Type 2 | Worthington Biochemical | LS004176 |
| Protease from Streptomyces griseus | Sigma-Aldrich | P6911 |
| DNase I | AppliChem | A3778 |
| Critical Commercial Assays | ||
| MICROFLUORAL Mikroalbumin-Test | Progen | PR2005 |
| Creatinine PAP LT-SYS | LT-Sys | LT-CR 0106 |
| Deposited Data | ||
| Raw and analyzed mass spectrometry proteomics data | This paper | ProteomeXchange Consortium via PRIDE partner repository with the dataset identifier PXD010626 and PXD010601 For reviewer access: Username: reviewer06821@ebi.ac.uk Password: K0FQ12Zk And Username: reviewer05438@ebi.ac.uk Password: 2NoS9SGe |
| Experimental Models: Cell Lines | ||
| Immortalized human podocytes (AB8/13) | M. Saleem, Bristol University | N/A |
| Primary mouse podocytes (isolated from Arp3 and N-WASP conditional mice) | this study | N/A |
| Experimental Models: Organisms/Strains | ||
| mouse: N-WASPflox/flox | Cotta-de-Almeida et al. (2007) | N/A |
| mouse: Arp3flox/flox | Arnold HH, TU Braunschweig | N/A |
| mouse: Six2-Cre | Kobayashi et al. (2008) | N/A |
| mouse: Nphs2∗Cre | Moeller et al. (2003) | N/A |
| mouse: hNphs2∗rtTA∗TetOCre | Shigehara et al. (2003) | N/A |
| mouse: Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | Muzumdar et al. (2007) | N/A |
| mouse: PAX8∗rtTA∗TetOCre | Traykova-Brauch et al. (2008) | N/A |
| Oligonucleotides | ||
| Genotyping PCR primers (Arp3) Fw: AGGGACAGTCTGTGTTTCTCCTAATATC Rev: AGGTGTTGACAACATGCTAAGAGCT |
Eurofins | N/A |
| Genotyping PCR primers (NWASP) Fw: CACTTCTTTGCCCACATACA Rev: TAAAATGGCAGTGACAATGATGAC |
Eurofins | N/A |
| Genotyping PCR primers (Nphs2Cre, Six2Cre) Fw: GCATAACCAGTGAAACAGCATTGCTG Rev: GGACATGTTCAGGGATCGCCAGGCG |
Eurofins | N/A |
| Genotyping PCR primers (Pax8rtTA) Fw: CCATGTCTAGACTGGACAAGA Rev: CTCCAGGCCACATATGATTAG |
Eurofins | N/A |
| Genotyping PCR primers (Nphs2rtTA) Fw: CGCACTTCAGTTACTTCAGGTCCTC Rev: GCTTATGCCTGATGTTGATGATGC |
Eurofins | N/A |
| Genotyping PCR primers (Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) Fw: CTCTGCTGCCTCCTGGCTTCT Rev(wt): CGAGGCGGATCACAAGCAATA Rev(mut): TCAATGGGCGGGGGTCGTT |
Eurofins | N/A |
| Software and Algorithms | ||
| Graph Pad Prism 6 Software | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Fiji Image Analyzer | FIJI | https://fiji.sc/ |
| Cytoscape 3.2.1 | Cytoscape | http://www.cytoscape.org/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tobias B. Huber (t.huber@uke.de).
Experimental Model and Subject Details
Animals
Mice were housed in a SPF facility with free access to chow (from Kliba-NAFAG, standard chow - 3807.PM.L15) and water, according to the NIH guide for the care and use of Laboratory animals as well as the German law for the welfare of animals (kept at 12 hour day/night cycle). Genotyping and breeding of the animals was performed according to standard procedures. To generate deletion of N-WASP in the metanephric mesenchyme N-WASP flox/flox mice (Cotta-de-Almeida et al., 2007) were crossed to Six2-Cre animals (Park et al., 2012, Kobayashi et al., 2008). The Arp3flox/flox conditional knock-out mouse strain was generated by Ines Lahmann and H.H. Arnold (TU Braunschweig, Germany). It will be described in detail elsewhere. Arp3flox/flox mice were crossed to Nphs2∗Cre animals (generously provided by L. Holzmann, University of Pennsylvania, School of Medicine, Philadelphia, USA) for generation of a podocyte specific knockout model (Moeller et al., 2002). Further, Arp3flox/flox mice were bred to hNphs2∗rtTA∗TetOCre (generously provided by S. Quaggin, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, University of Toronto, Canada) to generate an inducible podocyte specific knockout system (Shigehara et al., 2003). At 4 weeks of age inducible animals and respective control animals (either lacking TetOCre or hNphs2rtTA) received doxycycline hydrochloride (Fagron, Barsbuettel, Germany) via drinking water (2mg/ml with 5% sucrose) for 14 days. For isolation of primary podocytes, a reporter strain was established using Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (purchased from JAX mice; (Muzumdar et al., 2007)), resulting in either N-WASPfl/l∗Nphs2Cre;tomato<EGFPfl/+ or Arp3fl/fl ∗Nphs2Cre;tomato<EGFPfl/+mice. For deletion of N-WASP in the tubular compartment N-WASP flox/flox were bred to the PAX8∗rtTA∗TetOCre system (purchased from JAX Mice; (Traykova-Brauch et al., 2008)), induction was performed as for podocyte specific deletion approaches. Age/developmental stage of animals used for respective experiments are stated in the figures and/or figure legends (male and female animals showed similar phenotypes and combined analysis is presented). All animal experiments were approved by local authorities (Regierungspräsidium Freiburg - G11/51 and G17/127).
Human Kidney Samples
Use of human kidney material (samples were derived from unaffected areas of tumor nephrectomies; no information available concerning age and sex of affected patient) was approved by the Scientific-Ethical Committee of the University Medical Center of Freiburg.
Method Details
Genotyping
Animals were genotyped using DNA extracted from ear biopsies. DNA was analyzed and amplified by PCR using ReddyMix genotyping mix (ThermoFisher) and visualized via gel electrophoresis.
Histology and Immunofluorescence Staining on Cryosections
Dissection and fixation of kidney specimens was essentially performed as described previously (Schell et al., 2013, Bechtel et al., 2013). In brief, kidneys were dissected under a binocular light microscope and the kidney capsule was removed. Fixation was performed using 4% PFA in PBS at 4° degrees overnight. Kidneys from older animals (older 7 days postpartum) were perfusion fixated: using small silicone catheters a total volume of about 4-5 ml per kidney 4%PFA in PBS was applied via A. renales. After perfusion, capsules were removed and kidneys were immersion fixated in 4% PFA at 4° overnight. Dehydration and embedding in paraffin was performed using an automated system (Histokinette, Leica, Germany). Paraffin embedded kidneys were cut using a Leica microtome (Leica, Germany) and 2 μm sections were transferred on glass slides. Hematoxylin-Eosin and Periodic-acid-Schiff staining procedures were performed in the Department of Pathology, University Hospital of Freiburg. Images of respective WT and KO kidneys were acquired using an Axioplan 2 microscope (Zeiss, Germany) equipped with 10x, 20x and 63x objectives and an AxioCam camera (Zeiss, Germany). Immunofluorescence studies were essentially performed as previously described (Hartleben et al., 2013). In brief, cryosections (3-4 μm) of snap frozen tissue samples were generated using a cryotome (Leica, Wetzlar, Germany). Fixation of cryosections was performed at room temperature using 4% PFA in PBS solution for 3 minutes. Blocking of cryosections was performed with bovine serum albumin (Sigma, Schnelldorf, Germany) diluted in PBS to a final concentration of 5% for 45 min. Primary antibodies were applied to the sections at room temperature for 1-2 hour and were diluted in 5% BSA/PBS. Secondary antibodies were diluted in PBS (all from Invitrogen - Alexa-fluor labeled) were incubated for 45 minutes and after final wash, slides were mounted using Prolong Gold Antifade (Invitrogen, Darmstadt, Germany). Image acquisition of cryo-sections was done either using a conventional wide field epifluorescence imaging or a confocal laser scanning imaging setup (WF: Zeiss Axioscope 40FL microscope, equipped with an Axiocam MRc5 digital video camera and conventional HBO lamp – Carl Zeiss, Oberkochen, Germany; CLSM: the laser scanning setup was built up of Zeiss LSM 510 upright microscope equipped with a Plan-Apochromat 63x/1.4 Oil M27 objective – Carl Zeiss, Oberkochen, Germany). Image recording was performed via Axiovision 4.3 or the ZEN black software, analysis was done using Fiji Lifeline 2014 software bundle.
Electronmicroscopy & Scanning Electron Microscopy (SEM)
Specimens for conventional transmission electron microscopy were fixated using 4% PFA in PB. Kidneys were perfused either via A. renales using small silicone catheters or via the heart. After initial fixation small pieces of the renal cortex were dissected using razor blades. Cubes of about 2x2x2 mm were transferred into glass vials and immersion fixated in 4% PFA at 4° Celsius overnight. Embedding, semi-thin sections and electron microscopy of fixated kidney samples were performed at the Institute of Pathology, Vienna (Schell et al., 2013). For preparation of samples for scanning electron microscopy fixation was performed using 4% Glutaraldehyde (Sema) diluted in PBS. Small 5x5x5 mm samples were prepared and immersion fixated in 4% GA at 4° for 3 days. Further dehydration was done in Ethanol (50-100% - each step for 1h at RT), following transfer to HMDS (Sigma, Schnelldorf, Germany). Kidney samples were dissected using syringe needles under a binocular microscope. Sputtering with gold was performed using a Polaron Cool Sputter Coater E 5100. Samples were visualized using a scanning electron microscope (Leo 1450 VP scanning). Freshly isolated kidneys of adult mice were dissected into thin slices and cultured in RPMI medium at 37°C, 5% CO2 and controlled humidity. Slice cultures were treated with DMSO as control or with 100μM CK869 for 3 hours. After treatment, kidney slices were fixated and processed for SEM.
Super Resolution Microscopy (SIM)
For evaluation of the slit diaphragm density as a direct marker for podocyte effacement, we used the recently established super resolution microscopy-based podocyte effacement measurement procedure (PEMP) (Siegerist et al., 2017). Briefly, 2 μm paraffin sections were performed and cooked in 10 mM citrate buffer (pH 6) in a pressure cooker. After blocking in 1% normal goat serum, 1% bovine serum albumin, 1% fetal calf serum and 0.05 % fish gelatin for 1 h at room temperature, guinea pig anti-nephrin serum (gpN2, Progene, Heidelberg, Germany, diluted 1:300 in blocking solution) was incubated at 4°C overnight. After washing steps (PBS) followed by a second blocking step, primary antibodies were detected by Cy3-conjugated donkey antiguinea pig antibodies (Jackson Immuno, diluted 1:600). Sections were mounted in Mowiol (Carl Roth, Karlsruhe, Germany) for microscopy and imaged by a Zeiss Elyra PS.1 (Carl Zeiss, Jena, Germany) super resolution structured illumination microscopy system. For measurement of the DSD custom-built Fiji plugin was used. DSD values of 28-30 glomeruli of at least two mice per group were quantified.
Measurement of Urinary Albumin and Creatinine
Levels of proteinuria (expressed as albumin to creatinine ratio) were quantified by measuring albumin and creatinine in spot urine from wild type and knockout mice at defined time points. Measurement of urinary albumin was performed using a mouse specific albumin fluorescent based kit (Progene, Germany). Assessment of creatinine was done in a similar way using an enzymatic creatinine kit (Creatinine PAP LT-SYS, Labor&Technik, Eberhard Lehmann GmbH, Germany).
Glomerular and Podocyte Preparation for Western Blot and MS Applications
In principle isolation was performed as previously described (Boerries et al., 2013). In brief, kidneys were dissected together with the abdominal aorta and transferred into dishes filled with 37 °C prewarmed Hank’s buffered salt solution (HBSS). Each kidney was perfused slowly through the renal artery with 4 ml 37 °C warm bead solution and 1 ml bead solution plus enzymatic digestion buffer (containing: collagenase 300 U/ml (Worthington, Collagenase Type II, Lakewood, NJ), 1 mg/ml pronase E (Sigma P6911, Schnelldorf, Germany), DNase I 50 U/ml (AppliChem A3778, Darmstadt, Germany). Kidneys were minced, further digested and then after several mechanical dissociation steps inserted into a magnetic particle concentrator and the separated glomeruli were washed twice. Isolation of podocytes followed several steps of mechanical dissociation and final FACS sorting for GFP+ cells only. Isolated protein fractions were furthermore analyzed using unlabeled mass spectrometry as previously described (Boerries et al., 2013). Samples were taken up in SDS–PAGE loading buffer. Disulfide bonds were reduced with 1 mM DTT and alkylated using 5.5 mM iodoacetamide (Sigma–Aldrich) for 30 min at 25°C. Proteins were separated by 4–12% Bis–Tris gradient SDS–PAGE gels (Invitrogen). Gel lanes were cut into 10 fractions and proteins therein were digested with trypsin (Promega) (Shevchenko et al., 2006), and resulting peptides were desalted on STAGE Tips (Rappsilber et al., 2007).
Mass Spectrometry and MaxQuant-Based Identification
Peptides were analysed with a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific). An Agilent 1200 nanoflow–HPLC with a fused silica column, 75 mm inner diameter, packed with Reprosil–Pur 120 ODS–3 (Dr. Maisch) was used for peptide separation prior MS analysis. Samples were loaded with 500 nl/min, and the gradient was conducted at 250 nl/min. A gradient of Buffer A (0.5% acetic acid in deionized water) and Buffer B (0.5% acetic acid, 80% ACN in deionized water) with increasing ACN proportion was used for peptide separation. Samples were loaded onto the column with 2% Buffer B; a 100 min separation gradient was run up to 35% buffer B. The ion–transfer tube temperature was set to 200°C and the spray voltage was 2.3 kV with no sheath and auxiliary gas flow. Data was acquired in the data–dependent mode and switched automatically between MS (max. of 1x106 ions) and MS/MS (max. of 5000 ions). In the linear ion trap the top five abundant peptides were isolated and fragmented with 35% collision energy. Parent ions with a charge of z=1 and unassigned charge states were excluded from fragmentation. The resolution was set to 60 000 with a mass range of m/z=350–2000. MaxQuant software (Cox and Mann, 2008) was used for data search for identification and quantification of peptides and proteins. A full length UNIPROT mouse database was used. Carbamidomethylation of cysteines was defined as fixed modification, whereas methionine oxidations and protein amino–terminal acetylations were defined as variable modifications. At least one peptide had to be unique per protein group and a minimum ratio count of two was needed for quantification. For the enzymatic cleavage with trypsin/P a maximum of three missed cleavages was allowed. The MS/MS tolerance was set to 0.5 Da. Mass precision of identified peptides had to be less than 20 ppm. A FDR of 0.01 was set for peptide and protein identification. Minimum peptide length had to be seven amino acids and identified proteins were re-quantified. Furthermore the match-between-run option was used with a matching time window of 2 min and an alignment time window of 20 minutes.
Focal Adhesion (Adhesome) Complex Isolation
Isolation of focal adhesion complexes was performed as previously described (Horton et al., 2015, Kuo et al., 2011). In brief, human immortalized podocytes were seeded to a 15 cm cell culture dish and grown on 33°C. Then podocytes were differentiated by incubation on 37°C for 10 days. Cells were incubated with the protein cross-linker DSP (3,3'-Dithiobis(sulfosuccinimidylpropionate); 100mM; Sigma-Aldrich) and DPPB (1,4-Bis[3-(2-pyridyldithio)propionamido]butane; 10mM; Sigma-Aldrich) for 10 minutes. Cross-linker treated cells were washed with PBS and cross-linkers were quenched using 1 M Tris-HCl (pH 8, 10 min). Cell bodies were removed by hydrodynamic force by washing the cell culture dish using a waterpik (2x 10s; PBS). Proteins and focal adhesion complexes left bound to the ECM were recovered by scraping in 100 μl scraping buffer (125 mM Tris-HCl (pH 6.8), 1% (w/v) SDS, 15% (v/v) β-mercaptoethanol) and denatured at 70 °C for 10 min. Western blotting and MS analysis was performed subsequently (as described above).
Isolation of Primary Podocytes and Culture Conditions
To study podocyte phenotypes in vitro we made use of a previously established isolation and culture method (Schell et al., 2013). In brief, glomeruli were isolated from p7-p10 old mice of the respective genotypes. Kidneys were removed and a digest protocol was applied (Worthington Collagenase I 1mg/ml, Pronase 1mg/ml, DNAse I 100 U/ml all diluted in HBSS – all from Sigma, Schnelldorf, Germany) for about 8 minutes. The digest solution was then filtered using cell strainers with 3 different pore sizes, starting with 100 microns, followed by 70 and finally 40 microns (BD Falcon). Glomeruli were harvested from the last filter and transferred after centrifugation to culture flasks. After an outgrowth phase of about 4-6 days, cells were sorted using an Aria Cell Sorter (Aria BD Biosciences). GFP-positive cells were collected in individual sorting approaches, depending on the individual isolation procedure and reseeded again on collagen IV (Sigma-Aldrich, Schnelldorf, Germany) coated culture flasks (50 μg/ml). Cells after sorting were termed as passage 1 and cells for further experiments were only used up to passage 6-7 after the sorting procedure. Primary podocytes were cultured in RPMI 1640 (Invitrogen, Schnelldorf, Germany), supplemented with 10% FCS (Invitrogen), Penicillin/Streptomycin (0.5 mg/ml) and a combination supplement of ITS (Insulin, Selenite and Transferrin – Roche).
All drugs were stored, dissolved for stock solutions and used according to the manufacturer's recommendations. Solvents for stock solutions or inactive inhibitors were used as negative control in respective treatments experiments. All experiments were performed at least 3 times. Due to availability of higher cell numbers human immortalized podocytes were used for treatment studies in Figures 5K, 5Q, 5R, 6P, 6Q, and S6E. Human immortalized podocytes were cultivated in collagen IV coated Ibidi 8-well cambers (ibidi, Martinsried, Germany) and serum-starved for 12h before and while application of indicated drugs (in Figure 5K: NSC23766 100μM for 2h; Nocodazole 15μM for 4h; Calyculin A 2nM for 20min; CK869 100μM for 3h; Blebbistatin 10μM for 1h); (in Figures 5G and 5R: 1μM Cytochalasin D pretreatment for 1h followed by 4x wash with culture medium; subsequent inhibitor treatment times and concentrations are indicated in Figure 5); (in Figure S6E: CK312/CK689/CK869/CK666/CK636/CK548 100μM for 4h; SMIFH2 15μM for 3h; Y27632 10μM for 3h; Blebbistatin 30μM for 1h; Cytochalasin D 0.1 and 1μM for 30 minutes; Latrunculin A 0.1 and 1μM for 30 minutes). Primary podocytes for NSC23766 treatment (Figures S5G–S5I) were cultured on collagen 4 coated glass cover slips for 24h, serum starved for 2h and treated with NSC23766 100μM for 2h in serum free cell culture medium. All other treatment experiments were performed by use of primary podocytes from indicated WT or KO mice models. Primary podocytes were pre-cultivated on collagen IV coated glass cover slips (2D cell culture) in 10% FBS supplemented culture medium. Indicated drugs were applied in 10% FBS supplemented culture medium. (Figures 5D–5J and S6 a: Blebbistatin 5 and 20μM for 2hours; Figures 5M–5P: SMIFH2 15μM for 1h; Figures S6B–S6D: CK666 100μM for 6h; Figures S6F–S6J: Blebbistatin 20μM for 2 h and Calyculin A 10nM for 10 minutes). Treatment of 3D cultured podocytes was performed by application of indicated drugs in 10% FBS supplemented cell culture medium (Blebbistatin 20μM – 2h, CK666 and CK548 100 and 50μM respectively – 4h, SMIFH2 15μM – 1h and Latrunculin A 1μM); (Figure S5Q: SMIFH2 5 and 15μM for 50 minutes, 2h after seeding of primary podocytes on thin collagen gels (PureCol, Advanced BioMatrix, San Diego, CA).
3D Protrusion Formation of Primary Podocytes and Cortical Actin Fluorescence Measurement
3D culture assays were adapted for primary podocyte culture (Fraley et al., 2010). For generating 3D growth matrices matrigel (Corning) was used and prepared according to manufacturer's instructions. A final concentration of 3mg/ml of matrigel was used in the experiments. In brief, cells were trypsinized and 20.000 cells were diluted in a final amount of 120μl of the appropriate matrigel solution (everything ice-cold to prevent polymerization of matrigel). Cells were seeded in pre-coated 8 well Labtek growth chamber slides (pre-coated with 30μl of undiluted 9mg/ml matrigel solution) and cultivated at 37° Celsius for 24 hours. Quantification of cells with formed protrusions was made on fixated samples. After fixation in 4% PFA and 0.1 % GA in PBS for 30 minutes, cells were stained for phalloidin and mounted with Prolong Gold antifade. Samples were imaged using a confocal LSM set up and z-stacks of 0.39 μm were generated. Reconstruction was performed using the 3D image viewer plugin of ImageJ and measurements of protrusions and branches were done using the Simple Neurite Tracer plugin from the Fiji image analysis software package.
Immunofluorescence on Primary Podocytes
Staining of primary podocytes was performed after seeding cells on coated glass coverslips. Cells were seeded one day prior to the staining procedure in sub-confluent conditions (otherwise indicated). Pre-warmed 4% PFA diluted in PBS was used for fixation and after 10 minutes treatment with PFA quenching with 50mM NH4CL was applied, followed by brief permeabilization with Triton-X-100 0.1% in PBS for 3 minutes. Blocking and incubation with primary as well as secondary antibodies was performed in 5% bovine serum albumin/PBS, each step for 1 hour. The staining procedure was followed by final mounting in Prolong Gold antifade (Invitrogen, Schnelldorf).
Isometric Cycling Stress (Flexcell)
Primary and immortalized human podocytes were seeded on collagen IV coated flexible biomembranes (Flexcell International Corporation; Burlington, USA) and after a 12 hour cultivation period transferred to the Flexcell manipulator. According to previous studies (Schordan et al., 2011) stretch of 7% and 0.5 Hz cycling were applied for a total duration of 24 hours. After stress application cells were fixed and further analyzed. For detachment quantification, at least 15 low power fields were randomly chosen and automatically analyzed for attached nuclei; experiments were repeated 3 times independently.
Traction Force Microscopy
Traction force microscopy is an established method for quantification of cellular forces exerted on the underlying substratum (Schwarz and Gardel, 2012). Previously established protocols for preparation of substratum matrices were adapted for primary podocytes (Plotnikov et al., 2012). For all experiments gels with a Young's modulus of 16kPA PAA (poly-acrylamide) were prepared. After pouring preparation of the gels, crosslinking for fibronectin was performed with the SulfO-SANPAH crosslinker (under UV light for 5 minutes) and indicated concentrations of respective ECM components were applied. 20.000 cells per cover slip were seeded and cultivated for 12-16 hours before measurement of forces. Coverslips were placed in flow chamber and images of respective cells were recorder before and after removal of the cell. Manipulation of the cells was performed with a micromanipulator (Eppendorf). Image analysis was done as previously described (Sabass et al., 2008).
Random Single Cell Migration Assays
Migration behavior of respective primary podocytes was assessed using a Nikon Biosstation imaging setup (20 x magnification - Nikon biosstation, Nikon, Duesseldorf, Germany). Cells were seeded one day prior to the experiment on ibidi μ-treat culture chambers (ibidi, Martinsried, Germany). Using the implemented software 15-20 fields per experimental condition were selected and focus as well as exposure times for the camera was set individually. Recording was performed over 12 hours while capturing images every 10 minutes.
Quantification and Statistical Analysis
Statistical Analysis
If not stated otherwise, data are expressed as mean ± SEM. Paired Student's t-test, ONE-WAY ANOVA (multiple comparison test - Tukey), non-parametric two-tailed Mann Whitney test or Newman-Keuls multiple comparisons test were used based on data distribution. Statistical significance was defined as ∗ p<0.05, ∗∗ p<0.01, ∗∗∗ p<0.001 and ∗∗∗∗ p<0.0001, n.s. - not significant. Number of independent experiments and total amount of analyzed cells or animals were stated in the figure legends.
Glomerluar Sclerosis Index
To quantify glomerular sclerosis a previously established scoring method was adapted and modified (el Nahas et al., 1991). The degree of sclerosis and alteration of glomeruli was categorized according to following criteria: grade I – normal appearing glomeruli, grade II – mild to moderate alterations, at least one feature like mesangial hypercellularity or partial sclerosis (<50% of total glomerulus), grade III – totally sclerosed glomerulus. 50-60 glomeruli per animal were assessed and counted in a blinded manner. A total number of 3-4 animals per time point and genotype were analyzed.
Immunofluorescence Quantification
ppMLC intensity/area per glomerulus was quantified by segmentation of the podocyte compartment using Nephrin immunofluorescence as a specific marker. >20 glomeruli per animal were analyzed and the mean podocyte ppMLC intensity/area per animal was calculated. The percentage of YAP positive podocyte nuclei was analyzed by immunofluorescence staining for YAP and the podocyte specific nucleus marker WT1. >20 glomeruli per animal were analyzed and the mean percentage of YAP positive podocyte nuclei per animal was calculated.
Quantification of Foot Process Morphology and Branching Index
Image analysis was performed using Image J 4.17 and Adobe Photoshop CS6. Measurement of foot process width was essentially performed as described before (Ma et al., 2010). Briefly, numbers of podocyte processes were divided by the length of the GBM region in every image. Branching levels of podocyte major processes were calculated from semi-planar SEM areas. Therefore branches per major trunk were counted and the ratio of branches per μm length was calculated (10-23 major processes per animal were analyzed and the mean branching ratio per animal was calculated).
Quantification of Focal Adhesion Morphology and Analysis of IF Intensities
Evaluation of focal adhesion size and distribution was basically performed as described previously, with some modifications (Berginski et al., 2011). Primary podocytes were trypsinized and seeded on either fibronectin or collagen IV coated coverslips (50mikrog/mikrol). According to the experimental setup cells were allowed to attach and spread for the respective time, and finally fixated using pre-warmed 4% PFA in PBS for 10 minutes. Image acquisition was performed using a Zeiss Axioscope 40FL microscope, equipped with a 63x objective and individual cells of each experimental condition were recorded using standardized exposure times. The analysis algorithm was composed of following steps: 1.) high pass filtering with averaging filter (radius of 11 pixels), 2.) adjusting the threshold of the image and binarization, 3.) measurement of area, major and minor axis using the particle analyzer function of ImageJ NIH (lower cutoff 0.2 Pixels). Output data were collected for individual cells and analyzed using Microsoft Excel and GraphPad Prism. Quantification of IF signal intensity was basically performed as described previously (Bechtel et al., 2013) : Images were background subtracted and the area of individual cells was outlined manually. Fluorescence intensity in the outlined area was recorded and background intensity was subtracted. Cellular immunofluorescence intensities were normalized for cell area and plotted using GraphPad Prism. Line scans were performed using the plot profile tool of ImageJ/Fiji NIH software (PlotProfile).
Cell Migration
Tracking of individual cells was performed using ImageJ 1.47 NIH software package and the manual tracking plugin (http://rsbweb.nih.gov/ij/plugins/track/track.html). Resulting xy coordinates were further analyzed with the ibidi ChemoTaxis Plugin (ibidi, Martinsried, Germany) and resulting data were plotted using Graphpad Prism (accumulated distance and velocity). Each experimental condition was at least repeated 3 times.
Protein Enrichment and Functional Annotation Analysis
Detected proteins (podocyte fingerprint podocyte proteome) were identified as enriched/upregulated with a “>2 fold enrichment podocytes to non-podocytes. Gene ontology term (GO-Term) annotation was implemented using the MGI database (UniProt Consortium, 2015, Eppig, 2017). Analysis and categorization of detected proteins was performed according to enrichment scores and GO-Term annotation and is in detail shown in Table S1. Proteins with >2 fold enrichment in podocytes were filtered into GO-Term based groups (focal adhesion, actin binding and actin driven cell projection) (see Table S1 for details). To generate a visualized protein network out of this sub-analysis (see Figures 1 and S1), the dataset was imported into Cytoscape (version 3.2.1) (Shannon et al., 2003). This network was merged with a human protein-protein interaction network from the BioGRID (release 3.2.105) database (Chatr-Aryamontri et al., 2017). Interaction and protein duplications were removed. Detailed analysis of the podocyte focal adhesion proteome (FAP) is shown in Figure S1 and Table S1. The podocyte FAP was analyzed by GO-Term annotation using the UniProt database (UniProt Consortium, 2015) and by use of a recently published, proteomic based, consensus focal adhesome (Horton et al., 2015). Protein network visualization was achieved as described above.
Data and Software availability
The accession number for proteomic datasets reported in this paper is PDX010626 and PDX010601.
Acknowledgments
We would like to thank Charlotte Meyer and Betina Kiefer for their expert technical assistance. In addition, we would like to express our gratitude to all members of our laboratories, to Sebastian Arnold, and to the Life Imaging Center (LIC) of the University of Freiburg for helpful discussions and support. This study was supported by the German Research Foundation (DFG): CRC 1140 (to T.B.H.), CRC 1192 (to T.B.H.), and CRC 992 (to T.B.H.), Heisenberg program (to T.B.H.), HU 1016/5-1 and HU 1016/8-1 (to T.B.H.); by the European Research Council (ERC grant 616891 to T.B.H.) and by the H2020-IMI2 consortium BEAt-DKD (115974; to T.B.H.); by the BMBF (Bundesministerium für Bildung und Forschung) STOP-FSGS 01GM1518B (to N.E.) and BMBF STOP-FSGS 01GM1518C (to T.B.H.); by the Excellence Initiative of the German Federal and State Governments (BIOSS to T.B.H. and the Freiburg Institute for Advanced Studies [FRIAS] to T.B.H.); by the Alexander von Humboldt Foundation (to A.A.); by the German Society of Nephrology (DGFN to C.S.); by the Else Kröner Fresenius Stiftung, NAKSYS (to C.S., M.R., and T.B.H.) and Matriglom A_09 (to C.S.); and by the Berta-Ottenstein Programme, Faculty of Medicine, University of Freiburg (to C.S.).
Author Contributions
C.S., M.R., F. Geist, J.D., B.S., and T.B.H. conceived and analyzed experiments. C.S., M.R., and T.B.H. supervised the study. C.S., M.R., F. Geist, M.H., A.A., M.Y.-Y., J.I.M., A.S., F.S., N.A., N.E., and D.K. performed experiments. H.H.A., F. Grahammer. and J.D. provided critical reagents. C.S. and M.R. prepared figures and tables. C.S., M.R., and T.B.H. wrote the manuscript with input and discussion from all authors.
Declaration of Interests
T.B.H. declares to be a member of the scientific advisory board of Goldfinch Bio and to have a research grant from Fresenius Medical Care/ Unicyte. N.E. declares that PEMP (podocyte exact morphology measurement procedure) that has been used for this manuscript is registered for a patent. N.E. and F.S. are among the founders of the Start-UP NIKOKA, which will commercialize PEMP. All other authors declare no competing interests.
Published: November 29, 2018
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.devcel.2018.11.011.
Supplemental Information
Analysis of podocyte enriched proteins of isolated mouse podocytes measured by mass spectrometry. Index of each tab is included.
Analysis of podocyte enriched focal adhesions (isolated FA complexes) derived from immortalized human podocytes measured by mass spectrometry. Index of each tab is included.
References
- Bechtel W., helmstädter M., Balica J., Hartleben B., Kiefer B., Hrnjic F., Schell C., Kretz O., Liu S., Geist F. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J. Am. Soc. Nephrol. 2013;24:727–743. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berginski M.E., Vitriol E.A., Hahn K.M., Gomez S.M. High-resolution quantification of focal adhesion spatiotemporal dynamics in living cells. PLoS One. 2011;6:e22025. doi: 10.1371/journal.pone.0022025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisi S., Disanza A., Malinverno C., Frittoli E., Palamidessi A., Scita G. Membrane and actin dynamics interplay at lamellipodia leading edge. Curr. Opin. Cell Biol. 2013;25:565–573. doi: 10.1016/j.ceb.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Boerries M., Grahammer F., Eiselein S., Buck M., Meyer C., Goedel M., Bechtel W., Zschiedrich S., Pfeifer D., Laloë D. Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int. 2013;83:1052–1064. doi: 10.1038/ki.2012.487. [DOI] [PubMed] [Google Scholar]
- Bravo-Cordero J.J., Hodgson L., Condeelis J. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 2012;24:277–283. doi: 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown E.J., Schlöndorff J.S., Becker D.J., Tsukaguchi H., Tonna S.J., Uscinski A.L., Higgs H.N., Henderson J.M., Pollak M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 2010;42:72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke T.A., Christensen J.R., Barone E., Suarez C., Sirotkin V., Kovar D.R. Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers. Curr. Biol. 2014;24:579–585. doi: 10.1016/j.cub.2014.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone K.G., Welch M.D. A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010;11:237–251. doi: 10.1038/nrm2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N.K., O'donnell L., Oster S., Theesfeld C., Sellam A. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–D379. doi: 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C.K., Vicente-Manzanares M., Zareno J., Whitmore L.A., Mogilner A., Horwitz A.R. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorev D.S., Moscovitz O., Geiger B., Sharon M. Regulation of focal adhesion formation by a vinculin-Arp2/3 hybrid complex. Nat. Commun. 2014;5:3758. doi: 10.1038/ncomms4758. [DOI] [PubMed] [Google Scholar]
- Cotta-de-Almeida V., Westerberg L., Maillard M.H., Onaldi D., Wachtel H., Meelu P., Chung U.I., Xavier R., Alt F.W., Snapper S.B. Wiskott Aldrich syndrome protein (WASP) and N-WASP are critical for T cell development. Proc. Natl. Acad. Sci. USA. 2007;104:15424–15429. doi: 10.1073/pnas.0706881104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Dang I., Gorelik R., Sousa-Blin C., Derivery E., Guérin C., Linkner J., Nemethova M., Dumortier J.G., Giger F.A., Chipysheva T.A. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 2013;503:281–284. doi: 10.1038/nature12611. [DOI] [PubMed] [Google Scholar]
- Di Martino J., Henriet E., Ezzoukhry Z., Goetz J.G., Moreau V., Saltel F. The microenvironment controls invadosome plasticity. J. Cell Sci. 2016;129:1759–1768. doi: 10.1242/jcs.182329. [DOI] [PubMed] [Google Scholar]
- el Nahas A.M., Bassett A.H., Cope G.H., Le Carpentier J.E. Role of growth hormone in the development of experimental renal scarring. Kidney Int. 1991;40:29–34. doi: 10.1038/ki.1991.175. [DOI] [PubMed] [Google Scholar]
- Endlich N., Endlich K. Stretch, tension and adhesion - adaptive mechanisms of the actin cytoskeleton in podocytes. Eur. J. Cell Biol. 2006;85:229–234. doi: 10.1016/j.ejcb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Eppig J.T. Mouse genome informatics (MGI) resource: genetic, genomic, and biological KnowledgeBase for the Laboratory Mouse. ILAR J. 2017;58:17–41. doi: 10.1093/ilar/ilx013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D., Notbohm J., Benjamin A., He S., Wang M., Ang L.H., Bantawa M., Bouzid M., Del Gado E., Krishnan R. Disease-causing mutation in alpha-actinin-4 promotes podocyte detachment through maladaptation to periodic stretch. Proc. Natl. Acad. Sci. USA. 2018;115:1517–1522. doi: 10.1073/pnas.1717870115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley S.I., Feng Y., Krishnamurthy R., Kim D.H., Celedon A., Longmore G.D., Wirtz D. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat. Cell Biol. 2010;12:598–604. doi: 10.1038/ncb2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahammer F., Schell C., Huber T.B. The podocyte slit diaphragm–from a thin grey line to a complex signalling hub. Nat. Rev. Nephrol. 2013;9:587–598. doi: 10.1038/nrneph.2013.169. [DOI] [PubMed] [Google Scholar]
- Hartleben B., Widmeier E., Suhm M., Worthmann K., Schell C., Helmstädter M., Wiech T., Walz G., Leitges M., Schiffer M. aPKCλ/ι and aPKCζ contribute to podocyte differentiation and glomerular maturation. J. Am. Soc. Nephrol. 2013;24:253–267. doi: 10.1681/ASN.2012060582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Has C., Spartà G., Kiritsi D., Weibel L., Moeller A., Vega-Warner V., Waters A., He Y., Anikster Y., Esser P. Integrin α 3 mutations with kidney, lung, and skin disease. N. Engl. J. Med. 2012;366:1508–1514. doi: 10.1056/NEJMoa1110813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton E.R., Byron A., Askari J.A., Ng D.H.J., Millon-Frémillon A., Robertson J., Koper E.J., Paul N.R., Warwood S., Knight D. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 2015;17:1577–1587. doi: 10.1038/ncb3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter M.V., Fernandez-Gonzalez R. Coordinating cell movements in vivo: junctional and cytoskeletal dynamics lead the way. Curr. Opin. Cell Biol. 2017;48:54–62. doi: 10.1016/j.ceb.2017.05.005. [DOI] [PubMed] [Google Scholar]
- Inagaki N., Katsuno H. Actin waves: origin of cell polarization and migration? Trends Cell Biol. 2017;27:515–526. doi: 10.1016/j.tcb.2017.02.003. [DOI] [PubMed] [Google Scholar]
- Kaplan J.M., Kim S.H., North K.N., Rennke H., Correia L.A., Tong H.Q., Mathis B.J., Rodríguez-Pérez J.C., Allen P.G., Beggs A.H. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- Kim I.H., Racz B., Wang H., Burianek L., Weinberg R., Yasuda R., Wetsel W.C., Soderling S.H. Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J. Neurosci. 2013;33:6081–6092. doi: 10.1523/JNEUROSCI.0035-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I.H., Rossi M.A., Aryal D.K., Racz B., Kim N., Uezu A., Wang F., Wetsel W.C., Weinberg R.J., Yin H. Spine pruning drives antipsychotic-sensitive locomotion via circuit control of striatal dopamine. Nat. Neurosci. 2015;18:883–891. doi: 10.1038/nn.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A., Valerius M.T., Mugford J.W., Carroll T.J., Self M., Oliver G., Mcmahon A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M., Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014;15:577–590. doi: 10.1038/nrm3861. [DOI] [PubMed] [Google Scholar]
- Kriz W., Shirato I., Nagata M., Lehir M., Lemley K.V. The podocyte's response to stress: the enigma of foot process effacement. Am. J. Physiol. Renal Physiol. 2013;304:F333–F347. doi: 10.1152/ajprenal.00478.2012. [DOI] [PubMed] [Google Scholar]
- Kuo J.C., Han X., Hsiao C.T., Yates J.R., 3rd, Waterman C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for beta-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoux B., Mège R.M., Trepat X. Front-rear polarization by mechanical cues: from single cells to tissues. Trends Cell Biol. 2016;26:420–433. doi: 10.1016/j.tcb.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leijnse N., Oddershede L.B., Bendix P.M. An updated look at actin dynamics in filopodia. Cytoskeleton. 2015;72:71–79. doi: 10.1002/cm.21216. [DOI] [PubMed] [Google Scholar]
- Liapis H., Romagnani P., Anders H.J. New insights into the pathology of podocyte loss: mitotic catastrophe. Am. J. Pathol. 2013;183:1364–1374. doi: 10.1016/j.ajpath.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin A.J., Lee K.C., Han S.J., Bui D.A., Davidson M., Mogilner A., Danuser G. Competition for actin between two distinct F-actin networks defines a bistable switch for cell polarization. Nat. Cell Biol. 2015;17:1435–1445. doi: 10.1038/ncb3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Togawa A., Soda K., Zhang J., Lee S., Ma M., Yu Z., Ardito T., Czyzyk J., Diggs L. Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J. Am. Soc. Nephrol. 2010;21:1145–1156. doi: 10.1681/ASN.2009090991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller M.J., Sanden S.K., Soofi A., Wiggins R.C., Holzman L.B. Two gene fragments that direct podocyte-specific expression in transgenic mice. J. Am. Soc. Nephrol. 2002;13:1561–1567. doi: 10.1097/01.asn.0000015614.68893.0b. [DOI] [PubMed] [Google Scholar]
- Moeller M.J., Sanden S.K., Soofi A., Wiggins R.C., Holzman LB. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis. 2003;35:39–42. doi: 10.1002/gene.10164. [DOI] [PubMed] [Google Scholar]
- Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Park J.S., Ma W., O’Brien L.L., Chung E., Guo J.J., Cheng J.G., Valerius M.T., Mcmahon J.A., Wong W.H., Mcmahon A.P. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell. 2012;23:637–651. doi: 10.1016/j.devcel.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavenstädt H., Kriz W., Kretzler M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- Perico L., Conti S., Benigni A., Remuzzi G. Podocyte-actin dynamics in health and disease. Nat. Rev. Nephrol. 2016;12:692–710. doi: 10.1038/nrneph.2016.127. [DOI] [PubMed] [Google Scholar]
- Plotnikov S.V., Pasapera A.M., Sabass B., Waterman C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell. 2012;151:1513–1527. doi: 10.1016/j.cell.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi A., Jarad G., Moeckel G.W., Coffa S., Zhang X., Gewin L., Eremina V., Hudson B.G., Borza D.B., Harris R.C. Beta1 integrin expression by podocytes is required to maintain glomerular structural integrity. Dev. Biol. 2008;316:288–301. doi: 10.1016/j.ydbio.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Reginensi A., Scott R.P., Gregorieff A., Bagherie-Lachidan M., Chung C., Lim D.S., Pawson T., Wrana J., Mcneill H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013;9:e1003380. doi: 10.1371/journal.pgen.1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotty J.D., Wu C., Bear J.E. New insights into the regulation and cellular functions of the Arp2/3 complex. Nat. Rev. Mol. Cell Biol. 2013;14:7–12. doi: 10.1038/nrm3492. [DOI] [PubMed] [Google Scholar]
- Rotty J.D., Wu C., Haynes E.M., Suarez C., Winkelman J.D., Johnson H.E., Haugh J.M., Kovar D.R., Bear J.E. Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell. 2015;32:54–67. doi: 10.1016/j.devcel.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabass B., Gardel M.L., Waterman C.M., Schwarz U.S. High resolution traction force microscopy based on experimental and computational advances. Biophys. J. 2008;94:207–220. doi: 10.1529/biophysj.107.113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N., Kreft M., Van Den Bergh Weerman M.A., Beynon A.J., Peters T.A., Weening J.J., Sonnenberg A. Kidney failure in mice lacking the tetraspanin CD151. J. Cell Biol. 2006;175:33–39. doi: 10.1083/jcb.200603073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell C., Baumhakl L., Salou S., Conzelmann A.C., Meyer C., Helmstädter M., Wrede C., Grahammer F., Eimer S., Kerjaschki D. N-wasp is required for stabilization of podocyte foot processes. J. Am. Soc. Nephrol. 2013;24:713–721. doi: 10.1681/ASN.2012080844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell C., Rogg M., Suhm M., Helmstädter M., Sellung D., Yasuda-Yamahara M., Kretz O., Küttner V., Suleiman H., Kollipara L. The FERM protein EPB41L5 regulates actomyosin contractility and focal adhesion formation to maintain the kidney filtration barrier. Proc. Natl. Acad. Sci. USA. 2017;114:E4621–E4630. doi: 10.1073/pnas.1617004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell C., Wanner N., Huber T.B. Glomerular development–shaping the multi-cellular filtration unit. Semin. Cell Dev. Biol. 2014;36:39–49. doi: 10.1016/j.semcdb.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Schordan S., Schordan E., Endlich K., Endlich N. AlphaV-integrins mediate the mechanoprotective action of osteopontin in podocytes. Am. J. Physiol. Renal Physiol. 2011;300:F119–F132. doi: 10.1152/ajprenal.00143.2010. [DOI] [PubMed] [Google Scholar]
- Schwarz U.S., Gardel M.L. United we stand: integrating the actin cytoskeleton and cell-matrix adhesions in cellular mechanotransduction. J. Cell Sci. 2012;125:3051–3060. doi: 10.1242/jcs.093716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrels B., Serrels A., Brunton V.G., Holt M., Mclean G.W., Gray C.H., Jones G.E., Frame M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007;9:1046–1056. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Shigehara T., Zaragoza C., Kitiyakara C., Takahashi H., Lu H., Moeller M., Holzman L.B., Kopp J.B. Inducible podocyte-specific gene expression in transgenic mice. J. Am. Soc. Nephrol. 2003;14:1998–2003. doi: 10.1681/ASN.V1481998. [DOI] [PubMed] [Google Scholar]
- Siegerist F., Ribback S., Dombrowski F., Amann K., Zimmermann U., Endlich K., Endlich N. Structured illumination microscopy and automatized image processing as a rapid diagnostic tool for podocyte effacement. Sci. Rep. 2017;7:11473. doi: 10.1038/s41598-017-11553-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling S.H. Grab your partner with both hands: cytoskeletal remodeling by Arp2/3 signaling. Sci. Signal. 2009;2:pe5. doi: 10.1126/scisignal.255pe5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez C., Carroll R.T., Burke T.A., Christensen J.R., Bestul A.J., Sees J.A., James M.L., Sirotkin V., Kovar D.R. Profilin regulates F-actin network homeostasis by favoring formin over Arp2/3 complex. Dev. Cell. 2015;32:43–53. doi: 10.1016/j.devcel.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez C., Kovar D.R. Internetwork competition for monomers governs actin cytoskeleton organization. Nat. Rev. Mol. Cell Biol. 2016;17:799–810. doi: 10.1038/nrm.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian B., Sun H., Yan P., Charoonratana V.T., Higgs H.N., Wang F., Lai K.V., Valenzuela D.M., Brown E.J., Schlöndorff J.S. Mice with mutant Inf2 show impaired podocyte and slit diaphragm integrity in response to protamine-induced kidney injury. Kidney Int. 2016;90:363–372. doi: 10.1016/j.kint.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleiman H.Y., Roth R., Jain S., Heuser J.E., Shaw A.S., Miner J.H. Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. JCI Insight. 2017;2 doi: 10.1172/jci.insight.94137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Schlondorff J.S., Brown E.J., Higgs H.N., Pollak M.R. Rho activation of mDia formins is modulated by an interaction with inverted formin 2 (INF2) Proc. Natl. Acad. Sci. USA. 2011;108:2933–2938. doi: 10.1073/pnas.1017010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney K.F., Li R. Function and regulation of the Arp2/3 complex during cell migration in diverse environments. Curr. Opin. Cell Biol. 2016;42:63–72. doi: 10.1016/j.ceb.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traykova-Brauch M., Schönig K., Greiner O., Miloud T., Jauch A., Bode M., Felsher D.W., Glick A.B., Kwiatkowski D.J., Bujard H. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat. Med. 2008;14:979–984. doi: 10.1038/nm.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh G.I., Saleem M.A. The podocyte cytoskeleton–key to a functioning glomerulus in health and disease. Nat. Rev. Nephrol. 2011;8:14–21. doi: 10.1038/nrneph.2011.151. [DOI] [PubMed] [Google Scholar]
- Wu C., Asokan S.B., Berginski M.E., Haynes E.M., Sharpless N.E., Griffith J.D., Gomez S.M., Bear J.E. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Plotnikov S.V., Moalim A.Y., Waterman C.M., Liu J. Two distinct actin networks mediate traction oscillations to confer focal adhesion mechanosensing. Biophys. J. 2017;112:780–794. doi: 10.1016/j.bpj.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero J.B., Fu M.M., Sloan S.A., Ibrahim A., Olson A., Zaremba A., Dugas J.C., Wienbar S., Caprariello A.V., Kantor C. CNS myelin wrapping is driven by actin disassembly. Dev. Cell. 2015;34:152–167. doi: 10.1016/j.devcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of podocyte enriched proteins of isolated mouse podocytes measured by mass spectrometry. Index of each tab is included.
Analysis of podocyte enriched focal adhesions (isolated FA complexes) derived from immortalized human podocytes measured by mass spectrometry. Index of each tab is included.