

Abstract
The human 2-oxoglutaric acid dehydrogenase complex (hOGDHc) plays a pivotal role in the tricarboxylic acid (TCA) cycle, and its diminished activity is associated with neurodegenerative diseases. The hOGDHc comprises three components, hE1o, hE2o, and hE3, and we recently reported functionally active E1o and E2o components, enabling studies on their assembly. No atomic-resolution structure for the hE2o component is currently available, so here we first studied the interactions in the binary subcomplexes (hE1o–hE2o, hE1o–hE3, and hE2o–hE3) to gain insight into the strength of their interactions and to identify the interaction loci in them. We carried out multiple physico-chemical studies, including fluorescence, hydrogen–deuterium exchange MS (HDX-MS), and chemical cross-linking MS (CL-MS). Our fluorescence studies suggested a strong interaction for the hE1o–hE2o subcomplex, but a much weaker interaction in the hE1o–hE3 subcomplex, and failed to identify any interaction in the hE2o–hE3 subcomplex. The HDX-MS studies gave evidence for interactions in the hE1o–hE2o and hE1o–hE3 subcomplexes comprising full-length components, identifying: (i) the N-terminal region of hE1o, in particular the two peptides 18YVEEM22 and 27ENPKSVHKSWDIF39 as constituting the binding region responsible for the assembly of the hE1o with both the hE2o and hE3 components into hOGDHc, an hE1 region absent in available X-ray structures; and (ii) a novel hE2o region comprising residues from both a linker region and from the catalytic domain as being a critical region interacting with hE1o. The CL-MS identified the loci in the hE1o and hE2o components interacting with each other.
Keywords: tricarboxylic acid cycle (TCA cycle) (Krebs cycle); hydrogen exchange mass spectrometry; isothermal titration calorimetry (ITC); dehydrogenase; mass spectrometry (MS); chemical cross-linking MS; human 2-oxoglutarate dehydrogenase complex; neurodegeneration, oxidative stress; protein complex interaction loci; site-selected fluorescent labeling
Introduction
The human 2-oxoglutarate dehydrogenase complex (hOGDHc)4 is a key enzyme in the tricarboxylic acid (TCA) cycle. The diminished physiological activity of the hOGDHc and oxidative stress had been correlated with multiple neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease (1, 2); however, a link between diminished OGDHc activity and impaired glucose metabolism is not well established. At present, hOGDHc is recognized as a significant source of the reactive oxygen species (superoxide and H2O2) that could lead to oxidative stress in mitochondria (3, 4). Another function suggested for hOGDHc is that it could be involved in histone modifications by lysine succinylation that represents an important mechanism for regulation of gene expression (5). Also, reversible post-translational succinylation of the cytosolic and mitochondrial proteins by hOGDHc in neuronal cell lines and in cultured neurons was suggested (1).
Recently, we have reported findings in vitro suggesting both functional and regulatory cross-talk between the hOGDHc and a novel human 2-oxoadipate dehydrogenase complex from the final degradation pathway of l-lysine, l-hydroxylysine, and l-tryptophan, demonstrating that the two complexes share the same dihydrolipoyl succinyltransferase (hE2o) and dihydrolipoyl dehydrogenase (hE3) components (7). These findings raised important questions about assembly of the individual components in these two complexes and regarding the physiological relevance of such assemblies, information which is missing to date.
The hOGDHc is a macromolecular assembly comprising multiple copies of three enzyme components, similar to other 2-oxo acid dehydrogenase complexes, which display a remarkable active-site coupling mechanism (8, 9). The thiamin diphosphate (ThDP)-dependent 2-oxoglutarate dehydrogenase component (hE1o, EC 1.2.4.2) catalyzes the decarboxylation of 2-oxoglutarate, releasing CO2, followed by the reductive succinylation of the dihydrolipoamide succinyltransferase component (hE2o, EC 2.3.1.6), which has a single lipoyl domain, carrying a covalently amidated lipoic acid as a redox cofactor. The reductive succinylation reaction of hE2o by hE1o is followed by trans-thiol esterification of the succinyl group onto CoA in the active centers of hE2o, generating succinyl-CoA. The dihydrolipoamide dehydrogenase component (hE3, EC 1.8.1.4), with a tightly but noncovalently bound FAD as cofactor, re-oxidizes dihydrolipoamidated hE2o to commence the next turnover, while concomitantly reducing an equivalent of NAD+ to NADH (10–17).
Historically, the oligomeric E2 component has been assumed to be the core of the 2-oxo acid dehydrogenase multienzyme complexes, to which multiple copies of the E1 and E3 components (sometimes called peripheral components) are noncovalently attached (18–21). As to the OGDHc, both the human E2o and the Escherichia coli E2o components have a single N-terminal lipoyl domain (LDo) connected by a long linker to the core domain (CDo) (22). An amino acid sequence alignment of the hE2o with known E2o components from bacterial and mammalian sources has failed to identify any sequence corresponding to a peripheral subunit-binding domain (PSBD) for E1o and E3 binding (23–26) nor is there a fourth component present corresponding to E3-binding protein (E3BP), an additional component found in the mammalian pyruvate dehydrogenase complex (PDHc) (11, 27–31).
To date, there is no X-ray crystal structure for any of the 2-oxo acid dehydrogenase complexes in their intact state, possibly due to their size (Mr of 4–10 million) and flexible attachment of the E1 and E3 components to the core (25, 32, 33). The numbers and distribution of E1s and E3s around the core surface also vary from complex to complex (34). X-ray structural data have been published for truncated Escherichia coli E1o lacking its N-terminal region but providing information about the E1o active centers (35). However, no information about E1o contacts with the E2o and E3 components was apparent in this structure (35). Earlier, based on studies with mammalian OGDHc subjected to tryptic digestion, it was suggested that the N-terminal region of the mammalian E1o could form contacts with the E2o and E3 components (36–38). Additionally, an X-ray structure of the Mycobacterium smegmatis α-ketoglutarate decarboxylase lacking 115 N-terminal amino acid residues had been reported (39), once more missing information regarding the flexible N-terminal region. Structural approaches have been successfully applied to the E. coli E2o lipoyl domain (40), to the E. coli E2o truncated cubic core (41), and to the E. coli E3-binding domain of the E2o (42); however, no high-resolution X-ray structures for any full-length E2o component or E2o-core assembly are available at present.
These observations served as our hypothesis and motivation to establish the loci and strength of interaction in the binary hE1o–hE2o, hE1o–hE3, and hE2o–hE3 subcomplexes. Our principal structural tool was H/D exchange MS (HDX-MS), which the Rutgers' group has already applied (among other studies) to the following. (i) Elucidation of the interaction loci of the human pyruvate dehydrogenase complex E2·E3BP core with the human pyruvate dehydrogenase kinases 1 and 2 (43). (ii) An understanding of the structural alterations induced by disease-causing mutations in the human E3 component (44). Taken together, our data reported here suggest that both hE2o and hE3 interact with the same peptides located at the N-terminal region of hE1o, revealing that the N terminus of hE1o is pivotal for mediating assembly of the entire hOGDHc and suggesting a new mode of assembly in the superfamily of 2-oxo acid dehydrogenase complexes. We complement the HDX-MS studies with chemical cross-linking mass spectrometric studies. These cross-linking mass spectrometric studies provide strong evidence for which locus on hE1o interacts with which locus on hE2o and are in agreement with all other results.
Results and discussion
Probing the interactions between the hE1o, hE2o, and hE3 components in binary subcomplexes
The ability to produce active, full-length hE1o, hE2o, and hE3 components and to assemble them into functionally active hOGDHc enabled us to evaluate the strength and loci of interaction in the binary hE1o–hE2o, hE1o–hE3, and hE2o–hE3 subcomplexes. Experiments using size-exclusion chromatography (SEC) and isothermal titration calorimetry (ITC) revealed formation of a binary hE1o–hE2o subcomplex (Fig. 1). On measuring heats of interaction of hE1o with hE2o, formation of the binary subcomplex was found to be endothermic (positive peaks in the ITC input, ΔH = 12.1 kcal/mol) and displayed saturation with an estimated Kd of 0.95 ± 0.10 μm (Ka = 1.057 × 106 m−1) for their binding (Fig. 1A). Binding was also accompanied by a large positive entropy change (TΔS = 20.6 kcal/mol). However, a low N value of 0.1999 ± 0.0069 was estimated from the fit of the experimental data. The reduction in N value is likely due to self-assembly of the hE2o subunits to form a 24-meric cube core as in hOGDHc or even larger E2o assemblies. Thus, although the ITC experiment suggests strong interaction between the hE1o and hE2o, this method could not provide an accurate assessment of the stoichiometry of their binding.
Figure 1.

Isothermal titration calorimetry and size-exclusion chromatography to probe the formation of the binary hE1o–hE2o subcomplex. A, ITC probing of the hE1o–hE2o subcomplex formation. Top panel, raw data obtained from a series of 10-μl injections of hE1o (25 μm subunit concentration) into the cell containing hE2o (450 μm subunit concentration) and plotted as heat change versus time. Bottom panel: plot of the areas under peaks in A against the molar ratio of hE1o injected. The Kd of 0.95 ± 0.10 μm for the hE1o–hE2o subcomplex formation was calculated from the fit of the experimental data to a single site–binding model. B, SEC demonstration of the hE1o–hE2o binary subcomplex formation. The hE1o and hE2o were preincubated at 2:1 molar ratio of subunits in 50 mm KH2PO4 (pH 7.5) containing 0.15 m NaCl, 0.5 mm ThDP, and 1.0 mm MgCl2 for 1 h at room temperature followed by elution on an analytical Superose 6 column with 1 ml/min flow rate (black). In control experiments, the hE1o by itself was eluted with 14.4 ml; the ThDP by itself was eluted with 18–20 ml (both in green); the hE2o by itself was eluted in the void volume (brown). C, SDS-PAGE analysis of the eluted protein peaks (from B). Lanes 1–5 correspond to hE1o–hE2o binary subcomplex, eluted with 6–11 ml. Lane 6 corresponds to hE1o by itself eluted with 14–16 ml.
Formation of the hE1o–hE2o subcomplex was also suggested by a 1H-15N TROSY-HSQC NMR experiment using uniformly 15N-labeled hE1o as shown in Fig. S1. The superimposed 1H-15N TROSY-HSQC spectra of 15N-labeled hEo in the absence and in the presence of hE2o revealed multiple differences in the spectrum of 15N-labeled hE1o on interaction with hE2o. While at this level of analysis (no sequence-specific assignments were made on these large enzymes with mass of >200,000 Da for the hE1o homodimer and hE2o oligomer of >45,000 Da per subunit), these NMR results do not provide information about the loci of interaction, but they do support the presence of interaction in the binary hE1o–hE2o subcomplex.
In contrast, the hE3, which is shared by all members of the 2-oxo acid dehydrogenase complex family in a mammalian cell, forms unstable binary subcomplexes with hE1o and hE2o according to SEC experiments, where each protein in the preformed binary hE1o–hE3 and hE2o–hE3 subcomplex was eluted as an individual peak (Fig. S2). A similar conclusion could also be drawn from ITC titration experiments where hE1o or hE2o were titrated by hE3 (Fig. S3). The titration plots did not exhibit saturation (Fig. S3), more probably due to a weak and transient binding between components (34). According to the accepted OGDHc mechanism, with each turnover, the reduced lipoyl domain (dihydrolipoamide) on hE2o must be re-oxidized by the FAD on hE3 (45, 46).
The exact stoichiometry of the assembly of the hE1o, hE2o, and hE3 components into hOGDHc is unknown. Earlier it was reported that the isolated E. coli E2o core (octahedral (24-mer)) could bind six E1o dimers (α2) compared with the E. coli E2p (octahedral (24-mer)) that could bind 12 E1p (α2) dimers and six E3 (α2) dimers (14). On assembly of the hOGDHc from its individual components followed by removal of the excess of hE1o and hE3 by passing their mixture through the analytical column, a molar ratio of the hE1o/hE2o/hE3 subunits of 10.2:13.4:2.7 was estimated from three independent experiments. This subunit composition corresponds to an association of 5.1 hE1o dimers, 4.5 hE2o trimers, and 1.4 hE3 dimers corresponding to an approximate stoichiometry of 3(hE1o)2/3(hE2o)3/1(hE3)2. These data suggest that each (hE2o)3 binds one (hE1o)2, and this stoichiometry is unaffected by the hE3 component (Fig. S4).
Strength of hE1o–hE2o interactions as quantified by site-specifically introduced external fluorophores into hE2o-derived proteins
To help identify the loci of interaction between the hE1o, hE2o, and hE3 components of the hOGDHc, we created truncated versions of the hE2o:hE2o(1–95) (lipoyl domain, LDo, comprising the lipoyl domain and part of the linker region), the hE2o(1–173) di-domain, comprising the LDo, the linker region and part of the hE2o core domain, and the C-terminal hE2o(144–386) core domain, consisting of the hE2o core domain and part of the linker region (see Fig. 2 for domain structure of the hE2o). Two approaches were developed to introduce an external fluorophore site-specifically onto the truncated versions of the hE2o. In the first approach, the lipoamide carrying hE2o proteins, the hE2o(1–95) lipoyl domain, and the hE2o(1–173) di-domain were dansylated (Scheme S1). The lipoamide in both proteins was first reduced to dihydrolipoamide using TCEP, then it was labeled with a dansyl group that had been tethered to the amino group of 4-aminophenyl arsenoxide, resulting from a reaction of the trivalent arsenoxide with the reduced dihydrolipoamide (Scheme S1) (47). In the second approach, N-(1-pyrene) maleimide was used to label the unique Cys37 in the hE2o(1–173) di-domain and Cys178 in the hE2o(144–386) core domain. Titration of DANS-As–labeled hE2o(1–95) lipoyl domain by hE1o resulted in quenching of the fluorescence of dansylamide, as was also observed on titration of DANS-As–labeled hE2o(1–173) di-domain, suggesting that on interaction with hE1o, a more hydrophilic microenvironment was created around the fluorophore (Fig. 3) (48). The calculated Kd values were as follows: 0.135 μm (with hE2o(1–95) lipoyl domain) and 0.039 μm (with hE2o(1–173) di-domain), providing the first hint that the residues from the linker region, which connects the lipoyl domain and the hE2o catalytic domain and is present in the hE2o(1–173) di-domain, could be an important determinant for the hE1o–hE2o interaction.
Figure 2.
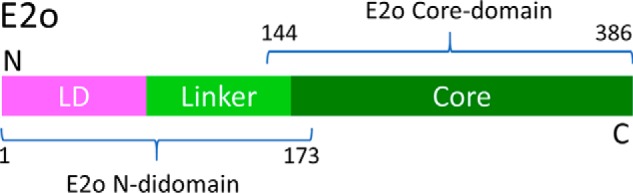
Domain structure of the hE2o. The following E2o-derived proteins were created: hE2o(1–95) (lipoyl domain, LDo, encompassing the lipoyl domain and the linker region partially); hE2o(1–173) di-domain, encompassing the LDo, the linker region and the core domain partially; and hE2o(144–386), hE2o core domain (CDo, encompassing the core domain and the linker region partially). The hE2o(1–173) di-domain and hE2o(144–386) core domain have 30-amino acid overlapping sequence.
Figure 3.
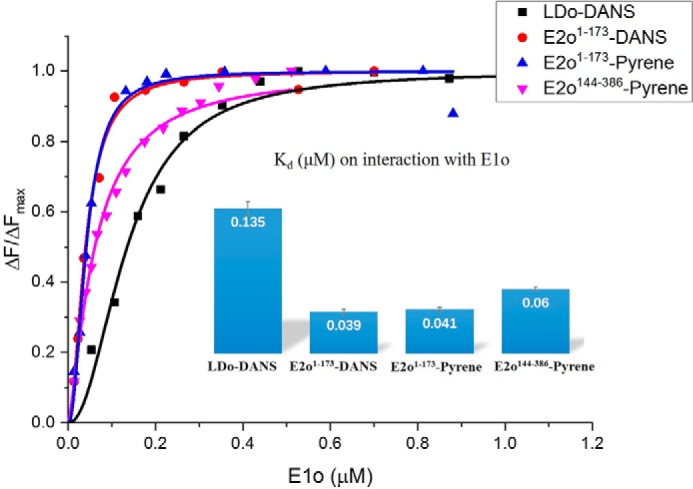
Fluorescence titration curves for DANS-As–labeled. The hE2o(1–95) lipoyl domain and hE2o(1–173) di-domain and for pyrene labeled hE2o(1–173) di-domain and hE2o(144–386) core domains by hE1o are shown. In all cases, the fluorescence intensity of the fluorophore-labeled hE2o-derived protein was quenched on titration by hE1o. In a typical experiment, the fluorophore-labeled hE2o-derived protein (1.0–2.0 μm concentration of subunits) in 30 mm KH2PO4 (pH 7.5) was titrated by hE1o (0.01–1.5 μm) at room temperature. The excitation wavelength was 338 nm for DANS and 340 nm for pyrene labeled hE2o-derived proteins. The fluorescence titration curves were fitted by using the Hill equation (Equation 5). Inset: calculated Kd values are presented.
With the N-(pyrene) maleimide-labeled hE2o(1–173) di-domain and hE2o(144–386) core domain proteins, on excitation at 340 nm, three major fluorescence emission bands were in evidence (374, 395, and 416 nm) (Fig. 4). The maximum emission intensities at 374 nm were used to calculate binding parameters. In the spectrum of the hE2o(1–173) di-domain, there was an additional broad peak centered around 470 nm, which is likely due to excimer formation (Fig. 4) according to the literature (49).
Figure 4.

Fluorescence titration of pyrene-labeled hE2o(1–173) di-domain and pyrene-labeled hE2o(144–386) core domain by hE1o. A, quenching of the fluorescence of pyrene-labeled hE2o(1–173) di-domain by hE1o. Pyrene-labeled hE2o(1–173) di-domain (1.5 μm active centers) in 30 mm KH2PO4 (pH 7.5) was titrated by hE1o (0.013–0.261 μm). B, enhancement of the fluorescence of the pyrene-labeled hE2o(144–386) core domain on hE1o binding. Pyrene-labeled hE2o(144–386) core domain (1.5 μm) in 30 mm KH2PO4 (pH 7.5) was titrated by hE1o (0.013–0.261 μm).
Addition of hE1o to the pyrene-labeled hE2o(1–173) di-domain quenched the pyrene fluorescence (Kd = 0.041 μm), whereas addition of hE1o to the pyrene-labeled hE2o(144–386) core domain enhanced the pyrene fluorescence intensity (Kd = 0.06 μm), without any blue or red shift of the peak position (Fig. 3). The results suggested that interactions of hE1o with hE2o(1–173) di-domain or with the hE2o(144–386) core domain induce different changes in the microenvironment around the pyrene fluorophore; interaction between hE1o and labeled hE2o(1–173) leads the pyrene fluorophore to a relatively more hydrophilic environment, whereas interaction of hE1o with labeled hE2o(144–386) leads the pyrene fluorophore to a more hydrophobic environment. The calculated Kd values are in good accord when the interaction with the same protein is measured with the two different fluorophores (Fig. 3). The fluorescence binding experiments enabled us to conclude that the linker region in hE2o located between the lipoyl domain and hE2o catalytic domain participates in the interaction with hE1o.
Deuterium uptake changes detected by HDX-MS in hE1o on interaction with hE2o and its truncated proteins
As a first step, HDX-MS experiments were conducted with full-length hE1o and hE2o components. On-line digestion by pepsin, followed by LC-MS analysis, resulted in 99 peptides for hE1o (Table S1) and 19 peptides for hE2o (Table S2), many of them partially overlapping, providing 94% sequence coverage for hE1o and 66.8% sequence coverage for hE2o. The high-sequence coverage on hE1o, including the N-terminal flexible region corresponding to residues 1–77, which was missing (residues 1–77 removed by proteolysis) from the previous crystallographic study of the E. coli E1o (35), enabled us to study the structural changes on assembly of the binary subcomplexes. On digestion of hE2o with pepsin, almost 100% sequence coverage resulted for its core domain, corresponding to residues 144–386. However, the information about many peptides from the lipoyl domain and linker region was missing due to being relatively sparse in hydrophobic residues, proline residues (total of 26 prolines in these two regions), and charged residues, leading to poor digestion by pepsin. Because of all the above considerations, we also examined interaction of hE1o with the hE2o(1–173) di-domain and the hE2o(144–386) core domain.
A summary of the HDX-MS analysis of the peptic peptides originating from hE1o in the absence and in the presence of hE2o is provided in Fig. 5. Below, we focus the discussion on that subset of hE1o peptides, which displayed statistically significant difference in deuterium uptake upon interaction with hE2o. The major findings on hE1o are as follows.
Figure 5.

Comparative HDX-MS analysis of the hE1o by itself and of the hE1o on interaction with hE2o. A, Butterfly plot representing relative deuterium incorporation percentage (ΔD%, y axis), deuterons exchanged/maximum exchangeable amides × 100% of peptic peptides from E1o (x axis, listed peptic peptides from the N to C terminus) in the absence of hE2o (right) versus in the presence of hE2o (left) based on three independent experiments. B, difference plot showing the changes in deuterium incorporation of peptic fragments of hE1o in the absence and presence of hE2o (ΔΔD, y axis), (deuterons exchanged in the absence and presence of hE2o).
(i) The HDX-MS experiments indicate that the interaction of hE1o with hE2o did not induce large-scale conformational changes in hE1o (Fig. 5B). However, local backbone amide proton perturbations were indeed observed.
(ii) Importantly, two regions from the N-terminal end of hE1o comprising residues 18YVEEM22 and 27LENPKSVHKSWDIF40 were the unique regions, which experienced significant deuterium uptake retardation on interaction with full-length hE2o (Fig. 5), and also on interaction with the hE2o(1–173) di-domain and hE2o(144–386) core domain at all exchange time points analyzed (Fig. 6). Thus, the hE1o peptides comprising residues 18YVEEM22 and 27LENPKSVHKSWDIF40 represent the most likely candidates for binding with hE2o.
Figure 6.

A, deuterium uptake difference plot for peptide encompassing residues 18–22 in hE1o on interaction with hE2o, hE2o(1–173) di-domain, and hE2o(144–386) core domain. B, deuterium uptake difference plot for peptide encompassing residues 27–40 in hE1o on interaction with hE2o, hE2o(1–173) di-domain, and hE2o(144–386) core domain.
(iii) The HDX-MS results, together with the predicted secondary structure of the N-terminal region of hE1o presented in Fig. 7, suggested that upon binding of the hE2o to hE1o, the two predicted helical N-terminal regions of hE1o consisting of residues 18YVEEM22 and 27LENPKSVHKSWDIF40 became less flexible.
Figure 7.

Predicted secondary structure for the N-terminal region of the hE1o encompassing residues 1–152. The calculations were performed using the JPred program (http://www.compbio.dundee.ac.uk/jpred/) (65, 66). (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.) The highlighted two α-helices (in red) were found to have significant decreases in the level of deuterium uptake when hE1o was incubated with hE2o. The tertiary structure prediction was performed using I-TASSER and SWISS-MODEL (http://swissmodel.expasy.org/) was employed.
(iv) In addition to the N-terminal region of hE1o, some peptides near the putative ThDP- and Mg2+-binding sites displayed modestly increased deuterium uptake (peptide consisting of residues 407IGFTTDPRM415 and two peptides encompassing residues 529EAFARSKDEKILHIKHWLDSPWPGF553 and 554FTLDGQPRSMSCPSTGLTE572). The magnitude of these small perturbations could be related to perturbations in the hE1o catalytic center in response to hE2o binding. These results lead us to a major conclusion that two N-terminal regions of hE1o comprising residues 18YVEEM22 and 27LENPKSVHKSWDIF40 are, most likely, candidates for binding with hE2o.
Deuterium uptake changes identified in hE2o by HDX-MS on interaction with hE1o
The Butterfly plot of the peptic peptides originating from hE2o by itself and in the presence of hE1o (Fig. 8) revealed that there were no major changes in deuterium uptake upon hE1o binding. The lipoyl domain was too rigid to be digested by pepsin, resulting in low peptide coverage as was mentioned above. Also, the hE2o region comprising residues from the linker region and from the beginning of the core domain were not covered due to a lack of pepsin cleavage in these regions. The Butterfly plot also revealed that the following regions in the hE2o core domain were highly protected even in the absence of hE1o: 181LTTFNE186, 217FVKASAF223, and 369LRKIKAAVEDPRVLL383, all three of which had almost no deuterium uptake at all times of exchange used (Table S2 and Fig. 8B).
Figure 8.

Comparative HDX-MS analysis of the hE2o by itself and in the presence of hE1o. Top panel, Butterfly plot representing average relative deuterium incorporation percentage (ΔD%, y axis) (deuterons exchanged/maximum exchangeable amides × 100%) of peptic peptides from the hE2o, (x axis, listed peptic peptides from the N to C terminus)) in the absence of hE1o (top part) versus in the presence of hE1o (bottom part). Bottom panel, difference plot showing the change in deuterium incorporation of peptic fragments originated from hE2o in the absence and presence of hE1o (ΔΔD, y axis) (deuterons exchanged in the absence minus deuterons exchanged in the presence of hE1o).
In view of the above uncertainties, we also carried out experiments with the hE2o(1–173) di-domain and hE2o(144–386) core domain, which provided complementary coverage of the linker region (Tables S3 and S4). Upon binding of the hE2o(1–173) di-domain to hE1o, the peptide encompassing residues 134AVKPTVAPPLAEAGAGKGLRSEHREKMNRMRQRIAQRLK172 mostly from the hE2o linker region, and also including the N-terminal region of the core domain, revealed a significant decrease in deuterium uptake during the first 3 min of the exchange time (Fig. 9A). However, this deuterium uptake retardation quickly faded at a longer exchange time indicating a weaker protection by hE1o at a longer time scale. The data suggest involvement of the hE2o stretch of residues 134AVKPTVAPPLAEAGAGKGLRSEHREKMNRMRQRIAQRLK172 in its interaction with hE1o. In a complementary experiment, it was shown that upon binding of hE2o(144–386) core domain to hE1o, the N-terminal peptide comprising residues 144AEAGAGKGLRSEHREKMNRMRQRIAQRLKEAQNTCAM180 from the linker region and including the first 33 N-terminal residues of the hE2o(144–386) core domain also displayed a significant decrease in deuterium uptake. The peptide comprising residues 134–172 in hE2o(1–173) di-domain and the peptide comprising the first 33 residues in the hE2o(144–386) core domain overlap significantly, having 30 amino acids in common, corresponding to the following residues: 144AEAGAGKGLRSEHREKMNRMRQRIAQRLKE173. Because this is the only hE2o region that displayed a significant decrease in the level of the deuterium uptake upon hE1o binding, this region could be critical for the hE1o–hE2o interaction. A sequence alignment of this 30-amino acid–long region with some known peripheral subunit-binding domains in E2 components (Fig. 9B) allowed us to identify conserved residues, suggesting that hE2o shares some but not all sequence features of other subunit-binding domains that are believed to be important for E1o binding. This experiment allowed us to conclude that in contrast to other E2s, the binding region of hE2o is extended to its core domain with corresponding residues (157REKMNRMRQRIAQRLKE173) that represent a unique hE2o-binding mode in the human OGDHc (remark: the residues are not unique but the hE2o-binding mode is unique). The observation that the linker-core region of hE2o is the critical region interacting with hE1o does raise the possibility that this region on hE2o might also serve as a binding site for the hE3 component.
Figure 9.

A, deuterium uptake difference plot for peptide encompassing residues 134–172 in hE2o on interaction with hE1o. B, sequence alignment of the human hE2o linker-core region with sequences of the known peripheral subunit-binding domains. The abbreviations are denoted: human (h), Escherichia coli (Ec), Bacillus stearothermophilus (Bst), Pseudomonas putida (Pp), and Azotobacter vinelandii (Av), 2-oxoglutarate dehydrogenase complex (o), pyruvate dehydrogenase complex (p), and branched chain 2-oxoacid dehydrogenase complex (b). Alignment of multiple sequences was carried out using the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/) (64) with default settings. (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.)
HDX-MS identification of the binding loci in the E1o–E3 and E2o–E3 binary subcomplexes
Digestion of hE3 by pepsin followed by LC-MS analysis resulted in 27 peptides, with some of them partially overlapping, and provided 90.9% sequence coverage (Table S5). Upon binding of hE1o to hE3, many regions on both hE1o and hE3 were observed to undergo changes in deuterium uptake (Figs. S5 and S6). (i) On the hE1o, significant changes were identified in the N-terminal region and in the ThDP-binding fold (Fig. S5). In particular, peptides encompassing residues 18YVEEM22, 27LENPKSVHKSWDIF40, and 119DSSVPADIISSTDKLGF135 from the N-terminal region of hE1o and peptides encompassing residues 299EAADEGSGDVKYHLGM314, 315YHRRINRVTDRNIT328, 505LVSQGVVNQPEYEEE519, 529EAFARSKDEKILHIKHWLDSPWPGF553, and 554FTLDGQPRSMSCPSTGLTE572 from the ThDP-binding fold of hE1o displayed a decrease in deuterium uptake during the first 3 min of exchange (Fig. S5). Notably, the two peptides with residues 18YVEEM22 and 27LENPKSVHKSWDIF40 from the N-terminal region of hE1o displayed the most significant decrease in deuterium uptake while associated with hE3. These two peptides were also found to be significantly protected in the hE1o–hE2o subcomplex (Fig. 5). This finding, together with the fact that the N-terminal region of hE1o showed limited similarity to the corresponding sequence in the hPDHc E3-binding protein, suggested that the N-terminal region of hE1o, in particular the two peptides 18YVEEM22 and 27ENPKSVHKSWDIF39, constitute the binding region responsible for the assembly of the hE1o with both the hE2o and hE3 components into hOGDHc.
On the hE3 partner, upon binding to hE1o, two peptides encompassing residues 35EKNETLGGTCLNVGCIPSKALLNNSHYYHMAHGKD69 and 107FKQNKVVHVNGYGKITGKNQVTATKADGGTQVIDTKNIL145 in the FAD-binding domain of hE3, and one peptide 381YKVGKFPFAANSRAKTNADTDGM403 from the interface domain of hE3, showed the most significant decrease in deuterium uptake (Fig. S6). This result is consistent with our data reported recently (43) where the same hE3 peptides showed a decrease in deuterium uptake on interaction with the hE3-binding domain of the human PDHc, indicating a similar binding mode in both.
In contrast to the evidence for the hE1o–hE3 interaction, the HDX-MS data did not provide clear evidence for direct binding between hE2o and hE3. Even though during the catalytic cycle of hOGDHc, the reduced lipoic acid covalently attached to hE2o must be re-oxidized by FAD on hE3, this transient interaction could be too weak to be captured by the method used here, suggesting that the presence of hE1o is needed for assembly of hE3 into hOGDHc.
Chemical cross-linking of hE1o and hE2o
The HDX-MS studies above provide strong evidence for the loci of inter-component interactions on both the hE1o and hE2o components at a peptide resolution. But, when multiple interaction loci are identified on each component, as is the case here, the analysis needs to be taken to the next higher level, i.e. which site on hE1o interacts with which site on hE2o? To address this question, we carried out chemical cross-linking analysis of full-length hE1o and hE2o components by using two cross-linkers to react with the ϵ-amino side chain of the lysine residues: 1,1′-carbonyldiimidazole (CDI, Cα–Cα distance ∼16 Å to be bridged) (50) and urea-based disuccinimidyl dibutyric urea (DSBU or BuUrBu, Cα–Cα distance ∼27 Å to be bridged) (51). These two cross-linkers have an advantage of being cleaved under mass spectrometric conditions. The covalent adducts were analyzed by MS. We identified 160 and 23 unique cross-linked residue pairs (cross-links) with CDI and BuUrBu, respectively, at p < 0.01. The majority of cross-links identified are intra-component cross-links, i.e. two lysines from the same component are being cross-linked. Of the several inter-component cross-linked peptides (60 cross-linked residue pairs), we selected the ones with p < 0.001 (Table 1). The following cross-links were identified.
Table 1.
Inter-component cross-links between the hE1o and hE2o components identified by LC-MS-MS for the CDI and BuUrBu cross-linkers

a CDI is 1,1′-carbonyldiimidazole (Cα–Cα distance ∼16 Å to be bridged).
b BuUrBu is disuccinimidyl dibutyric urea (Cα–Cα distance ∼27 Å to be bridged).
On hE1o, the cross-linked Lys31 is located in the N-terminal region, and the other cross-linked lysines are located on the hE1o surface near the putative ThDP- and Mg2+-binding sites (see Table 1 and Fig. 10 for cross-linked lysines mapped onto the hE1o structure). Importantly, the cross-link between hE1o Lys31 and hE2o Lys85 from the linker region was identified by both CDI and BuUrBu providing complementary information on similar hE1o–hE2o interaction. As identified by HDX-MS above, Lys31 is located on a unique peptide comprising residues 27LENPKSVHKSWDIF40 in hE1o that experienced significant deuterium uptake retardation on interaction with the full-length hE2o and also on interaction with the hE2o(1–173) di-domain and the hE2o(144–386) core domain (see Fig. 10). A film (see Fig. S7 a film in mpg) clearly indicates that the hE1o lysines participating in the cross-links are all on the surface.
Figure 10.
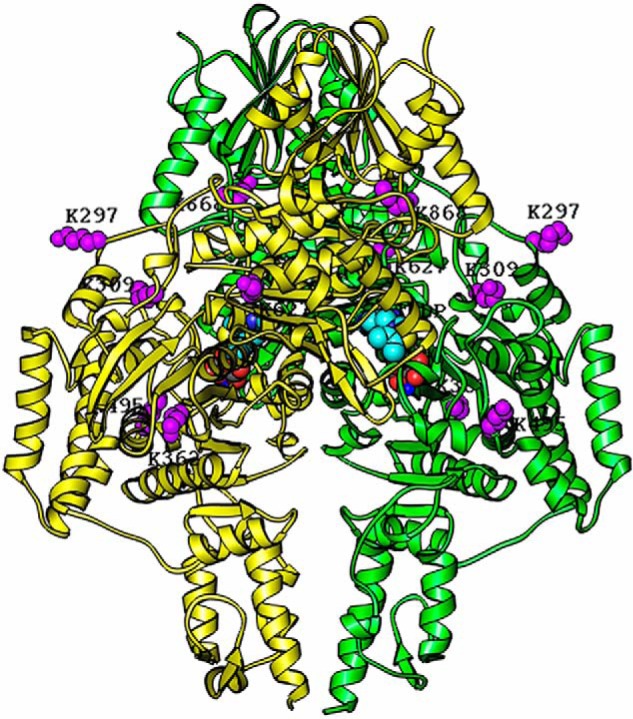
Homology modeling of the hE1o dimer structure with lysine residues involved in inter-component cross-linking highlighted in purple. The structure of the hE1o dimer was modeled based on amino acid sequence homology with the E. coli E1o (PDB code 2JGD (35)) and M. smegmatis E1o (PDB code 2YIC (39)) whose X-ray structures are available. The hE1o homodimer is colored in yellow and green. The identified inter-component cross-linked lysine residues (Lys297, Lys309, Lys362, Lys495, Lys627, and Lys868) are highlighted in purple and were positioned by sequence alignment of the hE1o with E. coli E1o. The identified lysine residues and ThDP cofactors are shown as space-filling representations. The program RIBBONS (6) was used to create this figure.
On hE2o, the cross-linked lysines identified were as follows: Lys78 from the lipoyl domain; Lys150 from the linker region; and Lys172, Lys289, and Lys371 from the hE2o core domain. The Lys172 is part of the hE2o(1–173) di-domain that displayed ∼3.5-fold weaker binding constant for hE1o compared with lipoyl domain by itself. Importantly, the hE2o region comprising residues 144AEAGAGKGLRSEHREKMNRMRQRIAQRLKE173 from the linker region (residues 144–156) and from the core domain (residues 157–173) displayed a significant decrease in deuterium uptake upon hE1o binding. The cross-linking experiments provide strong support for the involvement of the hE2o core domain in the hE1o–hE2o subcomplex, not realized previously for any of the E1–E2 subcomplexes.
Conclusions
Strong interactions were identified between the hE1o- and hE2o-derived truncated proteins. The fluorescence experiments using the hE2o(1–173) di-domain and hE2o(144–386) core domain, which revealed strong interactions with hE1o (Kd in the 0.04–0.14 μm range), implied that the hE2o linker region connecting the hE2o lipoyl domain and hE2o core domain is involved in the interaction with hE1o.
In the hE1o–hE2o binary subcomplex, two peptides from the N-terminal region of hE1o comprising residues 18YVEEM22 and 27LENPKSVHKSWDIF40 were identified, which experienced significant deuterium uptake retardation on complexation with hE2o and its derived hE2o(1–173) di-domain and hE2o(144–386) core domain proteins. Therefore, these two hE1o peptides represent the most likely candidates for binding with hE2o.
On hE2o, the region comprising residues from the core domain (in bold) and linker region (AEAGAGKGLRSEHREKMNRMRQRIAQRLKE) displayed a significant decrease in the level of deuterium uptake upon complexation with hE1o, and this could be critical for the hE1o–hE2o interaction. In contrast to other E2s, part of the hE2o core domain also participates in the interaction with hE1o, suggesting a unique subunit-binding mode in human OGDHc assembly.
For the hE1o–hE3 binary subcomplex, only weak interactions could be observed. According to HDX-MS, similarly to the finding with the hE1o–hE2o subcomplex, two peptides spanning residues 18YVEEM22 and 27LENPKSVHKSWDIF40 from the N-terminal region of hE1o displayed the most significant decrease in deuterium uptake upon assembly with hE3. On the hE3 partner, two peptides comprising residues 35–70 and 107–145 from the FAD-binding domain and one peptide comprising residues 381–403 from the interface domain of hE3 displayed a decrease in deuterium uptake on complexation with hE1o.
The HDX-MS data failed to provide convincing evidence for direct interaction between hE2o and hE3 in the absence of hE1o. In view of the obligatory interaction of the dihydrolipoyl domain of hE2o with the FAD-binding site of hE3, this result suggests a weak and short-lived interaction.
Given the fact that the N-terminal region of hE1o displays only limited similarity to corresponding sequences in the E3-binding protein, residues 18–40 in hE1o may constitute a unique dual-subunit–binding domain (DSBD) in hOGDHc. Similar to the PSBD in the bacterial E2 enzymes, in the hOGDHc assembly both the hE2o and hE3 components bind to hE1o via this novel DSBD. It is worth noting that the dimeric hE1o component could bind hE2o and hE3 simultaneously.
Finally, two cross-linking agents were used to identify the following. (a) The most prominent loci on hE1o interacting with hE2o was indeed the N-terminal region once more, and the thiamin diphosphate environments were identified. (b) On hE2o, the lipoyl domain and the linker-core region were found to participate in cross-links with hE1o. These two powerful mass spectrometric methods provided complementary results in excellent agreement with each other.
In summary, this study identified unique sites for inter-component interactions in this important human TCA enzyme complex. On the hE1o component, the N-terminal region was identified as a binding locus for both hE2o and hE3. An initial formation of the uniquely strong hE1o–hE2o interaction facilitates assembly with hE3 to form the human OGDHc assembly.
Experimental procedures
Materials
The materials and sources used in this study are as follows. ThDP, dl-α-lipoic acid, IPTG, thiamin·HCl, imidazole, and isopropyl 1-thio-β-d-galactopyranoside (IPTG) were purchased from Affymetrix. DNase, micrococcal nuclease, and E. coli BL21(DE3) were purchased from New England Biolabs. Deuterium oxide (D2O) and 15NH4Cl (99%) were from Cambridge Isotope Laboratories. Dansyl chloride, p-arsanilic acid, benzamidine hydrochloride hydrate, and 2,6-dichlorophenolindophenol sodium salt hydrate were from Sigma. N-(1-Pyrene) maleimide was purchased from AnaSpec, Inc. KOH, HEPES, KCl, MgCl2, and phenylmethylsulfonyl fluoride (PMSF) were from ThermoFisher Scientific. Nickel-Sepharose 6 Fast flow was from GE Healthcare.
Construction of plasmids for expression of C-terminally truncated hE2o proteins
The pET-15b-hE2o plasmid encoding the His6 tag hE2o from the N-terminal end was used as a template, and the amplification primers and their complements were used for site-directed mutagenesis to introduce a TAA stop codon in place of Lys88, Glu174, and Gln176 (the numbers include Met as the first amino acid). The following C-terminally truncated hE2o proteins were created: hE2o(1–87), so-called lipoyl domain comprising the lipoyl domain (residues 1–77) and part of the linker region (residues 78–87); hE2o(1–173) di-domain comprising the LDo and part of the linker region. The C-terminally truncated hE2o proteins were expressed in BL21 (DE3) cells (New England Biolabs) in LB medium supplemented with ampicillin (50 μg/ml) and 0.3 mm dl-α-lipoic acid (Affymetrix). Protein expression was induced by 0.5 mm IPTG at 30 °C for 15 h. Cells were collected and were stored at −20 °C.
Construction of plasmid for expression of N-terminally truncated hE2o(144–386) core domain
The gene encoding C-terminally His6-tagged hE2o(144–386) core domain was synthesized by ATUM (Newark, CA). The gene was optimized for expression in E. coli cells and was inserted into the pET-22b(+) vector through the NdeI and XhoI restriction sites, and the resulting plasmid was expressed in BL21(DE3) cells. Protein expression was induced by 0.5 mm IPTG for 15 h at 18 °C. The harvested cells were stored at −20 °C.
Expression and purification of hE1o, hE2o, and hE3 components
Expression and purification of hE1o and hE3 was as reported earlier (52). Purification of hE2o was carried out as reported by us earlier for purification of the human E2p·E3BP core (43).
Purification of C- and N-terminally truncated hE2o proteins
Purification of C- and N-terminally truncated hE2o proteins was as reported by us earlier for the E. coli E2o di-domain (53, 54). Cells were dissolved in 50 mm KH2PO4 (pH 7.5) containing 1 mm benzamidine·HCl, 1 mm PMSF, 0.30 m NaCl, and 20 mm imidazole. Lysozyme was added to a final concentration of 0.60 mg/ml, and cells were incubated for 20 min at 4 °C. Next, 1000 units of DNase I (New England Biolabs) and micrococcal nuclease (New England Biolabs) were added, and the cells were incubated for an additional 40–60 min at 4 °C. Cells were then disrupted using a sonic dismembrator. For more complete removal of nucleic acids, the clarified lysate was additionally treated by streptomycin sulfate (0.8% w/w final concentration) for 20 min at 4 °C. The clarified lysate was applied to the Ni-Sepharose 6 Fast Flow column equilibrated with 50 mm KH2PO4 (pH 7.5) containing 0.3 m NaCl (buffer A) and 20 mm imidazole. The column was washed with 700 ml of buffer A containing 20 mm imidazole and then with 300 ml of buffer A containing 35 mm imidazole. The protein was eluted with 300 mm imidazole in buffer A. Next, the protein was dialyzed against 50 mm KH2PO4 (pH 7.5) containing 0.5 mm EDTA, 1 mm benzamidine·HCl, and 0.3 m NaCl for 15 h at 4 °C, followed by protein concentration using a concentrating unit with 10,000 MWCO, and finally the buffer was exchanged to 50 mm KH2PO4 (pH 7.5) containing 0.5 mm EDTA, 1 mm benzamidine·HCl, and 0.15 m NaCl. Proteins were stored at −80 °C.
Labeling of the lipoyl domain with DANS-As
The in vitro lipoylated LDo (50 μm concentration, mass of 11,257.70 Da) in 30 mm KH2PO4 (pH 7.5) containing 0.15 m NaCl was incubated with 100 μm TCEP for 5 min at room temperature, resulting in fully reduced LDo according to FT-MS (mass of the reduced LDo = 11,259.72 Da), which was then reacted with 150 μm DANS-As for 2 h at room temperature to form chemically modified LDo. The excess of TCEP and DANS-As was removed using a centrifugal filter unit (Vivaspin 500, 10,000 MWCO). The DANS-As-labeled LDo was diluted to 1 μm with the ESI buffer (50:50:0.1 v/v/v mixture of methanol, water, and formic acid) and was analyzed by FT-MS in the positive ion mode as reported by our group earlier (47). The mass of the DANS-As labeled LDo was estimated to be 11,658.76 Da, in good accord with the expected theoretical mass of 11,646.2 Da.
Labeling of the hE2o(1–173) di-domain and hE2o(144–386) core domain with N-(1-pyrene) maleimide
The hE2o(1–173) di-domain and hE2o(144–386) core domain (50 μm), each containing a single cysteine residue at positions 38 and 179, respectively, were incubated with 100 μm TCEP in 30 mm KH2PO4 (pH 7.5) containing 0.15 m NaCl for 5 min at room temperature. Then, 100 μm N-(1-pyrene) maleimide was added, and the reaction mixture was incubated for 2 h at room temperature. The excess of TCEP and N-(1-pyrene) maleimide were removed from the reaction mixture using a centrifugal filter unit (Vivaspin 500, 10,000 MWCO). The protein labeling was confirmed by LC-MS. The labeled proteins were first digested with trypsin. The LC-MS was carried out to identify the peptides derivatized with the fluorophore. The pyrene-labeled peptide originated from the hE2o(144–386) core domain (EAQNTCAMLTTFNEIDMSNIQEMR), and the mass of 3090.32 Da was identified in accord with the expected theoretical mass of 3090.32 Da. The mass of the pyrene-labeled peptide (AVGDTVAEDEVVCEIETDK) from the hE2o(1–173) di-domain with a mass of 2510.06 Da was also in good accord with the theoretical mass of 2510.05 Da.
Preparation of the 15N-labeled hE1o for NMR studies
A single colony from the plate was inoculated into 20 ml of the LB medium supplemented with 50 μg/ml ampicillin, and the cells were grown at 37 °C overnight. Cells were collected by centrifugation, dissolved in 20 ml of the minimal medium supplemented with 15NH4Cl (1.0 g/liter), and inoculated into 800 ml of the minimal medium supplemented with 15NH4Cl (1.0 g/liter). Cells were grown to an A600 of 0.5–0.6 at 37 °C, and then the temperature was lowered to 20 °C, and the protein expression was induced by the addition of 0.50 mm IPTG. Cells were grown for 16 h. The 15N-labeled hE1o was purified using a Ni-Sepharose 6 Fast Flow column with 10–300 mm imidazole gradient in 50 mm K2HPO4 (pH 7.5) containing 500 mm NaCl, 0.5 mm ThDP, and 1.0 mm MgCl2. The purified 15N-labeled E1o was stored at −80 °C.
Size-exclusion chromatography
SEC was performed using a Varian ProStar HPLC system with a UV detector. A Yarra 3μ SEC-3000 column with a 20-μl sample loop was used at a flow rate of 1 ml/min. The column was equilibrated with 50 mm KH2PO4 (pH 7.5) containing 0.15 m NaCl, 0.5 mm ThDP, 1 mm MgCl2 and was calibrated with the following protein standards (mass in Da in parentheses): thyroglobulin (669,000), ferritin (440,000), catalase (232,000), aldolase (158,000), BSA (67,000), and ovalbumin (43,000). The elution of proteins from the column was at a flow rate of 1 ml/min and was monitored at 280 nm. The hE1o, hE2o (or hE2o truncated proteins), and hE3 were pre-incubated at various molar ratios for 1 h at room temperature. Samples were centrifuged at 17,500 × g for 5 min to remove any precipitated contaminations and were applied to the column.
Isothermal titration calorimetry
ITC measurements were performed using a VP-ITC microcalorimeter (MicroCal, Northampton, MA). Titration experiments were carried out in 50 mm KH2PO4 (pH 7.5), containing 100 mm KCl, 0.5 mm ThDP, and 2 mm MgCl2 at 15 °C. The proteins were dialyzed for 15 h against the above buffer prior to the experiment. For conditions of experiments, see the following: legend to Fig. 1 for the hE1o–hE2o interaction and Fig. S3 for the hE3–hE1o and hE3–hE2o interactions. In a typical experiment, the hE1o at 25–30 μm concentration was titrated by hE2o (or hE2o truncated proteins) or hE3 at 300–450 μm concentration. A single-site binding model was used for curve fitting, and the binding constant (1.0/Kd) was calculated using Origin version 7.0 software.
Fluorescence spectroscopy studies
All fluorescence spectra were recorded using a Varian Cary Eclipse fluorescence spectrophotometer. The titration experiments were conducted in 30 mm KH2PO4 (pH 7.5), containing 0.15 m NaCl.
For quenching the fluorescence of DANS-As–labeled hE2o(1–95) lipoyl domain, hE2o(1–173) di-domain, and hE2o(144–386) core domain by hE1o, the DANS-As–labeled hE2o protein (1–2 μm concentration of subunits) in 30 mm KH2PO4 (pH 7.5) was titrated by hE1o (0.05–1.05 μm) at room temperature. The excitation wavelength was 338 nm, and the emission spectra were recorded in the 400–650 nm range. To calculate Kd values, the fluorescence titration curves were fitted by using Hill Equation 1.
| (Eq. 1) |
where ΔF/ΔFmax is relative fluorescence; ΔF = Fmax − Fi, where Fmax is a maximum fluorescence intensity reached on titration by hE1o; and Fi is fluorescence intensity at a given concentration of hE1o; ΔFmax = Fmax − Fo, where Fo is the initial fluorescence before addition of hE1o; Kd, is the concentration of E1o at half-saturation; n is the Hill coefficient.
For the quenching of the fluorescence of N-(1-pyrene) maleimide-labeled hE2o(1–173) di-domain and hE2o(144–386) core domain by E1o, the experimental conditions were similar to those reported above for DANS-As-labeled hE2o-truncated proteins. An hE1o concentration in the 0.01–0.80 μm range was used for the titration. The excitation wavelength was 340 nm, and emission spectra were recorded in the 350–600 nm range. The maximum emission intensities at 374 nm were used to calculate binding parameters according to Hill Equation 1.
Hydrogen/deuterium exchange MS
HDX-MS analysis was conducted as described by us earlier using a 7T Bruker Daltonics FT-MS instrument (43, 44, 53–56). The hE1o, hE2o, hE3, hE2o(1–173) di-domain, and hE2o(144–386) core domain were analyzed individually and in their binary subcomplexes. For sample preparation, the hE1o was exchanged into 10 mm KH2PO4 (pH 7.5) containing 100 mm NaCl, 0.5 mm ThDP, and 1 mm MgCl2. The hE2o, hE2o(1–173) di-domain, and hE2o(144–386) core domain were exchanged into 10 mm KH2PO4 (pH 7.5) containing 100 mm NaCl. The hE3 was exchanged into 10 mm KH2PO4 (pH 7.5) containing 100 mm NaCl and 2 μm FAD. All binary subcomplexes were prepared by mixing an equal volume of each protein (160 μm concentration stock concentration) to a final concentration of 80 μm each. The samples were allowed to equilibrate for 1 h at 20 °C. The deuterium-labeling reaction was initiated by mixing 15 μl of the protein samples with 285 μl of labeling buffer (10 mm KH2PO4 (pH 7.5) containing 100 mm NaCl and 99.9% D2O) yielding a final concentration of 95% D2O. The samples were incubated for an additional 20 s at 20 °C. For studies of the time dependence of H/D exchange, incubation times of 1, 3, 10, 30, 90, and 270 min were used. After incubation for different times, a 30-μl aliquot from each labeling reaction mixture was rapidly quenched into 36 μl of ice-cold quench buffer (0.2 m KH2PO4 and 3 m guanidine hydrochloride (pH 2.1)) achieving a final pH of 2.5 to minimize the rate of H/D back-exchange. The samples were immediately frozen in liquid nitrogen and stored at −80 °C for no more than 2 weeks before analysis. Nondeuterated samples were generated following the same procedure except that protein samples were diluted into aqueous buffer instead of labeling buffer and were incubated for 5 min followed by the quench process. All experiments were carried out in triplicate. The frozen deuterated sample was quickly thawed and loaded with an ice-cold syringe into a 20-μl sample loop inside the refrigeration system. The sample analysis was similar to that reported by us previously (43, 53–55) using Bruker Daltonics Data Analysis 4.0 for spectrum analysis and data treatment. Peptides were identified from nondeuterated samples by a customized program Dxgest (57). H/D exchange data for each individual peptide at various time points were processed using HX-Express (58, 59). No back-exchange correction was needed for the purpose of comparative analysis. The number of exchangeable backbone amides (Dmax) of a peptide was calculated as the total number of residues excluding proline residues and two fast exchangeable N-terminal residues (59, 60). The percentage of deuterium incorporation (without back-exchange correction) of each peptide was calculated from the equation ΔD% = ΔD/(Dmax × 1.0063 × 0.948) × 100%, where 1.0063 is the atomic mass difference between deuterium and hydrogen, and 0.948 represents the fractional D2O content of the labeling reaction mixture. Butterfly and difference plots were produced with Microsoft Excel.
Chemical cross-linking analysis of hE1o and hE2o
The hE1o (2 nmol, 66 μm subunits) in 50 mm HEPES (pH 7.5) containing 0.5 mm ThDP, 1.0 mm MgCl2, 0.15 m NaCl, and 1 mm benzamidine·HCl and the hE2o (1 nmol, 33 μm subunits) in 50 mm KH2PO4 (pH 7.2) containing 0.50 mm EDTA, 1.0 mm DTT, 1.0 mm benzamidine·HCl, and 0.40 m NH4Cl were mixed at a 2:1 molar ratio of subunits in 30 μl of 20 mm HEPES (pH 7.5) containing 300 mm NaCl. A 50-fold molar excess of BuUrBu and 1000-fold molar excess of CDI over the concentration of hE2o subunits, both dissolved in DMSO, were added, and the cross-linking reactions were conducted at 15 °C for 30 min. To quench the reaction mixtures, 1 m Tris·HCl (pH 8.0) was added to a final concentration of 20 mm. All reactions were carried out in duplicate. Cross-linked hE1o–hE2o was identified by SDS-PAGE (5%). Next, cross-linked samples were processed with tryptic in-solution digestion. An aliquot containing 1 nmol of the total protein was withdrawn from each reaction mixture, and 5 μl of 2% sodium deoxycholate and 2 μl of 200 mm DTT were added, and then the samples were incubated at 60 °C for 30 min. Next, 2.5 μl of 200 mm iodoacetamide was added, and the samples were incubated for an additional 30 min at room temperature. The tryptic digestion was carried out at 1:35 protein/trypsin molar ratio at 37 °C. After overnight digestion, the reaction was terminated by addition of 1.5 μl of 95% formic acid. Cross-linked peptides were desalted on a SepPak SPE column (Waters) and were dried in a SpeedVac (Savant). Cross-linked peptides were analyzed by nano-LC-MS/MS (Dionex Ultimate 3000 RLSC nanosystem interfaced with Q Exactive HF (ThermoFisher Scientific, San Jose, CA)). Samples were loaded onto a Magic C18AQ capillary trap unit (5 μm particle size, 200-Å pore size, and 100 μm × 2 cm) (Michrom Bioresources, Inc.), and the trap unit was washed with mobile phase consisting of 0.1% TFA for 5 min at a flow rate of 10 μl/min. The trap was brought in-line with the analytical Magic C18AQ column (3-μm particle size, 200-Å pore size, and 75 μm ×50 cm), and cross-linked peptides were eluted using a segmented linear gradient (4–15% A, 30 min; 15–25% B, 40 min; 25–50% B, 44 min; 50–90% B, 11 min; where mobile phase A consisted of 0.2% formic acid, and mobile phase B consisted of 0.16% formic acid in 80% acetonitrile) at a flow rate of 300 nl/min. Mass spectrometric data were acquired using a data-dependent acquisition procedure with a cyclic series of a full scan with a resolution of 120,000, followed by MS/MS (higher-energy C-trap dissociation; relative collision energy, 27%) of the 20 most intense ions and a dynamic exclusion duration of 20 s. LC-MS/MS peak lists were generated using the ProteoWizard software package (61) and searched against the SwissProt database (using the MS-GF+ search engine). Search parameters were as follows: fragment mass error, 20 ppm; parent mass error, 5 ppm; fixed modification, carbamidomethylation on cysteine; potential modifications during initial search, methionine oxidation, and acetylation on protein N termini and up to one missed tryptic cleavage. This search followed by extracted ion chromatogram (XIC)-based quantitation showed that no contaminant protein above 5% was present in the sample. Cross-links were evaluated using software tool MeroX 1.5.1 (62) with the following parameters: precursor 5 ppm; fragment 20 ppm mass accuracy; CDI or BuUrBu set as cross-linker. With both cross-linkers, only the Lys–Lys cross-linked peptides were analyzed. The automatically identified cross-linked peptides were manually analyzed, and the results with p < 0.001 limit were exported. The results were visualized on line using the XVIS website (63).
Two-dimensional 1H-15N HSQC NMR
The NMR experiments were performed on a Varian INOVA 600 MHz spectrometer at 25 °C. For the HSQC NMR experiment, a sample containing 200 μm 15N-labeled hE1o was exchanged into 50 mm K2HPO4 (pH 7.5) containing 100 mm NaCl, 0.5 mm ThDP, 1.0 mm MgCl2, and 7% D2O for a lock signal before the NMR spectrum was recorded.
Bioinformatics tools employed
Alignment of multiple sequences was carried out using the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/)5 (64) with default settings. The secondary structure of the E1o N-terminal region was calculated and predicted with the JPred program (http://www.compbio.dundee.ac.uk/jpred/)5 (65, 66). To predict the possible 3D structure of the E1o N-terminal region, I-TASSER and SWISS-MODEL (http://swissmodel.expasy.org/) were employed.
Author contributions
J. Z., O. O., X. Z., and R. B. resources; J. Z., L. Y., and X. Z. investigation; J. Z., L. Y., O. O., X. Z., J. W., P. A., R. B., and N. S. N. methodology; J. Z., O. O., P. A., N. S. N., W. F., and F. J. writing-original draft; L. Y., O. O., and J. W. data curation; L. Y. and O. O. formal analysis; O. O., J. W., A. A., W. F., and F. J. conceptualization; O. O., P. A., and W. F. software; O. O., X. Z., P. A., and W. F. visualization; A. A. supervision; P. A. validation; N. S. N., W. F., and F. J. writing-review and editing; F. J. funding acquisition; F. J. project administration.
Supplementary Material
Acknowledgments
The research at Semmelweis Medical University was supported by the following grants, all awarded to Professor Veronika Adam-Vizi (Dept. of Medical Biochemistry, MTA-SE Laboratory for Neurobiochemistry, Semmelweis University, H-1094 Budapest, Hungary), Hungarian Academy of Sciences MTA Grant 02001, Hungarian Scientific Research Fund OTKA Grant 112230, and Hungarian Brain Research Program Grants KTIA_13_NAP-A-III/6 and 2017-1.2.1-NKP-2017-00002.
This work was supported by National Institutes of Health Grant R15GM116077-01 (to F. J.), National Science Foundation Grant CHE-1402675 (to F. J.), a Rutgers-Newark Chancellor's SEED Grant (to F. J.), and National Institutes of Health Grant R01GM121469 (to W. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7, Tables S1–S5 and supporting Scheme S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- hOGDHc
- human 2-oxoglutaric acid dehydrogenase complex
- OGDHc
- oxoglutarate dehydrogenase complex
- HDX-MS
- hydrogen–deuterium exchange MS
- PSB
- Protein Data Bank
- ITC
- isothermal titration calorimetry
- SEC
- size-exclusion chromatography
- ThDP
- thiamin diphosphate
- TCEP
- tris(2-carboxyethyl)phosphine
- E3BP
- E3-binding protein
- PSBD
- peripheral subunit–binding domain
- PDHc
- pyruvate dehydrogenase complex
- h
- human
- LDo
- lipoyl domain
- CDo
- core domain
- CDI
- 1,1′-carbonyldiimidazole
- BuUrBu
- disuccinimidyl dibutyric urea
- DSBD
- dual-subunit-binding domain
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- PMSF
- phenylmethylsulfonyl fluoride
- TCA
- tricarboxylic acid
- HSQC
- heteronuclear single quantum coherence
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl
- FT-MS
- Fourier transform-MS
- H/D
- hydrogen–deuterium exchange
- DANS-As
- (4-((5-(dimethylamino)naphthalene)-1-sulfonalido)phenyl)arsenous acid.
References
- 1. Gibson G. E., Xu H., Chen H.-L., Chen W., Denton T. T., and Zhang S. (2015) α-ketoglutarate dehydrogenase complex-dependent succinylation of proteins in neurons and neuronal cell lines. J. Neurochem. 134, 86–96 10.1111/jnc.13096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen H., Denton T. T., Xu H., Calingasan N., Beal M. F., and Gibson G. E. (2016) Reduction in the mitochondrial enzyme α-ketoglutarate dehydrogenase complex in neurodegenerative disease-beneficial or detrimental? J. Neurochem. 139, 823–838 10.1111/jnc.13836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quinlan C. L., Goncalves R. L., Hey-Mogensen M., Yadava N., Bunik V. I., and Brand M. D. (2014) The 2-oxoacid dehydrogenase complex in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J. Biol. Chem. 289, 8312–8325 10.1074/jbc.M113.545301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tretter L., and Adam-Vizi V. (2005) α-Ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360, 2335–2345 10.1098/rstb.2005.1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y., Guo Y. R., Liu K., Yin Z., Liu R., Xia T., Tan L., Yang P., Lee J.-H., Li X.-J., Hawke D., Zheng Y., Qian X., Lyu J., He J., Xing D., Tao Y. J., and Lu Z. (2017) KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 552, 273–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carson M. (1991) RIBBONS 2.0. J. Appl. Crystallogr. 24, 958–961 10.1107/S0021889891007240 [DOI] [Google Scholar]
- 7. Nemeria N. S., Gerfen G., Yang L., Zhang X., and Jordan F. (2018) Evidence for functional and regulatory cross-talk between the tricarboxylic acid cycle 2-oxoglutarate dehydrogenase complex and 2-oxoadipate dehydrogenase on the l-lysine, l-hydroxylysine and l-tryptophan degradation pathways from studies in vitro. Biochim Biophys Acta Bioenerg. 1859, 932–939 10.1016/j.bbabio.2018.05.001 [DOI] [PubMed] [Google Scholar]
- 8. Perham R. N., Jones D. D., Chauhan H. J., and Howard M. J. (2002) Substrate channeling in 2-oxo acid dehydrogenase multienzyme complexes. Biochem. Soc. Trans. 30, 47–51 10.1042/bst0300047 [DOI] [PubMed] [Google Scholar]
- 9. Marrott N. L., Marshall J. J., Svergun D. I., Crennell S. J., Hough D. W., van den Elsen J. M., and Danson M. J. (2014) Why are the 2-oxoacid dehydrogenase complexes so large? Generation of an active trimeric complex. Biochem. J. 463, 405–412 10.1042/BJ20140359 [DOI] [PubMed] [Google Scholar]
- 10. Yeaman S. J. (1986) The mammalian 2-oxoacid dehydrogenases: a complex family. Trends Biochem. Sci. 11, 293–296 10.1016/0968-0004(86)90033-2 [DOI] [Google Scholar]
- 11. Patel M. S., and Roche T. E. (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 4, 3224–3233 10.1096/fasebj.4.14.2227213 [DOI] [PubMed] [Google Scholar]
- 12. Perham R. N. (1991) Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochemistry 30, 8501–8512 10.1021/bi00099a001 [DOI] [PubMed] [Google Scholar]
- 13. Harris R. A., Hawes J. W., Popov K. M., Zhao Y., Shimomura Y., Sato J., Jaskiewicz J., and Hurley T. D. (1997) Studies on the regulation of the mitochondrial α-ketoacid dehydrogenase complexes and their kinases. Adv. Enzyme Regul. 37, 271–293 10.1016/S0065-2571(96)00009-X [DOI] [PubMed] [Google Scholar]
- 14. Perham R. N. (2000) Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu. Rev. Biochem. 69, 961–1004 10.1146/annurev.biochem.69.1.961 [DOI] [PubMed] [Google Scholar]
- 15. Brautigam C. A., Wynn R. M., Chuang J. L., Naik M. T., Young B. B., Huang T. H., and Chuang D. T. (2011) Structural and thermodynamic basis for weak interactions between dihydrolipoamide dehydrogenase and subunit-binding domain of the branched-chain α-ketoacid dehydrogenase complex. J. Biol. Chem. 286, 23476–23488 10.1074/jbc.M110.202960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kikuchi G., Motokawa Y., Yoshida T., and Hiraga K. (2008) Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 84, 246–263 10.2183/pjab.84.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mattevi A., Obmolova G., Kalk K. H., van Berkel W. J., and Hol W. G. (1993) Three-dimensional structure of lipoamide dehydrogenase from Pseudomonas fluorescence at 2.8 Å resolution: analysis of redox and thermostability properties. J. Mol. Biol. 230, 1200–1215 10.1006/jmbi.1993.1236 [DOI] [PubMed] [Google Scholar]
- 18. Hunter A., and Lindsay J. G. (1986) Immunological and biosynthetic studies on mammalian 2-oxoglutarate dehydrogenase multienzyme complex. Eur. J. Biochem. 155, 103–109 10.1111/j.1432-1033.1986.tb09464.x [DOI] [PubMed] [Google Scholar]
- 19. Reed L. J. (1974) Multienzyme complexes. Acc. Chem. Res. 7, 40–56 10.1021/ar50074a002 [DOI] [Google Scholar]
- 20. Reed L. J., Pettit F. H., Eley M. H., Hamilton L., Collins J. H., and Oliver R. M. (1975) Reconstitution of the Escherichia coli pyruvate dehydrogenase complex. Proc. Natl. Acad. Sci. 72, 3068–3072 10.1073/pnas.72.8.3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reed L. J. (2001) A trial of research from lipoic acid to α-keto acid dehydrogenase complexes. J. Biol. Chem. 276, 38329–38336 10.1074/jbc.R100026200 [DOI] [PubMed] [Google Scholar]
- 22. Koike K., Suematsu T., and Ehara M. (2000) Cloning, overexpression and mutagenesis of cDNA encoding dihydrolipoamide succinyltransferase component of the porcine 2-oxoglutarate dehydrogenase complex. Eur. J. Biochem. 267, 3005–3016 10.1046/j.1432-1033.2000.01320.x [DOI] [PubMed] [Google Scholar]
- 23. Bradford A. P., Aitken A., Beg F., Cook K. G., and Yeaman S. J. (1987) Amino acid sequence surrounding the lipoic acid cofactor of bovine kidney 2-oxoglutarate dehydrogenase complex. FEBS Lett. 222, 211–214 10.1016/0014-5793(87)80221-1 [DOI] [PubMed] [Google Scholar]
- 24. Packman L. C., and Perham R. N. (1987) Limited proteolysis and sequence analysis of the 2-oxoacid dehydrogenase complexes from Escherichia coli. Cleavage sites and domains in the dihydrolipoamide acyltransferase components. Biochem. J. 242, 531–538 10.1042/bj2420531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wagenknecht T., and Grassucci R., and Schaak D. (1990) Cryoelectron microscopy of frozen-hydrated α-ketoacid dehydrogenase complexes from Escherichia coli. J. Biol. Chem. 265, 22402–22408 [PubMed] [Google Scholar]
- 26. Nakano K., Takase C., Sakamoto T., Nakagawa S., Inazawa J., Ohta S., and Matuda S. (1994) Isolation, characterization and structural organization of the gene and pseudogene for the dihydrolipoamide succinyltransferase component of the human 2-oxoglutarate dehydrogenase complex. Eur. J. Biochem. 224, 179–189 10.1111/j.1432-1033.1994.tb20010.x [DOI] [PubMed] [Google Scholar]
- 27. Behal R. H., DeBuysere M. S., Demeler B., Hanson J. C., and Olson M. S. (1994) Pyruvate dehydrogenase multienzyme complex. Characterization of assembly intermediates by sedimentation velocity analysis. J. Biol. Chem. 269, 31372–31377 [PubMed] [Google Scholar]
- 28. Sanderson S. J., Khan S. S., McCartney R. G., Miller C., and Lindsay J. G. (1996) Reconstitution of mammalian pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase complexes: analysis of protein X involvement and interaction of homologous and heterologous dihydrolipoamide dehydrogenases. Biochem. J. 319, 109–116 10.1042/bj3190109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smolle M., Prior A. E., Brown A. E., Cooper A., Byron O., and Lindsay J. G. (2006) A new level of architectural complexity in the human pyruvate dehydrogenase complex. J. Biol. Chem. 281, 19772–19780 10.1074/jbc.M601140200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vijayakrishnan S., Kelly S. M., Gilbert R. J., Callow P., Bhella D., Forsyth T., Lindsay J. G., and Byron O. (2010) Solution structure and characterization of the human pyruvate dehydrogenase complex core assembly. J. Mol. Biol. 399, 71–93 10.1016/j.jmb.2010.03.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hiromasa Y., Fujisawa T., Aso Y., and Roche T. E. (2004) Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J. Biol. Chem. 279, 6921–6933 10.1074/jbc.M308172200 [DOI] [PubMed] [Google Scholar]
- 32. Wagenknecht T., Grassucci R., Berkowitz J., and Forneris C. (1992) Configuration of interdomain linkers in pyruvate dehydrogenase complex of Escherichia coli as determined by cryoelectron microscopy. J. Struct. Biol. 109, 70–77 10.1016/1047-8477(92)90069-M [DOI] [PubMed] [Google Scholar]
- 33. Murphy G. E., and Jensen G. J. (2005) Electron cryotomography of the E. coli pyruvate and 2-oxoglutarate dehydrogenase complexes. Structure 13, 1765–1773 10.1016/j.str.2005.08.016 [DOI] [PubMed] [Google Scholar]
- 34. Byron O., and Lindsay J. G. (2017) The pyruvate dehydrogenase complex and related assemblies in health and disease. Subcell. Biochem. 83, 523–550 10.1007/978-3-319-46503-6_19 [DOI] [PubMed] [Google Scholar]
- 35. Frank R. A., Price A. J., Northrop F. D., Perham R. N., and Luisi B. F. (2007) Crystal structure of the E1 component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J. Mol. Biol. 368, 639–651 10.1016/j.jmb.2007.01.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rice J. E., Dunbar B., and Lindsay J. G. (1992) Sequences directing dihydrolipoamide dehydrogenase (E3) binding are located on the 2-oxoglutarate dehydrogenase (E1) component of the mammalian 2-oxoglutarate dehydrogenase multienzyme complex. EMBO J. 11, 3229–3235 10.1002/j.1460-2075.1992.tb05400.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCartney R. G., Rice J. E., Sanderson S. J., Bunik V., Lindsay H., and Lindsay J. G. (1998) Subunit interactions in the mammalian α-ketoglutarate dehydrogenase complex. Evidence for direct association of the α-ketoglutarate dehydrogenase and dihydrolipoamide dehydrogenase components. J. Biol. Chem. 273, 24158–24164 10.1074/jbc.273.37.24158 [DOI] [PubMed] [Google Scholar]
- 38. Al-Alaway A. I. A. (2013) Subunit Organisation and Assembly of the 2-Oxoglutarate Dehydrogenase Multienzyme Complex (OGDC). Ph.D. Thesis, University of Glasgow, Glasgow, Scotland, UK [Google Scholar]
- 39. Wagner T., Bellinzoni M., Wehenkel A., O'Hare H. M., and Alzari P. M. (2011) Functional plasticity and allosteric regulation of α-ketoglutarate decarboxylase in central mycobacterial metabolism. Chem. Biol. 18, 1011–1020 10.1016/j.chembiol.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 40. Ricaud P. M., Howard M. J., Roberts E. L., Broadhurst R. W., and Perham R. N. (1996) Three-dimensional structure of the lipoyl domain from the dihydrolipoyl succinyltransferase component of the 2-oxoglutarate dehydrogenase multienzyme complex of Escherichia coli. J. Mol. Biol. 264, 179–190 10.1006/jmbi.1996.0632 [DOI] [PubMed] [Google Scholar]
- 41. Knapp J. E., Mitchell D. T., Yazdi M. A., Ernst S. R., Reed L. J., and Hackert M. L. (1998) Crystal structure of the truncated cubic core component of the Escherichia coli 2-oxoglutarate dehydrogenase multienzyme complex. J. Mol. Biol. 280, 655–668 10.1006/jmbi.1998.1924 [DOI] [PubMed] [Google Scholar]
- 42. Robien M. A., Clore G. M., Omichinski J. G., Perham R. N., Appella E., Sakaguchi K., and Gronenborn A. M. (1992) Three-dimensional solution structure of the E3-binding domain of the dihydrolipoamide succinyltransferase multienzyme complex of Escherichia coli. Biochemistry 31, 3463–3471 10.1021/bi00128a021 [DOI] [PubMed] [Google Scholar]
- 43. Wang J., Kumaran S., Zhou J., Nemeria N. S., Tao H., Kakalis L., Park Y. H., Birkaya B., Patel M. S., and Jordan F. (2015) Elucidation of the interaction loci of the human pyruvate dehydrogenase complex E2·E3BP core with pyruvate dehydrogenase kinase 1 and kinase 2 by H/D exchange mass spectrometry and nuclear magnetic resonance. Biochemistry 54, 69–82 10.1021/bi5013113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ambrus A., Wang J., Mizsei R., Zambo Z., Torocsik B., Jordan F., and Adam-Vizi V. (2016) Structural alterations induced by ten disease-causing mutations of human dihydrolipoamide dehydrogenase analyzed by hydrogen/deuterium-exchange mass spectrometry: implications for structural basis of E3 deficiency. Biochim. Biophys. Acta 1862, 2098–2109 10.1016/j.bbadis.2016.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carothers D. J., Pons G., and Patel M. S. (1989) Dihydrolipoamide dehydrogenase: functional similarities and divergent evolution of the pyridine nucleotide-disulfide oxidoreductases. Arch. Biochem. Biophys. 268, 409–425 10.1016/0003-9861(89)90309-3 [DOI] [PubMed] [Google Scholar]
- 46. Mande S. S., Sarfaty S., Allen M. D., Perham R. N., and Hol W. G. (1996) Protein–protein interactions in the pyruvate dehydrogenase multienzyme complex: dihydrolipoamide dehydrogenase complexed with the binding domain of dihydrolipoamide acetyltransferase. Structure 4, 277–286 10.1016/S0969-2126(96)00032-9 [DOI] [PubMed] [Google Scholar]
- 47. Guevara E. L., Yang L., Birkaya B., Zhou J., Nemeria N. S., Patel M. S., and Jordan F. (2017) Global view of cognate kinase activation by the human pyruvate dehydrogenase complex. Sci. Rep. 7, 42760 10.1038/srep42760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schuldiner S., Weil R., Robertson D. E., and Kaback H. R. (1977) Microenvironment of the binding site in the lac carrier protein. Proc. Natl. Acad. Sci. U.S.A. 74, 1851–1854 10.1073/pnas.74.5.1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Han M. K., Lin P., Paek D., Harvey J. J., Fuior E., and Knutson J. R. (2002) Fluorescence studies of pyrene maleimide-labeled translin: excimer fluorescence indicates subunits associate in a tail-to-tail configuration to form octamer. Biochemistry 41, 3468–3476 10.1021/bi015901e [DOI] [PubMed] [Google Scholar]
- 50. Hage C., Iacobucci C., Rehkamp A., Arlt C., and Sinz A. (2017) The first zero-length mass spectrometry-cleavable cross-linker for protein structure analysis. Angew. Chem. Int. Ed. Engl. 56, 14551–14555 10.1002/anie.201708273 [DOI] [PubMed] [Google Scholar]
- 51. Müller M. Q., Dreiocker F., Ihling C. H., Schäfer M., and Sinz A. (2010) Cleavable cross-linker for protein structure analysis: reliable identification of cross-linking products by tandem MS. Anal. Chem. 82, 6958–6968 10.1021/ac101241t [DOI] [PubMed] [Google Scholar]
- 52. Nemeria N. S., Ambrus A., Patel H., Gerfen G., Adam-Vizi V., Tretter L., Zhou J., Wang J., and Jordan F. (2014) Human 2-oxoglutarate dehydrogenase complex E1 component forms a thiamin-derived radical by aerobic oxidation of the enamine intermediate. J. Biol. Chem. 289, 29859–29873 10.1074/jbc.M114.591073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang J., Nemeria N. S., Chandrasekhar K., Kumaran S., Arjunan P., Reynolds S., Calero G., Brukh R., Kakalis L., Furey W., and Jordan F. (2014) Structure and function of the catalytic domain of the dihydrolipoyl acetyltransferase component in Escherichia coli pyruvate dehydrogenase complex. J. Biol. Chem. 289, 15215–15230 10.1074/jbc.M113.544080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang J. (2014) Interaction of the Components in Human and Escherichia coli Pyruvate Dehydrogenase Multienzyme Complexes. Ph.D. Thesis, Rutgers, University-Newark [Google Scholar]
- 55. Chandrasekhar K., Wang J., Arjunan P., Sax M., Park Y. H., Nemeria N. S., Kumaran S., Song J., Jordan F., and Furey W. (2013) Insight to the interaction of the dihydrolipoamide acetyltransferase (E2) core with the peripheral components in the Escherichia coli pyruvate dehydrogenase complex via multifaceted structural approaches. J. Biol. Chem. 288, 15402–15417 10.1074/jbc.M113.466789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Arjunan P., Wang J., Nemeria N. S., Reynolds S., Brown I., Chandrasekhar K., Calero G., Jordan F., and Furey W. (2014) Novel binding motif and new flexibility revealed by structural analyses of a pyruvate dehydrogenase-dehydrolipoyl acetyltransferase subcomplex from the Escherichia coli pyruvate dehydrogenase complex. J. Biol. Chem. 289, 30161–30176 10.1074/jbc.M114.592915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hamuro Y., Coales S. J., Molnar K. S., Tuske S. J., and Morrow J. A. (2008) Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 22, 1041–1046 10.1002/rcm.3467 [DOI] [PubMed] [Google Scholar]
- 58. Weis D. D., Engen J. R., and Kass I. J. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 1700–1703 10.1016/j.jasms.2006.07.025 [DOI] [PubMed] [Google Scholar]
- 59. Kan Z. Y., Mayne L., Chetty P. S., and Englander S. W. (2011) ExMS: data analysis for HX-MS experiments. J. Am. Mass Spectrom. 22, 1906–1915 10.1007/s13361-011-0236-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bai Y., Milne J. S., Mayne L., and Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 10.1002/prot.340170110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., et al. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 10.1038/nbt.2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Götze M., Pettelkau J., Fritzsche R., Ihling C. H., Schäfer M., and Sinz A. (2015) Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass Spectrom. 26, 83–97 10.1007/s13361-014-1001-1 [DOI] [PubMed] [Google Scholar]
- 63. Grimm M., Zimniak T., Kahraman A., and Herzog F. (2015) xVis: a web server for the schematic visualization and interpretation of crosslink-derived spatial restraints. Nucleic Acids Res. 43, W362–W369 10.1093/nar/gkv463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li W., Cowley A., Uludag M., Gur T., McWilliam H., Squizzato S., Park Y. M., Buso N., and Lopez R. (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W58–W584 10.1093/nar/gkv316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Scott M. S., Boisvert F. M., McDowall M. D., Lamond A. I., and Barton G. J. (2010) Characterization and prediction of protein nucleolar localization sequences. Nucleic Acids Res. 38, 7388–7399 10.1093/nar/gkq653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Scott M. S., Troshin P. V., and Barton G. J. (2011) NoD: a nucleolar localization sequence detector for eukaryotic and viral proteins. BMC Bioinformatics 12, 317 10.1186/1471-2105-12-317 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.