

Abstract
Eukaryotic genomes are packaged into linker–oligonucleosome assemblies, providing compaction of genomic DNA and contributing to gene regulation and genome integrity. To define minimal requirements for initial steps in the transition of compact, closed chromatin to a transcriptionally active, open state, we developed a model in vitro system containing a single, unique, “target” nucleosome in the center of a 25-nucleosome array and evaluated the accessibility of the linker DNA adjacent to this target nucleosome. We found that condensation of H1-lacking chromatin results in ∼60-fold reduction in linker DNA accessibility and that mimics of acetylation within all four core histone tail domains of the target nucleosome synergize to increase accessibility ∼3-fold. Notably, stoichiometric binding of histone H1 caused >2 orders of magnitude reduction in accessibility that was marginally diminished by histone acetylation mimics. Remarkably, a nucleosome-free region (NFR) in place of the target nucleosome completely abrogated H1-dependent restriction of linker accessibility in the immediate vicinity of the NFR. Our results suggest that linker DNA is as inaccessible as DNA within the nucleosome core in fully condensed, H1-containing chromatin. They further imply that an unrecognized function of NFRs in gene promoter regions is to locally abrogate the severe restriction of linker DNA accessibility imposed by H1s.
Keywords: chromatin, chromatin structure, chromatin regulation, histone, histone acetylation, nucleosome, epigenetics, gene regulation, histone H1, linker histone, nucleosome free region
Introduction
The genomes of eukaryotes are packaged into chromosome-sized oligonucleosome assemblies, which provide orderly compaction of genomic DNA and contribute to the regulation of transcription and other processes. These functions are due in part to the propensity of oligonucleosomes (referred to as nucleosome arrays) to spontaneous fold and condense into higher-order chromatin structures within the ionic environment of the nucleus (1–3). Such condensation requires internucleosome interactions mediated through the core histone tail domains (4–8). Importantly, reconstituted arrays containing only core histones and defined DNA templates recapitulate the behavior of native chromatin with regard to salt-dependent folding and compaction (1, 3, 9). Oligonucleosome arrays lacking H1 exist as extended “beads on a string” structures (defined as primary chromatin structure (10)) in low ionic strength buffers in vitro but equilibrate between moderately compacted and fully folded secondary structures such as the chromatin fiber in 0.5–2 mm MgCl2. In slightly higher concentrations of divalent salts (>2–3 mm MgCl2), the arrays oligomerize (self-associate) into large globular tertiary structures that resemble the size, shape, and appearance of interphase chromosomes (3). Arrays containing H1 undergo similar transitions but at lower concentrations of multivalent or monovalent salts (1, 11). Therefore, the core histones are sufficient to direct formation of chromatin fibers and higher-order structures in vitro (1, 12).
Nucleosomes impose a severe restriction to the accessibility of DNA that is exploited by gene regulatory mechanisms. For example, pioneering experiments showed that even in the absence of activating conditions or upstream activating sequence elements appropriate repression of several yeast genes requires nucleosomes (13–15). In vitro experiments have demonstrated that the accessibility of trans-acting factors to DNA within the ∼147-bp nucleosome core region is restricted 102–105-fold, depending on proximity to the nucleosome dyad, an extent of inhibition sufficient to elicit a regulatory effect (16, 17). The core histones compete with transcription factors for binding to cognate sites, thereby providing a thermodynamic barrier to binding that must be overcome by nucleosome remodeling, increased factor concentration, or increased binding activity (18).
In contrast, linker DNA, which extends between successive nucleosome core regions in nucleosome arrays, is not in tight association with the core histones and is nearly as accessible as naked DNA in mononucleosomes (19, 20). However, within condensed nucleosome arrays lacking linker histones, accessibility to linker DNA is reduced compared with naked DNA about 15–50-fold, consistent with a role of condensed higher-order chromatin structures in restricting access to cognate sites regardless of their position in chromatin (16, 21). In addition, linker histones (H1s) are present in similar abundance to the core histone, bind to the exterior of the nucleosome, and neutralize the charge of linker DNA to stabilize the salt-dependent folding of nucleosome arrays (22, 23). Despite the occurrence of a single canonical H1-binding site per nucleosome, a range of H1:nucleosome ratios has been observed in cells with significantly less than one H1 per nucleosome in more transcriptionally active and pluripotent cell types to ≥1 in quiescent cell types, suggesting a general role of H1s in gene repression (24, 25). However, although H1 drastically inhibits transcription of chromatin templates in vitro (26–28), H1 knockdowns and knockouts in vivo do not result in genome-wide increases in transcription, consistent with additional overlapping mechanisms of gene repression (29–31). Moreover, stable reduction or elimination of H1 results in reduced nucleosome spacing, which may also contribute to repression (30, 31). However, the precise effect of H1 on linker DNA accessibility in condensed chromatin and the extent to which histone acetylation or other chromatin modifications mitigate these effects have not been precisely quantified.
The activation of a silent gene locus is thought to involve initial binding of a transcription factor(s) that initiates chromatin remodeling events such as histone acetylation and displacement of H1s to open chromatin (32). This transition typically involves recruitment of histone acetyltransferases to target nucleosomes to direct local histone acetylation (32, 33). Importantly, acetylation of the core histone tail domains directly destabilizes higher-order condensed chromatin structures (11, 34, 35). In addition, active promoters have a nucleosome-free region (NFR)3 immediately upstream of the transcription start site (TSS), generated by a combination of the binding of general transcription factors, ATP-dependent nucleosome remodeling, and, in some cases, DNA sequences that exhibit reduced affinity for binding core histones (15, 36–41). The NFR and associated activities serve to order nucleosomes upstream and downstream of the TSS with the regularity of spacing decaying with distance from the TSS (37, 42, 43). Although the NFR allows space for binding of the preinitiation complex, the lack of a canonical nucleosome may disrupt stable folding/condensation of higher-order chromatin structure and therefore contribute to promoter accessibility (21, 44).
To better understand the effects of factors involved in locus activation on chromatin accessibility, we developed an in vitro model system wherein a single “designer” nucleosome is located in the center of an otherwise unmodified 25-nucleosome array (21). Independent reconstitution of the central target nucleosome allows installation of any single or combination of histone modifications or other alteration associated with locus activation. In initial experiments with this system, we showed that acetylation mimics within the histone H4 tail domain of the target nucleosome increased accessibility of the surrounding linker DNA about 2-fold, whereas acetylation mimics within the H3 tail had little effect but were able to synergize with H4 tail acetylation mimics to further increase accessibility. Here, we assess the effects of acetylation mimics in histones H2A and H2B, linker histones, and an NFR on linker DNA accessibility. We found that acetylation mimics within all four core histones increase accessibility beyond that when only H3 and H4 contain acetylation mimics. Importantly, we found that stoichiometric association of H1 with nucleosomes in the array results in a drastic (>100-fold) reduction in accessibility that is minimally abrogated by acetylation. However, installation of a NFR completely abolished the effect of H1.
Results
To investigate the effects of chromatin remodeling events that initiate opening closed chromatin, we generated 25-nucleosome arrays in which a single “target” nucleosome (TNuc) was ligated between two 12-mer nucleosome arrays (Fig. 1A). In previous work, the array templates contained tandem repeats of the Lytechinus 5S nucleosome positioning sequence (21, 45). However, nucleosome positioning on these arrays is degenerate (46). Although the 5S arrays recapitulate the somewhat loose positioning found in some regions of native chromatin, arrays based on the 601 nucleosome positioning sequence exhibit precisely positioned nucleosomes and have slightly altered salt-dependent folding properties in vitro compared with 5S arrays (5, 47). Indeed, the latter might better model selected regions of native chromatin in which nucleosome positioning is highly ordered.
Figure 1.

Generation and analysis of 25-nucleosome arrays. A, the TNuc and 12-mer arrays are independently reconstituted and then ligated via directional DraIII half-sites (arrowheads) to generate 25-mer arrays in which the TNuc is flanked by linker regions containing the DraIII sites (red arrows). REA, restriction enzyme accessibility. B, representative self-association assay showing that arrays remain saturated with nucleosomes after ligation. Arrays were incubated with increasing MgCl2, then samples were centrifuged, and supernatants were loaded on SDS-agarose gels (see “Experimental procedures”). Loss of solubility in the range of ∼3 mm MgCl2 indicates saturation of the array. C, example of quantitative DraIII digestion assay. Arrays were mixed with an equal mass of a naked control DNA containing a single DraIII site and then digested, and the products were analyzed on SDS-agarose gels followed by ethidium bromide staining. Bands corresponding to the 5.3-kb 25-mer array template, the 4.5-kb naked DNA control, and residual 12/13-mer arrays and other digestion products (bracket) are indicated. D, plot of digestion data quantified as described under “Experimental procedures.” Lines represent linear regression fits with R2 indicated. krel was calculated with correction for inherent rate of digestion of chromatin template versus naked DNA control (see “Experimental procedures”). The small fraction of the chromatin that was digested with naked DNA kinetics (typically 10–20%) was excluded from the fits.
To determine whether the precision of nucleosome positioning significantly affects accessibility in condensed chromatin, we compared accessibility within arrays based on the 601 sequence with prior results with 5S arrays (21). We first assessed the impediment to accessibility in maximally condensed nucleosome arrays containing unmodified core histones in 10 mm MgCl2 (high-Mg2+) buffer. In such buffers, the 25-mer nucleosome arrays spontaneously condense and self-associate into large aggregates resembling the size and density of individual interphase chromosomes (1, 3). Indeed, >90% of the arrays are insoluble in 3 mm MgCl2 buffer, indicating complete saturation of the template with nucleosomes (Fig. 1B and Ref. 1). A 4.5-kb naked DNA fragment containing a single DraIII site was included in every reaction as an internal control, allowing normalization for variations in enzyme activity (21). Digestion was initiated by addition of DraIII, and the ratio of the rates of disappearance of 25-mer DNA and 4.5-kb DNA bands (k relative (krel)) was determined from plots of digestion kinetics (Fig. 1, C and D) with correction for inherent target-specific differences in cleavage rates (see “Experimental procedures”). Plots showed that the vast majority of both the DNA and chromatin digests contained a single kinetic component (Fig. 1D). Our analysis revealed that DraIII sites in the TNuc linker DNA were about 60-fold less accessible in the condensed chromatin compared with the naked DNA template (krel = 0.015 ± 0.001) (Table 1), commensurate with previous measurements for 601 arrays (16). Thus, the TNuc linker DNA is about 5-fold less accessible in the 601 arrays compared with 5S arrays (21), suggesting that precise nucleosome spacing generates a more stable and less accessible condensed chromatin structure. However, similar to the 5S nucleosome arrays (21), acetylation mimics within the H3 and H4 tail domains of the target nucleosome locally increased DNA accessibility about 1.5-fold within the 601-based arrays (Table 1).
Table 1.
Accessibilities of TNuc linker DNA relative to control DNA (krel) in arrays containing the indicated histones assembled on the TNuc template flanked by 12-mer oligonucleosomes containing WT core histones
Accessibilities normalized to arrays in which the TNuc contains unmodified histones are shown (krel/WT). Errors are ±S.E.; n ≥ 3 for all determinations.
| krel | krel/WT | |
|---|---|---|
| WT | 0.015 ± 0.001 | 1 |
| H3acm/H4acm | 0.023 ± 0.002 | 1.50 ± 0.21 |
| H2Aacm/H2Bacm | 0.021 ± 0.002 | 1.41 ± 0.12 |
| All-acm | 0.051 ± 0.005 | 3.33 ± 0.10 |
In addition to the H3 and H4 tail domains, the N-terminal tail domains of H2A/H2B dimers also contribute to the stability of nucleosome packing in higher-order chromatin structures and are acetylated in association with gene expression (34, 48, 49). To determine whether acetylation of the H2A and H2B N-terminal tails in a single nucleosome can open chromatin structure alone or in combination with H3/H4 acetylation, we assembled 25-mer arrays in which the TNuc contained unmodified histones (WT), acetylation mimics (acm) only within H2A/H2B (H2Aacm/H2Bacm), or mimics within all four core histones (All-acm) (Fig. 2A). Comparison of krel values indicated that acetylation mimics within H2A/H2B resulted in an ∼1.4-fold increase in the accessibility of TNuc linker DNA, comparable with that observed for H3acm/H4acm (Fig. 2B and Table 1). Importantly, installation of acetylation mimics in all four core histones resulted in significantly more enhancement in linker DNA accessibility (krel = 3.3 ± 0.10) than the predicted combined effects of the individual sets of mimics (1.5 ± 0.21 × 1.4 ± 0.12 = 2.1 ± 0.24; p < 0.001), indicating that acetylation mimics within the H2A/H2B dimers and the H3/H4 tetramer of a single nucleosome cooperatively increase accessibility of the adjoining linker DNA.
Figure 2.
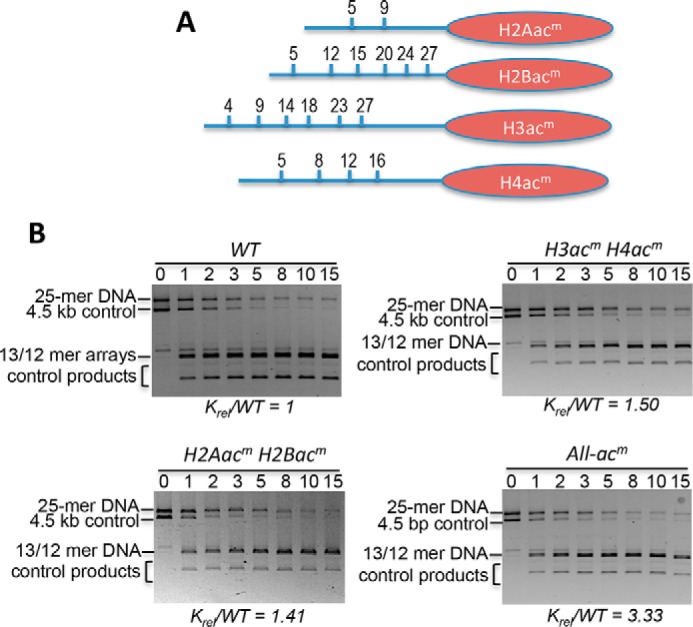
Acetylation mimics in all core histone tail domains increase accessibility of central target nucleosome linker DNA within a condensed 25-mer array. A, location of Lys → Gln substitutions as acetylation mimics (acm) in each of the core histone N-terminal tail domains (blue lines). B, representative SDS-agarose gels showing the DraIII digestion time course for 25-mer arrays with TNuc containing the indicated modified histones. Digestions were carried out for the times indicated above the lanes, and then products were separated on 0.7% SDS-agarose gels and stained with ethidium bromide. Gels were quantified, and rates of digestions were determined as described under “Experimental procedures.” The relative rate of digestion for each construct, normalized to the WT TNuc, is shown below the gel. See also Table 1. Bands corresponding to the 5.3-kb 25-mer array template (array) and the 4.5-kb naked control DNA (DNA) are indicated.
Linker histones (H1s) bind to the exterior of nucleosomes and stabilize higher-order chromatin structures (50, 51). Thus, linker histones restrict the accessibility of DNA to trans-acting factors, and many (or all) H1 subtypes, including H1.0, are depleted in the vicinity of active promoters (52–57). However, quantitative determinations of the effect of linker histones on DNA accessibility in chromatin are lacking, hampered by the difficulty in precisely controlling the stoichiometry of H1 binding, as slight excesses of H1 can cause aggregation of chromatin samples, whereas undersaturation can lead to drastically different results compared with saturated conditions. To overcome these issues, we used Nap1 as an H1 chaperone to precisely control H1 deposition (58, 59). Incubation of increasing amounts of preformed H1–Nap1 complex results in a small but detectable reduction in the electrophoretic migration of 12-mer nucleosome arrays until a point where a uniquely migrating species is apparent over a range of H1–Nap1 concentrations expected to deposit one H1 per nucleosome (Fig. 3A, white line). Importantly, the H1-bound arrays are completely soluble in 0.5 mm Mg2+ (see “Experimental procedures”), indicating that the arrays are not overloaded with H1 (Fig. 3B).
Figure 3.

Stoichiometric binding of H1 drastically decreases linker DNA accessibility. A, deposition of H1 onto nucleosome arrays via Nap1. Arrays were incubated with increasing amounts of H1–Nap1 complex, and binding was analyzed on a native agarose gel stained with ethidium bromide. The white lines indicate a species with a distinct mobility that arises over a range of H1–Nap1:nucleosome ratios (numbers above the lanes) consistent with one H1 bound to each nucleosome (white lines). B, self-association assay showing H1-bound arrays are soluble in 0.5 mm Mg2+. Nucleosome arrays were incubated as in A in H1 binding buffer (50 mm KCl) and 0.5 mm Mg2+ and centrifuged, and the DNA content of the supernatant was assessed on SDS-agarose gels. C, stoichiometric binding of H1 drastically reduces accessibility of TNuc linker DNA. 25-mer arrays containing a WT nucleosome at the central (TNuc) position were digested with DraIII either in the absence or presence of a 1.3:1 ratio of H1–Nap1:nucleosome. Digestions were carried out in buffer containing 0.5 mm Mg2+. Note that chromatin in the absence or presence of H1 was digested with either 10 or 100 units of DraIII, respectively. Digestion rates were determined as described, and relative rates were calculated and adjusted for amount of enzyme in each reaction. D, as in C except that the central nucleosome contained histones with acetylation mimics in all four histone proteins (All-acm). E, data from C and D were plotted as described in the text. The relative digestion rates in the absence/presence of H1 for this experiment are shown. See also Table 1. F, rates of DraIII digestion were determined for WT and All-acm arrays in the absence or presence of increasing H1–Nap1. Plotted is the rate of digestion normalized to the rate in the absence of H1 (100%).
We next determined the extent to which H1 restricts access to the TNuc linker DNA. Note that in these experiments we directly compared rates of digestion as calculation of krel based on inclusion of a naked DNA control was not possible because linker histones bind cooperatively to naked DNA with affinities only a few-fold less than to nucleosomes (60, 61). We first determined the effect of H1 binding to arrays in buffer containing 0.5 mm MgCl2 where the arrays are only partially condensed and not self-associated (21). Remarkably, we found that stoichiometric association of H1 to arrays (one per nucleosome) resulted in an ∼50-fold reduction in accessibility of the WT TNuc linker DNA (Fig. 3, C and E, and Table 2). Moreover, acetylation mimics within all four histones in the TNuc (All-acm) had only a small effect on the H1-dependent reduction in accessibility (Fig. 3, D and E, and Table 2). Importantly, titration of H1 into both WT and All-acm nucleosome arrays yielded a reduction in TNuc linker DNA accessibility, as measured by DraIII digestion, until the same apparent point of saturation as indicated by the gel-shift binding assay (Fig. 3, compare F with A), further indicating that the arrays in the above experiments are saturated with H1 over a range of H1–Nap1 inputs. Moreover, substoichiometric ratios of H1:nucleosomes, such as are found in many cell types, result in significantly less restriction in access to linker DNA (see “Discussion”).
Table 2.
Relative accessibilities of TNuc linker DNA for the indicated samples in the absence and presence of H1
The values indicate the -fold reduction in accessibility caused by the binding of H1 in each case. Errors are ±S.E.
| Relative rates |
||
|---|---|---|
| 10 mm Mg2+ | 0.5 mm Mg2+ | |
| WT/WT + H1 | 280 ± 62 | 48 ± 13 |
| All-acm/All-acm + H1 | 68 ± 12 | 25 ± 7 |
| NFR/NFR + H1 | 0.97 ± 0.07 | 0.99 ± 0.08 |
We next assessed the effect of H1 association on linker DNA accessibility when the arrays are fully condensed and self-associated (21). Remarkably, we found that binding of H1 to arrays in 10 mm MgCl2 resulted in an even more drastic, ∼200-fold reduction in linker DNA accessibility compared with that observed in the low-Mg2+ buffer (Fig. 4, A and B, and Table 2). Given that accessibility of the linker DNA is already ∼60-fold inhibited in such buffers in the absence of H1, these results suggest that the restriction of linker DNA accessibility approaches that found within the nucleosome core region (see “Discussion”).
Figure 4.

The NFR overrides the H1-dependent decrease in linker DNA accessibility. A, digests of nucleosome arrays containing WT TNuc in 10 mm MgCl2 in the absence and presence of H1. Units of DraIII used in each digestion are indicated above the gels. B, plot of data taken from gel shown in A. C and D, as in A and B except arrays contained an NFR in place of the TNuc.
Active genes in metazoans contain an NFR upstream of the transcription start site that generates a gap in the ordered array of nucleosomes and leads to a 3–4-fold increase in linker DNA accessibility in H1-lacking nucleosome arrays (21). Because an NFR also results in the loss of a canonical H1-binding site on the nucleosome, we determined the effect of replacing the target nucleosome with an NFR on observed restriction of linker DNA accessibility by H1. Remarkably, in stark contrast to the effect of acetylation mimics, installation of a single NFR completely reversed the effect of H1 on DraIII accessibility for both partially and maximally condensed arrays (Fig. 4, C and D, and Table 2). Indeed, equivalent amounts of DraIII can be used to achieve the same extent of digestion in NFR arrays in the presence or absence of H1, in contrast to WT arrays. To determine whether the drastic effect of the NFR on H1 repression extended throughout the entire 25-mer arrays, we digested arrays with EcoRV, which cuts between every nucleosome in the flanking 12-mer arrays except for the TNuc–linker DNA (Fig. 5A). We found that the effect of the NFR is largely localized to the vicinity of the TNuc as the remainder of the array exhibits roughly equivalent, inhibited rates of digestion with EcoRV in the presence of H1, regardless of whether the central position was a WT nucleosome or an NFR (Fig. 5B). These results indicate that H1 imposes a dominant constraint on DNA accessibility in native chromatin and suggests that the NFR, which lacks a canonical H1-binding site, locally counteracts H1-mediated condensation. These results are consistent with whole-genome mapping studies, which show that H1 is widely distributed across genomes except for in the immediate vicinity of NFRs of active genes (54–57).
Figure 5.
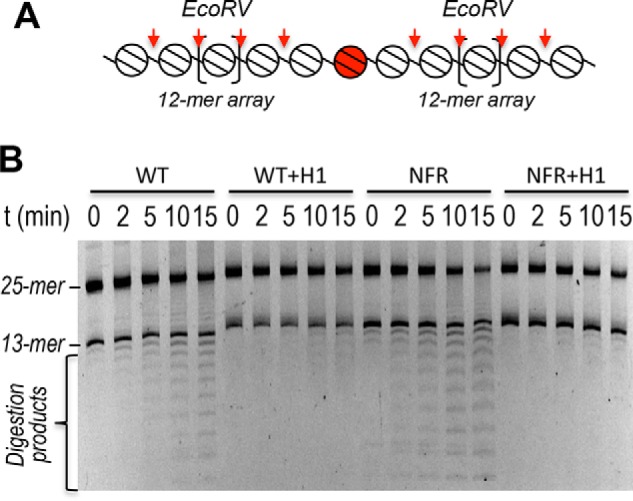
The effect of the NFR on H1-dependent restriction of linker DNA accessibility does not extend to the entire array. A, schematic showing the 25-mer array, EcoRV sites (vertical arrows) and the TNuc (center; shaded). B, 25-mer arrays containing either a WT nucleosome or an NFR at the central (TNuc) position were incubated with or without H1 in buffer containing 10 mm Mg2+ as indicated and digested with EcoRV (2 units) for the indicated times, and then products were separated on SDS-agarose gels.
Discussion
Our data show that chromatin condensation can drastically affect linker DNA accessibility in model nucleosome arrays. We observed a 60-fold reduction in linker DNA accessibility in condensed chromatin lacking H1 compared with naked DNA. Moreover, we found an additional 50–200-fold reduction in accessibility in an H1-containing over H1-lacking chromatin, depending on whether the chromatin was moderately or fully condensed, respectively. Interestingly, acetylation had a modest 2–4-fold effect in both the presence and absence of H1. Therefore, our results indicate that displacement of H1 either locally or globally will result in a much larger effect than acetylation on accessibility and binding of transcription factors and associated gene activation (Fig. 6).
Figure 6.
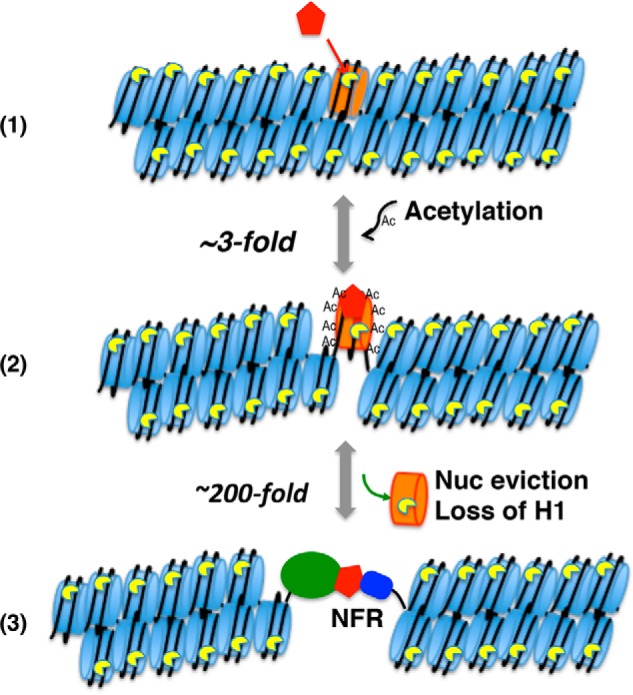
Factors affecting linker DNA accessibility. A pioneer factor (red) binds the target nucleosome (orange) in closed chromatin and recruits histone acetyltransferases (1), resulting in ∼3–4-fold increase in accessibility to the linker DNA (2). The acetylation and increased accessibility allow the recruitment of additional factors, resulting in nucleosome (Nuc) and H1 displacement (3) and a ∼200-fold increase in accessibility to the DNA. Note that some pioneer factors displace H1 directly, resulting in accessible nucleosomes (68). Note that the NFR was still digested 4 times slower than naked DNA or a nucleosome ligated to two naked 25-mer templates, indicating that the folding of the remainder of the nucleosome arrays still provides significant impediment (21).
Our results indicate that acetylation of all four core histone N-terminal tail domains of a single nucleosome contributes to opening chromatin. Indeed, acetylation mimics within either H3/H4 or H2A/H2B tails resulted in a modest but significant increase in accessibility, whereas mimics within all four tail domains in a single nucleosome cooperate to increase accessibility of linker DNA. These results indicate that acetylation within a single nucleosome brought about by an activating pioneer factor is sufficient to initiate a transition to an open, active chromatin structure (Fig. 6).
Previous work showed that installation of a single NFR results in a 3–4-fold increase in accessibility in fully condensed H1-lacking arrays (21). In contrast, we found that the NFR completely abrogates the large repressive effect of H1, resulting in 50- and ∼200-fold increases in accessibility in partially and fully condensed arrays, respectively. Our results suggest that the effect of the NFR is primarily local as little effect on H1-dependent reduction in accessibility was observed when arrays were digested with EcoRV, which cuts between the nucleosomes in the regions of the nucleosome array flanking the TNuc (Fig. 5). Importantly, whole-genome ChIP studies indicate that many H1 subtypes, including H1.0, the subtype used in the current studies, are drastically depleted in the immediate vicinity of the NFR of active genes but only modestly reduced upstream and downstream of the NFR (54–56), consistent with the idea that the lack of a single nucleosome creates a discontinuity in the H1-stablized condensed chromatin structure (Fig. 6). Although it is difficult to determine from these data whether the reduction of H1 in the promoter region is due to the NFR or transcription factors that bind to the DNA, our data indicate that the lack of a single nucleosome and canonical H1-binding site plays a primary role in counteracting H1-dependent repression. (Note that although H1 may bind to the NFR, binding occurs with a lower affinity to naked DNA than to nucleosomes and is not expected to have an equivalent chromatin-condensing effect.) Thus, our data suggest that recruitment of chromatin remodeling factors that ultimately evict histones contributes a much larger enhancement to promoter accessibility than acetylation of the core histones per se, although acetylation may be required for recruitment or efficient function of such activities.
Many cell types contain less than one linker histone per nucleosome. For example, mouse embryonic stem cells contain about 0.5 H1s per nucleosome, which appears to contribute to maintaining a transcriptionally permissive environment in these pluripotent cells (25, 30, 62). Moreover, an increase in the H1:nucleosome ratio is observed in embryonic stem cell differentiation along with chromatin condensation (25). Interestingly, our data show that chromatin containing less than one H1 per nucleosome is much more accessible than when saturated with H1. Indeed, a reduction in H1 stoichiometry approximately from one to 0.5 per nucleosome resulted in an ∼10-fold increase in accessibility of the TNuc linker DNA (Fig. 3F). These results provide strong support to the idea that a similarly reduced H1 content will result in a much more transcriptionally permissive chromatin environment.
Even in the absence of chromatin folding, nucleosomes impart a 103–105-fold restriction to DNA sites within the ∼147-bp nucleosome core region, depending on location with respect to the nucleosome dyad. Thus, the vast majority of cognate sites within the nucleosome core would remain unoccupied by trans-acting factors at concentrations typically found within the nucleus (16). (Exceptions include pioneer transcription factors that have the ability to bind to the nucleosome surface (32, 33). However, whether chromatin condensation reduces binding of pioneer factors remains an open question.) Interestingly, compaction of nucleosome arrays causes only a modest 3–8-fold additional restriction on accessibility within the nucleosome core region (16). However, we found that although the linker DNA separating core regions remains relatively accessible in expanded chromatin containing only the core histones, complete condensation of the chromatin imparts an ∼60-fold impediment on linker DNA accessibility in the absence of H1 (Table 1 and see also Ref. 21), consistent with prior published work (16). Moreover, binding of H1 causes an additional 50–200-fold reduction in accessibility, resulting in an overall restriction in linker DNA accessibility approaching that found in the nucleosome core region (Figs. 3 and 4). These results suggest that linker DNA in a fully condensed H1-bound chromatin is not appreciably occupied by sequence-specific DNA-binding factors responsible for driving gene activation, similar to that in the nucleosome core region (16). The large dynamic range of this effect suggests that linker DNA accessibility represents a potential H1-dependent switch within chromatin.
Our data indicate that precisely spaced 601 nucleosome arrays form a more tightly condensed chromatin structure than the 5S arrays (21), which have a more heterogeneous nucleosome spacing. We observed a krel of 0.015 ± 0.001 for the WT 601 arrays, about 5-fold less than the krel observed for the 5S arrays (21) (Table 1). However, we note that the inherent rate of digestion of the ligated 601 array DNA template is 2-fold greater than that of the 5S templates (see “Experimental procedures” and Ref. 21). These differences are likely due to sequence-dependent DNA structure, which results in modest effects in the rate of two-dimensional diffusion and cognate site recognition by the restriction enzyme. It is possible that these effects are not relevant when the templates are assembled into nucleosomes; however, even if corrections for DNA sequence effects are ignored, the 601 arrays are ∼3-fold less accessible than the 5S arrays (krel of 0.17 ± 0.02 versus 0.06 ± 0.01, respectively), indicating that arrays with more regularly spaced nucleosomes form a more stable and compact structure. These results are supported by EM of long 601 arrays (35) and folding studies (63) and suggest that more regular spacing of nucleosomes in vivo results in a more regular and stable condensed chromatin structure.
We found that the accessibility of linker DNA is severely restricted by stoichiometric binding of linker histones. The extent of this restriction likely depends on the linker H1 subtypes as they vary in their degree of condensation, length of DNA, and potentially the type of enzymes (length of recognition sequence) being used. It will be interesting in the future to test subtypes that have a range of chromatin-condensing capabilities (57).
Experimental procedures
Expression and purification of core and linker histone proteins
WT and mutant recombinant Xenopus core histones were expressed and purified as described previously (21). Briefly, coding sequences for Xenopus core histones H2A, H2B, H3, and H4 containing specific lysine-to-glutamine substitutions were obtained using the Stratagene QuikChange site-directed mutagenesis kit. Mutants were confirmed by Sanger sequencing. Escherichia coli BL21(DE3) cells were transformed with the pET3a expression plasmids harboring the WT or mutant core histone gene sequences. Cultures (3 ml of LB) containing ampicillin (100 μg/ml) were inoculated from single colonies and grown overnight, then 1 ml was used to inoculate 100 ml of LB, and the cultures were allowed to grow at 37 °C in a shaker incubator until 0.6 A600. Expression of histones was induced by addition of 0.4 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h at 37 °C. Cells were harvested, lysed, and sonicated, and H3/H4 tetramers and H2A/H2B dimers were purified as described (64). Xenopus H1.0 protein was expressed and purified as described (65). Briefly, E. coli BL21(DE3) cells transformed with the plasmid pET3aH1(0) were grown in 100 ml of LB containing ampicillin (50 μg/ml) until 0.6 A600, expression was induced as described above, cells were harvested, and H1 was purified as described (65). Protein concentrations were determined by quantitative analysis of samples and standards run on 15% SDS-polyacrylamide gels stained with Coomassie Blue. The concentrations of standards were determined by amino acid analyses (66). Proteins were stored in a −80 °C freezer.
Preparation of DNA templates
A 204-bp DNA fragment containing the 601 nucleosome positioning sequence and asymmetric DraIII overhangs (DC template) was prepared as described (21). A complementary array DNA template containing 12 tandem repeats of a 207-bp 601 nucleosome positioning element was created by conversion of the XbaI site in the plasmid pSub601-12 to an asymmetric DraIII site to create plasmid PD6. A 2.5-kb DNA fragment containing the array template was generated by digestion of 200 μg of PD6 plasmid with 200 units of HindIII-HF (High Fidelity) and DraI enzymes in CutSmart buffer (New England Biolabs) in a 300-μl reaction for 6 h. The digested plasmid was incubated with 50 units of calf intestinal alkaline phosphatase at 37 °C for 30 min, extracted with phenol/chloroform, ethanol-precipitated, dried, resuspended in 200 μl of TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA), and digested with 200 units of DraIII-HF at 37 °C for 2 h. The ∼2.5-kb band was isolated on a preparative 0.7% agarose gel, electroeluted, precipitated, and resuspended in TE buffer. Note that the 2.5-kb 12 × 207 array template has a DraIII (3′) overhang complementary to the DC template overhangs at one end and a dephosphorylated 5′ terminus at the opposite (HindIII) end to block self-ligation. A ∼4.5-kb DNA fragment, derived by digestion of p12-5S-C1 with SalI and AlwN1 (21), was also prepared for use as an internal control in the DraIII digestion assays.
Preparation of nucleosomes and nucleosome arrays
Reconstitution of DC mononucleosomes and 12-mer nucleosome arrays was independently carried out via the salt dialysis method as described previously (21). Briefly, 25 μg of DC DNA or 12 × 207 array template was mixed with 12.5 μg each of H2A/H2B dimer and H3/H4 tetramer in 2 m NaCl, TE buffer containing 20 mm DTT; incubated on ice for 30 min; and then transferred into 6–8-kDa–cutoff Spectrapore dialysis tubing. The sample was dialyzed into 1.2 m NaCl, TE buffer in the cold room followed by addition of TE buffer every 90 min to bring the NaCl concentration to 1, 0.8, and 0.6 m. Finally, the mixture was dialyzed overnight into T10E0.1 (10 mm Tris-HCl, pH 8.0, 0.1 mm EDTA) buffer. Mononucleosomes were purified by sedimentation through 7–20% sucrose gradients (10 ml) with ultracentrifugation at 197,868 × g for 18 h in a Beckman SW41 Ti rotor at 4 °C. Nucleosome fractions (∼0.4 ml) were collected in BSA-treated 0.6-ml Eppendorf tubes, and 20 μl of each was analyzed on a 0.7% native agarose gel with 0.5× TBE (90 mm Tris borate, pH 8.3, 2.5 mm EDTA). Array reconstitutions were carried out identically except that dialysis was for 4 h in TE buffer, 1 m NaCl; 3 h in TE buffer, 0.75 m NaCl; and then overnight in T10E0.1 buffer. The extent of saturation of the array template with nucleosomes was assessed by magnesium-dependent self-association assays (67).
Ligation of DC mononucleosomes and 12-mer arrays was carried out with empirically optimized ratios of components to maximize formation of 25-mer arrays (Fig. 1). The ligation reactions typically contained 50 ng of sucrose gradient–purified DC nucleosomes, 1.5 μg of 12-mer arrays, and 2 μl (800 units) of T4 DNA ligase in ligation buffer (0.2 μg of BSA, 1 mm ATP, 10 mm MgCl2, 0.25 mm EDTA, 10 mm Tris-HCl, pH 8.0) incubated in a final volume of 50 μl for 30 min at room temperature and then overnight at 4 °C. The 25-mer arrays were subjected to buffer exchange into TEN buffer (10 mm Tris, 0.25 mm EDTA, 2.5 mm NaCl) via microfiltration (48). To generate arrays containing a central NFR, the naked 204-bp DC DNA (601 nucleosome positioning sequence) was ligated with the 12 × 207 arrays. After ligation, all nucleosome dyads in the 25-nucleosome arrays are separated by 207 bp. Self-association assays and DraIII digestion kinetics support minimal loss of nucleosomes through the ligation process (see below).
Binding of linker histone to nucleosome arrays
Xenopus NapI and H1 were mixed in 2:1 ratio in binding buffer containing 10 mm Tris, pH 8.0, 1 mm EDTA, and 50 mm NaCl (58). The H1–NapI complex was mixed with 12- or 25-mer nucleosome arrays in the ratios stated in the figure legends in binding buffer containing 10 mm Tris-HCl, pH 8.0, 0.1 mm EDTA, and 50 mm NaCl and incubated on ice for 1 h, and binding was assessed by loading arrays with 5% glycerol onto a 0.7% native agarose gel (0.5× TBE).
Quantification of 25-mer array DNA digestion and identification of krel rates of cleavage
Analytical DraIII digestions were performed with 25-mer arrays in which the central DC nucleosome contained either all native histones (WT) or combinations of H3/H4 tetramers and H2A/H2B dimers in which acetylatable lysines were swapped for glutamines (H4 K5Q,K8Q,K12Q,K16Q (H4acm); H3 K4Q,K9Q,K14Q,K18Q,K23Q,K27Q (H3acm); H2A K5Q,K9Q (H2Aacm); and H2B K5Q,K12Q,K15Q,K20Q,K24Q,K27Q (H2Bacm) as stated in the text and figure legends. For digestions, 20 μl of each array preparation (∼1 μg) was mixed with an equal amount of p12 DNA control in 80 μl of 1× New England Biolabs CutSmart digestion buffer (50 mm potassium acetate, 20 mm Tris acetate, 10 or 0.5 mm magnesium acetate (as indicated), 1 mm DTT, pH 7.9). Nine microliters of the solution was removed as an undigested control (t = 0 min) and added to 2 μl of 6× SDS gel loading solution (10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 30% glycerol, 1% SDS, 0.25% bromphenol blue dye). The remaining 71 μl was mixed with DraIII-HF (units as indicated in the figure legends) followed by rapid mixing and further incubation at 37 °C. Nine microliters of digested product was removed at time points as indicated in the figures legends and immediately mixed with 2 μl of 6× SDS gel loading solution. Samples were run on 0.7% SDS-agarose gels, the gels were imaged on a Bio-Rad imager (Gel DocTM XR+), and the 25-mer array DNA bands and p12 control DNA bands were quantified with the volume tools in Image Lab. The rates of digestion were determined by plotting ln(fraction uncut) versus time for both the nucleosome array and naked DNA bands, and the data were fitted to a simple y = mx + b equation in Microsoft Excel as described previously (21). To allow comparison of the absolute rates of cleavage for chromatin and DNA templates within each sample, we also determined the relative rates of cleavage for the naked 25-mer array template and the naked 4.5-kb control DNA templates. We found that the naked 25-mer template, containing two DraIII sites, was cleaved 8 times faster than the 4.5-kb control template, which contains a single DraIII site, indicating that there is an inherent difference in the overall probability of a first-hit single cleavage event of about 4-fold between the two DNAs. The parameter krel was thus calculated as Slope chromatin/8x slope DNA for each of the 25-mer arrays, correcting for the differences in inherent rates between the two templates. For quantification of rates of cleavage for H1-bound arrays, the experiment was set up as described above except that naked DNA was not used as an internal control as the extent of H1 binding to the naked template and the effect on the control digestion rate were impossible to determine. Note that as originally shown by Widom and co-worker (19), under the conditions used for the digestions, the reactions are first-order in enzyme concentration, allowing correction of rates obtained at different restriction enzyme concentrations. Moreover, the rates reflect the probability of site exposure (equilibrium) rather than the rate at which sites become exposed.
Author contributions
L. N. M. and J. H. conceptualization; L. N. M. data curation; L. N. M. and J. H. formal analysis; L. N. M. and J. H. validation; L. N. M. investigation; L. N. M. visualization; L. N. M. and J. H. methodology; L. N. M. and J. H. writing-original draft; L. N. M. and J. H. project administration; L. N. M. and J. H. writing-review and editing; J. H. resources; J. H. supervision; J. H. funding acquisition.
Acknowledgment
We thank Prasoon Jaya for preparation of histone proteins.
This work was supported by National Institutes of Health Grant R01GM052426 (to J. J. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- NFR
- nucleosome-free region
- TSS
- transcription start site
- TNuc
- target nucleosome
- krel
- k relative
- acm
- acetylation mimics.
References
- 1. Hansen J. C. (2002) Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 31, 361–392 10.1146/annurev.biophys.31.101101.140858 [DOI] [PubMed] [Google Scholar]
- 2. Allahverdi A., Chen Q., Korolev N., and Nordenskiöld L. (2015) Chromatin compaction under mixed salt conditions: opposite effects of sodium and potassium ions on nucleosome array folding. Sci. Rep. 5, 8512 10.1038/srep08512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maeshima K., Rogge R., Tamura S., Joti Y., Hikima T., Szerlong H., Krause C., Herman J., Seidel E., DeLuca J., Ishikawa T., and Hansen J. C. (2016) Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J. 35, 1115–1132 10.15252/embj.201592660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fletcher T. M., and Hansen J. C. (1995) Core histone tail domains mediate oligonucleosome folding and nucleosomal DNA organization through distinct molecular mechanisms. J. Biol. Chem. 270, 25359–25362 10.1074/jbc.270.43.25359 [DOI] [PubMed] [Google Scholar]
- 5. Dorigo B., Schalch T., Bystricky K., and Richmond T. J. (2003) Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327, 85–96 10.1016/S0022-2836(03)00025-1 [DOI] [PubMed] [Google Scholar]
- 6. Gordon F., Luger K., and Hansen J. C. (2005) The core histone N-terminal tail domains function independently and additively during salt-dependent oligomerization of nucleosomal arrays. J. Biol. Chem. 280, 33701–33706 10.1074/jbc.M507048200 [DOI] [PubMed] [Google Scholar]
- 7. Kan P. Y., Caterino T. L., and Hayes J. J. (2009) The H4 tail domain participates in intra- and internucleosome interactions with protein and DNA during folding and oligomerization of nucleosome arrays. Mol. Cell. Biol. 29, 538–546 10.1128/MCB.01343-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kan P. Y., Lu X., Hansen J. C., and Hayes J. J. (2007) The H3 tail domain participates in multiple interactions during folding and self-association of nucleosome arrays. Mol. Cell. Biol. 27, 2084–2091 10.1128/MCB.02181-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zinchenko A., Berezhnoy N. V., Wang S., Rosencrans W. M., Korolev N., van der Maarel J. R. C., and Nordenskiöld L. (2018) Single-molecule compaction of megabase-long chromatin molecules by multivalent cations. Nucleic Acids Res. 46, 635–649 10.1093/nar/gkx1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woodcock C. L., and Dimitrov S. (2001) Higher-order structure of chromatin and chromosomes. Curr. Opin. Genet. Dev. 11, 130–135 10.1016/S0959-437X(00)00169-6 [DOI] [PubMed] [Google Scholar]
- 11. Allahverdi A., Yang R., Korolev N., Fan Y., Davey C. A., Liu C. F., and Nordenskiöld L. (2011) The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 39, 1680–1691 10.1093/nar/gkq900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Allan J., Harborne N., Rau D. C., and Gould H. (1982) Participation of the core histone tails in the stabilization of the chromatin solenoid. J. Cell Biol. 93, 285–297 10.1083/jcb.93.2.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grunstein M. (1992) Histones as regulators of genes. Sci. Am. 267, 68–74 [DOI] [PubMed] [Google Scholar]
- 14. Han M., and Grunstein M. (1988) Nucleosome loss activates yeast downstream promoters in vivo. Cell 55, 1137–1145 10.1016/0092-8674(88)90258-9 [DOI] [PubMed] [Google Scholar]
- 15. Jiang C., and Pugh B. F. (2009) Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 10, 161–172 10.1038/nrg2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poirier M. G., Bussiek M., Langowski J., and Widom J. (2008) Spontaneous access to DNA target sites in folded chromatin fibers. J. Mol. Biol. 379, 772–786 10.1016/j.jmb.2008.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vitolo J. M., Yang Z., Basavappa R., and Hayes J. J. (2004) Structural features of transcription factor IIIA bound to a nucleosome in solution. Mol. Cell. Biol. 24, 697–707 10.1128/MCB.24.2.697-707.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Polach K. J., and Widom J. (1996) A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J. Mol. Biol. 258, 800–812 10.1006/jmbi.1996.0288 [DOI] [PubMed] [Google Scholar]
- 19. Polach K. J., and Widom J. (1995) Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol. 254, 130–149 10.1006/jmbi.1995.0606 [DOI] [PubMed] [Google Scholar]
- 20. Chafin D. R., Vitolo J. M., Henricksen L. A., Bambara R. A., and Hayes J. J. (2000) Human DNA ligase I efficiently seals nicks in nucleosomes. EMBO J. 19, 5492–5501 10.1093/emboj/19.20.5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mishra L. N., Pepenella S., Rogge R., Hansen J. C., and Hayes J. J. (2016) Acetylation mimics within a single nucleosome alter local DNA accessibility in compacted nucleosome arrays. Sci. Rep. 6, 34808 10.1038/srep34808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thoma F., Koller T., and Klug A. (1979) Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 83, 403–427 10.1083/jcb.83.2.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clark D. J., and Kimura T. (1990) Electrostatic mechanism of chromatin folding. J. Mol. Biol. 211, 883–896 10.1016/0022-2836(90)90081-V [DOI] [PubMed] [Google Scholar]
- 24. Woodcock C. L., Skoultchi A. I., and Fan Y. (2006) Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 14, 17–25 10.1007/s10577-005-1024-3 [DOI] [PubMed] [Google Scholar]
- 25. Pan C., and Fan Y. (2016) Role of H1 linker histones in mammalian development and stem cell differentiation. Biochim. Biophys. Acta 1859, 496–509 10.1016/j.bbagrm.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Laybourn P. J., and Kadonaga J. T. (1991) Role of nucleosomal cores and histone H1 in regulation of transcription by RNA polymerase II. Science 254, 238–245 10.1126/science.1718039 [DOI] [PubMed] [Google Scholar]
- 27. Shimamura A., Sapp M., Rodriguez-Campos A., and Worcel A. (1989) Histone H1 represses transcription from minichromosomes assembled in vitro. Mol. Cell. Biol. 9, 5573–5584 10.1128/MCB.9.12.5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Neill T. E., Meersseman G., Pennings S., and Bradbury E. M. (1995) Deposition of histone H1 onto reconstituted nucleosome arrays inhibits both initiation and elongation of transcripts by T7 RNA polymerase. Nucleic Acids Res. 23, 1075–1082 10.1093/nar/23.6.1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen X., and Gorovsky M. A. (1996) Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell 86, 475–483 10.1016/S0092-8674(00)80120-8 [DOI] [PubMed] [Google Scholar]
- 30. Fan Y., Nikitina T., Zhao J., Fleury T. J., Bhattacharyya R., Bouhassira E. E., Stein A., Woodcock C. L., and Skoultchi A. I. (2005) Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212 10.1016/j.cell.2005.10.028 [DOI] [PubMed] [Google Scholar]
- 31. Hashimoto H., Takami Y., Sonoda E., Iwasaki T., Iwano H., Tachibana M., Takeda S., Nakayama T., Kimura H., and Shinkai Y. (2010) Histone H1 null vertebrate cells exhibit altered nucleosome architecture. Nucleic Acids Res. 38, 3533–3545 10.1093/nar/gkq076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zaret K. S., and Mango S. E. (2016) Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr. Opin. Genet. Dev. 37, 76–81 10.1016/j.gde.2015.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Soufi A., Garcia M. F., Jaroszewicz A., Osman N., Pellegrini M., and Zaret K. S. (2015) Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568 10.1016/j.cell.2015.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tse C., Sera T., Wolffe A. P., and Hansen J. C. (1998) Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol. 18, 4629–4638 10.1128/MCB.18.8.4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huynh V. A., Robinson P. J., and Rhodes D. (2005) A method for the in vitro reconstitution of a defined “30 nm” chromatin fibre containing stoichiometric amounts of the linker histone. J. Mol. Biol. 345, 957–968 10.1016/j.jmb.2004.10.075 [DOI] [PubMed] [Google Scholar]
- 36. Bernstein B. E., Liu C. L., Humphrey E. L., Perlstein E. O., and Schreiber S. L. (2004) Global nucleosome occupancy in yeast. Genome Biol. 5, R62 10.1186/gb-2004-5-9-r62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mavrich T. N., Jiang C., Ioshikhes I. P., Li X., Venters B. J., Zanton S. J., Tomsho L. P., Qi J., Glaser R. L., Schuster S. C., Gilmour D. S., Albert I., and Pugh B. F. (2008) Nucleosome organization in the Drosophila genome. Nature 453, 358–362 10.1038/nature06929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chereji R. V., and Clark D. J. (2018) Major determinants of nucleosome positioning. Biophys. J. 114, 2279–2289 10.1016/j.bpj.2018.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chereji R. V., Ocampo J., and Clark D. J. (2017) MNase-sensitive complexes in yeast: nucleosomes and non-histone barriers. Mol. Cell 65, 565–577.e3 10.1016/j.molcel.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen W., and Struhl K. (1985) Yeast mRNA initiation sites are determined primarily by specific sequences, not by the distance from the TATA element. EMBO J. 4, 3273–3280 10.1002/j.1460-2075.1985.tb04077.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bai L., and Morozov A. V. (2010) Gene regulation by nucleosome positioning. Trends Genet. 26, 476–483 10.1016/j.tig.2010.08.003 [DOI] [PubMed] [Google Scholar]
- 42. Ocampo J., Chereji R. V., Eriksson P. R., and Clark D. J. (2016) The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res. 44, 4625–4635 10.1093/nar/gkw068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang Z., Wippo C. J., Wal M., Ward E., Korber P., and Pugh B. F. (2011) A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 332, 977–980 10.1126/science.1200508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hansen J. C., and Lohr D. (1993) Assembly and structural properties of subsaturated chromatin arrays. J. Biol. Chem. 268, 5840–5848 [PubMed] [Google Scholar]
- 45. Simpson R. T., Thoma F., and Brubaker J. M. (1985) Chromatin reconstituted from tandemly repeated cloned DNA fragments and core histones: a model system for study of higher order structure. Cell 42, 799–808 10.1016/0092-8674(85)90276-4 [DOI] [PubMed] [Google Scholar]
- 46. Dong F., and van Holde K. E. (1991) Nucleosome positioning is determined by the (H3-H4)2 tetramer. Proc. Natl. Acad. Sci. U.S.A. 88, 10596–10600 10.1073/pnas.88.23.10596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hansen J. C., Ausio J., Stanik V. H., and van Holde K. E. (1989) Homogeneous reconstituted oligonucleosomes, evidence for salt-dependent folding in the absence of histone H1. Biochemistry 28, 9129–9136 10.1021/bi00449a026 [DOI] [PubMed] [Google Scholar]
- 48. Wang X., and Hayes J. J. (2008) Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol. Cell. Biol. 28, 227–236 10.1128/MCB.01245-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Z., Zang C., Rosenfeld J. A., Schones D. E., Barski A., Cuddapah S., Cui K., Roh T. Y., Peng W., Zhang M. Q., and Zhao K. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 10.1038/ng.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carruthers L. M., Bednar J., Woodcock C. L., and Hansen J. C. (1998) Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: mechanistic ramifications for higher-order chromatin folding. Biochemistry 37, 14776–14787 10.1021/bi981684e [DOI] [PubMed] [Google Scholar]
- 51. Cutter A. R., and Hayes J. J. (2017) Linker histones: novel insights into structure-specific recognition of the nucleosome. Biochem. Cell Biol. 95, 171–178 10.1139/bcb-2016-0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bresnick E. H., Bustin M., Marsaud V., Richard-Foy H., and Hager G. L. (1992) The transcriptionally-active MMTV promoter is depleted of histone H1. Nucleic Acids Res. 20, 273–278 10.1093/nar/20.2.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hill D. A., and Imbalzano A. N. (2000) Human SWI/SNF nucleosome remodeling activity is partially inhibited by linker histone H1. Biochemistry 39, 11649–11656 10.1021/bi001330z [DOI] [PubMed] [Google Scholar]
- 54. Cao K., Lailler N., Zhang Y., Kumar A., Uppal K., Liu Z., Lee E. K., Wu H., Medrzycki M., Pan C., Ho P. Y., Cooper G. P. Jr., Dong X., Bock C., Bouhassira E. E., et al. (2013) High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet. 9, e1003417 10.1371/journal.pgen.1003417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Izzo A., Kamieniarz-Gdula K., Ramírez F., Noureen N., Kind J., Manke T., van Steensel B., and Schneider R. (2013) The genomic landscape of the somatic linker histone subtypes H1.1 to H1.5 in human cells. Cell Rep. 3, 2142–2154 10.1016/j.celrep.2013.05.003 [DOI] [PubMed] [Google Scholar]
- 56. Millán-Ariño L., Islam A. B., Izquierdo-Bouldstridge A., Mayor R., Terme J. M., Luque N., Sancho M., López-Bigas N., and Jordan A. (2014) Mapping of six somatic linker histone H1 variants in human breast cancer cells uncovers specific features of H1.2. Nucleic Acids Res. 42, 4474–4493 10.1093/nar/gku079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mishra L. N., Shalini V., Gupta N., Ghosh K., Suthar N., Bhaduri U., and Rao M. R. S. (2018) Spermatid-specific linker histone HILS1 is a poor condenser of DNA and chromatin and preferentially associates with LINE-1 elements. Epigenetics Chromatin 11, 43 10.1186/s13072-018-0214-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Syed S. H., Goutte-Gattat D., Becker N., Meyer S., Shukla M. S., Hayes J. J., Everaers R., Angelov D., Bednar J., and Dimitrov S. (2010) Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc. Natl. Acad. Sci. U.S.A. 107, 9620–9625 10.1073/pnas.1000309107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bednar J., Garcia-Saez I., Boopathi R., Cutter A. R., Papai G., Reymer A., Syed S. H., Lone I. N., Tonchev O., Crucifix C., Menoni H., Papin C., Skoufias D. A., Kurumizaka H., Lavery R., et al. (2017) Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol. Cell 66, 384–397.e8 10.1016/j.molcel.2017.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Clark D. J., and Thomas J. O. (1988) Differences in the binding of H1 variants to DNA. Cooperativity and linker-length related distribution. Eur. J. Biochem. 178, 225–233 10.1111/j.1432-1033.1988.tb14447.x [DOI] [PubMed] [Google Scholar]
- 61. Hayes J. J., Tullius T. D., and Wolffe A. P. (1990) The structure of DNA in a nucleosome. Proc. Natl. Acad. Sci. U.S.A. 87, 7405–7409 10.1073/pnas.87.19.7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fan Y., Nikitina T., Morin-Kensicki E. M., Zhao J., Magnuson T. R., Woodcock C. L., and Skoultchi A. I. (2003) H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol. Cell. Biol. 23, 4559–4572 10.1128/MCB.23.13.4559-4572.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dorigo B., Schalch T., Kulangara A., Duda S., Schroeder R. R., and Richmond T. J. (2004) Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306, 1571–1573 10.1126/science.1103124 [DOI] [PubMed] [Google Scholar]
- 64. Pepenella S., Murphy K. J., and Hayes J. J. (2014) A distinct switch in interactions of the histone H4 tail domain upon salt-dependent folding of nucleosome arrays. J. Biol. Chem. 289, 27342–27351 10.1074/jbc.M114.595140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hayes J. J., Kaplan R., Ura K., Pruss D., and Wolffe A. (1996) A putative DNA binding surface in the globular domain of a linker histone is not essential for specific binding to the nucleosome. J. Biol. Chem. 271, 25817–25822 10.1074/jbc.271.42.25817 [DOI] [PubMed] [Google Scholar]
- 66. Caterino T. L., Fang H., and Hayes J. J. (2011) Nucleosome linker DNA contacts and induces specific folding of the intrinsically disordered H1 carboxyl-terminal domain. Mol. Cell. Biol. 31, 2341–2348 10.1128/MCB.05145-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schwarz P. M., Felthauser A., Fletcher T. M., and Hansen J. C. (1996) Reversible oligonucleosome self-association: dependence on divalent cations and core histone tail domains. Biochemistry 35, 4009–4015 10.1021/bi9525684 [DOI] [PubMed] [Google Scholar]
- 68. Iwafuchi-Doi M., and Zaret K. S. (2016) Cell fate control by pioneer transcription factors. Development 143, 1833–1837 10.1242/dev.133900 [DOI] [PMC free article] [PubMed] [Google Scholar]