

Abstract
Aims
Previous studies indicate that nitric oxide derived from endothelial nitric oxide synthase (eNOS) serves as both trigger and mediator in anaesthetic cardiac preconditioning. The mechanisms underlying regulation of eNOS by volatile anaesthetics have not been fully understood. Therefore, this study examined the role of vascular endothelial growth factor (VEGF) in isoflurane cardiac preconditioning.
Methods and results
Wistar rats underwent 30 min of coronary artery occlusion followed by 2 h of reperfusion. Isoflurane given prior to ischaemia/reperfusion significantly decreased myocardial infarct size from 60 ± 1% in control to 40 ± 3% (n = 8 rats/group, P < 0.05). The beneficial effects of isoflurane were blocked by neutralizing antibody against VEGF (nVEGF). Coronary arterial endothelial cells (ECs) alone or together with cardiomyocytes (CMs) were subjected to hypoxia/reoxygenation injury. The expression of VEGF and eNOS was analysed by western blot, and nitric oxide was measured by ozone-based chemiluminescence. In co-cultured CMs and ECs, isoflurane administered before hypoxia/reoxygenation attenuated lactate dehydrogenase activity and increased the ratio of phosphorylated eNOS/eNOS and nitric oxide production. The protective effect of isoflurane on CMs was compromised by nVEGF and after VEGF in ECs was inhibited with hypoxia inducible factor-1α short hairpin RNA (shRNA). The negative effect of hypoxia inducible factor-1α shRNA was restored by recombinant VEGF.
Conclusion
Isoflurane cardiac preconditioning is associated with VEGF regulation of phosphorylation of eNOS and nitric oxide production.
Keywords: Vascular endothelial growth factor, Endothelial nitric oxide synthase, Cardiac Preconditioning, Ischaemia/reperfusion
1. Introduction
Every year millions of cardiac surgeries are performed worldwide. Due to pre-existing coronary artery disease and clamping of coronary artery during surgery, the heart is inevitably subjected to ischaemia/reperfusion (I/R) injury. Perioperative myocardial I/R injury has been identified as the leading cause of morbidity and mortality in cardiac surgery.1 Reduction of myocardial I/R injury benefits numerous patients receiving cardiac surgery.
Volatile anaesthetics such as isoflurane (ISO), sevoflurane, enflurane, and desflurane are used to maintain the state of general anaesthesia during cardiac surgery.2 Clinical and experimental studies indicate that volatile anaesthetics attenuate myocardial I/R injury, when administered prior to ischaemia.3–6 This phenomenon is similar to ischaemic preconditioning, termed anaesthetic preconditioning.7,8 Previous studies indicate that anaesthetic preconditioning is triggered and mediated by endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO).8,9 However, the molecular mechanisms underlying the regulation of eNOS by volatile anaesthetics remain unclear.
Vascular endothelial growth factor (VEGF) is an angiogenic factor predominantly expressed in vascular endothelium.10 The VEGF promotes endothelial cell (EC) proliferation, migration, and differentiation in vitro and angiogenesis in vivo.11–13 These effects of VEGF are largely dependent on the activation of eNOS.14 A study from our laboratory found that VEGF was up-regulated by ISO preconditioning.15 Multiple lines of evidence indicate that VEGF regulates the phosphorylation or dephosphorylation of eNOS,16–19 ISO-elicited up-regulation of VEGF might alter phosphorylation of eNOS and the production of NO. Accordingly, the objective of this study was to examine the role of VEGF in regulation of eNOS phosphorylation in ISO-induced cardioprotection against I/R injury.
2. Methods
2.1 Animals
Male Wistar rats (weight 300–400 g; age 10–12 weeks) and pregnant female Wistar rats (age 9–12 weeks) were purchased from Charles River Laboratories International. Inc. (Wilmington, MA, USA). The animals were kept on a 12 h light-dark cycle in a temperature-controlled room. The experimental procedures were approved by the Animal Care and Use Committee of the Medical College of Wisconsin and conformed to the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research, National Academy of Sciences, USA, 8th edition, 2011).
2.2 Myocardial I/R injury in vivo
2.2.1 Surgical preparation
We have previously described the rat in vivo model of myocardial I/R injury.20,21 Briefly, sodium thiobutabarbital (150 mg/kg, i.p.)-anaesthetized rats were ventilated with room air supplemented with 100% oxygen. The right carotid artery and the left jugular vein were cannulated with heparin-filled catheters for continuous measurement of arterial blood pressure and fluid or drug administration, respectively. A left thoracotomy was performed at the 4th intercostal space, and the heart was suspended in a pericardial cradle.20 Myocardial ischaemia was produced by occluding the left anterior descending coronary artery, and reperfusion was initiated by loosening the snare. Area at risk (AAR) and infarct size were delineated by intravenous injection of patent blue dye and incubation of myocardium at risk with 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich, St. Louis, MO, USA), respectively. Arterial blood gas tensions, acid–base status, and body temperature were monitored and maintained within a physiological range throughout the experiment.
2.2.2 Experimental protocol
Wistar rats were randomly assigned to the following six experimental groups (6–9 rats/group): control (CON), ISO, neutralizing antibody against VEGF (nVEGF), nVEGF + ISO, IgG, and IgG + ISO (Figure 1A). After instrumentation was completed, all rats were stabilized for 30 min and subjected to 30 min of coronary occlusion followed by 2 h of reperfusion. ISO at a concentration of 1.4% was administered via an ISO-specific vaporizer (Ohio Medical Instruments, Madison, WI, USA) for 30 min followed by a period of 15 min washout prior to coronary artery occlusion. Rats in CON, nVEGF, and IgG groups received no ISO. To study the role of VEGF in the cardioprotective effect of ISO, rats were injected intravenously with a bolus of the rat antibody against VEGF (affinity purified goat IgG, 0.3 mg/kg) (R&D, Minneapolis, MN, USA) or goat IgG (0.3 mg/kg) as control 60 min before coronary artery occlusion.
Figure 1.

Schematic representation of the experimental protocols. (A) Effect of neutralizing antibody against vascular endothelial growth factor (nVEGF) on isoflurane (ISO)-induced myocardial protection against ischaemia/reperfusion injury in vivo; (B) effect of nVEGF on ISO-induced CM protection against hypoxia/reoxygenation injury in vitro; (C) effect of hypoxia-induced factor 1α (HIF1α) shRNA in ECs on ISO-induced CM protection against hypoxia/reoxygenation injury in vitro. EC + CM, endothelial cell-cardiomyocyte co-culture; L-NMMA, L-NG-monomethyl arginine; rVEGF, recombinant VEGF; SCR, scrambled HIF1α shRNA sequence.
2.3 Endothelial cell-cardiomyocyte co-culture
Human coronary artery ECs (Cell Applications, San Diego, CA, USA) were cultured in MesoEndo cell growth medium (Cell Applications) and used at 4th passage when reaching 80–90% confluent.22 Cardiomyocytes (CMs) were isolated from hearts of 1- to 2-day-old Wistar rats, as described.23 Briefly, animals were anaesthetized with 100% carbon dioxide and sacrificed by cervical dislocation. The heart was removed, washed in ice-cold phosphate buffered saline (pH 7.4, GIBCO, St. Louis, MO, USA) without Ca2+ and Mg2+, and minced into pieces of approximately 1 mm3 using micro-dissecting scissors and washed with cold phosphate buffered saline. Cardiac tissues were resuspended in ADS buffer (116 mM NaCl, 20 mM HEPES, 1 mM NaH2PO4, 5.5 mM Glucose, 5.4 mM KCl, 0.8 mM MgSO4, pH 7.35) containing 0.15 mg/mL collagenase II (Worthington CLS II, Lakewood, NJ, USA) and 0.52 mg/mL pancreatin (Sigma-Aldrich) and incubated at 37°C for 20 min at 100 rpm.22 The supernatant was collected and centrifuged at 1000 ×g for 6 min, and the pellet was collected in 1 mL of calf serum. The procedure was repeated till all heart tissue was completely digested. The pellet from each digestion was collected in 30 mL Dulbecco’s modified DMEM medium containing 17% medium 199, 10% horse serum, 5% foetal bovine serum, 0.5% penicillin-streptomycin and 20 mM HEPES (pH 7.4) with 100 μM proliferation inhibitor bromodeoxyuridine (Sigma Aldrich). CMs were used 3–5 days after isolation when demonstrating rhythmic contractions.
ECs were pre-cultured for 72 h, mixed with CMs at a ratio of 1:6, and cultured in MesoEndo cell growth medium for 12 h, as described. Culture medium was subsequently replaced, and the cells were exposed to 2 h of hypoxia (0.1% O2, Biospherix hypoxia chamber, Lacona, NY, USA) in glucose-free DMEM medium followed by 2 h of reoxygenation. The release of lactate dehydrogenase (LDH) into the culture medium as a marker of cell damage was quantified with a commercially available kit (Genzyme Diagnostics, Cambridge, MA, USA).22
2.4 Effect of VEGF inactivation on I/R injury in EC-CM co-culture
To investigate the role of VEGF in ISO-induced CM protection, co-cultured ECs, and CMS were assigned to the following eight experimental groups (n = 6): normoxia, CON, ISO, nVEGF, nVEGF + ISO, IgG, IgG + ISO, and L-NG-monomethyl arginine (L-NMMA, an inhibitor of nitric oxide synthase) + ISO (Figure 1B). ECs and CMS were co-cultured for 12 h and subsequently treated with 5 µg/mL nVEGF, 5 µg/mL IgG, 1 mM L-NMMA, or vehicle for 60 min prior to administration of ISO. All cells, except normoxic group, were exposed to hypoxia for 2 h followed by 2 h of reoxygenation.
2.5 Immunoblotting
Total soluble protein from cell lysates was prepared, as previously described.22 Forty microgram of protein was resolved on a 7.5% SDS-polyacrylamide gel and transferred to polyvinylidene fluoride membranes. The membranes were blocked in Tris-buffered saline containing 5% milk and subsequently incubated with primary antibodies against rabbit VEGF (1:1000) (Abcam, Cambridge, MA, USA), VEGF receptor 2 (VEGFR2) (1:500) (Cell signaling, MA, USA), rabbit Akt (Cell Signaling), phosphorylated Akt at the activation site Serine 473 (Cell Signaling), rabbit eNOS (1:200) (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), phosphorylated eNOS at the activation site Serine1177 (p-eNOS) (1:100) (Cell Signaling), and mouse β-actin (1:5000) (Cell signaling) overnight at 4°C. The membrane was washed and then incubated with the appropriate anti-rabbit and anti-mouse secondary antibody. Immunoreactive bands were visualized by enhanced chemiluminescence followed by densitometric analysis using image acquisition and analysis software (Image J, NIH, USA).
2.6 Ozone chemiluminescence
The concentration of NO was measured as the amount of nitrite formed when NO reacts with oxygen using iodide and acetic acid at room temperature by using a Sievers NO gas analyser (Model 280, GE Analytical Instruments, Boulder, CO, USA), as described.24,25 Briefly, 1 mL of medium sample was taken 60 min after ISO exposure (control medium was collected at the same time) as basal NO production. Thirty microlitre of medium was sampled in NO gas analyser, and nitrite concentration was calculated after subtraction of background of MesoEndo cell growth medium and normalized to total cell proteins from cell lysates.
2.7 Generation of HIF1α short hairpin RNA
Lentiviral particles containing HIF1α short hairpin RNA (shRNA) and its scrambled sequence were produced, as described.26 Briefly, 10 μg HIF1α or CON shRNA plasmid, 6.5 μg porcine cytomegalovirus (p-CMV) R8.74 as packing construct, and 3.5 μg vesicular stomatitis virus glycoprotein-G (VSV-G) carrying the sequence for the envelope, were co-transfected into 293T cells using the calcium-phosphate co-precipitation method. Media were replaced after 12–14 h. After 36–48 h, the replication-defective lentiviral vectors were harvested, cleared by low-speed centrifugation and filtered through 0.45 μm cellulose acetate filters. The viral titer was determined by FACS analysis of GFP-positive 293T cells. Pilot experiments using a GFP expressing lentiviral vector were performed to determine the optimal multiplicity of infection and were found to be 10 (95–100% GFP-positive EC). ECs were infected with HIF1α shRNA or scramble shRNA at a dose of 4 × 105 transducing unit per well in 24-well plate for three consecutive days when it reached 50–60% confluency.
2.8 Effects of VEGF knockdown with HIF1α short hairpin RNA on the cardioprotective effect of ISO in vitro
The effects of HIF1α knockdown in ECs on ISO-induced CM protection were studied in EC-CM co-culture divided into eight experimental groups (n = 6): CON, ISO, shRNA, shRNA + ISO, SCR, SCR + ISO, shRNA + recombinant VEGF (rVEGF), and shRNA + rVEGF + ISO (Figure 1C). ECs in shRNA, shRNA + ISO, SCR, SCR + ISO, shRNA + rVEGF, and shRNA + rVEGF + ISO groups were infected with HIF1α shRNA or scramble shRNA for 72 h, whereas ECs in CON and ISO groups underwent all culture procedures without shRNA or scramble sHRNA infection. ECs and CMs were co-cultured for 12 h and subjected to 2 h of hypoxia followed by 2 h of reoxygenation. ISO was administered for 60 min via an ISO-specific vaporizer (Ohio Medical Instruments) prior to hypoxia, and the ISO concentration was monitored by a gas analyser (POET IQ, Criticare System, Waukesha, WI, USA). In some experiments, human recombinant VEGF165 (rVEGF) (Calbiochem, San Diego, CA, USA) at a final concentration of 100 ng/mL was added into the cultured cells at the start of the co-culture. The release of LDH into the media was determined at the conclusion of reoxygenation.
2.9 Statistics
All data are expressed as mean ± standard error of mean (SEM). Kruskal–Wallis test followed by Dunn’s test was used to analyse multiple group comparisons. Repeated-measures analysis of variance followed by Bonferroni multiple comparison test was used to evaluate differences in heart rate and mean arterial blood pressure. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, Inc.). A two-tailed P-value of <0.05 was considered statistically significant.
3. Results
3.1 ISO-induced decrease in infarct size was blocked by VEGF inactivation in rats
Heart rate and mean arterial blood pressure during in vivo rat experiment were listed in Table 1. There were no significant differences in both heart rate and mean arterial blood pressure at baseline among six experimental groups (n = 6–9 rats/group): CON, ISO, nVEGF, nVEGF + ISO, IgG, and IgG + ISO. Coronary artery occlusion did not significantly change heart rate in all groups but decreased mean arterial blood pressure in nVEGF + ISO and IgG + ISO groups in comparison with baseline blood pressure. In all six groups, mean arterial blood pressure was significantly lower at 60 and 120 min after reperfusion as compared to the baseline (P < 0.05).
Table 1.
Systemic haemodynamics during in vivo rat experiments
| Group | Baseline | Coronary occlusion | Reperfusion |
||
|---|---|---|---|---|---|
| 60 min | 120 min | ||||
| Heart rate (min−1) | |||||
| CON | 373 ± 17 | 373 ± 21 | 352 ± 18 | 335 ± 17 | |
| ISO | 372 ± 12 | 357 ± 11 | 336 ± 11 | 329 ± 14 | |
| nVEGF | 346 ± 15 | 354 ± 14 | 351 ± 12 | 334 ± 8 | |
| nVEGF + ISO | 370 ± 13 | 355 ± 13 | 317 ± 15* | 297 ± 13*,** | |
| IgG | 347 ± 15 | 343 ± 20 | 306 ± 21 | 302 ± 20 | |
| IgG + ISO | 363 ± 9 | 337 ± 9 | 322 ± 13* | 310 ± 12* | |
| Mean arterial pressure (mmHg) | |||||
| CON | 110 ± 5 | 111 ± 6 | 86 ± 6*,** | 77 ± 5*,** | |
| ISO | 118 ± 6 | 111 ± 4 | 82 ± 7*,** | 73 ± 7*,** | |
| nVEGF | 122 ± 6 | 128 ± 6 | 106 ± 7** | 91 ± 6*,** | |
| nVEGF + ISO | 127 ± 2 | 112 ± 5* | 89 ± 5*,** | 73 ± 3*,**,*** | |
| IgG | 117 ± 4 | 115 ± 11 | 79 ± 7*,** | 71 ± 5*,** | |
| IgG + ISO | 124 ± 4 | 106 ± 4* | 86 ± 5*,** | 74 ± 5*,** | |
CON, control; ISO, isoflurane; IgG, Immunoglobulin G; nVEGF, neutralizing VEGF antibody.
P < 0.05 vs. baseline.
P < 0.05 vs. coronary occlusion.
P < 0.05 vs. 60 min after reperfusion with Kruskal–Wallis test followed by Dunn’s test (n = 6–9 rats/group).
AAR and myocardial infarct size in rat experiment are shown in Figure 2. There were no significant differences in AAR among the six experimental groups (Figure 2A). Coronary occlusion followed by reperfusion resulted in infarct size of 60 ± 1% of AAR (n = 8 rats/group) in control rats, which was significantly decreased to 40 ± 3% (n = 8 rats/group, P < 0.05) by ISO (Figure 2B). Neither nVEGF nor IgG itself significantly changed infarct size compared with CON. ISO-induced decrease in infarct size was blocked by nVEGF (n = 6 rats/group, P < 0.05) but not IgG (Figure 2B).
Figure 2.
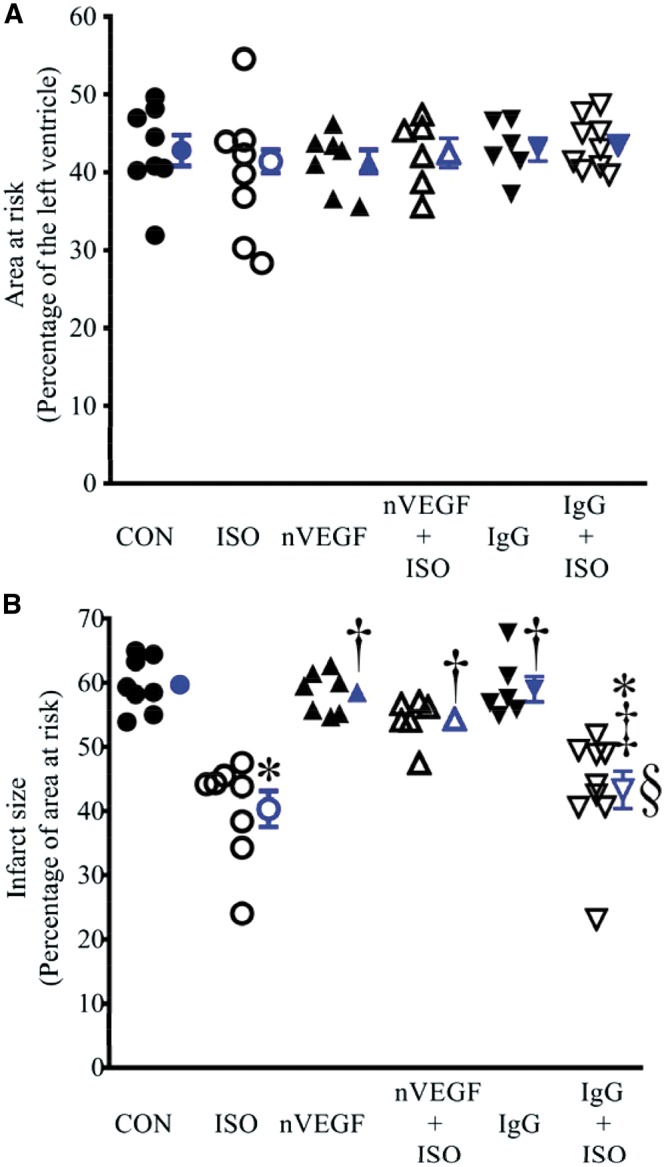
Isoflurane (ISO)-induced decrease in infarct size was blocked by neutralizing anti-vascular endothelial growth factor (nVEGF) in rats subjected to myocardial ischaemia/reperfusion injury. (A) Area at risk expressed as a percentage of the left ventricle; (B) infarct size expressed as a percentage of area at risk. The rats were subjected to 30 min of coronary artery occlusion followed by 2 h of reperfusion (control, CON). Isoflurane (ISO) was administered for 30 min followed by a period of 15 min washout prior to coronary artery occlusion. Data are presented as mean ± SEM. Kruskal–Wallis test followed by Dunn’s test was used to analyse multiple group comparisons. *P < 0.05 vs. CON (control); †P < 0.05 vs. ISO; ‡P < 0.05 vs. nVEGF + ISO; §P < 0.05 vs. IgG (n = 6–9 rats/group).
3.2 VEGF inactivation blocked ISO-induced CM protection in vitro
Figure 3 demonstrates LDH activity in CMs, ECs, and co-cultured ECs and CMs. Hypoxia/reoxygenation significantly increased LDH activity in both CMs and co-cultured CMs and ECs but not ECs. ISO pre-treatment significantly decreased LDH activity in co-cultured CMs and ECs (n = 6, P < 0.05 vs. CON) (Figure 3C) but not in either CMs or ECs (Figures 3A and B) after hypoxia/reoxygenation injury. Neither nVEGF nor IgG significantly changed LDH activity in co-cultured CMs and ECs compared with CON group. ISO-induced decrease in LDH activity was blocked by either nVEGF or L-NMMA but not IgG (Figure 3C).
Figure 3.

Isoflurane (ISO)-induced decrease in lactate dehydrogenase (LDH) activity was blocked by neutralizing antibody against vascular endothelial growth factor (nVEGF) and L-NG-monomethyl arginine (L-NMMA) in co-cultured endothelial cells (ECs) and cardiomyocytes (CMs) subjected hypoxia/reoxygenation injury. (A) Effect of ISO on LDH activity in CMs; (B) effect of ISO on LDH activity in ECs; (C) LDH activity in co-cultured ECs and CMs. The values of the normoxia groups were arbitrarily defined as one in each batch of experiments. Data are presented as mean ± SEM. Kruskal–Wallis test followed by Dunn’s test was used to analyse multiple group comparisons. *P < 0.05 vs. normoxia; †P < 0.05 vs. CON (control); ‡P < 0.05 vs. ISO; §P < 0.05 vs. IgG (n = 6).
3.3 VEGF inactivation decreased ISO-induced increase in NO production
The pre-treatment of ECs and co-cultured ECs and CMs with ISO resulted in a significant increase in NO production (Figure 4). This increase in NO production was not significantly altered by either IgG or scrambled HIF1α shRNA sequence (n = 5, P > 0.05). nVEGF, L-NMMA, HIF1α shRNA did not significantly affect NO production but blocked ISO-induced increases in NO production from ECs and co-cultured ECs and CMs (n = 5, P < 0.05).
Figure 4.

Isoflurane (ISO)-induced increase in nitric oxide (NO) production were blocked by either neutralizing antibody against vascular endothelial growth factor (nVEGF) or hypoxia-inducible factor (HIF1α) shRNA in endothelial cells (ECs) and co-cultured ECs and cardiomyocytes (CMs). (A) Effects of IgG, nVEGF, and L-NMMA (L-NG-monomethyl arginine) on ISO-induced increases in NO production in ECs; (B) effects of IgG, nVEGF, and L-NMMA on ISO-induced increases in NO production in co-cultured ECs and CMS; (C) effects of HIF1α shRNA, scrambled HIF1α shRNA sequence (SCR), and recombinant VEGF (rVEGF) on ISO-induced increases in NO production in ECs; (D) effects of HIF1α shRNA, SCR, and rVEGF on ISO-induced increases in NO production in co-cultured ECs and CMS. The values of the control groups were arbitrarily defined as one in each batch of experiments. Data are presented as mean ± SEM. Kruskal–Wallis test followed by Dunn’s test was used to analyse multiple group comparisons. *P < 0.05 vs. CON (control); †P < 0.05 vs. ISO; ‡P < 0.05 vs. shRNA + ISO (n = 5).
3.4 VEGF inactivation reduced ISO-induced increase in p-eNOS in ECs
As shown in Figures 5A andB, the ratios of p-Akt/Akt and p-eNOS/eNOS in ECs were significantly increased by ISO. The pre-treatment of ECs with nVEGF significantly decreased ISO-induced increases in the ratios of p-Akt/Akt and p-eNOS/eNOS (n = 4). In contrast, IgG pretreatment did not significantly change ISO-induced increases in the ratio of p-Akt/Akt and p-eNOS/eNOS. Furthermore, the Akt inhibitor, wortmannin (WM), was used to determine if Akt was involved in eNOS phosphorylation by VEGF. WM did not affect the NO production in ECs but blocked the ISO-induced increases in NO (Figure 5C).
Figure 5.

Isoflurane (ISO) induced increases in phosphorylated Akt (p-Akt) and endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) production in endothelial cells. (A) Blockade of ISO-induced Akt phosphorylation by neutralizing anti-vascular endothelial growth factor (nVEGF) in endothelial cells. Top: western blot bands showing the expression of p-Akt, Akt, and β-actin as a loading control in endothelial cells; bottom: the ratio of p-Akt/Akt; (B) blockade of ISO-induced eNOS phosphorylation by nVEGF in endothelial cells. Top: representative western blot bands showing the expression of phosphorylated eNOS (p-eNOS) at the activation site Serine1177, eNOS, and β-actin; bottom: the ratio of p-eNOS/total eNOS. In A and B, endothelial cells were pre-treated with nVEGF or IgG prior to ISO and subsequently subjected to hypoxia/reoxygenation. *P < 0.05 vs. control; †P < 0.05 vs. ISO (n = 4). (C) Blockade of ISO-induced increases in NO production by wortmannin (WM) in endothelial cells. The values of the control groups were arbitrarily defined as one in each batch of experiments. Data are presented as mean ± SEM. Kruskal–Wallis test followed by Dunn’s test was used to analyse multiple group comparisons. *P < 0.05 vs. control; †P < 0.05 vs. WM, ‡P < 0.05 vs. ISO (n = 7).
3.5 HIF1α short hairpin RNA down-regulated VEGF and VEGFR2 in ECs
The expression of VEGF and VEGFR2 proteins was not significantly altered in ECs infected with scrambled HIF1α shRNA sequence compared with CON (n = 5, P > 0.05) (Figure 6). In contrast, the expression of both VEGF and its receptor proteins was significantly decreased in ECs infected with lentiviral vector expressing the HIF1α shRNA compared with CON and the scrambled HIF1α shRNA sequence (n = 5, P < 0.05).
Figure 6.

Hypoxia-inducible factor 1α (HIF1α) knockdown decreased the expression of vascular endothelial growth factor (VEGF) and VEGF receptor 2 (VEGFR2) proteins in endothelial cells. (A) expression of VEGF protein in endothelial cells (top: representative western blot bands of VEGF); (B) expression of VEGFR2 protein in endothelial cells (top: representative bands of western blot bands of VEGFR2). Endothelial cells were infected with lentiviral vectors containing HIF1α shRNA (shRNA) or scrambled HIF1α shRNA sequence (SCR). Data are presented as mean ± SEM. *P < 0.05 vs. control; †P < 0.05 vs. SCR analysed by Kruskal–Wallis test followed by Dunn’s test (n = 5).
3.6 HIF1α short hairpin attenuated the protective effect of ISO on CMs
EC infection with lentiviral vector expressing the HIF1α shRNA results in 80–90% decrease in the expression of HIF1α protein (n = 5, P < 0.05 vs. CON) (Figure 7A). Neither HIF1α shRNA nor scrambled HIF1α shRNA sequence significantly altered LDH activity in co-cultured CMs and ECs subjected to hypoxia/reoxygenation injury (Figure 7B). The treatment of co-cultured ECs and CMs with ISO caused a significant decrease in LDH activity (n = 6, P < 0.05). This protective effect of ISO was abolished by HIF1α shRNA (n = 6, P < 0.05) but not scrambled HIF1α shRNA sequence. The detrimental effect of HIF1α shRNA was restored by rVEGF administered prior to CM-EC co-culture (Figure 7B).
Figure 7.

Hypoxia-induced factor 1α (HIF1α) knockdown blocked isoflurane (ISO)-induced decrease in lactate dehydrogenase (LDH) activity in co-cultured endothelial cells (ECs) and cardiomyocytes (CMs) subjected to hypoxia/reoxygenation injury. (A) The expression of HIF1α protein in ECs treated with lentiviral vector containing either HIF1α shRNA (shRNA) or scrambled sequence (SCR) (left: representative western blot bands of HIF1α from the extract of cultured ECs). *P < 0.05 vs. CON (control); †P < 0.05 vs. SCR (n = 5). (B) LDH activity in media released from co-cultured ECs and CMs. All cells were subjected to 2 h of hypoxia followed by 2 h of reoxygenation (CON); whereas the cells in normoxia group underwent all culture procedures without hypoxia/reoxygenation. The values of the control groups were arbitrarily defined as one in each batch of experiments. Data are presented as mean ± SEM. Kruskal–Wallis test followed by Dunn’s test was used to analyse multiple group comparisons. *P < 0.05 vs. normoxia; †P < 0.05 vs. CON; ‡P < 0.05 vs. ISO; §P < 0.05 vs. shRNA (n = 6). rVEGF, recombinant vascular growth factor.
4. Discussion
The results of the present study demonstrate that ISO attenuates myocardial I/R injury in rats and in co-cultured ECs and CMs when administered prior to either I/R or hypoxia/reoxygenation injury. These protective effects of ISO are associated with increases in p-eNOS and protection of NO. Interestingly, either VEGF inactivation or knockdown with HIF1α shRNA in ECs blocks these beneficial effects of ISO. These results not only suggest involvement of the interaction between ECs and CMs in ISO cardiac preconditioning but also indicate the role of VEGF-regulated phosphorylation of eNOS in the cardioprotective effect of ISO.
We showed that ISO decreased infarct size in rats when applied for 30 min prior to myocardial ischaemia. This result is consistent with previous studies by Baotic et al.20 Myocardial infarct size reflects the degree of severity of I/R injury.27 Thus, ISO administered prior to ischaemia potently reduces myocardial I/R injury. The treatment of rats with IgG antibody prior to myocardial ischaemia neither altered AAR and infarct size nor impacted ISO-induced decreases in infarct size. Intriguingly, inactivation of VEGF with its specific antibody blocked ISO-induced decreases in infarct size. Multiple previous studies indicate that VEGF favourably regulates myocardial I/R injury.28,29 Our results indicate that VEGF is improtant in ISO-induced cardioprotection against I/R injury in rats.
In the heart, CMs are intimately related to ECs in a web-like network of capillaries.30 Communication between ECs and CMs is important in maintaining physiological regulation of CMs by releasing multiple paracrine factors.30,31 In the present study, ISO pretreatment promotes CM survival in co-cultured ECs and CMs but not CMs or ECs alone following hypoxia/reoxygenation injury. These results suggest that EC-CM interaction plays an important role in ISO-induced CM protection from hypoxia/reoxygenation injury. It has been known that in response to ischaemia and reperfusion, either ECs or CMs produce and release multiple endogenous mediators such as NO, reactive oxygen species, adenosine, cyclic ADP-ribose, opioids, neuregulin, and succinate.22,24,32,33 These mediators possess strong biological effects on adjacent cells and can alter cell survival and injury.34–37 Thus, interactions between ECs and CMs are involved in ISO-induced cardioprotection against I/R injury.
The current study demonstrated that ISO increased the phosphorylation of eNOS in ECs. These results are consistent with previous studies by Baotic et al.20 and Qiao et al.6 Intriguingly, this beneficial effect of ISO on eNOS was blocked by VEGF inactivation. Furthermore, our study shows that VEGF binds to its receptors, VEGFR2 (KDR/Flk-1), which induces the phosphorylation of AKT and eNOS. These results are consistent with previous studies.38,39 Taken together, VEGF plays an important role in the phosphorylation of eNOS in ISO-induced cardioprotection.
Phosphorylation of eNOS modulates the production of NO.40,41 In ECs alone or co-cultured ECs and CMs, ISO elevated the production of NO, whereas either VEGF inactivation or knockdown blocked the favourable effect of ISO on NO. It is clear that NO derived from eNOS acts as a paracrine factor to trigger the cardioprotective effect of ISO.42 Thus, VEGF may mediate the production of NO.
We demonstrated that HIF1α shRNA decreased the expression of not only HIF1α but also VEGF in ECs. HIF1α is a key regulator responsible for the induction of genes that facilitate the adaptation and survival of cells from normoxia to hypoxia.43,44 Previous studies by Wang et al.15 and Leucker et al.42 reports that the expression of HIF1α protein in rat hearts or cultured CMs is significantly enhanced following ISO treatment. HIF1α has a general role in multiple physiological responses to ischaemia or hypoxia such as energy metabolism, iron homeostasis, vascular remodelling, cell proliferation, and angiogenesis via activating numerous target genes.45 Interestingly, HIF1α shRNA also decreased the expression of VEGF and VEGFR2 in ECs, suggesting that VEGF is a down-stream mediator of HIF1α. It is likely that HIF1α shRNA decreases the expression of VEGF and VEGFR2 through inhibiting their RNAs.
We have previously demonstrated that co-culture of CMs with ECs can mimic myocardial I/R injury in vivo.22 Indeed, hypoxia/reoxygenation resulted in significant cell injury (increased LDH activity) in co-culture of CMs with ECs. ISO decreased LDH activity following hypoxia/reoxygenation injury. Furthermore, HIF1α shRNA eliminated the protective effect of ISO on CMs, whereas rVEGF the impaired cardioprotective effect of ISO resulting from HIF1α knockdown can be restored by rVEGF. Collectively, the inhibitory effect of HIF1α shRNA on the cardioprotective effect of ISO depends on reduction of VEGF expression as well as VEGF inactivation.
Based on our findings and previous data, we proposed the signalling transduction pathway of ISO cardiac preconditioning (Figure 8). ISO preconditioning activates VEGF in coronary vasculature.15 VEGF binding to the receptor kinase VEGFR2 triggers multiple transduction pathways, which leads to an increase in p-Akt. Phosphorylated Akt specifically phosphorylates the Serine1177 of eNOS proteins. The p-eNOS proteins catalyse the substrate, L-arginine, to form NO and L-citrulline. NO produced from eNOS in coronary artery endothelium exerts paracrine effects on CMs and produces cytoprotective effects, in addition to vasodilatory effect.
Figure 8.
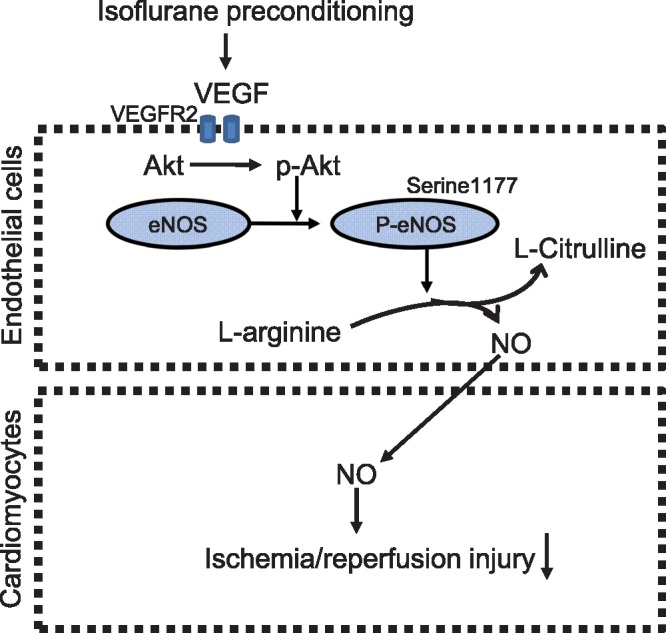
A schematic view of the signalling transduction pathway of isoflurane (ISO) cardiac preconditioning. Vascular endothelial growth factor (VEGF) in coronary artery endothelium is up-regulated following isoflurane preconditioning. VEGF activates VEGF receptor 2 (VEGFR2) and induces the phosphorylation of Akt. Phosphorylated Akt (p-Akt) specifically phosphorylates the Serine1177 of endothelial nitric oxide synthase (eNOS), which enhances the eNOS activity. The phosphorylated eNOS (p-eNOS) catalyses the substrate L-arginine to form nitric oxide (NO) and L-citrulline. Coronary artery endothelium-derived NO acts as a paracrine mediator to produce the cytotective effect in cardiomyocytes.
Volatile anaesthetic agents such as ISO, sevoflurane, enflurane, and desflurane are widely used in human and animal cardiac and non-cardiac surgery. Cardiac as well as non-cardiac surgery may evoke perioperative adverse events including myocardial ischaemia, diverse arrhythmias, and reperfusion injury. The findings in this study indicate that administration of volatile anaesthetics prior to myocardial ischaemia reduces the degree of I/R injury to the heart. This favourable effect of volatile anaesthetics has been verified in some studies of patients undergoing coronary artery bypass graft surgery.4,5 Furthermore, we demonstrate that the cardioportective effect of ISO preconditioning is associated with increases in VEGF-mediated phosphorylation of Akt and eNOS and the production of NO. It is clear that VEGF, Akt, and eNOS have multiple favourable effects on ischaemic/reperfused myocardium such as enhancement of myocardial tolerance to ischaemia, inhibition of the mitochondrial permeability transition pore opening, reduction of necrosis and apoptosis, and improvements in cardiac function.39,46,47 Moreover, VEGF is a well-known pro-angiogenetic factor, which stimulates neo-vascularization and increases microvascular permeability.12,39 Higher levels of VEGF are beneficial to cardiac remodelling after infarction.
A limitation of this study is incomplete mechanistic link between VEGF and the phosphorylation of eNOS. Previous studies have shown that VEGF activates Akt, AMPK, PKA, PKC, PKG, and CaMK-2, all of which can phosphorylate eNOS.16,48 The present study demonstrates that Akt contributes to the eNOS phosphorylation in ISO cardiac preconditioning. Whether non-Akt kinases are implicated in the phosphorylation of eNOS is not examined. Another limitation is incomplete phosphorylation sites of eNOS by VEGF. Previous studies have indicated that the phosphorylation of Serine114, 116, 615, 633, and 1177 of eNOS proteins elevates the enzyme activity, whereas the phosphorylation of Threonine495 inhibits the activity.48 Our present study only examined the phosphorylation of Serine1177 in eNOS proteins. Lastly, this study lacks the study of the essential co-factor of eNOS, tetrahydrobiopterin, which is important in the determination of the eNOS product, NO or superoxide.49 Our previous studies demonstrated that tetrahydrobiopterin levels, eNOS dimerization, and NO production were significantly elevated following ISO preconditioning.8,22 Thus, ISO preconditioning not only enhances the phosphorylation of eNOS but improves its dimerization via increasing tetrahydrobiopterin bioavailability.
In summary, the present investigation demonstrates that endothelial-myocardial interaction is involved in the cardioprotective effect of ISO. VEGF in ECs plays an important role in ISO-induced cardioprotection against I/R injury. In the signal transduction pathway of ISO-induced cardioprotection, eNOS may serve as the down-stream mediator of VEGF. VEGF regulation of phosphorylation of eNOS is a key step for ISO-induced cardioprotection against I/R injury.
Acknowledgements
The authors thank David Schwabe, John Tessmer, and John G. Krolikowski for excellent technical assistance.
Conflict of interest: none declared.
Funding
This work was supported, in part, by a National Institutes of Health research [P01GM 066730 to Z.J.B.] from the United States Public Health Services, Bethesda, MD, USA.
Footnotes
Time for primary review: 31 days
References
- 1. Bosnjak ZJ, Ge ZD.. The application of remote ischemic conditioning in cardiac surgery. F1000Res 2017;6:928.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uhlig C, Bluth T, Schwarz K, Deckert S, Heinrich L, De Hert S, Landoni G, Serpa Neto A, Schultz MJ, Pelosi P, Schmitt J, Gama de Abreu M.. Effects of volatile anesthetics on mortality and postoperative pulmonary and other complications in patients undergoing surgery: a systematic review and meta-analysis. Anesthesiology 2016;124:1230–1245. [DOI] [PubMed] [Google Scholar]
- 3. Kersten JR, Orth KG, Pagel PS, Mei DA, Gross GJ, Warltier DC.. Role of adenosine in isoflurane-induced cardioprotection. Anesthesiology 1997;86:1128–1139. [DOI] [PubMed] [Google Scholar]
- 4. De Hert SG, ten Broecke PW, Mertens E, Van Sommeren EW, De Blier IG, Stockman BA, Rodrigus IE.. Sevoflurane but not propofol preserves myocardial function in coronary surgery patients. Anesthesiology 2002;97:42–49. [DOI] [PubMed] [Google Scholar]
- 5. Fräßdorf J, Borowski A, Ebel D, Feindt P, Hermes M, Meemann T, Weber R, Müllenheim J, Weber NC, Preckel B, Schlack W.. Impact of preconditioning protocol on anesthetic-induced cardioprotection in patients having coronary artery bypass surgery. J Thorac Cardiovasc Surg 2009;137:1436–1442, 1442.e1–2. [DOI] [PubMed] [Google Scholar]
- 6. Qiao S, Olson JM, Paterson M, Yan Y, Zaja I, Liu Y, Riess ML, Kersten JR, Liang M, Warltier DC, Bosnjak ZJ, Ge ZD.. MicroRNA-21 mediates isoflurane-induced cardioprotection against ischemia-reperfusion injury via Akt/nitric oxide synthase/mitochondrial permeability transition pore pathway. Anesthesiology 2015;123:786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kersten JR. Anesthetic preconditioning: an anesthesiologist's tale. 1997. Anesthesiology 2011;114:162–166. [DOI] [PubMed] [Google Scholar]
- 8. Ge ZD, Li Y, Qiao S, Bai X, Warltier DC, Kersten JR, Bosnjak ZJ, Liang M.. Failure of isoflurane cardiac preconditioning in obese type 2 diabetic mice involves aberrant regulation of microRNA-21, endothelial nitric oxide synthase, and mitochondrial complex I. Anesthesiology 2018;128:117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiari PC, Bienengraeber MW, Weihrauch D, Krolikowski JG, Kersten JR, Warltier DC, Pagel PS.. Role of endothelial nitric oxide synthase as a trigger and mediator of isoflurane-induced delayed preconditioning in rabbit myocardium. Anesthesiology 2005;103:74–83. [DOI] [PubMed] [Google Scholar]
- 10. Ferrara N, Adamis AP.. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov 2016;15:385–403. [DOI] [PubMed] [Google Scholar]
- 11. Wang S, Li X, Parra M, Verdin E, Bassel-Duby R, Olson EN.. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc Natl Acad Sci USA 2008;105:7738–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moens S, Goveia J, Stapor PC, Cantelmo AR, Carmeliet P.. The multifaceted activity of VEGF in angiogenesis—implications for therapy responses. Cytokine Growth Factor Rev 2014;25:473–482. [DOI] [PubMed] [Google Scholar]
- 13. Huang M, Qiu Q, Xiao Y, Zeng S, Zhan M, Shi M, Zou Y, Ye Y, Liang L, Yang X, Xu H.. BET bromodomain suppression inhibits VEGF-induced angiogenesis and vascular permeability by blocking VEGFR2-mediated activation of PAK1 and eNOS. Sci Rep 2016;6:23770.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC.. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest 1997;100:3131–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang C, Weihrauch D, Schwabe DA, Bienengraeber M, Warltier DC, Kersten JR, Pratt PF Jr, Pagel PS.. Extracellular signal-regulated kinases trigger isoflurane preconditioning concomitant with upregulation of hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression in rats. Anesth Analg 2006;103:281–288. [DOI] [PubMed] [Google Scholar]
- 16. Gentile C, Muise-Helmericks RC, Drake CJ.. VEGF-mediated phosphorylation of eNOS regulates angioblast and embryonic endothelial cell proliferation. Dev Biol 2013;373:163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blanes MG, Oubaha M, Rautureau Y, Gratton JP.. Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. J Biol Chem 2007;282:10660–10669. [DOI] [PubMed] [Google Scholar]
- 18. Kou R, Greif D, Michel T.. Dephosphorylation of endothelial nitric-oxide synthase by vascular endothelial growth factor. Implications for the vascular responses to cyclosporin A. J Biol Chem 2002;277:29669–29673. [DOI] [PubMed] [Google Scholar]
- 19. Feliers D, Chen X, Akis N, Choudhury GG, Madaio M, Kasinath BS.. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int 2005;68:1648–1659. [DOI] [PubMed] [Google Scholar]
- 20. Baotic I, Weihrauch D, Procknow J, Vasquez-Vivar J, Ge ZD, Sudhakaran S, Warltier DC, Kersten JR.. Isoflurane favorably modulates guanosine triphosphate cyclohydrolase-1 and endothelial nitric oxide synthase during myocardial ischemia and reperfusion injury in rats. Anesthesiology 2015;123:582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu Y, Jin J, Qiao S, Lei S, Liao S, Ge ZD, Li H, Wong GT, Irwin MG, Xia Z.. Inhibition of PKCβ2 overexpression ameliorates myocardial ischaemia/reperfusion injury in diabetic rats via restoring caveolin-3/Akt signaling. Clin Sci 2015;129:331–344. [DOI] [PubMed] [Google Scholar]
- 22. Leucker TM, Ge ZD, Procknow J, Liu Y, Shi Y, Bienengraeber M, Warltier DC, Kersten JR.. Impairment of endothelial-myocardial interaction increases the susceptibility of cardiomyocytes to ischemia/reperfusion injury. PLoS One 2013;8:e70088.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rutering J, Ilmer M, Recio A, Coleman M, Vykoukal J, Alt E.. Improved method for isolation of neonatal rat cardiomyocytes with increased yield of C-Kit+ cardiac progenitor cells. J Stem Cell Res Ther 2015;5:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, Baumgardt SL, Fang J, Shi Y, Qiao S, Bosnjak ZJ, Vasquez-Vivar J, Xia Z, Warltier DC, Kersten JR, Ge ZD.. Transgenic overexpression of GTP cyclohydrolase 1 in cardiomyocytes ameliorates post-infarction cardiac remodeling. Sci Rep 2017;7:3093.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu HE, Baumgardt SL, Fang J, Paterson M, Liu Y, Du J, Shi Y, Qiao S, Bosnjak ZJ, Warltier DC, Kersten JR, Ge ZD.. Cardiomyocyte GTP cyclohydrolase 1 protects the heart against diabetic cardiomyopathy. Sci Rep 2016;6:27925.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Canfield SG, Sepac A, Sedlic F, Muravyeva MY, Bai X, Bosnjak ZJ.. Marked hyperglycemia attenuates anesthetic preconditioning in human-induced pluripotent stem cell-derived cardiomyocytes. Anesthesiology 2012;117:735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hausenloy DJ, Yellon DM.. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 2013;123:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li GH, Luo B, Lv YX, Zheng F, Wang L, Wei MX, Li XY, Zhang L, Wang JN, Chen SY, Tang JM, He X.. Dual effects of VEGF-β on activating cardiomyocytes and cardiac stem cells to protect the heart against short- and long-term ischemia-reperfusion injury. J Transl Med 2016;14:116.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang L, Zhang Y, Zhu M, Zhang Q, Wang X, Wang Y, Zhang J, Li J, Yang L, Liu J, Liu F, Yang Y, Kang L, Shen Y, Qi Z.. Resveratrol attenuates myocardial ischemia/reperfusion injury through up-regulation of vascular endothelial growth factor B. Free Radic Biol Med 2016;101:1–9. [DOI] [PubMed] [Google Scholar]
- 30. Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev 2003;83:59–115. [DOI] [PubMed] [Google Scholar]
- 31. Fountoulaki K, Dagres N, Iliodromitis EK.. Cellular communications in the heart. Card Fail Rev 2015;1:64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ge ZD, Li PL, Chen YF, Gross GJ, Zou AP.. Myocardial ischemia and reperfusion reduce the levels of cyclic ADP-ribose in rat myocardium. Basic Res Cardiol 2002;97:312–319. [DOI] [PubMed] [Google Scholar]
- 33. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP.. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014;515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Black RG Jr, Guo Y, Ge ZD, Murphree SS, Prabhu SD, Jones WK, Bolli R, Auchampach JA.. Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ Res 2002;91:165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA.. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5'-n-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther 2006;319:1200–1210. [DOI] [PubMed] [Google Scholar]
- 36. Ge ZD, van der Hoeven D, Maas JE, Wan TC, Auchampach JA.. A3 adenosine receptor activation during reperfusion reduces infarct size through actions on bone marrow-derived cells. J Mol Cell Cardiol 2010;49:280–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garbayo E, Gavira JJ, de Yebenes MG, Pelacho B, Abizanda G, Lana H, Blanco-Prieto MJ, Prosper F.. Catheter-based intramyocardial injection of FGF1 or NRG1-loaded MPS improves cardiac function in a preclinical model of ischemia-reperfusion. Sci Rep 2016;6:25932.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koch S, Claesson-Welsh L.. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2012;2:a006502.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simons M, Gordon E, Claesson-Welsh L.. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 2016;17:611–625. [DOI] [PubMed] [Google Scholar]
- 40. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC.. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999;399:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM.. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999;399:601–605. [DOI] [PubMed] [Google Scholar]
- 42. Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK, Warltier DC, Kersten JR, Pratt PF Jr.. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J Mol Cell Cardiol 2011;51:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang GL, Jiang BH, Semenza GL.. Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem Biophys Res Commun 1995;212:550–556. [DOI] [PubMed] [Google Scholar]
- 44. Bellanti F. Hypoxia-inducible factor-1 in myocardial ischaemia/reperfusion injury. Acta Physiol (Oxf) 2017;221:93–94. [DOI] [PubMed] [Google Scholar]
- 45. Ke Q, Costa M.. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 2006;70:1469–1480. [DOI] [PubMed] [Google Scholar]
- 46. Schwarz ER, Speakman MT, Patterson M, Hale SS, Isner JM, Kedes LH, Kloner RA.. Evaluation of the effects of intramyocardial injection of DNA expressing vascular endothelial growth factor (VEGF) in a myocardial infarction model in the rat–angiogenesis and angioma formation. J Am Coll Cardiol 2000;35:1323–1330. [DOI] [PubMed] [Google Scholar]
- 47. Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K.. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med 2000;6:1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kukreja RC, Xi L.. Enos phosphorylation: a pivotal molecular switch in vasodilation and cardioprotection? J Mol Cell Cardiol 2007;42:280–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vladic N, Ge ZD, Leucker T, Brzezinska AK, Du JH, Shi Y, Warltier DC, Pratt PF Jr, Kersten JR.. Decreased tetrahydrobiopterin and disrupted association of HSP90 with eNOS by hyperglycemia impair myocardial ischemic preconditioning. Am J Physiol Heart Circ Physiol 2011;301:H2130–H2139. [DOI] [PMC free article] [PubMed] [Google Scholar]