

Abstract
Aims
Myocardial ischaemia followed by reperfusion (IR) causes an oxidative burst resulting in cellular dysfunction. Little is known about the impact of oxidative stress on cardiomyocyte lipids and their role in cardiac cell death. Our goal was to identify oxidized phosphatidylcholine-containing phospholipids (OxPL) generated during IR, and to determine their impact on cell viability and myocardial infarct size.
Methods and results
OxPL were quantitated in isolated rat cardiomyocytes using mass spectrophotometry following 24 h of IR. Cardiomyocyte cell death was quantitated following exogenously added OxPL and in the absence or presence of E06, a ‘natural’ murine monoclonal antibody that binds to the PC headgroup of OxPL. The impact of OxPL on mitochondria in cardiomyocytes was also determined using cell fractionation and Bnip expression. Transgenic Ldlr−/− mice, overexpressing a single-chain variable fragment of E06 (Ldlr−/−-E06-scFv-Tg) were used to assess the effect of inactivating endogenously generated OxPL in vivo on myocardial infarct size. Following IR in vitro, isolated rat cardiomyocytes showed a significant increase in the specific OxPLs PONPC, POVPC, PAzPC, and PGPC (P < 0.05 to P < 0.001 for all). Exogenously added OxPLs resulted in significant death of rat cardiomyocytes, an effect inhibited by E06 (percent cell death with added POVPC was 22.6 ± 4.14% and with PONPC was 25.3 ± 3.4% compared to 8.0 ± 1.6% and 6.4 ± 1.0%, respectively, with the addition of E06, P < 0.05 for both). IR increased mitochondrial content of OxPL in rat cardiomyocytes and also increased expression of Bcl-2 death protein 3 (Bnip3), which was inhibited in presence of E06. Notably cardiomyocytes with Bnip3 knock-down were protected against cytotoxic effects of OxPL. In mice exposed to myocardial IR in vivo, compared to Ldlr−/− mice, Ldlr−/−-E06-scFv-Tg mice had significantly smaller myocardial infarct size normalized to area at risk (72.4 ± 21.9% vs. 47.7 ± 17.6%, P = 0.023).
Conclusions
OxPL are generated within cardiomyocytes during IR and have detrimental effects on cardiomyocyte viability. Inactivation of OxPL in vivo results in a reduction of infarct size.
Keywords: Oxidized phospholipids • Coronary artery disease • Myocardial infarction • Ischaemic heart disease • Ischaemia–reperfusion injury • Mass spectrometry
Introduction
Coronary artery disease remains the leading cause of morbidity and mortality worldwide. Major advances in the treatment of acute coronary syndromes and myocardial infarction (MI) have occurred over the past 20 years. In particular, the ability to rapidly restore blood flow to the myocardium during ischaemia, using thrombolytic or percutaneous interventional approaches has resulted in improved outcomes.1 However, despite timely revascularization and contemporary medical therapies, one-quarter of patients presenting with an anterior ST-elevation MI (STEMI) can experience death or heart failure.2 Restoration of blood flow to the culprit vessel can paradoxically induce cardiomyocyte death, a phenomenon termed reperfusion injury, which can account for up to 50% of the final infarct size3 and contribute to poor outcomes. Despite intense investigation, there have been multiple failures of pharmacologic attempts to reduce ischaemia–reperfusion (IR) injury.4,5 Currently, there are no effective pharmacologic therapies, and there is an urgent need to further understand this process.6
Reperfusion is followed by an intense burst of reactive oxygen species (ROS) that results in cellular dysfunction, inflammation, and ultimately cardiomyocyte death by modifying intracellular molecules, including lipids.7 One target for ROS is the abundant phosphatidylcholine-containing phospholipids molecules that comprise the cellular bilayer within mammalian cells.8 Oxidation of phosphocholine containing phospholipids results in the formation of OxPL [‘OxPL’ as used in this manuscript refers exclusively to phosphocholine (PC) containing OxPL]. This results in fragmentation of polyunsaturated fatty acids at the sn-2 position, altering conformation of the PC head group, allowing it to be recognized by innate pattern recognition receptors (PRRs) such as macrophage scavenger receptors CD-36 and SR-B1 and TLRs, soluble proteins such as CRP and natural antibodies (NAbs), such as E06.9–11 OxPL potentiate oxidative stress and trigger sterile inflammation and have been broadly implicated in many inflammatory diseases including atherosclerosis, lung injury, and age-related macular degeneration.11–15
As a specific example of the biological activity of OxPL, a brief exposure of human aortic endothelial cells (HAECs) or murine macrophages to OxPL results in changes in transcription of >1000 genes involved in inflammation, pro-coagulant activity, redox reaction, sterol metabolism, cell cycle, unfolded protein response, and angiogenesis.16,17 Furthermore, the murine IgM NAb E06, which recognizes and bind OxPL but not unoxidized PL,18,19 neutralize its proinflammatory effects.20 In culture, E06 prevented the uptake of oxidized LDL (OxLDL) by macrophages,20,21 and attenuated the inflammatory effects of OxPL on endothelial cells and monocytes,22 while immunization-induced increased titres of E06 were associated with attenuated atherosclerosis in a mouse model of atherosclerosis.23 Because OxPL are products of lipid peroxidation, it might be predicted that OxPL would adversely contribute to IR injury and cell death. Despite the well-established role of mitochondria in cardiomyocyte cell death, little is known about the role of OxPL and mitochondrial dysfunction in cardiomyocytes. Bcl-2-like 19 kDa-interacting protein 3 (Bnip3) has been shown to play a critical role in regulating mitochondrial function and cell death during hypoxic injury in cardiomyocytes. Activation of Bnip3 leads to mitochondrial perturbations that include loss of mitochondrial membrane potential, increased ROS and permeability transition pore (mPTP) opening, culminating in cell death. Using both in vivo and in vitro models of IR injury, we examine the effects of OxPL on cardiac myocyte viability through a Bnip3-mediated pathway and whether neutralization of OxPL with E06 will suppress cardiac cell death and IR injury.
Materials and methods
Materials
Synthetic standards 1,2-dinonanoyl-sn-glycero-3-phosphocholine (DNPC), 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVPC), 1-palmitoyl-2-glutaroyl-sn-glycero-3-phosphocholine (PGPC), 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine (PAzPC), 1-palmitoyl-2-(9-oxo)nonanoyl-sn-glycero-3-phosphocholine (PONPC), and the IgM murine NAb E06, which is LPS free, were obtained from Avanti Polar Lipids (Alabaster, AL, USA). 1-(palmitoyl)-2-(5-keto-6-octene-dioyl)-3-phosphocholine (KOdiAPC) and, 1-palmitoyl-2-(4-keto-dodec-3-ene-dioyl)-sn-glycero-3-phosphocholine (KDdiAPC) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). All solvents were HPLC grade.
Cell culture of rat cardiomyocytes
All animal experiments performed conform to the NIH guidelines regarding animal experimentation and were approved by the Research Ethics Board of the University of Manitoba and/or the Institutional Animal Care and Use Committee (IACUC) at University of California San Diego. Neonatal rat cardiomyocytes (NNCM) were isolated from 1 to 2 day old Sprague-Dawley rat pups as previously described.24 Whole hearts were excised from rat pups by mid-line sternotomy after cervical dislocation. Hearts were washed and minced to adequately break up macroscopic structures before re-washing with cold filter sterilized phosphate buffered saline (PBS) containing 10 g/L of glucose to remove red blood cells and debris. Repeated enzymatic digestion of heart fragments was performed with collagenase (740U), trypsin (370U), and DNase (2880U) (Worthington Biochemical) agitating at 35°C for three 10-min and three 7-min digestions. Digested supernatant solutions were centrifuged into a cell pellet and then separation of cell types using a Percoll® (GE Healthcare) gradient of 1.05, 1.06, and 1.082 g/mL allowed for a layer enriched with myocytes to be isolated. To remove fibroblasts, the cells were pre-plated on non-coated 150 mm culture plates for 45 min. Purified NNCM (95% pure by sarcomere staining) were then transferred to sterile tissue culture plates at a cell density of 1.75 × 106/35-mm plate. Collagen-coated glass coverslips in 24-well tissue culture plates were used for microscopy analysis, and NNCM were plated at 3.2 × 105/well. Cells were incubated overnight in Dulbecco’s Modified Eagle Medium/Ham’s nutrient mixture F-12 (1:1) containing 2 mM glutamine, 3 mM NaHCO3, 15 mM HEPES, and 50 mg/mL gentamycin (DMEM/F12) plus 10% foetal bovine serum (FBS). DMEM/F12 with 10% FBS was changed to serum-free DMEM/F12 (DFSF) the following day. Control solution consisted of 140 mM NaCl, 6 mM KCl, 1.25 mM CaCl2, 6 mM HEPES, and 10 mM D-glucose buffered to pH 7.4, ischaemic solution was buffered to pH 6.0, and consisted of the same components, except 8 mM KCl was added and no D-glucose was added. The ischaemic buffer was purged of oxygen by bubbling 95% N2 and 5% CO2 gas into the medium for 1 h before being applied to cells.25 A hypoxic chamber designed for storage in a 37°C water jacketed tissue culture incubator was used to maintain an atmosphere of 95% N2 and 5% CO2 gas conditions for 18 h overnight. Reperfusion was achieved by applying control buffer to cells for 4 h in a normal culture incubator. After the treatment, cells were scraped off into a small portion of PBS. Cardiomyocyte cell lysates were subjected to western Blot analysis.
Bnip3 knock-down
To knock-down Bnip3 expression in cardiac myocytes, we designed short hairpin RNA (shRNA) directed against Bnip3 targeted against exon 3 of Bnip3, with the sequence 5-CAC CGACA CCACA AGATA CCAAC AGCGA ACTGT TGGTA TCTTG TGGTG TC-3. Non-relevant sequences used for control shRNA was 5′-CAC CGCTA CACAA ATCAG CGATT TCGAA AAATC GCTGA TTTGT GTAG-3. Adenoviral vectors encoding shRNA-Bnip3 or non-relevant shRNA were constructed with BLOCK-iT U6 RNAi entry vector kit (Invitrogen) as we previously reported.26 Cells were infected with adenoviral vectors s encoding an shRNA directed against Bnip3 or control shRNA at a multiplicity of infection of 10 for 24 h in DMEMSF. This achieves a knock-down of endogenous Bnip3 > 85% compared to control as we reported.27 Twenty four hours following infection, cardiac myocytes without or with Bnip3 knock-down were treated with 5 µM PSPC, POVPC as detailed below.
Preparation of mitochondria and cell membrane fractionation
Mitochondria and cell membrane fractionation were carried out as previously described.25 Briefly, cardiomyocytes were scraped in treatment buffer and centrifuged for 5 min at 500 × g. The cell pellet was washed in PBS, and centrifugation was repeated. The cell pellet was gently resuspended in ice-cold buffer A (20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 MgCl2, 1 mM EDTA, 1 mM EGTA, and 250 mM sucrose) and kept on ice for 15 min. The cells were homogenized with the use of a teflon-on-glass homogenizer (Potter-Elvehjem), and incubated on ice for 15 min. Homogenization was repeated. Homogenates were centrifuged for 10 min at 500 × g at 4°C to remove cell debris. The cell debris pellet was resuspended in buffer B (10 mM Hepes-KOH, pH 7.5, 250 mM sucrose) for later analysis. The supernatant was centrifuged 2× for 10 min at 600 × g at 4°C to ensure removal of all cell debris. To obtain the mitochondrial pellet, the supernatant was centrifuged for 15 min at 10 000 × g, after which the mitochondrial pellet was resuspended in buffer A and the supernatant was kept as the cytosolic fraction for future analysis. The mitochondrial fraction was further purified by gently layering on the top of a sucrose pad solution (292 mM sucrose, 100 mM KCl, 2 mM MgCl2, 20 mM Hepes, and pH 7.4) followed by centrifugation for 10 min at 14 000 × g at 4°C. The supernatant was discarded, and the mitochondrial pellet was resuspended in buffer B. The purity of the mitochondrial fraction was confirmed via western blot using antibody against Cox IV (Rabbit mAb, #4850, Cell Signaling Technology, Danvers, MA, USA). To assess cytosolic contamination in the mitochondrial fraction, western blotting with α-tubulin (Mouse mAb, #7291, Abcam, Cambridge, MA, USA) was used as a marker of cytosol. No evidence of tubulin was observed indicating no detectable cytosolic contamination was present in the mitochondrial fraction. There is a small Cox IV band in the cytoplasmic fraction indicating a small amount of mitochondrial contamination in the cytosolic fraction. Therefore, the relative amount of OxPL recovered in the mitochondrial fraction is an underestimate (Supplementary material online, Figure).
Opening of the mitochondrial PTP after exposure to OxPL molecules was monitored by fluorescence microscopy using a solution of calcein-AM fluorescent dye with cobalt chloride (CoCl2) applied to ventricular cardiomyocytes cultured on glass coverslips as previously described.27 Unlike calcein-AM which can infiltrate the mitochondria, CoCl2 is unable to pass through the inner and outer membranes of the mitochondria. CoCl2 quenches the fluorescence of calcein, so the cytoplasmic fluorescence will be inhibited while still staining the mitochondria. After staining, NCMC were immediately washed with PBS−2 and treated with fresh serum-free DMEM/F12 cell culture medium. Reduced fluorescence compared to control cells will denote mitochondrial permeability.
Rat and mouse models
Generation and characterization of E06-scFv transgenic mice
The generation of transgenic C57BL/6 mice expressing the T15/E06 idiotype as a single-chain variable antibody fragment—termed E06-scFv-Tg mice—is reported in detail elsewhere in Que et al.28 In brief, the cDNAs encoding E06 variable regions of the heavy and light chains were linked with a 15-amino acid peptide by overlapping PCR and cloned into an expression vector pSecTag2A (Invitrogen) containing a murine Ig kappa-chain leader sequence for secretion and c-myc and polyHis as epitope tags. HEK293 cells were transfected, and the binding properties of E06-scFv secreted into culture supernatant were shown to mimic those of the intact E06. The same construct was then cloned into the liver-specific expression vector pLiv7 and used to generate transgenic (Tg) mice in the C57BL/6 background expressing the E06-scFv transgene driven by the apoE promoter, as previously reported.29 Offspring were screened both for plasma E06-scFv titre and integration of the transgene by PCR amplification of the tail DNA. The transgenic E06-scFv founder lines were bred with each other to generate ‘homozygous’ transgenic mice, and in turn, these were crossed into Ldlr−/− mice on the C57BL/6 background. All animals were genotyped for E06-scFv and Ldlr−/−, respectively and plasma assayed to confirm expression of the E06-scFv by immunoassay. The E06-scFv mRNA was strongly expressed in liver, peritoneal macrophages, and spleen, and to a lesser extent in heart. Plasma levels of the E06-scFv averaged 20–30 µg/mL in these studies.
Ischaemia–reperfusion surgery in rats and mice
Male Sprague-Dawley rats (200 g) underwent coronary artery ligation as previously described.30 The procedure was performed under anaesthesia with 3% isoflurane and oxygen at 2 L/min on a mask. Myocardial ischaemia was produced by occlusion of the left anterior descending (LAD) coronary artery. A 1–1.5 cm lateral thoracotomy incision is made to the left of and parallel to the sternum. The left coronary artery was tied with a 6-0 monofilament suture. In control animals, the suture was placed around the artery but not tied. After 60 min of ischaemia, the suture removed, and the heart allowed to reperfuse. The chest was closed in layers and de-aired, and the animals were allowed to recover. Post-operative analgesia was provided by subcutaneous buprenorphine at 0.03–0.05 mg/kg at 1 h post-surgery and every 8 h after for a total of 3 injections. After 24 h, the animals were sedated with 3% isoflurane and 2D echocardiographic studies were repeated, and subsequently the hearts were removed and 1 mm cross sections made for lipidomic analysis, and triphenyltetrazoliumchloride (TTC) staining as previously described.31 For rat studies, only hearts that demonstrated both ECG and 2D echocardiagraphic evidence of myocardial ischaemia were selected for the IR group. To minimize ex vivo lipid peroxidation, immediately after removal from the body, myocardial tissue was immediately flash frozen in liquid nitrogen. The entire heart tissue was pulverized with liquid nitrogen, and the resulting tissue underwent Folch extraction in the presence of DNPC (10 ng/100 µL) as internal standard and 0.01% BHT as antioxidant.32
Adult (18–20-week-old) Ldlr−/−-E06-scFv-Tg mice (n = 14) or Ldlr−/− mice (n = 15) weighing 16–40 g, were anaesthetized with a mixture of ketamine (50 mg/kg) and xylazine (5 mg/kg) and 1.0% isoflurane. Mice were intubated with a pressure ventilator (Kent Scientific, PhysioSuite). Peak inspiration pressure was ∼13 cm H2O and inspiration rate 100–110. The skin over the mid thorax was shaved and cleansed with betadine or chlorhexidine solution. A skin incision was made from the mid-sternal line toward the left armpit, and the chest opened with a 1 cm lateral cut along the left side of the sternum, cutting between the 3rd and 4th ribs to expose the left ventricle (LV) of the heart. The ascending aorta and main pulmonary artery were identified; the LAD coronary artery was located as it traverses the anterior wall of the heart, between the left and right ventricles. LAD coronary artery occlusion was performed by tying a 8-0 prolene suture ligature on a piece of 2-0 silk suture. Occlusion of the artery was assessed by blanching of the territory of perfusion of the LAD coronary artery, along with acute ST segment elevation on limb-lead electrocardiographic leads. Following an ischaemic period of 60 min, the suture was removed from around the LAD coronary artery. Reperfusion was confirmed by observing return of blood flow in the epicardial coronary arteries.
Histologic assessment of infarct size
One week following surgery, mice were euthanized with CO2 gas followed by thoracotomy to harvest the heart after intraperitoneal administration of 50 units of heparin. Hearts were immediately harvested, submerged in ice-cold relaxation solution A (120 mmol/L NaCl, 20 mmol/L NaHCO3, 11 mmol/L glucose, 5.4 mmol/L KCl, 1.2 mmol/L MgCl2, 10 mmol/L 2,3-butanedione 2-monoxime). The ascending aorta was cannulated and the hearts were perfused retrograde via a 21-gauge blunted needle connected to a Langendorff perfusion apparatus at 80 mmHg pressure. The suture left around the LAD coronary artery in situ after the IR surgery was religated and 1% (w/v) Evans blue (EB) dye in solution A was then injected to define area at risk (AAR). Failure in re-occlusion of the coronary artery resulted in exclusion of animals from the final analysis. The heart was then sectioned into 1 mm slices using a heart slicer (Zivic Instruments) and incubated in 1.5% (w/v) TTC in PBS. With TTC, viable myocardium stains brick red and the infarct appears pale white. The sizes of AAR and infarct area (IA) were quantified by computerized planimetry using ImageJ. The identity of animals remained anonymous to the examiner who carried out staining procedures and area measurements.
OxPL mass spectrometry
PC-containing PL were extracted from NNCM by modification of a previously described protocol.33–35 Cell media was removed, and cells were washed with PBS. Each well was scraped into 1 mL of methanol/acetic acid (3% v/v) solution containing 0.01% BHT and transferred to a 10 mL glass conical tube and capped under N2 (g). Ten nanograms of DNPC was added as internal standard into each sample for quantitation purposes. Two millilitres of hexane containing BHT was added to the tube, capped under N2 (g), vortexed for 5 s, and then centrifuged for 5 min at 3500 rpm at 4°C. The upper hexane layer was then siphoned off using a glass Pasteur pipette and discarded. The hexane/BHT wash was repeated three times, capping under N2 (g), vortexing for 5 s, and centrifuging after each wash. After the final hexane/BHT wash, 2 mL of chloroform containing BHT and 750 μL of PBS were added to the tube then vortexed and centrifuged as described above. The lower organic layer was removed using a glass Pasteur pipette and transferred to a clean 15 mL glass conical tube where the solution was aspirated off using a nitrogen evaporator, and then reconstituted into 300 μL of chloroform/methanol (2: 1 v/v) for storage at -80°C.
The separation of OxPLs was carried out using reverse-phase (RP) chromatography as reported previously.36 Extracted hearts were reconstituted in RP eluent consisting of 60:40 acetonitrile:water, 10 mM ammonium formate and 0.1% formic acid immediately prior to injection. Thirty microlitres of the sample were injected onto an Ascentis Express C18 HPLC column (15 cm × 2.1 mm, 2.7 µm; Supelco Analytical, Bellefonte, Pennsylvania, USA) with separation by a Prominence UFLC system from Shimadzu Corporation (Canby, OR, USA). Elution was performed using a linear gradient of solvent A (acetonitrile/water, 60:40 v/v) and solvent B (isopropanol/acetonitrile, 90:10, v/v) with both solvents containing 10 mM ammonium formate and 0.1% formic acid. The mobile phase composition that was used is as follows: initial solvent B at 32% until 4.00 min; switched to 45% B; 5.00 min 52% B; 8.00 min 58% B; 11.00 min 66% B; 14.00 min 70% B; 18.00 min 75% B; 21.00 min 97% B; 25.00 min 97% B; and 25.10 min 32% B. A flow rate of 260 µL/min was used for analysis, and the sample tray and column oven were held at 4°C and 45°C, respectively.
Detection of OxPL was carried out by mass spectrometry in positive polarity mode. MRM scans were performed on 6 transitions using a product ion of 184.3 m/z, corresponding to the cleaved phosphocholine moiety. Six commercially available standards of PONPC, POVPC, PGPC, PAzPC, KOdiAPC, and KDdiAPC were injected and accurate peak assignments were based upon retention times and mass transitions. The mass spectrometry settings were as follows: curtain gas, 26 psi; collision gas, medium; ion spray voltage, 5500 V; temperature, 500.0°C; ion source gas 1, 40.0 psi; ion source gas 2, 30.0 psi; declustering potential, 125 V, entrance potential, 10 V; collision energy, 53 V; collision cell exit potential, 9 V; and dwell time, 50 ms. External mass calibration was performed at regular intervals. For quantitation, multiple reaction monitoring (MRM) calibration curves were made for each of the six commercially available OxPL standards and peaks were normalized based on their relative responses. Ten nanograms of internal standard was added to all samples during extraction. A 4000 QTRAP® triple quadrupole mass spectrometer system with a Turbo V electrospray ion source from AB Sciex (Framingham, MA, USA) was coupled to the liquid chromatography system.
In vitro OxPL treatment and assessment of cell viability
Fresh DFSF was applied to NNCM on glass coverslips and supplemented with 1, 2, 5, and 10 μM concentrations of OxPL lipids sonicated into PBS, forming micelles. Four OxPL species, POVPC, PONPC, PGPC, and PAzPC were applied to NNCM plates for 4 h and compared to a non-oxidized PC standard PSPC. Plates were then washed with PBS before NNCM were analysed for cell viability using vital dyes, calcein-acetoxymethyl ester (AM) and ethidium homodimer-1 (Invitrogen) as previously described.37 The levels of total OxPL seen in rat myocardium following IR are 90-150 ng/mg protein, which represents a 3 µM concentration of OxPL. The range of OxPL used in these experiments—1–10 μM—are comparable to those measured in rat myocardium following IR.
To evaluate the effect of neutralizing OxPL on cell viability, the IgM E06 antibody was used at a concentration of 10 µg/mL in cell culture. The antibody was purchased as 100 µg suspended in 100 µL of PBS suitable for direct application into cell culture. Co-treatment with OxPL was at 5 µM of each OxPL molecule and PSPC for 2 h. Cell viability was assessed as described above. All microscopy was done on an Olympus AX70 microscope, with pictures taken using a CoolSnap® camera and Image Pro-Plus 5.1.2 Software. Images were assessed using Adobe Photoshop CS5.1 Software.
Statistical analysis
Multiple comparisons between groups were tested by one-way ANOVA. Bonferroni post hoc tests were used to determine difference among groups. Unpaired two-tailed Student’s t-test was used to compare mean difference from control. Differences were considered to be statistically significant to a level of P-value <0.05.
Results
OxPL are generated in rat cardiomyocytes following IR
NNCM exposed to control and IR conditions were analysed by LC/MS/MS with MRM for the six fragmented OxPL species for which standards are available. Five OxPL molecules POVPC (m/z 594), PGPC (m/z 610), PONPC (m/z 650), PAzPC (m/z 666), and KOdiAPC (m/z 664) were identified in cell extracts of both control and simulated IR conditions (Figure 1A–E). Significant increases in the mass of four of the identified OxPL were observed in NNCM exposed to simulated IR (Figure 1F), including PONPC (32.7 ± 3.2 to 53.0 ± 9.1 ng/mg protein, P < 0.001), POVPC (15.6 ± 1.5 to 23.1 ± 1.7 ng/mg protein, P < 0.05) PGPC (2.5 ± 1.2 to 6.1 ± 2.1 ng/mg protein, P < 0.05), and PAzPC (5.0 ± 1.5 to 9.9 ± 3.8 ng/mg protein P < 0.01). Levels of KOdiAPC were not different.
Figure 1.

Identification of fragmented OxPL molecules in NNCM during IR. (A–E) Single MRM ion chromatograms of fragmented OxPL identified in NNCM during IR. Cells were incubated under IR conditions are described in the Methods section. (F) Presence of most abundant OxPL in cultured NNCM after IR compared to control. The most abundant compound was PONPC, which had a significant increase after exposure to IR. n = 4 separate cultures, each done in triplicate (*P < 0.05, ANOVA).
We next investigated the changes in OxPL that occurred in vivo in rat myocardium during coronary IR. After 1 h of ischaemia and a subsequent 24 h of reperfusion (Figure 2A), there were significant increase in all five of the measured OxPL species, with PONPC again being the most abundant OxPL species, as observed in cardiomyocytes subjected to IR in vitro (Figure 2B). There was a relatively three-fold enrichment of POVPC and PONPC, and ∼1.5- to 2-fold enrichment of the other measured OxPL species in rat myocardium exposed to IR conditions compared to control.
Figure 2.
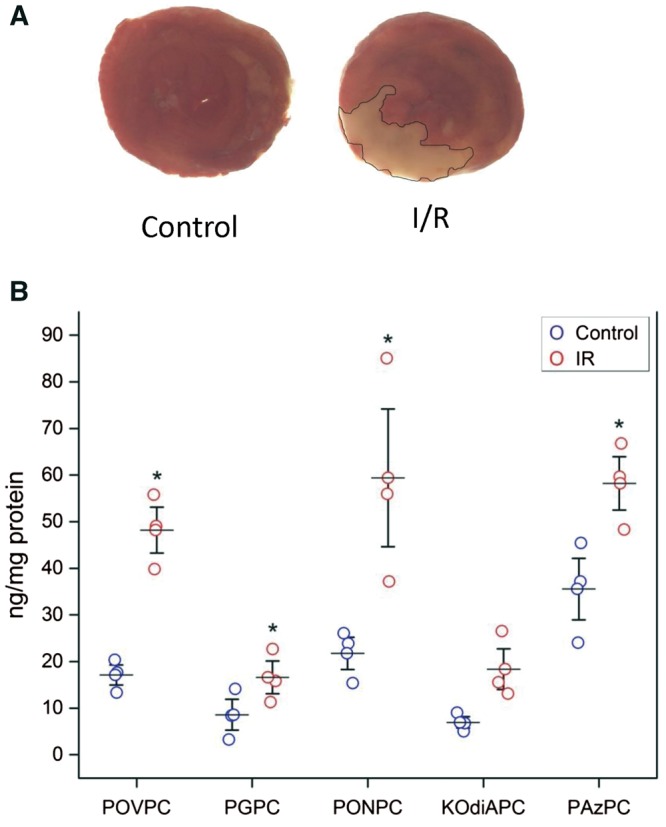
Changes in OxPL levels during rat coronary ligation IR injury. (A) TTC staining of cross section of rat myocardium in control and after 1 h of ischaemia with 24 h of reperfusion. (B) Significant increase in OxPL levels in rat myocardium after 24 h of IR injury, n = 4. Levels of the different OxPL (POVC, PGPC, PONPC, KOdiAPC, and PAzPC) have been each compared in controls vs. IR via the Student’s t-test, *P < 0.05).
OxPL causes rat cardiomyocyte cell death in vitro
It is known that adding sufficient amounts of certain OxPLs can result in cell death.38,39 However, we sought to define whether the specific OxPL species we identified in the extracts of myocardium subjected to IR would also be toxic at the identified concentrations measured, ∼1–10 μM. Therefore, we incubated varying concentrations (1.0 to 10 μM) of the identified OxPLs with NNCM in culture and evaluated their effect on cell viability, as compared to a non-oxidized PL (Figure 3A). With increasing doses of OxPL, there was a progressive decrease in the number of calcein-AM stained live cells (green), and correspondingly, an increase in ethidium homodimer-1 staining dead cells (red). Each of the OxPL species resulted in significant cell death in a concentration-dependant pattern (Figure 3B). PONPC, which was the most prominent OxPL identified in both the cardiomyocytes and in vivo myocardium exposed to IR, and which is also quantitatively the most dominant OxPL found in coronary and carotid atheroma,32 was also the most potent inducer of cardiomyocyte cell death when compared to PSPC control, resulting in percent cell death of 28.3 ± 8.9%, 42.0 ± 6.2%, 70.0 ± 18.6%, and 81.2.6 ± 15.1% for 1, 2, 5, and 10 µM respectively (P < 0.001). POVPC was also a potent inducer of cell death with 68.6 ± 14.8% at 10 µm (Figure 3B). The non-oxidized control PSPC treated NNCM showed non-significant increases in cell death at 1, 2, 5, and 10 µM, 7.6 ± 5.1%, 9.1 ± 10.5%, 10.5 ± 1.6% and 5.5 ± 1.6% respectively (vs. 7.0 ± 1.1% control).
Figure 3.
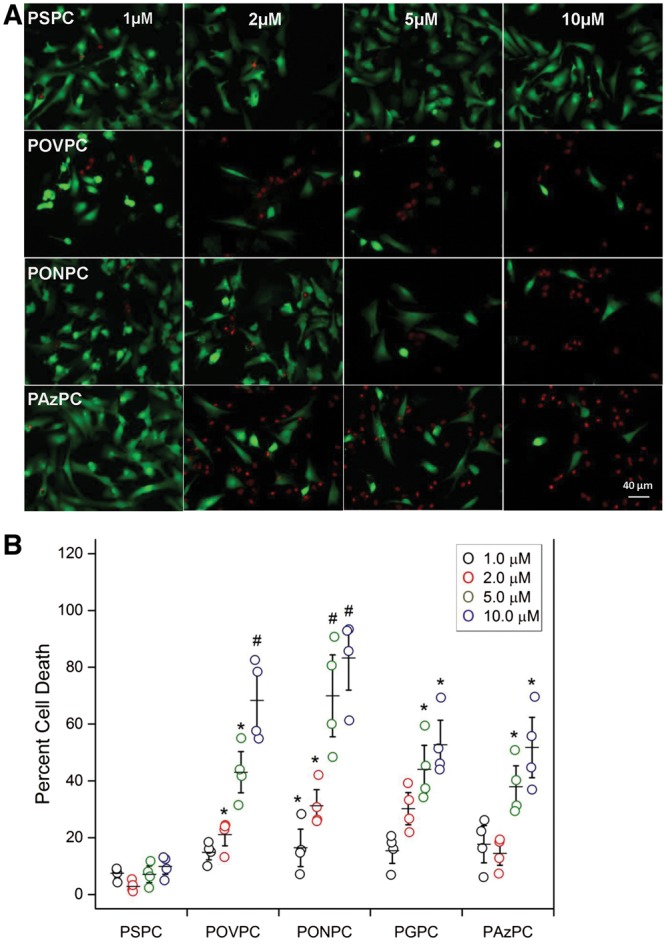
Cell viability of NNCM exposed to increasing concentrations of aldehyde and carboxylic acid based OxPL compared to cell viability after treatment with non-oxidized control PSPC. (A) Images of rat NNCM stained using the vital dyes, calcein-AM (green—live), and ethidium homodimer-1 (red—dead), exposed to the indicated concentrations of the control phospholipid PSPC, and the OxPL POVPC, PAzPC, and PONPC for 4 h at 37°C. (B) Cell viability measured as percent cell death after incubations with control PSPC and indicated OxPL (n = 4, separate cultures, each done in triplicate) (*P < 0.05, #P < 0.01, vs. PSPC at same concentration, ANOVA).
OxPL enrichment in mitochondria during IR
In cultured cardiomyocytes under IR conditions, the mitochondrial component was fractionated from other cell organelles and the levels of fragmented OxPL both in the mitochondrial fraction and also in the remainder of the cell organelles was measured. Figure 4A shows that after cardiomyocytes undergo IR, there is an increase in mitochondrial OxPL with significant increases in PONPC and POVPC when compared to control. When the mitochondrial fraction in IR was compared with non-mitochondrial cell membranes there was an enrichment of POVPC and PONPC, whereas the other cell membranes showed a trend towards a higher concentration of the other fragmented OxPCs (Figure 4B).
Figure 4.
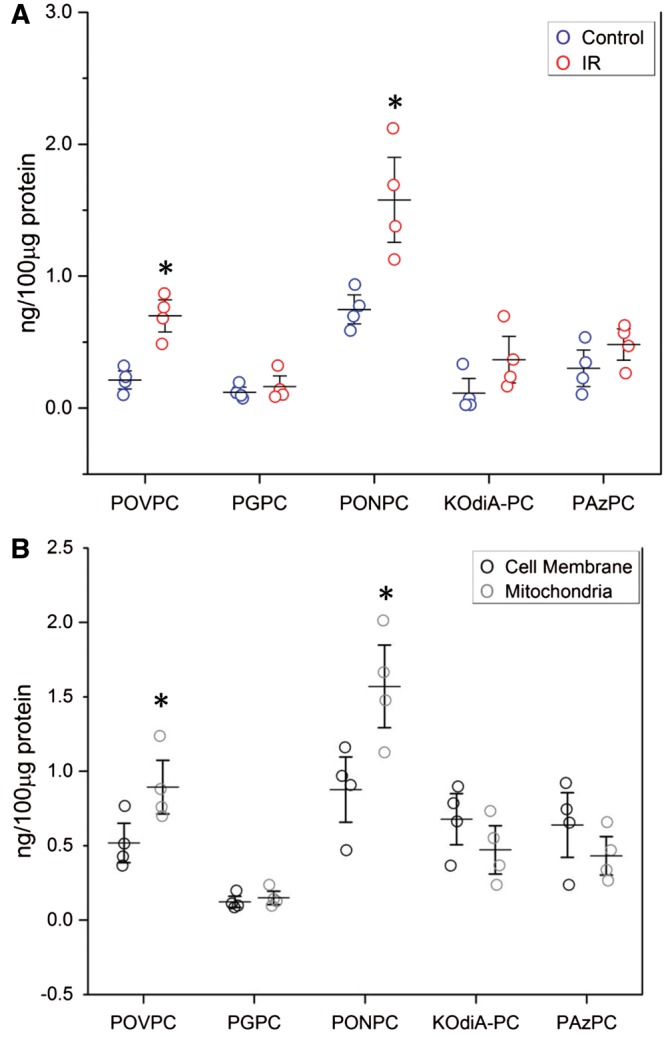
Cellular and mitochondrial fragmented OxPC in cardiomyocytes during IR injury. (A) Significant increase in PONPC and POVPC in mitochondria in cells undergoing IR compared to control cardiomyocytes. Level of POVPC and PONPC in control vs. IR *P < 0.05, Student’s t-test) (n = 4). (B) There is specific enrichment of POVPC and PONC in mitochondria undergoing IR when compared to cellular membrane. *P < 0.05, Student’s t-test (n = 4).
We also investigated the mitochondrial involvement of the two most potent inducers of cell death, POVPC and PONPC. There were significant changes to the mitochondrial permeability within cardiomyocytes when exposed to POVPC and PONPC at 2, 5, and 10 µM. Representative images as shown in Figure 5A. Increased mitochondrial permeability is demonstrated by a decrease in fluorescence and is represented as a fold reduction compared to non-oxidized PSPC (Figure 5B). A statistically significant fold reduction was observed at 2 µM for PONPC (0.83 ± 0.08, P < 0.05) and at 5 µM (POVPC 0.71 ± 0.08, PONPC 0.62 ±0.03, P < 0.05) and 10 µM (POVPC 0.33 ± 0.02, PONPC 0.18 ± 0.05 fold, P < 0.01).
Figure 5.
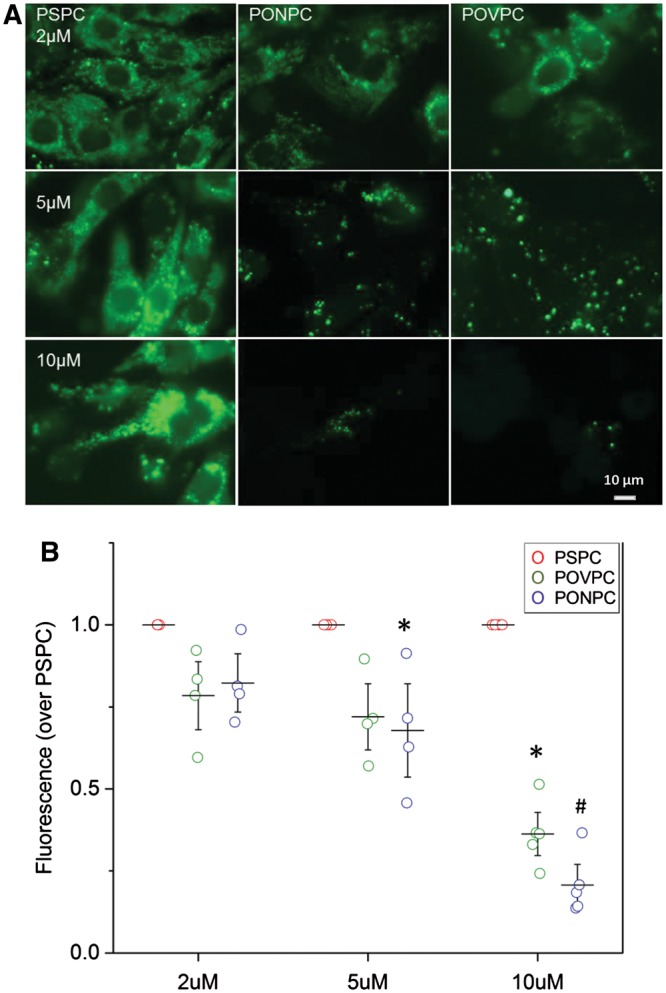
Mitochondrial permeability of NNMC in presence of OxPL. (A) Representative images of NNCM stained with calcein-AM and CoCl2 with exposure to increasing concentrations of POVPC and PONPC to determine mitochondrial permeability as compared to non-oxidized control PSPC. (B) Mitochondrial permeability of NNCM to increasing concentrations of PONPC and POVPC is depicted as the fold change over the fluorescence of non-oxidized control PSPC treated cardiomyocytes stained with calcein-AM and CoCl2 at equal concentrations OxPL’s significantly decreased fluorescence compared to PSPC if *P < 0.05, #P < 0.01, (n = 4) ANOVA.
Neutralization of OxPL by E06 attenuates cell death in vitro
To investigate whether inactivation of OxPL will prevent cell death, cardiomyocytes were exposed to 5 μM of PONPC and POVPC in the absence or presence of E06, at 10 µg/mL. In the presence of E06, OxPL mediated cardiomyocyte cell death was mitigated to near PSPC control treatment levels (Figure 6A). E06 inhibited POVPC induced cell death by 74.6% (22.6 ± 4.14% vs. 8.0 ± 1.6%, P < 0.05) and PONPC induced cell death by 74.7% (25.3 ± 3.4% vs. 6.4 ± 1.0%, P < 0.05) (Figure 6B). The amount of cell death observed with PSPC treatment was unaffected by E06 co-treatment (6.2 ± 1.6% without vs. 6.3 ± 1.1% with E06).
Figure 6.
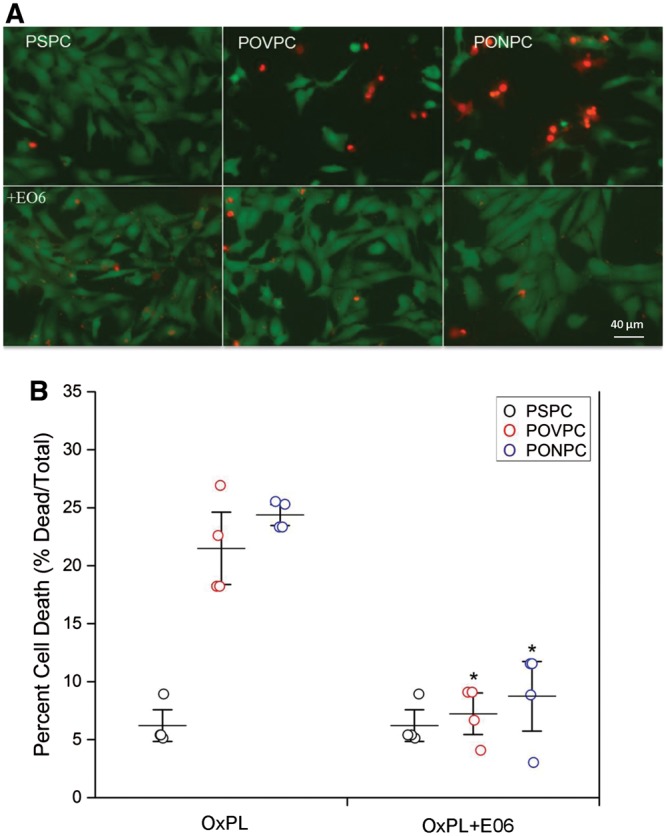
Attenuation of OxPL mediated cell death by E06. Representative images of NNCM treated with POVPC and PONPC, and in presence of E06. NNCM stained using the vital dyes, calcein-AM (green—live) and ethidium homodimer-1 (red—dead), co-treated with 5 μM of POVPC and PONPC and 10 μg/mL of OxLDL-specific E06 antibody for 2 h at 37°C (A). There was significant inhibition of OxPL-induced cell death at 5 μM concentration (B). (n = 4 separate cultures, each done in triplicate) (*P < 0.05 when compare to POVPC and PONPC without E06) ANOVA.
Given that Bnip3 is central to mitochondrial mediated reperfusion injury40 we investigated the change in Bnip3 expression in cardiomyocytes exposed to POVPC and PONPC. As shown in Figure 7A, a significant increase in Bnip3 expression was induced when cells were exposed to POVPC (2.7-fold ± 0.3, P < 0.05) and PONPC (3.2-fold ± 0.2, P < 0.05) when compared to PSPC. Pretreatment of the cardiomyocytes with E06 resulted in a significantly blunted response in Bnip3 expression when compared POVPC (1.3-fold ± 0.2, P < 0.05) and PONPC (1.4-fold ± 0.1, P < 0.05). To further investigate the role of Bnip 3 in OxPL mediated cell death, we tested the effects of Bnip3 knock-down in cells treated with POVPC (Figure 7B). As shown in Figure 7C, in contrast to wild-type cells, Bnip3 KD cells were resistant to the cytotoxic effects of POVPC displaying less cell death (Bnip KD 14.7% ± 5.4% vs. WT 35.8% ± 9.1%, P < 0.05).
Figure 7.
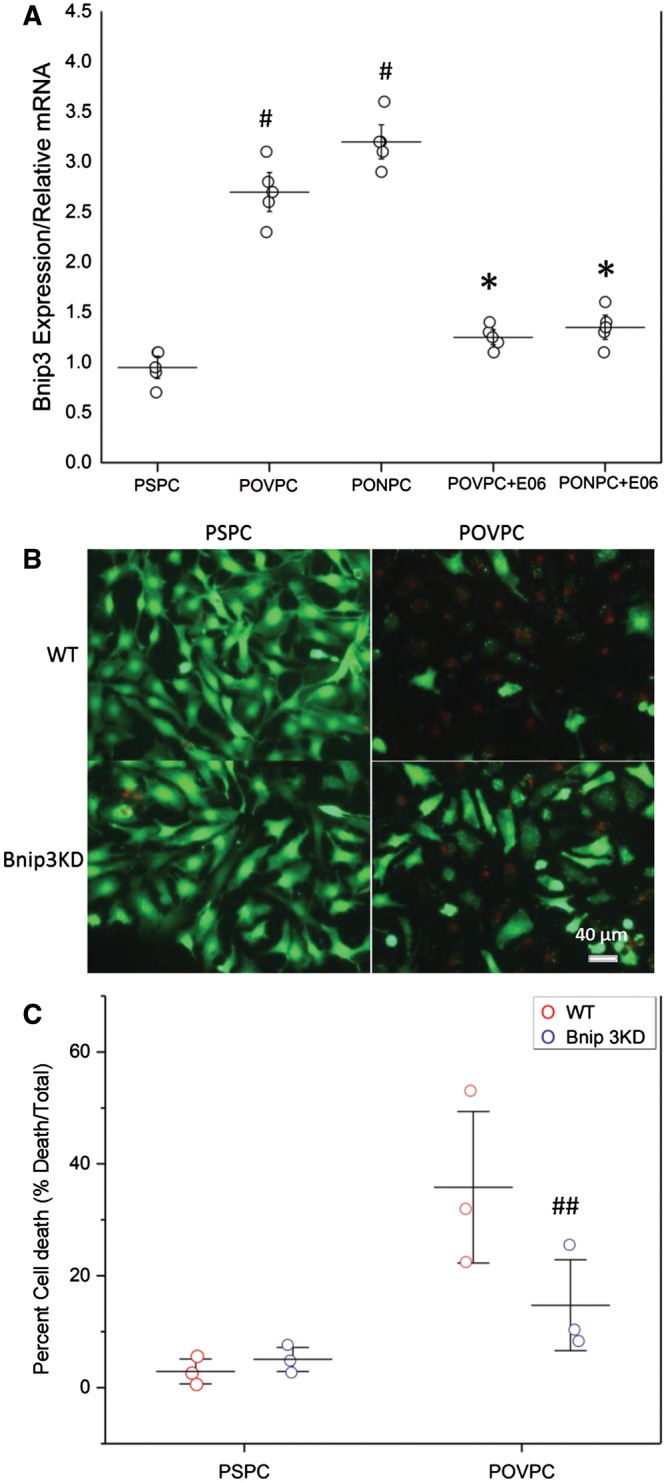
OxPL induce cell death through a Bnip3 pathway. (A) Representation of mRNA expression in neonatal cardiomyocytes when exposed to PONPC and POVPC in both the presence and absence of E06. (PSPC vs. POVPC, PSPC vs. PONPC #P < 0.05, Student’s t-test), (POVPC vs. POVPC + E06, *P < 0.05, Student’s t-test) (PONPC vs. PONPC + E06, *P < 0.05, Student’s t-test) (n = 4). (B) Cell viability of NNCM exposed to POVPC in wild-type and Bnip3KD cardiomyocytes. (C) Percent cell death in NNCM exposed to POVPC in wild-type and Bnip3KD cardiomyocytes (n = 3) (WT POVPC vs. Bnip KD POVPC, ##P < 0.05, Student’s t-test).
Neutralization of OxPL reduces IR infarct size in vivo
To evaluate the role of OxPL in vivo in mediating cell death under IR conditions, we evaluated whether in vivo neutralization of OxPL would attenuate IR infarct size. To accomplish this, we utilized transgenic mice that constitutively express a single-chain variable fragment of the E06 antibody (E06-scFv-Tg). The E06-scFv is under control of the apoE promoter and is expressed from hepatocytes and macrophages and is present in murine plasma at levels of 20–30 µg/mL.28 The E06-scFv-Tg mice were bred into the Ldlr−/− background. Ldlr−/− (n = 15) and E06-scFv-Tg/ Ldlr−/− (n = 14) mice were subjected to 60 min of LAD ischaemia by suture ligation, followed then by reperfusion. Seven days after IR, myocardial infarct size was evaluated histologically (Figure 8A). There was no significant difference in the AAR between the two groups, demonstrating that surgical ischaemic injury was equivalent between groups (Figure 8B). As measures of infarct size, we compared the ischaemic area (IA) as a percentage of the AAR (IA/ARR) or as a percentage of the LV (IA/LV). Compared to the Ldlr−/− mice, the E06-scFv-Tg/ Ldlr−/− mice had 65.9% smaller IA/AAR (47.7 ± 17.6% vs. 72.4 ± 21.9%, P = 0.023) (Figure 8C) and a 58.8% smaller IA/LV ratio (16.3 ± 7.5% vs. 27.7 ± 10.7%, P = 0.025) (Figure 8D).
Figure 8.

Mice expressing scFV-E06 have reduced infarct size following myocardial IR. (A) Representative myocardial TTC staining of LDLR−/− (n = 15) and scFvE06/LDLR−/− (n = 14) groups 7 days after 60 min of ischaemia. White indicates infarct; red, viable myocardium; and non-blue, AAR. (B) There were no significant difference in AAR normalized by LV mass distal to the ligation for ischaemia (AAR/LV). Compared to controls, E06-scFv/Ldlr−/− mice had 65.9% smaller IA/AAR (P = 0.0023) (C) and 58.5% smaller IA/LV (P = 0.0025) ANOVA. TTC, 2, 3, 5-triphenyltetrazolium chloride; AAR, area at risk; LV, left ventricle; IA, infarct area.
Discussion
In this report, we show that myocardial IR resulted in a significantly increased production of OxPL in primary cardiomyocyte cell culture in vitro as well as in myocardial tissue in vivo. In cell culture, exposure to exogenously added specific species of various OxPL, resulted in cardiomyocyte cell death in the absence of IR, particularly by PONPC and POVPC. PONPC and POVPC resulted in increased Bnip3 expression and mitochondrial permeability in cultured cardiomyocytes. E06, a well-characterized IgM NAb specifically recognizes the PC headgroup of OxPL, including those of fragmented OxPL such as PONPC and POVPC, but does not recognize unoxidized PC containing PL.18 E06 abolished OxPL mediated cell death in vitro and attenuated IR infarct size in vivo.
Myocardial IR results in generation of ROS via non-enzymatic mitochondrial dysfunction as well as enzymatic production by xanthine oxidase (from nearby endothelial cells) and NADPH (from infiltrating neutrophils and monocytes),41 which in turn mediate lipid peroxidation. PC-containing PL, the most abundant class of cellular membrane PL, are prone to oxidative damage from ROS as well as during apoptosis,22 resulting in formation of OxPL. In addition to direct lethal effects on cardiomyocytes demonstrated in this study, OxPL represents a subset of oxidation-specific epitopes (OSE) that are recognized by the innate immune system as danger associated molecular patterns (DAMPs) and promote sterile inflammation.11 Prior work has established the profound proinflammatory effect of OxPL on monocytes, macrophages, and endothelial cells resulting in release of chemokines (MCP-1, fibronectin, and CXCL8) and cytokines (IL-1β, IL-6, and IL-8), and that specific OxPL species (POVPC and PGPC) had direct pro-apoptotic effects on cells,42–48 all of which can contribute to IR injury. Fragmented OxPL species have been shown to increase mitochondrial permeability in other cells lines. In macrophages, PAzPC promotes permeabilization of mitochondrial membranes by Bax.49 In vascular smooth muscle cells, Chen et al.38 demonstrated that fragmented OxPL primarily targeted the mitochondria resulting in increased PTP opening and cell death. Concordantly the findings of the present study verify that that POVPC and PONPC are not only enriched in cardiac mitochondria during IR but mechanistically trigger mitochondrial damage resulting in PTP opening through a Bnip3 mediated pathway.
Moreover, our findings further show that E06 prevented OxPL mediated cell death in vitro and attenuated IR infarct size in vivo suggest that targeting OxPL in general, and specifically with this NAb, may be an effective pharmacologic therapy to reduce IR injury. E06 is an IgM NAb originally cloned from the spleens of Apoe−/− mice,19–21 and has the identical T15 idiotype as found in an IgA that was first identified for its ability to specifically recognize the PC moiety (not as a PL) of the cell wall polysaccharide of Streptococcus pneumoniae, and provides optimal protection against lethal infection with S. pneumoniae,50 a feature which likely strengthened positive selection for both antibodies. E06 neutralized the proinflammatory effects of OxPL, prevented cellular binding of OxLDL by CD36,51 inhibited OxLDL uptake by macrophages,21 and prevented OxPL containing apoptotic cells from activating endothelial cells to bind monocytes.22 Moreover, augmented levels of T15/E06 achieved by direct infusion52 or immunization with Streptococcal extract23 reduced atherosclerosis in mouse models.
Translational relevance
Epidemiologic studies have also suggested that IgM autoantibodies recognizing PC containing OSE (such as E06) are cardioprotective, with titres inversely associated with incident CVD,53 angiographic CAD,54 stroke,55 and carotid intimal thickness.56 IgM autoantibodies to OSE decline with age57 and loss of these protective antibodies may reflect a cause of increased risk of cardiovascular disease with advanced age.
An advantage of the E06-scFv present in the transgenic mice used in our in vivo experiment is that it can bind and neutralize OxPL, but lacks an Fc domain and therefore cannot bind and presumably activate complement. Considering the totality of the available pre-clinical and clinical evidence, the E06-scFv may represent an ideal candidate for further evaluation as a therapeutic for reducing IR injury.
In our current study, constitutive expression of the E06-scFv resulted in durable protection against IR injury, with reduced infarct size 1 week after IR, suggesting potential for clinical translation. In the clinical setting, the E06-scFv will need to be administered to the patient in an acute health care setting for an MI, and specifically, intravenously post thrombolytic therapy or by intracoronary route during PCI. Clinical endpoints would include measures of myocardial damage acutely and LV function subacutely, as well as traditional MACE endpoints in the longer term.
Limitations
We utilized a targeted approach to identify OxPL species generated following IR using mass spectrophotometry. While this approach offers precision in identifying specific OxPL using known standards, we cannot rule out additional contributions from OxPL species other than POVPC, PGPC, PAzPC, PONPC, KOdiAPC, and KDdiAPC towards IR injury. However, because the E06 binds to the PC headgroup of OxPLs, it binds a wide variety of different OxPL,18 including non-fragmented OxPL such as PEIPC (1-palmitoyl-2-(5, 6)-epoxyisoprostane E2-sn-glycero-3-phosphocholine), which has many proinflammatory properties.58 It would not bind to non-PC based oxidized PL. However, the fact that E06 attenuates IR injury in vivo suggests that PC based OxPL recognized by E06 are clinically relevant oxidized PL. Second, the approach used a transgenic mouse model with constitutive high expression of E06-scFv, which allowed establishment of the proof of principle. Future studies will require a more translational approach with purification of adequate amounts of E06-scFV and a control scFv antibody to assess the efficacy of acute administration of the antibody during an IR protocol.
Conclusion
In summary, these studies identify an important role for OxPL as mediators of cell death during myocardial IR and that E06-scFv can attenuate IR infarct size. The E06-scFv antibody represents a novel potential therapeutic avenue to prevent the detrimental effects of IR injury and improve cardiac outcomes during acute MI.
Funding
National Institutes of Health (R01-HL119828, P01-HL088093, P01-HL055798, and R01-HL093767 R01-HL086599 to S.T. and J.L.W.); Heart and Stroke Foundation of Canada (G-14-0006050, Research Manitoba 983 to A.R.).
Conflict of interest: S.T., X.C., and J.L.W. are named as co-inventors and receive royalties from patents owned by UCSD on oxidation-specific antibodies and of biomarkers related to oxidized lipoproteins. Dr Witztum is a consultant to Ionis Pharmaceuticals. S.T. currently has a dual appointment at UCSD and as an employee of Ionis Pharmaceuticals. The other authors have no relationships relevant to the contents of this paper to disclose.
Supplementary Material
Footnotes
Time for primary review: 25 days
References
- 1. O’Gara PT, Kushner FG, Ascheim DD, Casey DE, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, Morrow DA, Newby LK, Ornato JP, Ou N, Radford MJ, Tamis-Holland JE, Tommaso CL, Tracy CM, Woo YJ, Zhao DX.. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013;127:e362–e425. [DOI] [PubMed] [Google Scholar]
- 2. Cung TT, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guerin P, Elbaz M, Delarche N, Coste P, Vanzetto G, Metge M, Aupetit JF, Jouve B, Motreff P, Tron C, Labeque JN, Steg PG, Cottin Y, Range G, Clerc J, Claeys MJ, Coussement P, Prunier F, Moulin F, Roth O, Belle L, Dubois P, Barragan P, Gilard M, Piot C, Colin P, De Poli F, Morice MC, Ider O, Dubois-Rande JL, Unterseeh T, Le Breton H, Beard T, Blanchard D, Grollier G, Malquarti V, Staat P, Sudre A, Elmer E, Hansson MJ, Bergerot C, Boussaha I, Jossan C, Derumeaux G, Mewton N, Ovize M.. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med 2015;373:1021–1031. [DOI] [PubMed] [Google Scholar]
- 3. Yellon DM, Hausenloy DJ.. Myocardial reperfusion injury. N Engl J Med 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 4. Kloner RA, Hale SL, Dai W, Shi J.. Cardioprotection: where to from here? Cardiovasc Drugs Ther 2017;31:53–61. [DOI] [PubMed] [Google Scholar]
- 5. Gibson CM, Giugliano RP, Kloner RA, Bode C, Tendera M, Janosi A, Merkely B, Godlewski J, Halaby R, Korjian S, Daaboul Y, Chakrabarti AK, Spielman K, Neal BJ, Weaver WD.. EMBRACE STEMI study: a phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur Heart J 2016;37:1296–1303. [DOI] [PubMed] [Google Scholar]
- 6. Rezkalla SH, Stankowski RV, Hanna J, Kloner RA.. Management of no-reflow phenomenon in the catheterization laboratory. JACC Cardiovasc Interv 2017;10:215–223. [DOI] [PubMed] [Google Scholar]
- 7. Li CY, Jackson RM.. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol, Cell Physiol 2002;282:C227–C241. [DOI] [PubMed] [Google Scholar]
- 8. Van de Velde M, DeWolff M, Leather HA, Wouters PF.. Effects of lipids on the functional and metabolic recovery from global myocardial stunning in isolated rabbit hearts. Cardiovasc Res 2000;48:129–137. [DOI] [PubMed] [Google Scholar]
- 9. Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, Choi J, Perkmann T, Backhed F, Miller YI, Horkko S, Corr M, Witztum JL, Binder CJ.. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest 2009;119:1335–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang MK, Binder CJ, Torzewski M, Witztum JL.. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci USA 2002;99:13043–13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Binder CJ, Papac-Milicevic N, Witztum JL.. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol 2016;16:485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Samhan-Arias A, Ji J, Demidova O, Sparvero L, Feng W, Tyurin V, Tyurina Y, Epperly M, Shvedova A, Greenberger J, Bayır H, Kagan V, Amoscato A.. Oxidized phospholipids as biomarkers of tissue and cell damage with a focus on cardiolipin. Biochim Biophys Acta 2012;1818:2413–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glass CK, Witztum JL.. Atherosclerosis. the road ahead. Cell 2001;104:503–516. [DOI] [PubMed] [Google Scholar]
- 14. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM.. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008;133:235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steinberg D, Witztum JL.. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol 2010;30:2311–2316. [DOI] [PubMed] [Google Scholar]
- 16. Romanoski CE, Che N, Yin F, Mai N, Pouldar D, Civelek M, Pan C, Lee S, Vakili L, Yang WP, Kayne P, Mungrue IN, Araujo JA, Berliner JA, Lusis AJ.. Network for activation of human endothelial cells by oxidized phospholipids: a critical role of heme oxygenase 1. Circ Res 2011;109:e27–e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orozco Luz D, Bennett Brian J, Farber Charles R, Ghazalpour A, Pan C, Che N, Wen P, Qi Hong X, Mutukulu A, Siemers N, Neuhaus I, Yordanova R, Gargalovic P, Pellegrini M, Kirchgessner T, Lusis Aldons J.. Unraveling Inflammatory Responses using Systems Genetics and Gene-Environment Interactions in Macrophages. Cell 2012;151:658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Friedman P, Horkko S, Steinberg D, Witztum JL, Dennis EA.. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. Importance of Schiff base formation and aldol condensation. J Biol Chem 2002;277:7010–7020. [DOI] [PubMed] [Google Scholar]
- 19. Palinski W, Horkko S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, Witztum JL.. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996;98:800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, Witztum JL.. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest 2000;105:1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, Berliner JA, Friedman P, Dennis EA, Curtiss LK, Palinski W, Witztum JL.. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest 1999;103:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, Witztum JL.. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med 2004;200:1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Binder CJ, Horkko S, Dewan A, Chang MK, Kieu EP, Goodyear CS, Shaw PX, Palinski W, Witztum JL, Silverman GJ.. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 2003;9:736–743. [DOI] [PubMed] [Google Scholar]
- 24. Kirshenbaum LA, Schneider MD.. Adenovirus E1A represses cardiac gene transcription and reactivates DNA synthesis in ventricular myocytes, via alternative pocket protein- and p300-binding domains. J Biol Chem 1995;270:7791–7794. [DOI] [PubMed] [Google Scholar]
- 25. Maddaford TG, Hurtado C, Sobrattee S, Czubryt MP, Pierce GN.. A model of low-flow ischemia and reperfusion in single, beating adult cardiomyocytes. Am J Physiol Heart Circ Physiol 1999;277:H788–H798. [DOI] [PubMed] [Google Scholar]
- 26. Gang H, Dhingra R, Lin J, Hai Y, Aviv Y, Margulets V, Hamedani M, Thanasupawat T, Leygue E, Klonisch T, Davie JR, Kirshenbaum LA.. PDK2-mediated alternative splicing switches Bnip3 from cell death to cell survival. J Cell Biol 2015;210:1101–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, Dorn GW 2nd, Kirshenbaum LA.. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci USA 2014;111:E5537–E5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Que X, Hung M, Yeang C, Gonen A, Prohaska T, Sun X, Diehl C, Maatta A, Bowden K, Gaddis D, Pattison J, MacDonald J,SY-H, Melon P, Hedrick C, Ley K, Miller Y, Glass C,KLP, Binder C, Tsimikas S, Witztum J.. Oxidized phospholipids are proinflammatory and proatherogenic in cholesterol-fed Ldlr−/− mice. Nature;doi:10.1038/s41586-018-0198-8. Published online ahead of print 6 June 2018. [Google Scholar]
- 29. Schneider M, Witztum JL, Young SG, Ludwig EH, Miller ER, Tsimikas S, Curtiss LK, Marcovina SM, Taylor JM, Lawn RM, Innerarity TL, Pitas RE.. High-level lipoprotein [a] expression in transgenic mice: evidence for oxidized phospholipids in lipoprotein [a] but not in low density lipoproteins. J Lipid Res 2005;46:769–778. [DOI] [PubMed] [Google Scholar]
- 30. Philipp S, Cohen MV, Downey JM.. Animal models for the study of myocardial protection against ischemia. Drug Discov Today 2005;2:219–444. [Google Scholar]
- 31. Vivaldi MT, Kloner RA, Schoen FJ.. Triphenyltetrazolium staining of irreversible ischemic injury following coronary artery occlusion in rats. Am J Pathol 1985;121:522–530. [PMC free article] [PubMed] [Google Scholar]
- 32. Ravandi A, Leibundgut G, Hung MY, Patel M, Hutchins PM, Murphy RC, Prasad A, Mahmud E, Miller YI, Dennis EA, Witztum JL, Tsimikas S.. Release and capture of bioactive oxidized phospholipids and oxidized cholesteryl esters during percutaneous coronary and peripheral arterial interventions in humans. J Am Coll Cardiol 2014;63:1961–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bordun KA, Premecz S, daSilva M, Mandal S, Goyal V, Glavinovic T, Cheung M, Cheung D, White CW, Chaudhary R, Freed DH, Villarraga HR, Herrmann J, Kohli M, Ravandi A, Thliveris J, Pitz M, Singal PK, Mulvagh S, Jassal DS.. The utility of cardiac biomarkers and echocardiography for the early detection of bevacizumab- and sunitinib-mediated cardiotoxicity. Am J Physiol Heart Circ Physiol 2015;309:H692–H701. [DOI] [PubMed] [Google Scholar]
- 34. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, Nieuwdorp M, Joosten LA, Netea MG, Koschinsky ML, Witztum JL, Tsimikas S, Riksen NP, Stroes ES.. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 2016;134:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ganguly R, Hasanally D, Stamenkovic A, Maddaford TG, Chaudhary R, Pierce GN, Ravandi A.. Alpha linolenic acid decreases apoptosis and oxidized phospholipids in cardiomyocytes during ischemia/reperfusion. Mol Cell Biochem 2018;437:163–175. [DOI] [PubMed] [Google Scholar]
- 36. White CW, Ali A, Hasanally D, Xiang B, Li Y, Mundt P, Lytwyn M, Colah S, Klein J, Ravandi A, Arora RC, Lee TW, Hryshko L, Large S, Tian G, Freed DH.. A cardioprotective preservation strategy employing ex vivo heart perfusion facilitates successful transplant of donor hearts after cardiocirculatory death. J Heart Lung Transplant 2013;32:734–743. [DOI] [PubMed] [Google Scholar]
- 37. Weidman D, Shaw J, Bednarczyk J, Regula K, Yurkova N, Zhang T, Aguilar F, Kirshenbaum L.. Dissecting apoptosis and intrinsic death pathways in the heart. Methods Enzymol 2008;446:277–285. [DOI] [PubMed] [Google Scholar]
- 38. Chen R, Yang L, McIntyre TM.. Cytotoxic phospholipid oxidation products. Cell death from mitochondrial damage and the intrinsic caspase cascade. J Biol Chem 2007;282:24842–24850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I.. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab 2010;12:467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gang H, Hai Y, Dhingra R, Gordon JW, Yurkova N, Aviv Y, Li H, Aguilar F, Marshall A, Leygue E, Kirshenbaum LA.. A novel hypoxia-inducible spliced variant of mitochondrial death gene Bnip3 promotes survival of ventricular myocytes. Circ Res 2011;108:1084–1092. [DOI] [PubMed] [Google Scholar]
- 41. Bagheri F, Khori V, Alizadeh AM, Khalighfard S, Khodayari S, Khodayari H.. Reactive oxygen species-mediated cardiac-reperfusion injury: mechanisms and therapies. Life Sci 2016;165:43–55. [DOI] [PubMed] [Google Scholar]
- 42. Stemmer U, Dunai Z, Koller D, Pürstinger G, Zenzmaier E, Deigner H, Aflaki E, Kratky D, Hermetter A.. Toxicity of oxidized phospholipids in cultured macrophages. Lipids Health Dis 2012;11:110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S, Nelson SF, Horvath S, Berliner JA, Kirchgessner TG, Lusis AJ.. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA 2006;103:12741–12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qin J, Testai F, Dawson S, Kilkus J, Dawson G.. Oxidized phosphatidylcholine formation and action in oligodendrocytes. J Neurochem 2009;110:1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Loidl A, Sevcsik E, Riesenhuber G, Deigner HP, Hermetter A.. Oxidized phospholipids in minimally modified low density lipoprotein induce apoptotic signaling via activation of acid sphingomyelinase in arterial smooth muscle cells. J Biol Chem 2003;278:32921–32928. [DOI] [PubMed] [Google Scholar]
- 46. Scipione CA, Sayegh SE, Romagnuolo R, Tsimikas S, Marcovina SM, Boffa MB, Koschinsky ML.. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a). J Lipid Res 2015;56:2273–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, Nieuwdorp M, Joosten LA, Netea MG, Koschinsky ML, Witztum JL, Tsimikas S, Riksen NP, Stroes ES.. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 2016;134:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fruhwirth G, Moumtzi A, Loidl A, Ingolic E, Hermetter A.. The oxidized phospholipids POVPC and PGPC inhibit growth and induce apoptosis in vascular smooth muscle cells. Biochim Biophys Acta 2006;1761:1060–1069. [DOI] [PubMed] [Google Scholar]
- 49. Lidman M, Pokorna S, Dingeldein AP, Sparrman T, Wallgren M, Sachl R, Hof M, Grobner G.. The oxidized phospholipid PazePC promotes permeabilization of mitochondrial membranes by Bax. Biochim Biophys Acta 2016;1858:1288–1297. [DOI] [PubMed] [Google Scholar]
- 50. Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ.. B cells and humoral immunity in atherosclerosis. Circ Res 2014;114:1743–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, Witztum JL, Bae YS, Miller YI.. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res 2009;104:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Faria-Neto JR, Chyu KY, Li X, Dimayuga PC, Ferreira C, Yano J, Cercek B, Shah PK.. Passive immunization with monoclonal IgM antibodies against phosphorylcholine reduces accelerated vein graft atherosclerosis in apolipoprotein E-null mice. Atherosclerosis 2006;189:83–90. [DOI] [PubMed] [Google Scholar]
- 53. Tsimikas S, Willeit P, Willeit J, Santer P, Mayr M, Xu Q, Mayr A, Witztum JL, Kiechl S.. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol 2012;60:2218–2229. [DOI] [PubMed] [Google Scholar]
- 54. Tsimikas S, Brilakis ES, Lennon RJ, Miller ER, Witztum JL, McConnell JP, Kornman KS, Berger PB.. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res 2007;48:425–433. [DOI] [PubMed] [Google Scholar]
- 55. Sjöberg BG, Su J, Dahlbom I, Grönlund H, Wikström M, Hedblad B, Berglund G, de Faire U, Frostegård J.. Low levels of IgM antibodies against phosphorylcholine-A potential risk marker for ischemic stroke in men. Atherosclerosis 2009;203:528–532. [DOI] [PubMed] [Google Scholar]
- 56. Su J, Georgiades A, Wu R, Thulin T, de Faire U, Frostegård J.. Antibodies of IgM subclass to phosphorylcholine and oxidized LDL are protective factors for atherosclerosis in patients with hypertension. Atherosclerosis 2006;188:160–166. [DOI] [PubMed] [Google Scholar]
- 57. Prasad A, Clopton P, Ayers C, Khera A, de Lemos JA, Witztum JL, Tsimikas S.. Relationship of autoantibodies to MDA-LDL and ApoB-immune complexes to sex, ethnicity, subclinical atherosclerosis, and cardiovascular events. Arterioscler Thromb Vasc Biol 2017;37:1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA.. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem 1997;272:13597–13607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.