

Abstract
The cytoskeleton is a highly adaptive network of filamentous proteins capable of stiffening under stress even as it dynamically assembles and disassembles with time constants of minutes. Synthetic materials that combine reversibility and strain-stiffening properties remain elusive. Here, strain-stiffening hydrogels that have dynamic fibrous polymers as their main structural components are reported. The fibers form via self-assembly of bolaamphiphiles (BA) in water and have a well-defined cross-section of 9 to 10 molecules. Fiber length recovery after sonication, H/D exchange experiments, and rheology confirm the dynamic nature of the fibers. Cross-linking of the fibers yields strain-stiffening, self-healing hydrogels that closely mimic the mechanics of biological networks, with mechanical properties that can be modulated by chemical modification of the components. Comparison of the supramolecular networks with covalently fixated networks shows that the noncovalent nature of the fibers limits the maximum stress that fibers can bear and, hence, limits the range of stiffening.
Introduction
Biological fibrous networks are both adaptive and robust. These seemingly conflicting properties are encoded at different length scales. On the one hand, the constituent fibers (actin, microtubules, collagen) are held together by often quite weak supramolecular forces, enabling dynamic structural adaptation through assembly and disassembly.1−6 On the other hand, their networks, taken as a whole, are capable of “strain stiffening”,7−12 a strengthening that is prompted by external loading, without the need for structural changes. Combination of these antithetical properties provides biological functionality, which ideally one would like to reconstitute in synthetic materials. However, while both reversibility13−16 and strain stiffening17−20 have been repeatedly demonstrated separately, there is until now no single synthetic material that possesses both qualities. Here, we present biomimetic hydrogels composed of self-assembled semiflexible nanofibers with reversibility on a time scale of days that are simultaneously strain-stiffening and self-healing.
Bolaamphiphiles (BA), which are nonionic amphiphiles with two polar head groups (Figure 1A), have previously been used to create fully covalent strain-stiffening networks.20 In water, these molecules self-assemble into elongated fibers composed of 9 to 10 aggregated ribbons with an average contour length of 157 nm, 3.3 nm radius, and a persistence length of 280 nm. The assemblies are held together via intermolecular urea–urea hydrogen bonds and hydrophobic interactions. Photopolymerization of the diacetylenes produces fibers with a covalent polydiacetylene backbone.21 The system forms a covalent strain-stiffening hydrogel upon chemical cross-linking of reactive BAs with azide and acetylene moieties (Figure 1) using a Cu-catalyzed click reaction.
Figure 1.

Molecules and methods used to form strain-stiffening supramolecular hydrogels. (A) Molecular structure of the fiber-forming diacetylene bis-urea bolaamphiphile (BA), its cross-linkable analogues BA-AC and BA-N3, and the pentaazide 5-N3 cross-linker. (B) Hierarchical self-assembly through intermolecular H-bonding and hydrophobic interactions of functionalized (BA-AC) and unfunctionalized (BA) diacetylene bisurea bolaamphiphiles. The molecules self-assemble in water into fibers integrating 9 to 10 ribbons. Direct cross-linking via mixing of equal volumes of BA/BA-AC and BA/BA-N3 fibers results in the formation of exclusively interfiber triazole cross-links (upper, red). Multiarm cross-linking of BA/BA-AC fibers with 5-N3 results in the formation of inter- (network formation) and intrafiber (fiber covalent fixation) triazole cross-links (lower, blue).
In the current work, we investigate strain-stiffening in gels of supramolecular BA fibers that are not fixated via diacetylene cross-polymerization. We confirm with several experiments that the fibers are dynamically equilibrating. For instance, the fibers reassemble after being fragmented by ultrasound, with the fragments recombining completely after 2 days. Despite being in dynamic equilibrium, supramolecular BA hydrogels exhibit strain-stiffening and thus represent a self-assembling system with sufficient aggregation strength to withstand forces required to sustain a regime of nonlinear mechanical response. Furthermore, we demonstrate control over mechanical performance; by using a combined cross-linking-fixation strategy we significantly extend the stiffening range (i.e., the overall increase in modulus relative to the initial modulus before failure).
Results and Discussion
Hydrogel Formation
BA fibers were chemically cross-linked using two different strategies: direct cross-linking and multiarm cross-linking. In the first approach, BA fibers were functionalized by coassembling BA with a cross-linkable analogue BA-AC and in a separate solution with BA-N3 (Figure 1B, red). Direct cross-linking was then achieved by mixing solutions containing BA-AC-labeled fibers and BA-N3-labeled fibers (note that upon mixing, the concentration of each reactive bolaamphiphile is halved) followed by addition of a cross-linking catalyst. Assuming that the dynamics of monomer exchange is slower than the cross-linking reaction (see below), formation of intrafiber cross-links due to insertion of a BA-N3 molecule into a BA/BA-AC fiber is limited. Thus, every time a reaction between an acetylene and an azide group occurs, it effectively connects two different fibers, thereby contributing to the network’s modulus (see Figure 1B, red). In the second cross-linking strategy, multiarm cross-linking was performed by mixing BA/BA-AC with 5-N3, a pentaazide linker capable of forming up to five cross-links per molecule (Figure 1A).22,23 This linker can react with BA-AC groups of different fibers (i.e., network formation), although reaction of one molecule of 5-N3 with more than two fibers is unlikely due to steric reasons. 5-N3 can also react with BA-AC groups within a fiber, creating covalent bonds along the hydrophilic periphery of the fiber (i.e., intrafiber fixation) (Figure 1B, blue). The reaction between azide and acetylene groups in direct cross-linking and in the multiarm cross-linking strategy was initiated at room temperature by addition of a catalyst mixture containing Cu(I) and accelerating ligands.24 No diacetylene polymerization took place under these conditions. 1H NMR showed high conversions of the cross-linking reaction, in which 80% of the terminal acetylene groups from BA-AC reacted with 5-N3 during the gelation process (Figure 2).
Figure 2.
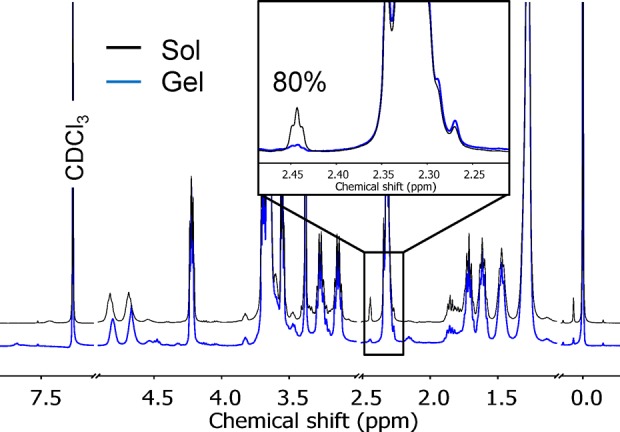
1H NMR (CDCl3, 25 °C) spectra of 20 mg mL–1 BA containing 20 mol % BA-AC, cross-linked with 5-N3 acquired before (blue line) and 24 h after addition of the catalyst (black line), showing 80% conversion (inset) calculated by integrating the area below the curve ascribed to the terminal acetylene protons of BA-AC. The known value of the integral corresponding to the −CH2COOCH2– protons was used as an internal standard.
Structural characterization of the system before (sol) and after (gel) a cross-linking-fixation reaction with 5-N3 was performed by small-angle X-ray scattering (SAXS). Except for small changes in slope at low q-values the scattering profiles were nearly overlapping (Figure 3). Fitting of the scattering profiles (Supporting Information and Figure S5) gave a value for the cross-sectional radius of 3.3 ± 0.2 nm before and after gelation, consistent with the radius of 3.3 nm measured in cryo-electron microscopy (cryo-EM) (Figure 4A). The absence of bundling is in marked contrast to the polyisocyanopeptide (PIC) strain-stiffening hydrogels reported by Rowan’s group, where gelation relies on physical aggregation of individual polymer chains with an associated increase in bundle dimensions measurable by SAXS.25
Figure 3.
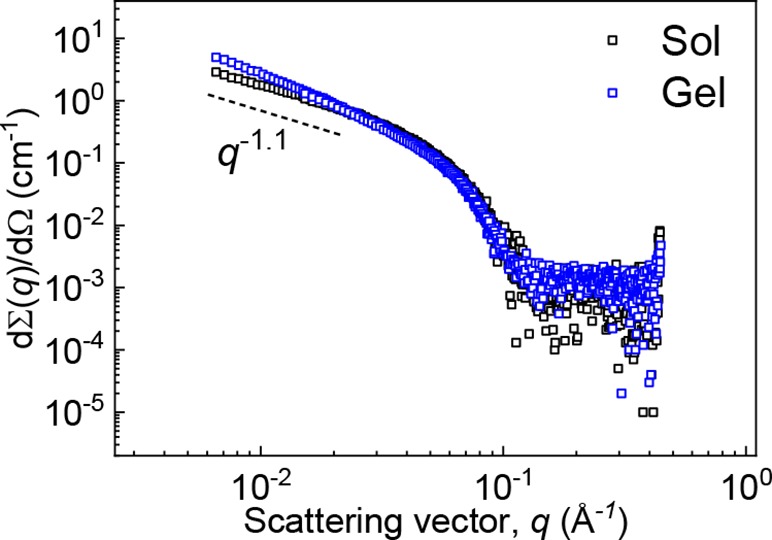
SAXS profiles recorded before (black squares) and 24 h after cross-linking (blue circles) of 15 mg mL–1 BA/BA-AC (20 mol % BA-AC) solutions with 5-N3.
Figure 4.
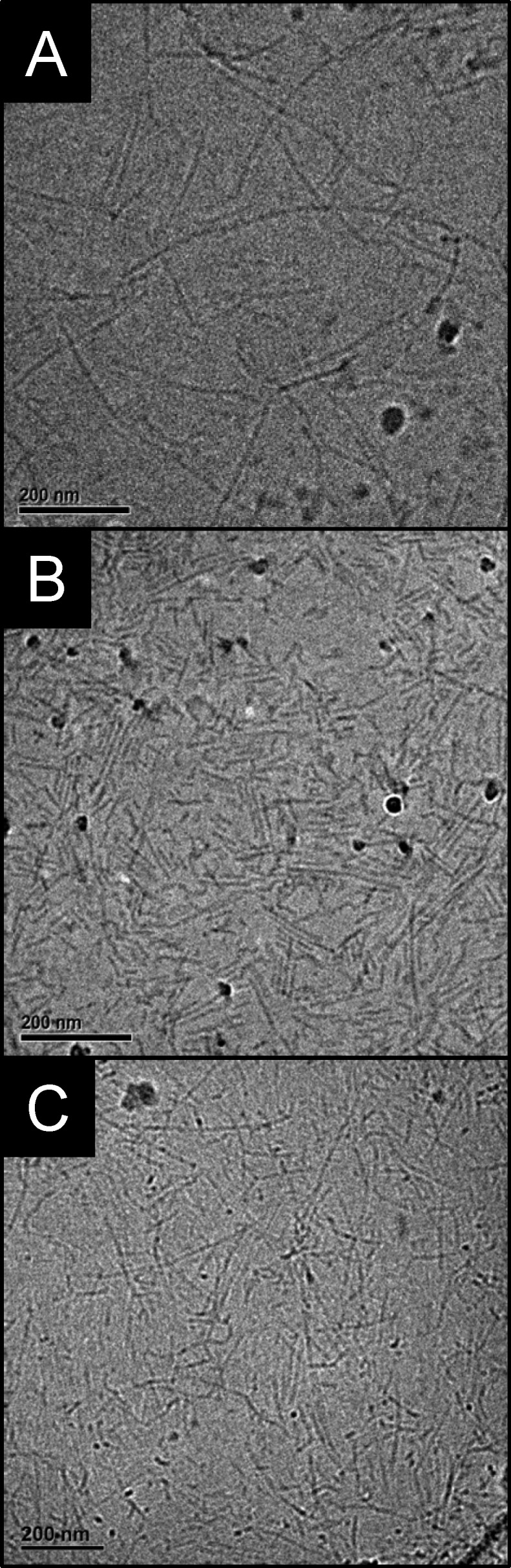
Cryo-EM micrographs of BA in water (1 mM). (A) Unperturbed system. (B) After 1 h of sonication. (C) After 1 h of sonication followed by 2 d of reassembly. Scale: 200 nm.
Dynamics from Sonication-Induced Fragmentation of BA Fibers and Recovery
The reversibility of BA aggregation was probed by sonication of un-cross-linked fiber solutions. Similar to experiments reported by Talens et al.26 involving analogous squaramide-based bolaamphiphiles, cryo-EM of fibers before and after sonication for 1 h shows that the average fiber contour length is reduced from 267 nm to 46 nm by sonication (Figure 4A and B, respectively) but regrow to 206 nm on average after equilibration for 2 days (Figure 4C). Similar reversible disassembly into much shorter fibers followed by recovery has been observed in solutions of amyloid fibrils subjected to sonication.27
H/D Exchange Kinetics of Un-Cross-Linked BA Fibers
Relevant insight into the solvent accessibility and dynamics of BA fibers was obtained by using hydrogen–deuterium exchange mass spectrometry (HDX-MS), which has been reported to be a powerful label-free method to unravel the dynamic processes of biological and synthetic supramolecular polymers.28,29 A 500 μM solution of BA in H2O was diluted 100-fold in D2O. Time dependence of the deuterium exchange is shown in the HDX-MS spectra (Figure 5A). The four hydrogen atoms of urea NH groups can undergo H/D exchange, increasing the molecular weight of the monomer by 1 Dalton for each exchanged proton. Solvent-accessible NH protons that are not involved in hydrogen bonding undergo rapid H/D exchange, while protons that are shielded from the surrounding water exchange more slowly or not at all. Two isotopic distributions corresponding to BA and BA-d4 were observed in the MS spectra, and their intensities were followed over time. In the first 10 h, 40% of the BA molecules exchanged their NH protons, after which exchange slowed down significantly (see Figure 5B), and nearly 40% of the urea protons had not exchanged after 7 d (Figure 5B).
Figure 5.
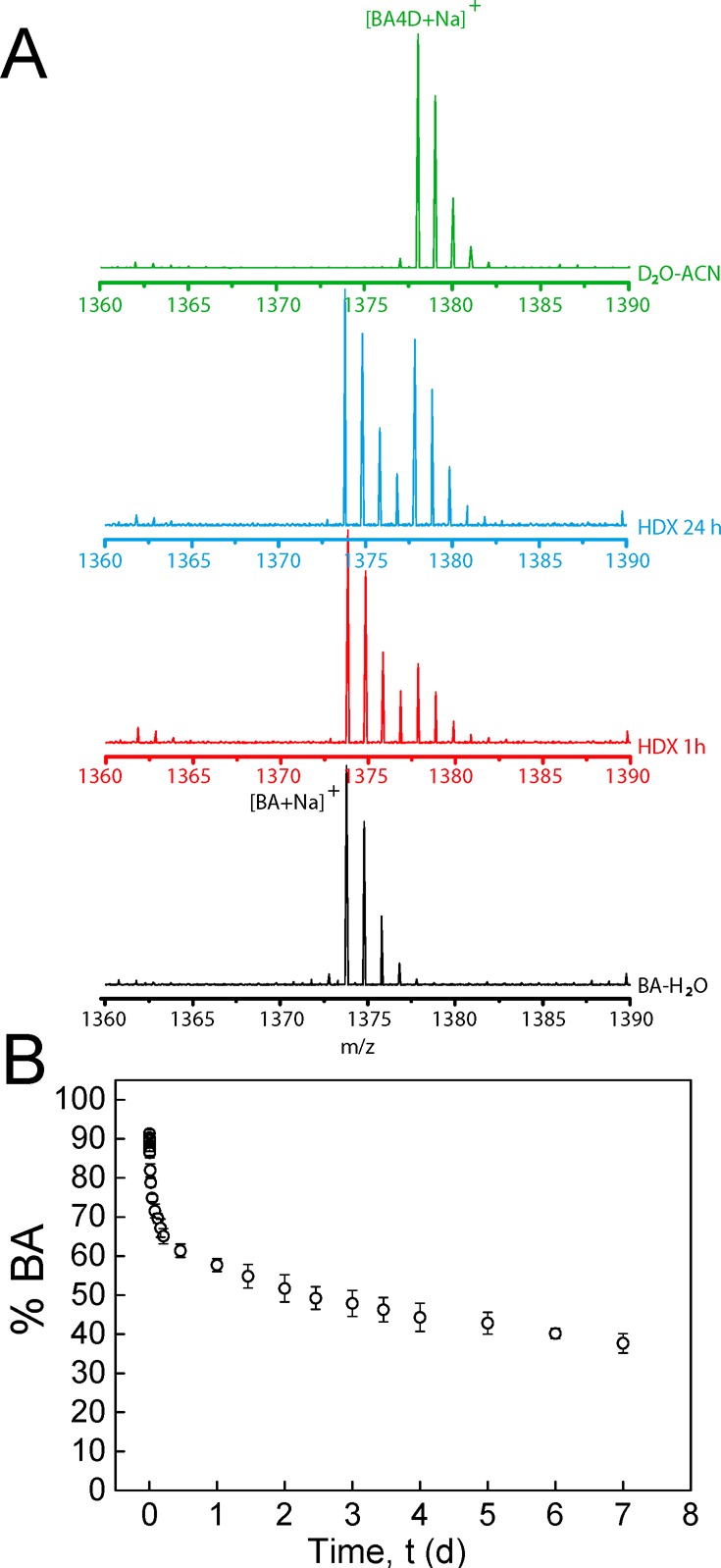
Time-dependent exchange experiments. (A) Mass spectra during the exchange reaction after 100-fold dilution of a 500 μM BA–H2O solution in D2O or H2O. The spectra at four different stages are presented in different colors: the black spectrum is a reference spectrum of BA diluted in H2O; the red spectrum and the blue spectrum are recorded after HDX for 1 and 24 h, respectively; the green spectrum is the spectrum of a control measurement where the polymers were diluted 100 times in a solvent mixture containing acetonitrile (ACN–D2O, 1:1 (v/v)) to break down the supramolecular assemblies. (B) Percentage of nondeuterated BA as a function of equilibration time. Error bars represent standard deviation of three separate experiments.
Proton exchange does not follow simple monoexponential kinetics, which excludes a mechanism in which all aggregated molecules have the same solvent accessibility and exchange with free molecules in solution at the same rate. Differences in solvent accessibility between molecules in fibers that contain 9 to 10 amphiphilic molecules in cross-section20,30 may explain the slow, nonexponential kinetics if recycling at chain ends is required for the protons of solvent-shielded molecules to exchange. In that sense, H/D exchange in BA fibers is reminiscent of H/D exchange in tightly packed biopolymer amyloid fibrils.31,32
BA Exchange Dynamics Measured by Rheology
Exchange dynamics of BA fibers at equilibrium were further investigated by equilibrating mixed aqueous solutions of BA fibers with and without 20 mol % reactive BA-AC amphiphiles for 0, 1, 4, and 7 d, after which time multiarm cross-linking with 5-N3 was initiated by addition of the catalyst mixture. After completion of the cross-linking reaction, the storage moduli G′ of the gels were measured. Without equilibration, only half of the fibers can contribute to the network formation, whereas the dynamic exchange of amphiphilic molecules will lead to a homogeneous distribution of reactive BA-AC in all fibers at full equilibration (i.e., all fibers contribute to the network’s modulus). After completion of the cross-linking reaction, the storage modulus G′ was nearly 3 times higher after equilibration for 1 week than without equilibration (Figure 4B and SIFigure S6), showing that redistribution of reactive BA-AC molecules takes place and leads to additional network formation. The increase in modulus is still incomplete after 1 and 4 d, indicating a slow dynamic equilibrium with a time constant slower than that found in the sonication/recovery experiment but in line with the rate found in the H/D exchange. It should be noted that the maximum cross-link density may already be obtained even when the reactive monomer has not yet fully redistributed over the fibers.
Nonlinear Mechanics of BA Hydrogels
In contrast to conventional synthetic hydrogels based on flexible polymers, biological networks are known to display a well-defined nonlinear response to deformation after a characteristic critical stress σc is imparted to the material.7,9−11,33,34 Networks of BA fibers were likewise found to respond to applied shear stress with an associated increase in modulus. To carefully capture linear and nonlinear regimes of cross-linked BA networks, a prestress protocol35 was used for the measurements, and the differential modulus K (the elastic part of which relates the change in stress with strain K′ = δσ/δγ) was measured by parallel superposition of an oscillatory and a steady prestress σ. By plotting K′ against σ, two distinct regimes arise: a low-stress regime where the elastic response is linear with K′ equal to the plateau modulus G0, and a high-stress regime above the critical stress σc where K′ becomes strongly dependent on σ and scales with stress as K′ = σm, m being the stiffening index. The combination of these parameters yields a direct measure of the network’s sensitivity to applied stress; a property commonly termed responsiveness.
To minimize the effect caused by the formation of intrafiber covalent bonds along the fiber axis, gels were prepared using the direct cross-linking approach depicted in Figure 1B (red). Given the slow kinetics of monomer exchange, formation of intrafiber cross-links due to insertion of, for instance, a BA-N3 molecule into a BA-AC-labeled fiber can be considered negligible within the experimental time scales (i.e., 14 h). Solutions (15–22 mg mL–1) containing equal amounts of BA-AC- and BA-N3-labeled fibers were cross-linked by adding the catalyst, and the differential moduli K′ of the so-formed hydrogels were measured as a function of σ. Gels formed via this procedure were found to strain-stiffen in spite of the absence of intrafiber covalent bonds reinforcing the fibers. The modulus of the material is characterized by a plateau storage modulus G0 at low σ, which increases beyond the critical stress σc (Figure 7A, red). The critical strain γc at which stiffening sets in is in the range of 0.14–0.21, similar to critical strains in biological networks.36 The relative increase in moduli prior to gel rupture K′max/G0 obtained after scaling of K′ to G0 and σ to σc is limited to values close to 2 (Figure 7B, red). Upon increasing the overall concentration of cross-links—by increasing the fraction of cross-linkable BA in the BA host—G0 increases, but the stiffening range does not change significantly (Figure 7B,C, red).
Figure 7.

Nonlinear mechanics of BA hydrogels using direct cross-linking (red symbols) and multiarm cross-linking (blue symbols) strategies (c BA in mg mL–1:mol % cross-linkable BA: 22:5% (red squares), 15:7.5% (red circles), 15:10% (red triangles), 15:5% (blue circles), 15:10% (blue squares), 15:15% (blue up triangles), 13:20% (blue down triangles)). (A) Differential modulus K′ vs stress σ obtained from BA gels formed through direct and multiarm cross-linking strategies. (B) Plot of K′ vs stress σ with K′ normalized to G0 and σ normalized to σc, showing collapse onto single master curve with K′ ∝ σ1 at high σ. (C) Stiffening factor K′max/G0 plotted against mol % percent of cross-linkable BA, with acetylene or azide groups. The yellow area indicates the K′max/G0 range of gels lacking fiber covalent reinforcement due to multiarm cross-linking. (D) Normalized data fitted to the Shung–Fung model (black line). (E) Differential modulus K′ vs stress σ obtained from gels of Figure 6B and Figure S7. (F) Stiffening ranges K′max/G0 as a function of fiber incubation time of the mixture of Figure 6. The dashed lines indicate the K′max/G0 values measured for networks containing 20 and 10 mol % BA-AC as obtained from normalized curves of Figure 7E.
Similarly, gels formed with the multiarm cross-linking strategy through reaction of BA/BA-AC fibers with 5-N3 (Figure 1B, blue) showed strain stiffening with a similar critical stress. However, K’max/G0 was higher than for the directly cross-linked system and displayed a clear upward trend with increasing functionalization of the fibers (Figure 7B,C, blue). These results can be rationalized by considering that the multiarm cross-linker may provide external reinforcement of the fibers because a single cross-linker molecule can react with multiple acetylene groups in a single fiber. For fibers incorporating 10% BA-AC, nearly two acetylene groups are present every urea–urea repeat distance (0.46 nm).37 This reaction provides fixation along the fiber axis and imparts enhanced axial strength, thereby extending the range of nonlinear deformation of the hydrogels prior to failure. Indeed, K′max/G0 reaches a value of 8.6 for a hydrogel with fibers labeled with 20 mol % BA-AC. Plotting of K′max/G0 vs mol % cross-linkable BA for both cross-linking strategies (in Figure 7C) shows that at 5% functionalization K′max/G0 is the same for direct cross-linking and for multiarm cross-linking, whereas at higher percentages, the stiffening range increases only for the multiarm strategy because external fiber fixation strengthens the fibers.
Normalizing of K′ and σ against G0 and σc in Figure 7B reduces all data to a single master curve featuring a well-defined linear relationship between K′ and σ (K′ ∝ σ1) above σc. A similar linear increase of the differential modulus with stress has been observed in various types of biopolymer gels reconstituted from extra- and intracellular proteins including type I collagen,11 neurofilament hydrogels,33 and, more recently, branched actin networks measured under compressive forces exerted by magnetic cylinders.38 These results highlight the common origins of elasticity in these systems where nonlinearity sets in due to enhanced flexural rigidity and backbone stiffness of the constituent biopolymers.7,39 To compare the nonlinear rheology of our gels to that of biological networks, we applied a model introduced by Shull and co-workers.40 This phenomenological hyperelastic model is equivalent to the Fung model41 commonly used to describe the mechanics of collagenous tissues such as those found in arteries.42,43 In the Fung–Shull model, the stress σ as a function of the strain γ is given by
with γc being the critical strain (at which nonlinearity sets in) and G0 the plateau modulus. Taking the strain derivative of this function, eliminating γ in favor of σ from the result, and normalizing G to G0 and σ to σc, respectively, yields a universal, zero-parameter stiffening curve that captures our data remarkably well (see Figure 7D). The stiffening exponent m in the Shull–Fung model is equal to 1, a direct consequence of an exponential stiffening. This agreement between biomaterial and synthetic performance again highlights the biomimetic nature of the hydrogels.
K′max/G0 values measured in the previously discussed equilibration experiment of Figure 6 gave additional confirmation of the dynamic nature of the self-assembled BA fibers. Figure 7E and F show that gels obtained without incubation exhibit a K′max/G0 of 6.7, similar to the value found in networks composed of fibers with 20 mol % BA-AC. K′max/G0 steadily decreases with increasing incubation times, after 4 to 7 days leading to a stiffening range remarkably close to the value for networks incorporating 10 mol % BA-AC (see Figure 7F). The gradual decrease in stiffening range reflects the dynamics of BA-AC redistribution across the fibers because without incubation, only fibers containing 20 mol % BA-AC form a network, while after equilibration, 10 mol % of BA-AC is distributed uniformly in all fibers (see Figure 6A).
Figure 6.
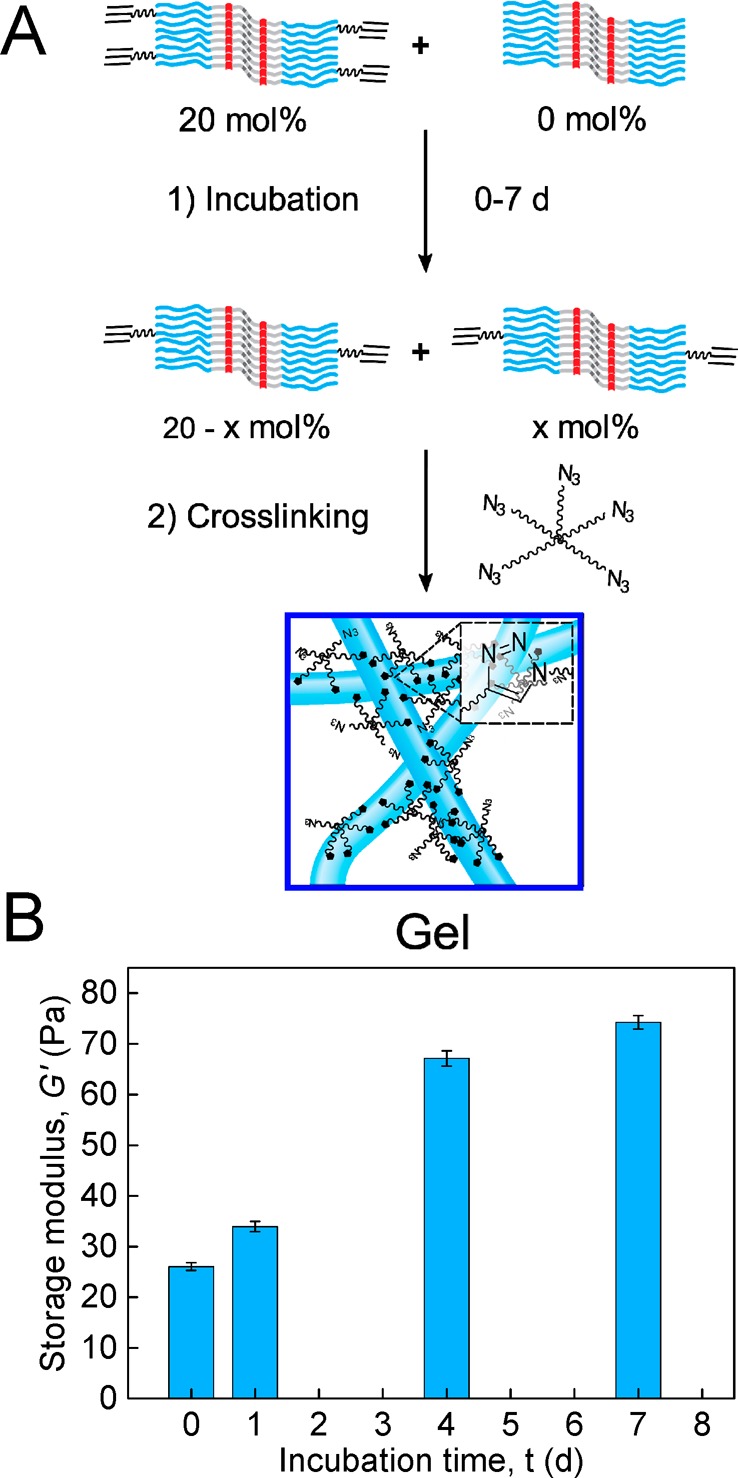
Reversibility of BA fibers in water measured by rheology. (A) Monomer exchange between labeled (BA/BA-AC) and unlabeled (BA) fibers followed by gelation via covalent cross-linking-fixation with 5-N3. (B) Storage modulus G′ measured in oscillatory shear (γ = 1% and ω = 6.28 rad s–1) of cross-linked solutions of 16 mg mL–1 BA/BA-AC (20 mol % BA-AC) mixed with 16 mg mL–1 BA after different incubation times. Error bars represent the standard deviation of G′ at the conclusion of the cross-linking reaction for hydrogels prepared from the same stock solution.
Nonlinear Mechanics of PDA Hydrogels
To further investigate the impact of fiber covalent reinforcement on the nonlinear mechanics of BA hydrogels, the experiments of Figure 7A–C were repeated with gels whose fibers were fixated prior to cross-linking via topochemical photopolymerization of the diacetylene groups to produce poly(diacetylene) (PDA) fibers (Figure 8A) in analogy to previous work.20 The presence of a covalent PDA backbone was found to have a strong effect on the stiffening range of the hydrogels. Thus, the K′max/G0 values of directly cross-linked hydrogels were raised from 1.5 to 2 to 8–11 upon photopolymerization of BA fibers regardless of the fraction cross-linkable BA (Figure 8B–D, red). Concomitantly, multiarm cross-linking of PDA fibers produced hydrogels characterized by K′max/G0 values ranging from 11.6 at 5 mol % BA-AC incorporation up to 68 at 20 mol % BA-AC incorporation (Figure 8B–D, blue). These values are much higher than for their nonfixated BA counterparts (2.5 to 8.6). Overall, these results showcase that, although the dynamic BA fibers are robust enough to exhibit strain-stiffening, covalent fixation results in a pronounced increase of the stiffening range from 1.5 up to 68 in a fully controlled fashion (in Figure 8D).
Figure 8.

Nonlinear mechanics of PDA hydrogels using direct (red solid symbols) and multiarm (blue solid symbols) cross-linking strategies (c PDA in mg mL–1:mol % cross-linkable BA: 22:5% (red squares), 15:7.5% (red circles), 15:10% (red triangles), 15:5% (blue circles), 15:10% (blue squares), 15:15% (blue up triangles), 13:20% (blue down triangles)). (A) Sample preparation method involving topochemical polymerization of self-assembled BA/BA-AC fibers by irradiating with UV light resulting in the formation of a π-conjugated PDA framework, followed by direct or multiarm cross-linking. (B) Differential modulus K′ vs stress σ obtained from BA gels formed through direct and multiarm cross-linking. (C) Plot of K′ vs stress σ with K′ normalized to G0 and σ normalized to σc, showing collapse onto a single master curve with K′ ∝ σ1 at high σ. (D) Stiffening factor K′max/G0 plotted against mol % percent of cross-linkable BA, with acetylene or azide groups.
Self-Healing Behavior of BA Hydrogels
To test the self-healing properties of BA hydrogels, a sample containing a high concentration of fibers (30 mg mL–1) and a low density (2.5 mol %) of cross-links was prepared using a direct cross-linking approach in order to facilitate recombination of chain-ends after disruption of the network structure. First, the cross-linking reaction was monitored until the moduli of the gel reached a plateau 40 h after addition of the catalyst (Figure S8), thereby ensuring that changes in moduli do not occur due to additional cross-linking.
The modulus of the so-formed hydrogel was continuously monitored with small-amplitude (0.1%) oscillatory shear to assess the extent and rate of recovery after cycles of 15 and 60 s oscillatory shear at increased high-amplitude shear strain ranging from 100% to 1000%. Figure 9 shows that the application of large strains resulted in a pronounced decrease of G′ with the material exhibiting fluid-like, viscous behavior at strains larger than 100%. Shearing for prolonged intervals at high strains resulted in accumulation of damage within the network structure, as inferred from a continuous decrease in moduli. After cessation of the strain, the material underwent a partial recovery of G′ within seconds that was dependent on the extent and duration of the damage sustained during the preceding cycle. The G′ of the material continued to increase more slowly thereafter, reaching its equilibrium G′ value after healing for approximately 18 h even as it was sheared at strains as high as 1000%. These results lead us to hypothesize a self-healing mechanism in which highly reactive chain-ends rapidly meet after rupture, leading to a fast recovery in modulus followed by a slower equilibration process likely involving interfiber monomer exchange.
Figure 9.
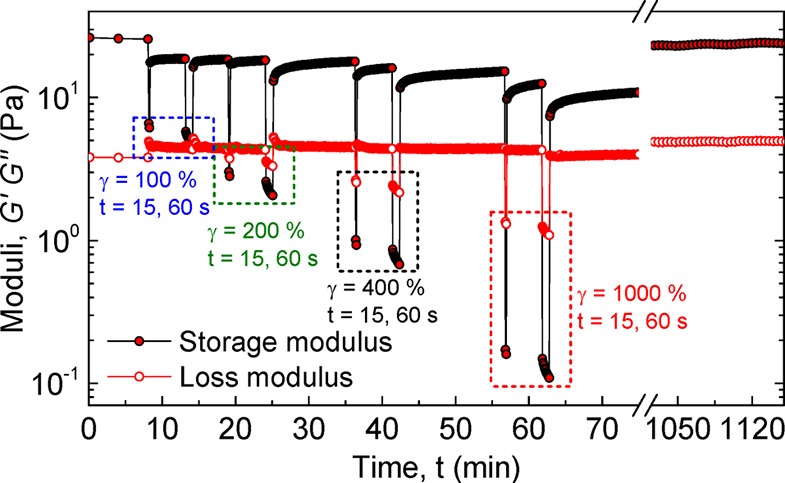
Self-healing of a directly cross-linked BA hydrogel (30 mg mL–1; 2.5 mol % cross-linkable BA) at 25 °C measured with oscillatory rheology. Moduli were recorded continuously at ω = 6.28 rad s–1 and γ = 1% with 15 and 60 s intervals of higher strain (denoted above).
The nonlinear mechanical properties of the healed material were tested with a prestress protocol to obtain the differential modulus as a function of the prestress. The healed material was found to exhibit strain-stiffening with a K′max/G0 = 1.34 (Figure S9), similar to the values found in the undamaged materials (1.5–2) (in Figure 7B and C). These results confirm that BA hydrogels simultaneously exhibit strain-stiffening and self-healing behavior, properties that are common to biopolymer networks but have remained elusive in synthetic materials up to now.
Conclusions
Strain-stiffening has been claimed to be inherent to any connected meshwork of semiflexible polymers.36 The observation of strain-stiffening and self-healing in the dynamic BA gels demonstrates that this behavior extends to supramolecular polymers, provided that the noncovalent interactions that hold the fibers together are sufficiently strong to support stresses larger than the characteristic network critical stress yet sufficiently dynamic to reassemble after rupture.
These results critically expand current knowledge about the origins of strain-stiffening in bio- as well as synthetic polymers, breaking ground for the rational design of genuinely supramolecular, biomimetic soft matter with mechanical properties that can be modulated by tuning the strength of the supramolecular interactions. These novel materials are expected to present cells with more dynamic environments and may open further possibilities for application in the biomedical field as injectable scaffolds to support cell growth or tissue engineering provided that alternative, bioorthogonal cross-linking strategies avoiding the use of cytotoxic copper are adopted, such as copper-free, strain-promoted reactions.44
In addition to the insights in the physics of strain-stiffening networks, the work advances chemical methodology to prepare such networks. A novel method to reinforce self-assembled fibers with covalent bonds has been introduced. The approach using a multiarm cross-linker offers a series of advantages compared to the use of diacetylene photopolymerization. The use of multiarm cross-linkers does not require a complex synthesis, and the extent of covalent fixation can be tuned. Thus, external fiber covalent fixation via nonselective 5-N3 cross-linking will likely provide the means to reinforce analogous fibrous assemblies without the complex geometrical requirements needed to undergo diacetylene topochemical fixation.
Acknowledgments
M.F.-C.R. was financially supported by the Marie Curie FP7 SASSYPOL ITN program (No. 607602). This work was supported by the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035). I.K.V. was financially supported by The Netherlands Organization for Scientific Research (NWO VIDI grant 723.014.006). We thank Dr. K. Pieterse (ICMS Animation Studio) for help with the graphics, Dr. A. W. Bosman of SupraPolix BV for support and fruitful discussions, and Dr. I. R. M. Cardinaels (Eindhoven University of Technology) for assistance with the rheological measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b09289.
Additional information (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Pollard T. D.; Borisy G. G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112 (4), 453–465. 10.1016/S0092-8674(03)00120-X. [DOI] [PubMed] [Google Scholar]
- Helfand B. T.; Mendez M. G.; Murthy S. N. P.; Shumaker D. K.; Grin B.; Mahammad S.; Aebi U.; Wedig T.; Wu Y. I.; Hahn K. M. Vimentin Organization Modulates the Formation of Lamellipodia. Mol. Biol. Cell 2011, 22 (8), 1274–1289. 10.1091/mbc.e10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner M.; Mitchison T. Beyond Self-Assembly: From Microtubules to Morphogenesis. Cell 1986, 45 (3), 329–342. 10.1016/0092-8674(86)90318-1. [DOI] [PubMed] [Google Scholar]
- Swaney K. F.; Huang C.-H.; Devreotes P. N. Eukaryotic Chemotaxis: A Network of Signaling Pathways Controls Motility, Directional Sensing, and Polarity. Annu. Rev. Biophys. 2010, 39 (1), 265–289. 10.1146/annurev.biophys.093008.131228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretschneider T.; The Three-Dimensional Dynamics of Actin Waves, a Model of Cytoskeletal Self-Organization. Biophys. J. 2009, 96 ( (7), ), 2888–2900 10.1016/j.bpj.2008.12.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windoffer R.; Beil M.; Magin T. M.; Leube R. E. Cytoskeleton in Motion: The Dynamics of Keratin Intermediate Filaments in Epithelia. J. Cell Biol. 2011, 194 (5), 669–678. 10.1083/jcb.201008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel M. L.; Shin J. H.; MacKintosh F. C.; Mahadevan L.; Matsudaira P.; Weitz D. A. Elastic Behavior of Cross-Linked and Bundled Actin Networks. Science 2004, 304 (5675), 1301–1305. 10.1126/science.1095087. [DOI] [PubMed] [Google Scholar]
- Lin Y.-C.; Yao N. Y.; Broedersz C. P.; Herrmann H.; MacKintosh F. C.; Weitz D. A. Origins of Elasticity in Intermediate Filament Networks. Phys. Rev. Lett. 2010, 104 (5), 058101. 10.1103/PhysRevLett.104.058101. [DOI] [PubMed] [Google Scholar]
- Yao N. Y.; Broedersz C. P.; Lin Y.-C.; Kasza K. E.; MacKintosh F. C.; Weitz D. A. Elasticity in Ionically Cross-Linked Neurofilament Networks. Biophys. J. 2010, 98 (10), 2147–2153. 10.1016/j.bpj.2010.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechocka I. K.; Jansen K. A.; Broedersz C. P.; Kurniawan N. A.; MacKintosh F. C.; Koenderink G. H. Multi-Scale Strain-Stiffening of Semiflexible Bundle Networks. Soft Matter 2016, 12 (7), 2145–2156. 10.1039/C5SM01992C. [DOI] [PubMed] [Google Scholar]
- Licup A. J.; Münster S.; Sharma A.; Sheinman M.; Jawerth L. M.; Fabry B.; Weitz D. A.; MacKintosh F. C. Stress Controls the Mechanics of Collagen Networks. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (31), 9573–9578. 10.1073/pnas.1504258112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. E. X.; Litvinov R. I.; Discher D. E.; Purohit P. K.; Weisel J. W. Multiscale Mechanics of Fibrin Polymer: Gel Stretching with Protein Unfolding and Loss of Water. Science 2009, 325 (5941), 741–744. 10.1126/science.1172484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadke A.; Zhang C.; Arman B.; Hsu C.-C.; Mashelkar R. A.; Lele A. K.; Tauber M. J.; Arya G.; Varghese S. Rapid Self-Healing Hydrogels. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (12), 4383–4388. 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncaboylu D. C.; Sari M.; Oppermann W.; Okay O. Tough and Self-Healing Hydrogels Formed via Hydrophobic Interactions. Macromolecules 2011, 44 (12), 4997–5005. 10.1021/ma200579v. [DOI] [Google Scholar]
- Cui J.; del Campo A. Multivalent H-Bonds for Self-Healing Hydrogels. Chem. Commun. 2012, 48 (74), 9302–9304. 10.1039/c2cc34701f. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Ye Q.; Lu X.; Lu Q. Self-Healing Polymers with PEG Oligomer Side Chains Based on Multiple H-Bonding and Adhesion Properties. Polym. Chem. 2015, 6 (28), 5086–5092. 10.1039/C5PY00621J. [DOI] [Google Scholar]
- Kouwer P. H. J.; Koepf M.; Le Sage V. A. A.; Jaspers M.; van Buul A. M.; Eksteen-Akeroyd Z. H.; Woltinge T.; Schwartz E.; Kitto H. J.; Hoogenboom R. Responsive Biomimetic Networks from Polyisocyanopeptide Hydrogels. Nature 2013, 493 (7434), 651–655. 10.1038/nature11839. [DOI] [PubMed] [Google Scholar]
- Jaspers M.; Dennison M.; Mabesoone M. F. J.; MacKintosh F. C.; Rowan A. E.; Kouwer P. H. J. Ultra-Responsive Soft Matter from Strain-Stiffening Hydrogels. Nat. Commun. 2014, 5, 5808. 10.1038/ncomms6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers M.; Vaessen S. L.; van Schayik P.; Voerman D.; Rowan A. E.; Kouwer P. H. J. Nonlinear Mechanics of Hybrid Polymer Networks That Mimic the Complex Mechanical Environment of Cells. Nat. Commun. 2017, 8, ncomms15478. 10.1038/ncomms15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Castano Romera M.; Lafleur R. P. M.; Guibert C.; Voets I. K.; Storm C.; Sijbesma R. P. Strain Stiffening Hydrogels through Self-Assembly and Covalent Fixation of Semi-Flexible Fibers. Angew. Chem., Int. Ed. 2017, 56 (30), 8771–8775. 10.1002/anie.201704046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A.; Voudouris P.; Koenigs M. M. E.; Besenius P.; Wyss H. M.; Degirmenci V.; Sijbesma R. P. Topochemical Polymerization in Self-Assembled Rodlike Micelles of Bisurea Bolaamphiphiles. Soft Matter 2014, 10 (7), 952–956. 10.1039/c3sm52605d. [DOI] [PubMed] [Google Scholar]
- Fransen P.; Pulido D.; Sevrin C.; Grandfils C.; Albericio F.; Royo M. High Control, Fast Growth OEG-Based Dendron Synthesis via a Sequential Two-Step Process of Copper-Free Diazo Transfer and Click Chemistry. Macromolecules 2014, 47 (8), 2585–2591. 10.1021/ma500166e. [DOI] [Google Scholar]
- Pulido D.; Albericio F.; Royo M. Controlling Multivalency and Multimodality: Up to Pentamodal Dendritic Platforms Based on Diethylenetriaminepentaacetic Acid Cores. Org. Lett. 2014, 16 (5), 1318–1321. 10.1021/ol500022n. [DOI] [PubMed] [Google Scholar]
- Hong V.; Presolski S. I.; Ma C.; Finn M. G. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angew. Chem., Int. Ed. 2009, 48 (52), 9879–9883. 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers M.; Pape A. C. H.; Voets I. K.; Rowan A. E.; Portale G.; Kouwer P. H. J. Bundle Formation in Biomimetic Hydrogels. Biomacromolecules 2016, 17 (8), 2642–2649. 10.1021/acs.biomac.6b00703. [DOI] [PubMed] [Google Scholar]
- Saez Talens V.; Englebienne P.; Trinh T. T.; Noteborn W. E. M.; Voets I. K.; Kieltyka R. E. Aromatic Gain in a Supramolecular Polymer. Angew. Chem., Int. Ed. 2015, 54 (36), 10502–10506. 10.1002/anie.201503905. [DOI] [PubMed] [Google Scholar]
- Carulla N.; Caddy G. L.; Hall D. R.; Zurdo J.; Gairí M.; Feliz M.; Giralt E.; Robinson C. V.; Dobson C. M. Molecular Recycling within Amyloid Fibrils. Nature 2005, 436 (7050), 554–558. 10.1038/nature03986. [DOI] [PubMed] [Google Scholar]
- Lou X.; Lafleur R. P. M.; Leenders C. M. A.; Schoenmakers S. M. C.; Matsumoto N. M.; Baker M. B.; van Dongen J. L. J.; Palmans A. R. A.; Meijer E. W. Dynamic Diversity of Synthetic Supramolecular Polymers in Water as Revealed by Hydrogen/Deuterium Exchange. Nat. Commun. 2017, 8, ncomms15420. 10.1038/ncomms15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheterpal I.; Zhou S.; Cook K. D.; Wetzel R. Aβ Amyloid Fibrils Possess a Core Structure Highly Resistant to Hydrogen Exchange. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (25), 13597–13601. 10.1073/pnas.250288897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obert E.; Bellot M.; Bouteiller L.; Andrioletti F.; Lehen-Ferrenbach C.; Boué F. Both Water- and Organo-Soluble Supramolecular Polymer Stabilized by Hydrogen-Bonding and Hydrophobic Interactions. J. Am. Chem. Soc. 2007, 129 (50), 15601–15605. 10.1021/ja074296l. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Rempel D. L.; Zhang J.; Sharma A. K.; Mirica L. M.; Gross M. L. Pulsed Hydrogen–Deuterium Exchange Mass Spectrometry Probes Conformational Changes in Amyloid Beta (Aβ) Peptide Aggregation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (36), 14604–14609. 10.1073/pnas.1309175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulla N.; Zhou M.; Arimon M.; Gairí M.; Giralt E.; Robinson C. V.; Dobson C. M. Experimental Characterization of Disordered and Ordered Aggregates Populated during the Process of Amyloid Fibril Formation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (19), 7828–7833. 10.1073/pnas.0812227106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee S.; Janmey P. A.; Bausch A. R. Mechanical and Structural Properties of in Vitro Neurofilament Hydrogels. Eur. Biophys. J. 2007, 36 (6), 661–668. 10.1007/s00249-007-0141-7. [DOI] [PubMed] [Google Scholar]
- Gardel M. L.; Shin J. H.; MacKintosh F. C.; Mahadevan L.; Matsudaira P. A.; Weitz D. A. Scaling of F-Actin Network Rheology to Probe Single Filament Elasticity and Dynamics. Phys. Rev. Lett. 2004, 93 (18), 188102. 10.1103/PhysRevLett.93.188102. [DOI] [PubMed] [Google Scholar]
- Broedersz C. P.; Kasza K. E.; Jawerth L. M.; Münster S.; Weitz D. A.; MacKintosh F. C. Measurement of Nonlinear Rheology of Cross-Linked Biopolymer Gels. Soft Matter 2010, 6 (17), 4120–4127. 10.1039/c0sm00285b. [DOI] [Google Scholar]
- Storm C.; Pastore J. J.; MacKintosh F. C.; Lubensky T. C.; Janmey P. A. Nonlinear Elasticity in Biological Gels. Nature 2005, 435 (7039), 191–194. 10.1038/nature03521. [DOI] [PubMed] [Google Scholar]
- Koevoets R. A.; Versteegen R. M.; Kooijman H.; Spek A. L.; Sijbesma R. P.; Meijer E. W. Molecular Recognition in a Thermoplastic Elastomer. J. Am. Chem. Soc. 2005, 127 (9), 2999–3003. 10.1021/ja0451160. [DOI] [PubMed] [Google Scholar]
- Bauër P.; Tavacoli J.; Pujol T.; Planade J.; Heuvingh J.; du Roure O. A New Method to Measure Mechanics and Dynamic Assembly of Branched Actin Networks. Sci. Rep. 2017, 7 (1), 15688. 10.1038/s41598-017-15638-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onck P. R.; Koeman T.; van Dillen T.; van der Giessen E. Alternative Explanation of Stiffening in Cross-Linked Semiflexible Networks. Phys. Rev. Lett. 2005, 95 (17), 178102. 10.1103/PhysRevLett.95.178102. [DOI] [PubMed] [Google Scholar]
- Seitz M. E.; Martina D.; Baumberger T.; Krishnan V. R.; Hui C.-Y.; Shull K. R. Fracture and Large Strain Behavior of Self-Assembled Triblock Copolymer Gels. Soft Matter 2009, 5 (2), 447–456. 10.1039/B810041A. [DOI] [Google Scholar]
- Erk K. A.; Henderson K. J.; Shull K. R. Strain Stiffening in Synthetic and Biopolymer Networks. Biomacromolecules 2010, 11 (5), 1358–1363. 10.1021/bm100136y. [DOI] [PubMed] [Google Scholar]
- Chuong C. J.; Fung Y. C. On Residual Stresses in Arteries. J. Biomech. Eng. 1986, 108 (2), 189–192. 10.1115/1.3138600. [DOI] [PubMed] [Google Scholar]
- Holzapfel G. A.; Gasser T. C.; Ogden R. W. Comparison of a Multi-Layer Structural Model for Arterial Walls With a Fung-Type Model, and Issues of Material Stability. J. Biomech. Eng. 2004, 126 (2), 264–275. 10.1115/1.1695572. [DOI] [PubMed] [Google Scholar]
- Dommerholt J.; Rutjes F. P. J. T.; van Delft F. L. Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. Top. Curr. Chem. 2016, 374 (2), 16. 10.1007/s41061-016-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.