

Abstract
Bone, nerve, and heart tissue engineering place high demands on the conductivity of three-dimensional (3D) scaffolds. Fibrous carbon-based scaffolds are excellent material candidates to fulfill these requirements. Here, we show that highly porous (up to 94%) hybrid 3D framework structures with hierarchical architecture, consisting of microfiber composites of self-entangled carbon nanotubes (CNTs) and bioactive nanoparticles are highly suitable for growing cells. The hybrid 3D structures are fabricated by infiltrating a combination of CNTs and bioactive materials into a porous (∼94%) zinc oxide (ZnO) sacrificial template, followed by the removal of the ZnO backbone via a H2 thermal reduction process. Simultaneously, the bioactive nanoparticles are sintered. In this way, conductive and mechanically stable 3D composites of free-standing CNT-based microfibers and bioactive nanoparticles are formed. The adopted strategy demonstrates great potential for implementing low-dimensional bioactive materials, such as hydroxyapatite (HA) and bioactive glass nanoparticles (BGN), into 3D carbon-based microfibrous networks. It is demonstrated that the incorporation of HA nanoparticles and BGN promotes the biomineralization ability and the protein adsorption capacity of the scaffolds significantly, as well as fibroblast and osteoblast adhesion. These results demonstrate that the developed carbon-based bioactive scaffolds are promising materials for bone tissue engineering and related applications.
Keywords: carbon nanotubes, 3D scaffolds, bioactive glass, hydroxyapatite, osteoblasts
1. Introduction
Bioactive materials are highly important for inducing specific cellular responses, particularly in tissue engineering.1 A highly promising bioactive material in bone tissue engineering is silicate bioactive glass (BG).2 In particular, the presence of CaO in silicate BG is decisive in the formation of a bone-like calcium phosphate layer on the material.3 This surface layer results in a strong connection between the biomaterial and both bone and soft tissue.4 To expand the interface between BG and tissue, BG can be prepared as scaffolds or incorporated into scaffolds. Depending on the microstructure and fabrication method of the BG, the Young’s modulus of porous scaffolds made of BG can be varied between 1 and 22 MPa.3 In addition, BG can be degraded by releasing ions into the environment so that a main component of bone, hydroxyapatite (HA), is formed.3,5 BG is also known to support the adhesion and proliferation of osteoblast cells, such as MC3T3-E16−9 and MLO-A5.1,10 In addition, many studies have shown that the ions released from BG, including Si, Ca, P, and Cu, play an important role in enhancing osteoblast proliferation and bone formation.4 Another important bioactive material is hydroxyapatite, as it is a main component of bone within the human body. It has frequently been used as a coating to promote bone ingrowth or as a bone graft substitute.11
BG is often implemented in biomaterials in the form of composites. Out of such composite systems, BG and carbon nanotubes (CNTs) have shown to be most promising due to a mutually beneficial modification of the features and functionality of each other.12 On the one hand, CNTs enhance the features of BG such as stiffness, electrical conductivity, and surface roughness in several ways: (i) the mechanical properties, such as stiffness and fracture toughness, are improved by the CNTs,13−15 (ii) the local and overall electrical conductivity is increased by CNTs,16,17 (iii) CNTs lead to a rougher surface on the nanoscale and consequently to an improvement of cell attachment and proliferation.18 On the other hand, BG makes CNTs more appropriate for biomedical applications by accelerating biomineralization19 and increasing the concentration of physiologically relevant ions, thus resulting in higher cell proliferation rates and faster regeneration of soft tissue.20
To exploit the unique properties of composites made of CNTs and bioactive materials (such as BG and HA) for tissue engineering, three-dimensional (3D) scaffold fabrication methods are needed that provide a mechanically stable 3D environment that supports cell proliferation.21 In such 3D environments, the pore architecture is one of the most decisive parameters as it affects cell growth by determining nutrient and oxygen diffusion, waste removal, as well as the growth rate of the cells. Open porous and interconnected pore architectures are strongly necessary to facilitate these parameters.22 Commonly used techniques to generate pores in materials, such as salt-leaching, freeze-drying, gas foaming, sol–gel crosslinking, and phase separation only provide very limited control of the internal pore architecture of the scaffold material for tissue engineering.23 Therefore, rapid prototyping techniques have been employed recently, which enable the fabrication of scaffolds with control over pore size and architecture together with good design reproducibility.24 Nevertheless, rapid prototyping techniques are limited with respect to the biomaterials that can be printed and they cannot be used to fabricate 3D structures from nanomaterials only.25
Recently, an infiltration-based synthesis method for the fabrication of porous, 3D scaffolds consisting of interconnected CNT-based microtubes has been reported.26 With this method, open porous scaffolds with total dimensions on the cm3 scale can be fabricated, resulting in a 3D hierarchical CNT tube (CNTT) structure with improved mechanical and electrical properties compared to other 3D assemblies prepared by wet chemistry. As the reported fabrication method relies on the infiltration of nanoparticle dispersions into sacrificial ZnO templates, in principle, also other nanoparticle types besides CNTs can be embedded. Therefore, this method is a great and versatile starting point for fabricating hybrid 3D scaffold materials with properties that can be tailored towards specific, electronic, catalytic, or biomedical applications, e.g., by the incorporation of bioactive nanoparticles.
We show here that low-dimensional bioactive materials in the form of bioactive glass and hydroxyapatite nanoparticles can be integrated into CNTT materials, to make it bioactive and supportive for cell growth. The bioactive nanoparticles incorporated into the scaffolds are sintered during a single step etching/sintering process. The structure of the resulting material was characterized with scanning electron microscopy (SEM), transmission electron microscopy (TEM), and X-ray diffraction (XRD). To prove the bioactivity of the scaffolds, we investigated biomineralization as well as their protein adsorption capacity. In vitro studies demonstrate that fibroblasts and osteoblasts can adhere to the fibrous structures of the hybrid CNT network, demonstrating the feasibility of this 3D composite material for bone tissue engineering.
2. Results and Discussion
2.1. Bioactive Carbon-Based Scaffolds
The successful fabrication of free-standing, fibrous 3D composite scaffolds, consisting of CNTs and a nanoscale bioactive ceramic biomaterial has been achieved via a very simple strategy based on a ceramic template. Figure 1A shows a schematic representation of the fabrication process, in which compressed and sintered ZnO tetrapods (Figure 1B) serve as sacrificial materials. To generate free-standing carbon-based and bioactive composite structures these 3D and porous (porosity ∼94%) ZnO templates were infiltrated with an aqueous dispersion of CNTs and either BGN, HA nanoparticles or both of them. Due to the strong capillary forces and the superhydrophilic properties of the ZnO template,27 the dispersion is rapidly soaked into the template during the infiltration process. Figure 1C shows an SEM image of a ZnO template after infiltration with CNTs and BGN. Figure 1E,F show SEM images of the templates after infiltration with CNTs and HA nanoparticles. In the experiments, it is in principle also possible to tailor the amount of CNTs as well as the amount of BGN and HA nanoparticles added on the template by controlling the volume of the injected nanoparticle dispersion into the ZnO network.
Figure 1.

(A) Sketch of the fabrication process: the porous ZnO templates are infiltrated with HA nanoparticles or BGN and CNT dispersions. Due to the strong capillary forces and superhydrophilic properties of ZnO, the micro-tetrapods are entirely covered with a layer of CNTs and BGN. Later, the sacrificial ZnO template is removed via H2 etching at 900 °C. The remaining structure consists of self-entangled CNTs as a backbone and sintered BGN as a bioactive material. (B–J) Scanning electron micrographs: (B) the 3D ZnO sacrificial template consisting of micron-sized ZnO tetrapods, (C) the ZnO–CNTT–BGN (entangled CNTs + BGN) structure before H2 etching/sintering process, (D) the CNTT–BGN (entangled CNTs + BGN) structure after H2 etching/sintering, (E, F) the CNTT–HA (entangled CNTs + HA) structure before H2 etching/sintering, (G, H) the CNTT–HA structure (entangled CNTs + HA) after the H2 etching/sintering. (H) The broken filament of the CNTT–HA scaffold showing that the structures are made of hollow tubes, (I, J) the CNTT–BGN/HA (entangled CNTs + BGN + HA) structure after H2 etching/sintering (BGN: bioactive glass nanoparticles, CNT: carbon nanotube, HA: hydroxyapatite).
To generate free-standing hybrid structures, we needed to remove the ZnO. This is also necessary to make the materials biocompatible, as a high amount of ZnO is toxic for cells.28 Most methods to remove ZnO are based on wet chemistry, e.g., using diluted HCl. However, since an acidic treatment would also dissolve the incorporated bioactive materials, a H2 etching process was used to remove the ZnO.29 At 900 °C, ZnO is completely reduced by H2, evaporated to Zn (gas) without the presence of a carbon source, and removed by the employed carrier gas (argon). In Figure 1D–J, SEM images of the resulting structures are shown. Figure 1D shows CNT and BGN scaffolds after etching of the ZnO template. Figure 1H,1J show that broken microtubes are hollow, proving that the entire ZnO template has been removed during the etching process. The free-standing composites are denoted as CNTT–BGN (CNT-based tubes containing BGN), CNTT–HA (CNT-based tubes containing HA nanoparticles), and CNTT–BGN/HA (CNT-based tubes containing both BGN and HA nanoparticles). In general, the process described here can be easily scaled-up as it is based on commercially available materials that can all be mass-produced.
Initially, the CNTs in our scaffolds form interwoven layers of self-entangled CNTs with high mechanical stability similar to the ones shown in previous work.26 Most importantly, the high temperature during the ZnO template removal fuses the ceramic nanoparticles to each other and leads to a physical interaction with the CNTs, resulting in a hollow free-standing entangled CNT architecture combined with sintered ceramic nanoparticles (Figure 2). These ceramic nanoparticles are believed to contribute to a further reinforcement of the mechanical properties of the scaffold (Table 1). Moreover, the geometry of the networks provides highly accessible interconnected pores from all sides, whereas the preferential alignment of CNTs in other 3D assemblies confines the accessibility of cavities.30
Figure 2.

TEM images of CNTT–BGN, CNTT–BGN/HA, and CNTT–HA scaffolds. (A, B) The CNTT–HA structure (entangled CNTs + HA), (C, D) CNTT–BGN structure (entangled CNTs + BGN), and (E, F) CNTT–BGN/HA structure (entangled CNTs + BGN and HA). Yellow and red arrows point at fused particles. BGN are partially fused to the particles in their vicinity (red arrows) and probably to the CNT matrix, whereas HA nanoparticles underwent a greater sintering deformation (yellow arrows). HA particles bridge the gaps between BG nanoparticles in the combined structure (CNTT–BGN/HA) during the etching/sintering process. HA nanoparticles and BGN in CNTT–BGN/HA scaffolds were distinguished by their shape in the CNTT–BGN and CNTT–HA scaffolds.
Table 1. Conductivity and Compressive Strength of CNTT–HA and CNTT–BGN Scaffoldsa.
| scaffold | specific conductivity (S/m) | compressive strength (kPa) | compressive strength/density (kPa cm3/g) | Young’s modulus (MPa) |
|---|---|---|---|---|
| CNTT–HA | ∼0.88 | ∼7.2 | ∼27 | ∼0.4 |
| CNTT–BGN | ∼12.7 | ∼32 | ∼118 | ∼1 |
The large discrepancy in conductivity between CNTT–HA and CNTT–BGN is most probably a result of clamping difficulties, as the surface of the material is covered with HA and BGN, respectively. Still, the conductivity is in the range of that of CNTTs without incorporated ceramic nanoparticles.26
In addition, the porous structure of our scaffolds leads to a large free volume, which should in turn result in higher bioactivity.31 The scaffolds possess up to 94% porosity (Table S1), thus being in the range of that of other highly porous BG-containing scaffolds.31,32 The porosity of the scaffolds presented is higher than that of 3D printed biomaterials that contain HA nanoparticles, including HA nanoparticle incorporated poly(lactide-co-glycolide) with up to 63.33% of porosity.33,34 Furthermore, due to the interwoven arrangement of CNTs in the microtubes, we assume that the scaffolds possess a similar conductivity (Table 1) as the recently reported self-entangled CNT assemblies,26 but with the additional feature that the incorporated ceramic nanoparticles make the material bioactive.
It is important to note that our scaffolds are not intended to replace bone in load-bearing areas, as their compressive strength (Table 1) is smaller than that of trabecular bone.35 Instead, the idea of tissue engineering in this context is to generate scaffolds, which induce the formation of new bone tissue based on the bioactivity of the scaffold.5,11 If the scaffold exhibits bioactivity, the mechanical properties will strengthen upon implantation so that a new bone is formed.36 The suitability of soft materials for bone tissue engineering has recently been shown by Huebsch et al.,37 who used soft hydrogels to induce bone formation. As our scaffolds are bioactive (see Section 2.5) and support osteoblast growth (see Section 2.6), we are convinced that they are suitable for bone tissue engineering.
2.2. Morphology and Composition of Ceramic Nanoparticles on CNTT Scaffolds
Figure 2 shows TEM images of CNTT–BGN, CNTT–HA, and CNTT–BGN/HA. The most striking result from these images is that all generated structures are free-standing and the ZnO template has been completely removed. Furthermore, the images show that the ceramic nanoparticles were fused to each other in the etching/sintering procedure. In addition, HA and BGN were fused to each other in CNTT–BGN/HA composites. Interestingly, BGN mainly retain their spherical shape and are only slightly fused together. In contrast, the initially also spherical HA nanoparticles were deformed to flakes or unevenly shaped particles during the etching/sintering procedure.
This allowed us to distinguish the particle types even in CNTT–BGN/HA scaffolds (Figure 2E,F). Furthermore, these images suggest that the HA particles bridged the gaps between the BGN in CNTT–BGN/HA samples (Figure 2E,F, yellow arrows). This difference in the sintering behavior is probably due to the lower effective sintering point of HA nanoparticles (700 °C) compared to silicate-based BGN (950–1000 °C),38,39 thus the deformation and fusion of HA particles is more pronounced. A further interesting result is that the CNTs in the structures do not seem to be altered by the sintering procedure, even in the presence of hydrogen.
To check the crystallinity of the ceramic nanoparticles after the fabrication process, we carried out an XRD study. The XRD pattern (Figure 3) of a CNTT–HA sample revealed diffraction peaks of HA. In contrast, the XRD pattern of a CNTT–BGN sample revealed no diffraction peaks related to BGN, but diffraction peaks related to α-quartz are recognized. Pristine BGN are amorphous and only possess broad peaks in the range of 2θ = 20–35°.40 Since the incorporated BGN had the composition 92SiO2–8CaO (in mol %),40 their crystallization behavior is similar to that of pure silica nanoparticles. However, in this study the processing temperature (900 °C) used for the fabrication of the scaffolds was high enough to cause the crystallization of silica-based nanoparticles,41 which explains the appearance of the diffraction peaks of α-quartz in CNTT–BGN. For the CNTT–BGN/HA structure, the diffraction pattern only correlates to that of crystallized HA. In addition, SiO2–CaO-based BGN sintered at 900 °C should be partially crystallized according to previous work.41 Importantly, Figure 3 also shows the effect of different amounts of infiltrated CNTs: the intensity of the ceramic peaks decreased if more CNTs had been infiltrated.
Figure 3.
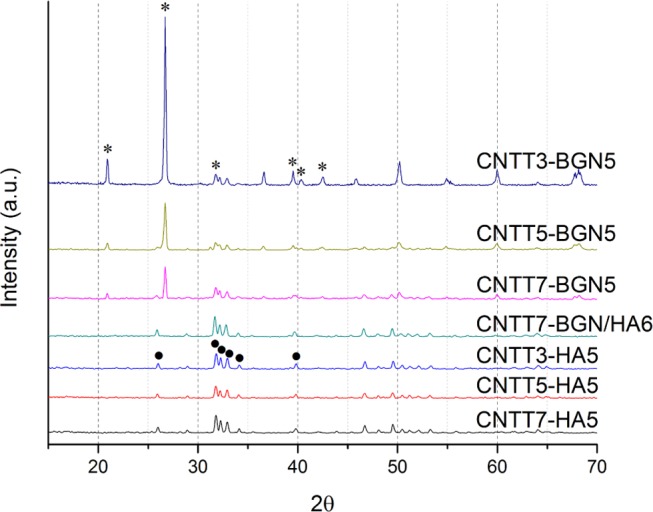
XRD patterns of CNTT–BGN, CNTT–HA, and CNT–BGN/HA scaffolds. CNTTy–BGNx: y and x correspond to the number of infiltrations of CNTs and ceramic (HA or BG) nanoparticles, respectively (as an example CNTT3–BGN5 means a ZnO template was infiltrated five times with BG nanoparticles and three times with CNT dispersion). Due to the smaller diffraction of X-rays by CNTs, the intensity of ceramic peaks is more pronounced in structures with a lower amount of infiltrated CNTs. The XRD patterns of the CNTT–BGN structures are in conformity with α-quartz peaks, and CNTT–HA structures mostly with hydroxyapatite (•hydroxyapatite, *α-quartz). Interestingly, the combined structure (CNTT–BGN/HA) only reveals the hydroxyapatite XRD pattern.
The XRD results of the HA-containing scaffolds show no change of the crystalline phase of HA. As the first phase transformation of HA occurs at 1000–1100 °C.42 we assume that our HA particles are not decomposed at our sintering temperature (900 °C). Furthermore, in a previous study, no reaction of multiwalled CNTs (MWCNTs) with glass matrices to form SiC or other reaction phases in response to sintering between 850 and 1000 °C was detected by powder X-ray diffraction.43
2.3. Protein Adsorption on Scaffolds
The adsorption of proteins on bioceramics is essential because it influences cell adhesion and can facilitate scaffold integration into tissues.44 To investigate the protein adsorption capacity of CNTT–BGN and CNTT–HA scaffolds, we used bovine serum albumin (BSA) as a model protein. The adsorption capacity of the scaffolds was quantified for 4, 8, 12, 24, 48, and 72 h of scaffold incubation with protein solution. The bicinchoninic acid (BCA) assay (Figure 4) shows that the protein adsorption is higher on CNTT–BGN scaffolds than on CNTT–HA scaffolds.
Figure 4.
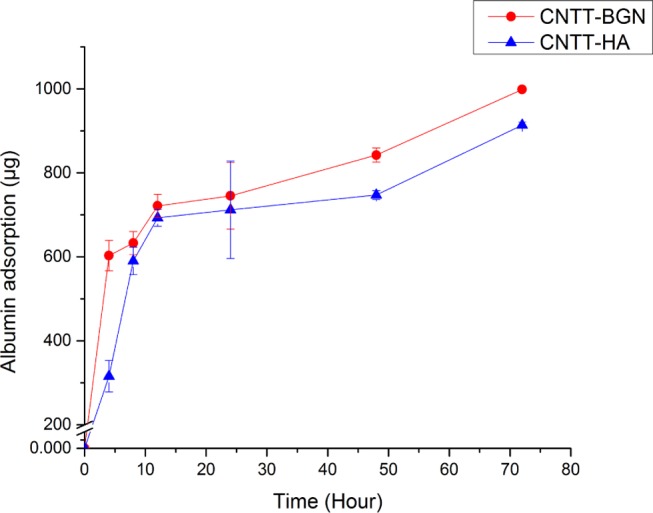
Bovine serum albumin adsorption (mean values) on CNTT–BGN and CNTT–HA scaffolds, measured with a BCA colorimetric assay. BGN containing structures have a slightly higher protein adsorption capacity compared to CNTT–HA scaffolds. (Each experiment was carried out on three samples and three replicates each. Error bars: standard deviation.)
This difference in adsorbing proteins is highest during the first 4 h of incubation with proteins and levels out after 8 h of incubation. Despite the fact that there was a slight difference regarding the protein adsorption ratio between the two scaffold types, both exhibited a similar temporal progression of protein adsorption. This can be explained by the fact that CNTs presumably play the main role in protein adsorption due to the high amount of CNTs in the matrix in both the CNTT–HA and CNTT–BGN scaffolds (Figure 1). It is also important to mention here that the protein adsorption capacity of scaffolds is a decisive parameter for osteoblast attachment.45 As the CNT matrix can serve as an attachment site for a variety of extracellular matrix molecules, biomolecules, proteins, and growth factors, it can further mediate cell proliferation and adhesion.46 Interestingly, in the present study CNTT–BGN has a higher adsorption capacity than CNTT–HA (Figure 4).
This result could be due to an electrostatic interaction between the highly polar BSA and the BGN surface,44 which might be a result of the etching/sintering process, as explained in the following: at the sintering temperature of 900 °C, H2 reacts with silica to form SiOx on the surface of BGN.47 The presence of SiOx on the surface of BGN can alter the surface charge density of BGN.48 Therefore, due to a change in surface charge, the BGN surface might have the potential to bind more BSA proteins. In addition, previous studies indicated that surface-modified bioactive glass adsorbs a higher amount of serum protein than hydroxyapatite.49
2.4. Ion Release from Scaffolds in Phosphate Buffered Saline (PBS)
To explore the ion release capability of the fabricated hybrid scaffolds within biologically relevant media, we measured the concentration of Ca, Si, and Zn ions in phosphate buffered saline (PBS) after 4, 8, 12, 24, 96, 158, 230, 302, and 398 h of incubation with the scaffolds using inductively coupled plasma-mass spectrometry (ICP-MS) (Figure 5). Clearly, the amount of ions released from the scaffolds increased with incubation time. The release of Zn ions is almost zero (∼5 μg after 400 h), even after 16 days of immersing CNTT–BGN or CNTT–HA scaffolds in PBS. This proves again that the sacrificial ZnO templates had been entirely removed during the H2 etching process. Hence, the hydrogen etching process at elevated temperatures provides a clear improvement compared to previously reported methods used for ZnO removal.50 As shown in Figure 5, a high amount of Ca2+ ions was released into the PBS from CNTT–HA and CNTT–BGN structures during the first 24 h of immersion (Figure 5). Afterwards, the release rate of Ca2+ ions was decreased. The high accessibility of Ca2+ ions on the surface of BG and HA nanoparticles leads to a high Ca2+ ion concentration gradient between PBS and the ceramic nanoparticles, presumably resulting in a high initial release rate and a smaller and more stable release rate for longer immersion times. As expected, we did not measure any Si ion release from the CNTT–HA scaffolds (Figure 5A). In contrast, CNTT–BGN scaffolds released Si ions (Figure 5B) and the release rate has a similar temporal progression as the one reported on pure BGN by Zheng et al.,40 though with a slightly lower release rate than our scaffolds. Zheng at al. carried out their experiments in Dulbecco’s modified Eagle’s medium (DMEM), whereas we used PBS. The ions in DMEM could cause the formation of an amorphous Ca-P layer on the surface of BGN,40 thus leading to a decreased release rate.
Figure 5.

Mean values of ion release from CNTT–HA (A) and CNTT–BGN (B), measured by ICP-mass spectrometry (mean values). The concentration of released Zn, Ca, and Si ions from immersed CNTT–HA and CNTT–BGN structures in phosphate buffer saline represents the degradation rate of the bioactive ceramics. The concentration of Zn ions was measured to quantify the amount of residual ZnO from the fabrication process. (Each experiment was carried out using three samples. Error bars: standard deviation.)
2.5. In Vitro Bioactivity
A highly important indicator for the ability of scaffolds to integrate with bone is the formation of hydroxyapatite on the scaffolds. On our samples, needle-shaped crystals are formed on CNTT–BGN scaffolds after immersion in a simulated body fluid (SBF) for 7 days (Figure 6A), whereas such crystals are neither observed on CNTT–HA nor on CNTT–BGN/HA scaffolds after immersion in the SBF. (Figure 6B,C). To explain this, we checked the results from energy dispersive X-ray spectroscopy (EDS). Our EDS results (Supporting Information, Figure S1) show that phosphorus is present on the surface of CNTT–BGN filaments after immersion in the SBF for 7 days, which indicates the formation of calcium phosphate species.
Figure 6.

SEM images of the scaffolds after immersion in the SBF. (A) CNTT–BGN in SBF for 7 days; (B) CNTT–HA in SBF for 7 days; (C) CNTT–BGN/HA in SBF for 7 days; (D) CNTT–BGN in SBF for 14 days. SEM images plus EDS results (Supporting Information, Figure S1) confirm the formation of biomineralized hydroxyapatite.
Additionally, silicon is still present according to the EDS spectrum of CNTT–BGN indicating that BGN have not completely dissolved yet after 7 days of immersion in SBF. After immersion in SBF for 14 days, cauliflower-like crystals are present on CNTT–BGN scaffolds (Figure 6D). This is a typical morphology of hydroxyapatite crystals formed on BG scaffolds after immersion in SBF.51 In addition, spherical BGN are still present on these scaffolds, showing that the time until full dissolution of the BGN is longer than 14 days. In general, BG is more surface reactive and has a higher dissolvability than other bioceramics (e.g., HA).52 Therefore, the formation of bone-like apatite crystals, which is a result of surface dissolution and ion exchange, occurs faster on BG surfaces than on HA surfaces in body fluids. In the study presented here, the apatite forming ability of the scaffolds was improved after coating of BGN in comparison with HA nanoparticles, considering that the apatite crystals were observed on CNTT–BGN scaffolds after 7 days of soaking in SBF, whereas no crystals were present on CNTT–HA. As shown in Figure 6C, the situation for CNTT–BGN/HA is similar as for CNTT–HA. Inhibited apatite formation occurs when BGN are mixed with non-reactive or less reactive materials.53 Our composite coating is like a glass-ceramic phase with less reactive surfaces, thus reducing the dissolution and ion exchange. The presence of HA in the coating also reduces the number of sites for apatite nucleation. Hence, apatite formation was inhibited in CNTT–BGN/HA scaffolds compared to CNTT–BGN. Interestingly, the formation of apatite crystals on our CNTT–BGN composite scaffolds took longer than when using pure BGN in SBF, where crystals had formed after 3 days of immersion.40 This result can be explained by the etching/sintering process that is necessary to remove the ZnO templates and caused the partial crystallization of BGN. The XRD result (Figure 3) shows the formation of α-quartz phase in BGN. Such a partial crystallization should not eliminate the biodegradability and bioactivity of BGN.54 BGN maintain their bioactivity and biodegradability, though these properties could be inhibited.55 Nevertheless, the BGN coating promotes the bioactivity and can therefore also be expected to improve the osteogenesis and osseointegration of our scaffolds.
2.6. Osteoblast and Fibroblast Growth on the Composite Scaffolds
The proliferation and adhesion of rat embryonic fibroblast and pre-osteoblastic cells (MC3T3-E1) on CNTT–HA, CNTT–BGN, and CNTT–BGN/HA were assessed individually using fluorescence microscopy. As the CNTT scaffolds absorb light almost completely,56 optical imaging was restricted to approximately the first 200 μm from the surface. A long-term (21 days) in vitro study of MC3T3-E1 cells with our fabricated scaffolds revealed a surprisingly low number of dead cells in comparison to live cells (Figure 7) and that the scaffolds support osteoblast growth and adhesion. In addition, pre-osteoblasts appear to be well supported by all the scaffolds as well.
Figure 7.

Fluorescence images of pre-osteoblast cells (MC3T3-e1) cultured for 21 days on CNTT–HA, CNTT–BGN, and CNTT–BGN/HA structures. The nucleus of the cells was stained with 4,6-diamidino-2-phenylindole (DAPI) (Hoechst), live cells with calcein AM (green), and dead cells with propidium iodide (red). All the scaffolds support osteoblast adhesion and proliferation. There is only a small number of dead cells after 21 days of cultivation (scale bars: 0.1 mm).
To investigate cell adhesion further, we also checked the adhesion of REF52 cells stably transfected with YFP-paxillin.57 Paxillin is an essential part of the fibroblast adhesion machinery.58 After 4 days of cultivation, fibroblast cells had well-adhered on all scaffold types (CNTT–BGN, CNTT–HA, CNTT–BGN/HA) (Figure 8). Imaging the paxillin fluorescence from adhesion clusters shows tiny and blurry spots (Figure 8A3–F3). Actin fibers are arranged in a meshed-form fibroblast cytoskeleton rather than stress fibers on all of our scaffolds (Figure 8A1–F1). Such a mesh-like arrangement of actin is typical for fibroblast adhesion in three dimensions.50,59−61
Figure 8.

Fluorescence images of rat embryonic fibroblasts cultivated for 4 days on CNTT–HA (row A, B), CNTT–BGN (row C, D), and CNT–BGN/HA (row E, F) scaffolds. YFP-paxillin transfected REF cells (focal adhesion sites; yellow) were stained with DAPI (nuclei; blue) and phalloidin (actin filament; red). Fluorescence imaging took place in optical sections of 200 μm from the surface inside the material. Small paxillin clusters are visible in each optical focal plane. Imaging of actin reveals spanned cells with actin meshes. To show more details, fluorescence images of two different regions of each scaffold are shown in two rows (scale bars: 10 μm).
To investigate cell morphology in our 3D scaffold materials, we studied cell shape and morphology via SEM (Figure 9). SEM micrographs reveal that the cells were stretched and spanned between filaments on all scaffolds. Interestingly, most of the fibroblast cells are stretched between two or three filaments, leading to large cell membrane extensions. A comparison of the SEM images with fluorescence images shows that the cells within the scaffolds are associated with scaffold microtubes and developed physical contact with scaffold filaments (Figure 9, right column), especially with the CNT matrix. In addition, the actin fiber meshes are elongated between microtubes of the scaffolds (Figure 8). This is in agreement with SEM images (Figure 9) of fibroblasts on all scaffold types, as membrane projections attach to the microtubes of the scaffold. Despite the ability of the fibroblasts to adhere to the scaffolds, an methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay (Figure 10) revealed a 5–20% lower growth rate for fibroblasts compared to the negative control after 1 day of incubation. Fibroblasts and fibroblast growth factors have an ambivalent impact on bone growth and osteogenesis. On the one hand, fibroblast growth factors increase the proliferation of immature osteoblasts,62 whereas fibroblast growth factors cause apoptosis in differentiated osteoblasts and block mineralization,62−64 and fibroblasts inhibit biomineralized bone nodule formation.65 As fibroblasts adhere very well to our materials (Figures 8 and 9), we assume that our material is feasible for tissue engineering applications.
Figure 9.

SEM micrographs of rat embryonic fibroblasts (colored in green) cultivated for 4 days on CNTT–BGN, CNTT–HA, and CNTT–BGN/HA scaffolds. Onto all types of structures cell membranes are extended and many adhesion sites are developed. The cells are mainly isolated and stretched. In the top right image, the fibroblast cell adhered preferentially to CNTs in the presence of CNT and BGN. Nevertheless, they adhered to HA even in those areas where CNTs are locally covered with HA nanoparticles (image to the right, middle row).
Figure 10.

Cell viability of rat embryonic fibroblasts (REF52wt) treated with extractions of CNTT–BGN, CNTT–HA, and CNTT–BGN/HA, measured via an MTT–formazan absorbance assay. Fresh and untreated culture medium served as control. The result of this MTT assay reveals a small decrease in the fibroblast proliferation rate on CNTT–BGN and CNTT–HA, which is more pronounced for the combined structure containing both HA and BGN (CNTT–BGN/HA). (Each experiment was carried out in five technical repeats with three extractions for each sample. Error bars: standard deviation.)
3. Conclusions
In summary, we have demonstrated a simple and efficient strategy to fabricate highly porous composite scaffolds made of self-entangled CNTs in microtube structures with incorporated bioactive ceramic nanoparticles (BGN and HA). These nanoparticles have been chosen to promote bone tissue ingrowth. To prepare the scaffolds, we have employed a H2 thermal reduction process to etch ZnO and sinter BG or HA nanoparticles in a single step. The preferential removal of the ZnO template by the H2 etching/sintering process offers the opportunity of implementing a variety of ceramic nanoparticles into the highly porous 3D networks for the fabrication of diverse hybrid composite structures. Moreover, we have proven that the incorporation of BGN and HA nanoparticles leads to biomineralization of the scaffolds. Apart from its bioactivity, the great advantage of our scaffolds compared to other 3D carbon-based porous structures is that its stiffness and porosity can in principle be tuned. The hollow microtubes lead to a low density of the scaffolds, and thus to an increase in ion release and electrical conductivity per scaffold weight. In addition, the hollow microtubes can serve as channels for nutrient transport and in future studies might also be filled with drugs or growth factors. The spatial architecture of fibers in the fabricated structures provides large free space for cell adhesion and both osteoblasts and fibroblasts were capable of adhesion to the scaffolds and stretched out along the fibers. The bioactivity of the scaffolds together with the electrical conductivity of the CNTT backbone makes them promising candidates for applications where porous 3D architectures are essential for cell growth, stimulation, function, or biomineralization, e.g., in bone tissue engineering.
4. Materials and Methods
4.1. Fabrication of Bioactive Scaffolds
The fabrication of the bioactive cell scaffold materials is based on infiltrating the bioactive material into a highly porous (∼94%) ceramic sacrificial template. For fabricating this template, tetrapodal-shaped ZnO microparticles (t-ZnO) were prepared using a flame transport approach as described by Mishra et al.66 To interconnect the microparticles, the loose powder was pressed into a cylindrical cast (diameter: 12 mm; height: 2 mm) and sintered at around 1150 °C for 5 h to form a 3D template.27
BGN with a nominal composition of 70SiO2–30CaO (in mol %) and a particle diameter of ∼400 nm were synthesized by a sol–gel method as described by Zheng et al.40 Hydroxyapatite (HA) nanoparticles were bought from Sigma (nanopowder, <200 nm). CNTs were purchased from Carbobyk (CARBOBYK-9810). The zinc oxide (ZnO) templates were coated with such nanoparticles using an infiltration process recently described by Schütt et al.26 The ceramic nanoparticles (BGN and HA) with a concentration of 214.28 ± 11.9 mg/cm3 were dispersed in absolute ethanol (Sigma, Germany). Due to the high porosity (∼94%) of the template and its superhydrophilic properties, nanoparticle dispersions are rapidly sucked into the template and a homogenous coverage is achieved after evaporation of the solvent. This process can be repeated several times to increase the amount of infiltrated nanoparticles. It is also possible to infiltrate different nanoparticle types in sequence so that a composite layer is formed around the template structure. Here, the templates were alternately infiltrated with a 1 wt % aqueous carbon nanotube (CNT) dispersion and a 4 wt % ceramic nanomaterial (HA or BGN) dispersion (in ethanol). The concentration of the ceramic nanoparticles was the same both for HA and BGN 214.28 ± 11.9 mg/cm3, whereas different CNT concentrations between 32.14 ± 1.78 and 75.00 ± 4.16 mg/cm3 were tested. After finishing the infiltration procedure, the ZnO template was removed by H2 etching. To do so, the samples were placed in a sealed quartz tube furnace and the air was replaced by pure argon. The pressure was adjusted to 200 mbar. Subsequently, the temperature was increased to 900 °C. Then, an evaporator (170 °C) was used to decompose urea into ammonia (NH3).67 At 900 °C ammonia decomposes to N and H2, thereby etching the ZnO template, resulting in free-standing, fibrous composite structures of CNTs with bioactive nanoparticles. Depending on the content of the composite scaffolds, they are called CNTT–BGN (free-standing CNT networks with BGN), CNTT–HA (free-standing CNT networks with hydroxyapatite nanoparticles), CNTT–BGN/HA (free-standing CNT networks with both BG and hydroxyapatite nanoparticles).
4.2. Characterization
The porosity of the scaffolds is obtained as follows: a defined amount of tetrapodal ZnO powder (here: 0.068 g) is pressed into a cylindrical shape (12 mm diameter; 6 mm height). The resulting volume is ∼0.226 cm3 and leads to a density of around 0.3 g/cm3. Since the bulk density of ZnO is 5.61 g/cm3, the porosity is ∼94% for the ZnO template. For the preparation of the bioactive scaffolds, a CNT dispersion of 1 wt % and a BGN or HA dispersion of 4 wt % were used and infiltrated into the ZnO templates. The amount of infiltrated dispersion was adjusted to the amount, which was needed to completely fill the free volume of the template (∼230 μL).
The porosities of the final scaffold materials were calculated as follows: by adding 230 μL of a 1 wt % CNT dispersion, around 2.3 mg of CNTs are added to the network. Considering the density for CNTs to be 1.4 g/cm3 (which is already quite a high value for MWCNTs) the additional volume is around 0.0016 cm3 for each infiltration. By the addition of 230 μL HA or BGN dispersion 9.2 mg are added. Considering the densities of BG and HA to be in the range of 2–6 g/cm3, we obtain an additional volume of 0.0015–0.0046 cm3. Therefore, in the case of a template that was infiltrated five times with a 1 wt % CNT dispersion and five times with a 4 wt % HA or BGN dispersion, we obtain a porosity between 86 and 93% (depending on the density of HA, as given by the manufacturer). More information is shown in the Supporting Information (Table S1).
Electrical characterization was performed using a Keithley 6400 source-meter, which is controlled by a self-written LabView program, capable of measuring IV curves. Therefore, a sample holder was used in which the flat sides of the cylindrical samples (d = 6 mm, h = 3 mm) were connected to copper plates. To ensure a good contact between the copper plates and the sample surface, conductive silver paste was used at the interface. To avoid any electrical contact between the sample holder and the copper plates, Kapton tape was used as an insulator (Figure S2). The current was measured as a function of applied voltage (from −1 to +1 V using a step size of 0.1 V).
Mechanical characterization was performed with a self-built setup consisting of a Märzhäuser Wetzlar HS 6.3 micromanipulator, which is driven by a stepper motor and a load cell (Burster präzisionsmesstechnik GmbH & Co KG, type 8523-5050). A self-written LabView program is used to control all components and measure the force. To avoid any vibration damping, the whole setup is located on a very rigid aluminum plate in a box filled with sand, which is mounted on a vibration isolated table. For the compression tests, the samples were placed in between the micromanipulator and the load cell. The samples were compressed by 2000 μm with a rate of ∼66.67 μm/s and the force was measured using the load cell. Finally, the stress–strain curves are evaluated and the compressive strength was determined (Figure S3).
4.3. Protein Adsorption Rate
Bovine serum albumin (Pierce; Thermo Fisher, Germany) was used to test the protein adsorption capacity of the scaffolds. Quantification was carried out using bicinchoninic acid (BCA; Thermo Fisher, Germany) in a colorimetric detection assay. For this, each scaffold type was immersed and incubated with 200 μL of bovine serum albumin (BSA) solution (1 mg/mL) for 4, 8, 12, 24, 48, and 72 h. Next, the supernatant was removed and mixed with 200 μL working reagent (Pierce; Thermo Fisher, Germany 23225). After 30 min of incubation, the protein concentration in the supernatant was quantified by a colorimetric microplate reader (BioTek uQuant) at 570 nm. Adsorption values were calculated by subtracting the measured protein concentration in the supernatant from the initial protein concentration in the protein solution before adding to the samples (1 mg/mL).
4.4. In Vitro Apatite Formation
The ability of the scaffolds to form apatite in vitro was evaluated by immersing the scaffolds into the simulated body fluid (SBF) according to the protocol proposed by Kokubo et al.68 Briefly, the scaffolds were incubated in the SBF at a ratio of 1 mg/mL at 37 °C and stirred at 90 rpm for up to 14 days. The SBF was replaced once a week during the incubation period. At every predetermined time point, the scaffolds were removed from the SBF, gently rinsed with deionized water, dehydrated with acetone, and dried at 60 °C for 12 h. The apatite formation on the scaffolds was assessed by field emission scanning electron microscopy (FE-SEM; Auriga, Carl Zeiss) and energy dispersive X-ray spectroscopy (EDS; X-MaxN Oxford Instruments).
4.5. Quantification of Ion Release
The concentration of ions released from the scaffolds during incubation in PBS (Biochrom, Germany) at different time points was analyzed with inductively coupled plasma mass spectrometry (ICP-MS). In brief, 15 mg of scaffold material was placed in 1× PBS for 4, 8, 12, and 24 h at 37 °C as a short-term experiment. In a long-term experiment the amount of released ions was measured every 3 days under the same conditions. For measuring ion release, PBS was extracted and the samples were immersed in fresh PBS. The extracts were acidified by 20 μL of nitric acid (Sigma, Germany). Acidified pure PBS was used as the control and the ion concentrations in pure PBS were subtracted from the ion concentrations measured in extractions to receive the ions released from the scaffolds.
4.6. Sample Preparation for Transmission Electron Microscopy (TEM)
A tiny piece of a scaffold was crushed and dispersed in butanol prior to dropping small crumbs on TEM grids. Then the micrographs of structures were taken by TEM (JEOL JEM-2100) at 200 kV.
4.7. Cell Adhesion Assays
4.7.1. Fibroblasts
The scaffolds (CNTT–BGN, CNTT–HA, CNTT–BGN/HA) were autoclaved at 121 °C and afterwards soaked in DMEM supplemented (Biochrom, Germany) with 10% fetal bovine serum (FBS, Biochrom, Germany) and 1% penicillin/streptomycin (Sigma, Germany). Approximately 10 000 rat embryonic fibroblast cells were seeded on each sample. The cells were incubated on the scaffolds for 4 days at 37 °C and 5% CO2. Afterwards, they were fixed by paraformaldehyde (Thermo Fisher, Germany) and dried using critical point drying (EMS 3000). Prior to scanning electron microscopy (Ultra Plus Zeiss SEM, 5 kV), the cells were coated with a thin sputtered gold layer (Bal-Tec SCD 050, 30 mA, 30 s).
Cell morphology, adhesion, and cytoskeleton were investigated by fluorescently staining cell nuclei and stress fibers. Cell nuclei were stained with DAPI (Thermo Fisher, Germany), which binds to DNA, and stress fibers were stained with Phalloidin (Alexa Fluor 647 Phalloidin, Thermo Fisher, Germany), which binds to the F-actin of the cytoskeleton. Images of the stained cells were recorded using a fluorescence microscope (Olympus IX81, camera: Hamamatsu, UV lamp: Lumencor).
4.7.2. Osteoblasts
The adhesion of osteoblast-like MC3T3-E1 to the scaffolds was visualized by detecting the filamentous actin of the cytoskeleton of cells on the scaffolds. Live/dead staining was carried out to assess the cytotoxicity of the scaffolds. In brief, the scaffolds were immersed into MEM (Biochrom, Germany) to stabilize the pH value prior to the seeding of cells on the scaffolds. After the pre-treatment, 0.2 mL of MC3T3-E1 cell suspension (2 × 105 cells/mL) was added on the scaffolds (in 24-well plates). After 3 h of incubation, an additional 1.8 mL of MEM was added. The culture medium was changed every 2 days. To minimize the influence of cells adhering to the bottom surface of the well during cultivation, the scaffolds were placed into new wells of a 24-well-plate when exchanging the medium. After 21 days of culture, cell adhesion on the scaffolds was visualized by staining. Cell nuclei were stained by 4,6-diamidino-2-phenylindole (DAPI, dilactate, Invitrogen), whereas the live/dead assay was carried out using calcein (Thermo Fisher, Germany) and propidium iodide (Thermo Fisher, Germany), according to the manufacturer’s protocol. Images of the fluorescently stained MC3T3-E1 cells were taken by a fluorescence microscope (Axio Scope A.1, Carl Zeiss Microimaging GmbH, Germany).
4.8. MTT Assay
The viability of fibroblast cells on the scaffolds was quantified according to the ISO 10993 norm. In brief, extractions were prepared by incubating the scaffolds at 37 °C in 1 mL culture medium (DMEM supplemented with 10% FBS and 1% penicillin/streptomycin) for 72 h. 10 000 REF52wt cells were cultured with 100 μL of medium for 24 h. The supernatant was replaced with 100 μL of extraction medium, then the cells were incubated for 24 h at 37 °C again. The number of vital cells incubated with the extraction medium was measured by adding 50 μL of methylthiazolyldiphenyl-tetrazolium bromide (MTT; Sigma-Aldrich, Germany) solution. After 4 h of incubation the absorbance was measured at 570 and 620 nm as a reference. Fresh and untreated culture medium was used as a negative control and medium containing 20% dimethyl sulfoxide as a positive control. The results were normalized to the absorbance measured in the controls. Five technical repeats were carried out in three independent experiments.
Acknowledgments
C.S.-U. and M.T. acknowledge funding from the European Research Council (Proof of Concept Grant 768740, Starting Grant 336104) and the German Research Foundation (SFB 1261, Project B7). M.T. was supported by the Deutscher Akademischer Austauschdienst (DAAD) through a research grant for doctoral candidates (91526555-57048249). In addition, R.A. gratefully acknowledges partial project funding by the Deutsche Forschungsgemeinschaft in the scope of contract Ad 183-17/1 and the research training group GRK 2154, as well. We gratefully acknowledge support from Prof. Dr. Dieter Garbe-Schönberg and Ulrike Westernströer in the ICP-mass spectrometry measurements. We also acknowledge Galen Ream for critical proofreading.
Glossary
Abbreviations
- 3D
three-dimensional
- CNT
carbon nanotube
- ZnO
zinc oxide
- HA
hydroxyapatite
- BGN
bioactive glass nanoparticles
- BG
bioactive glass
- CaO
calcium oxide
- CNTT
hierarchical carbon nanotube tube networks
- SEM
scanning electron microscopy
- TEM
transmission electron microscopy
- XRD
X-ray diffraction
- CNTT–BGN
CNT-based tubes containing bioactive glass nanoparticles
- CNTT–HA
CNT-based tubes containing hydroxyapatite nanoparticles
- CNTT–BGN/HA
CNT-based tubes containing both bioactive glass and hydroxyapatite nanoparticles
- t-ZnO
tetrapodal zinc oxide
- BSA
bovine serum albumin
- SBF
simulated body fluid
- ICP-MS
inductively coupled plasma mass spectrometry
- PBS
phosphate buffered saline
- REF52wt
rat embryonic fibroblasts 52 wild type
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.8b13631.
EDS results of the scaffolds after immersion in simulated body fluid, further data on the scaffolds (e.g., density, porosity) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gu Y.; Wang G.; Zhang X.; Zhang Y.; Zhang C.; Liu X.; Rahaman M. N.; Huang W.; Pan H. Biodegradable Borosilicate Bioactive Glass Scaffolds with a Trabecular Microstructure for Bone Repair. Mater. Sci. Eng., C 2014, 36, 294–300. 10.1016/j.msec.2013.12.023. [DOI] [PubMed] [Google Scholar]
- Gerhardt L.-C.; Boccaccini A. R. Bioactive Glass and Glass-Ceramic Scaffolds for Bone Tissue Engineering. Materials 2010, 3, 3867–3910. 10.3390/ma3073867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahaman M. N.; Day D. E.; Sonny Bal B.; Fu Q.; Jung S. B.; Bonewald L. F.; Tomsia A. P. Bioactive Glass in Tissue Engineering. Acta Biomater. 2011, 7, 2355–2373. 10.1016/j.actbio.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe A.; Güldal N. S.; Boccaccini A. R. A Review of the Biological Response to Ionic Dissolution Products from Bioactive Glasses and Glass-Ceramics. Biomaterials 2011, 32, 2757–2774. 10.1016/j.biomaterials.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Roether J. A.; Boccaccini A. R.; Hench L. L.; Maquet V.; Gautier S.; Jérôme R. Development and in Vitro Characterisation of Novel Bioresorbable and Bioactive Composite Materials Based on Polylactide Foams and Bioglass for Tissue Engineering Applications. Biomaterials 2002, 23, 3871–3878. 10.1016/S0142-9612(02)00131-X. [DOI] [PubMed] [Google Scholar]
- Turk M.; Deliormanlı A. M. Electrically Conductive Borate-Based Bioactive Glass Scaffolds for Bone Tissue Engineering Applications. J. Biomater. Appl. 2017, 32, 28–39. 10.1177/0885328217709608. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Xiao W.; Bal B. S.; Rahaman M. N. Effect of Copper-Doped Silicate 13–93 Bioactive Glass Scaffolds on the Response of MC3T3-E1 Cells in Vitro and on Bone Regeneration and Angiogenesis in Rat Calvarial Defects in Vivo. Mater. Sci. Eng., C 2016, 67, 440–452. 10.1016/j.msec.2016.05.073. [DOI] [PubMed] [Google Scholar]
- Axrap A.; Wang J.; Liu Y.; Wang M.; Yusuf A. Study on Adhesion, Proliferation and Differentiation of Osteoblasts Promoted by New Absorbable Bioactive Glass Injection in Vitro. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4677–4681. [PubMed] [Google Scholar]
- Yu C.; Zhuang J.; Dong L.; Cheng K.; Weng W. Effect of Hierarchical Pore Structure on ALP Expression of MC3T3-E1 Cells on Bioglass Films. Colloids Surf., B 2017, 156, 213–220. 10.1016/j.colsurfb.2017.05.011. [DOI] [PubMed] [Google Scholar]
- Bellucci D.; Sola A.; Anesi A.; Salvatori R.; Chiarini L.; Cannillo V. Bioactive Glass/Hydroxyapatite Composites: Mechanical Properties and Biological Evaluation. Mater. Sci. Eng., C 2015, 51, 196–205. 10.1016/j.msec.2015.02.041. [DOI] [PubMed] [Google Scholar]
- Suchanek W.; Yoshimura M. Processing and Properties of Hydroxyapatite-Based Biomaterials for Use as Hard Tissue Replacement Implants. J. Mater. Res. 1998, 13, 94–117. 10.1557/JMR.1998.0015. [DOI] [Google Scholar]
- Venkatesan J.; Kim S. K.. Carbon Nanotube for Bone Repair. In Handbook of Polymer Nanocomposites. Processing, Performance and Application; Springer: Berlin, 2015; pp 511–526. [Google Scholar]
- Schausten M. C.; Meng D.; Telle R.; Boccaccini A. R. Electrophoretic Deposition of Carbon Nanotubes and Bioactive Glass Particles for Bioactive Composite Coatings. Ceram. Int. 2010, 36, 307–312. 10.1016/j.ceramint.2009.09.008. [DOI] [Google Scholar]
- Touri R.; Moztarzadeh F.; Sadeghian Z.; Bizari D.; Tahriri M.; Mozafari M. The Use of Carbon Nanotubes to Reinforce 45S5 Bioglass-Based Scaffolds for Tissue Engineering Applications. Biomed. Res. Int. 2013, 2013, 465086 10.1155/2013/465086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C.; Feng P.; Peng S.; Shuai C. Carbon Nanotube, Graphene and Boron Nitride Nanotube Reinforced Bioactive Ceramics for Bone Repair. Acta Biomater. 2017, 61, 1–20. 10.1016/j.actbio.2017.05.020. [DOI] [PubMed] [Google Scholar]
- Porwal H.; Estili M.; Grünewald A.; Grasso S.; Detsch R.; Hu C.; Sakka Y.; Boccaccini A. R.; Reece M. J. 45S5 Bioglass–MWCNT Composite: Processing and Bioactivity. J. Mater. Sci.: Mater. Med. 2015, 26, 199. 10.1007/s10856-015-5529-9. [DOI] [PubMed] [Google Scholar]
- Doostmohammadi A.; et al. Fabrication of Chitosan/Poly (Vinyl Alcohol)/Carbon Nanotube/Bioactive Glass Nanocomposite Scaffolds for Neural Tissue Engineering. J. Nanomed. Res. 2016, 4, 00088 10.15406/jnmr.2016.04.00088. [DOI] [Google Scholar]
- Chen Q.; Garcia R. P.; Munoz J.; Pérez De Larraya U.; Garmendia N.; Yao Q.; Boccaccini A. R. Cellulose Nanocrystals-Bioactive Glass Hybrid Coating as Bone Substitutes by Electrophoretic Co-Deposition: In Situ Control of Mineralization of Bioactive Glass and Enhancement of Osteoblastic Performance. ACS Appl. Mater. Interfaces 2015, 7, 24715–24725. 10.1021/acsami.5b07294. [DOI] [PubMed] [Google Scholar]
- Han B.; Zhang X.; Liu H.; Deng X.; Cai Q.; Jia X.; Yang X.; Wei Y.; Li G. Improved Bioactivity of PAN-Based Carbon Nanofibers Decorated with Bioglass Nanoparticles. J. Biomater. Sci., Polym. Ed. 2014, 25, 341–353. 10.1080/09205063.2013.861169. [DOI] [PubMed] [Google Scholar]
- Miguez-Pacheco V.; Hench L. L.; Boccaccini A. R. Bioactive Glasses beyond Bone and Teeth: Emerging Applications in Contact with Soft Tissues. Acta Biomater. 2015, 13, 1–15. 10.1016/j.actbio.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Newman P.; Lu Z.; Roohani-Esfahani S. I.; Church T. L.; Biro M.; Davies B.; King A.; Mackenzie K.; Minett A. I.; Zreiqat H. Porous and Strong Three-Dimensional Carbon Nanotube Coated Ceramic Scaffolds for Tissue Engineering. J. Mater. Chem. B 2015, 3, 8337–8347. 10.1039/C5TB01052G. [DOI] [PubMed] [Google Scholar]
- Hollister S. J. Porous Scaffold Design for Tissue Engineering. Nat. Mater. 2005, 4, 518–524. 10.1038/nmat1421. [DOI] [PubMed] [Google Scholar]
- Loh Q. L.; Choong C. Three-Dimensional Scaffolds for Tissue Engineering Applications: Role of Porosity and Pore Size. Tissue Eng., Part B 2013, 19, 485–502. 10.1089/ten.teb.2012.0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota C.; Puppi D.; Chiellini F.; Chiellini E. Additive Manufacturing Techniques for the Production of Tissue Engineering Constructs. J. Tissue Eng. Regener. Med. 2015, 9, 174–190. 10.1002/term.1635. [DOI] [PubMed] [Google Scholar]
- Chia H. N.; Wu B. M. Recent Advances in 3D Printing of Biomaterials. J. Biol. Eng. 2015, 9, 4. 10.1186/s13036-015-0001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt F.; Signetti S.; Krüger H.; Röder S.; Smazna D.; Kaps S.; Gorb S. N.; Mishra Y. K.; Pugno N. M.; Adelung R. Hierarchical Self-Entangled Carbon Nanotube Tube Networks. Nat. Commun. 2017, 8, 1215 10.1038/s41467-017-01324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra Y. K.; Adelung R. ZnO Tetrapod Materials for Functional Applications. Mater. Today 2018, 21, 631–651. 10.1016/j.mattod.2017.11.003. [DOI] [Google Scholar]
- Papavlassopoulos H.; Mishra Y. K.; Kaps S.; Paulowicz I.; Abdelaziz R.; Elbahri M.; Maser E.; Adelung R.; Röhl C. Toxicity of Functional Nano-Micro Zinc Oxide Tetrapods: Impact of Cell Culture Conditions, Cellular Age and Material Properties. PLoS One 2014, 9, e84983 10.1371/journal.pone.0084983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecklenburg M.; Schuchardt A.; Mishra Y. K.; Kaps S.; Adelung R.; Lotnyk A.; Kienle L.; Schulte K. Aerographite: Ultra Lightweight, Flexible Nanowall, Carbon Microtube Material with Outstanding Mechanical Performance. Adv. Mater. 2012, 24, 3486–3490. 10.1002/adma.201200491. [DOI] [PubMed] [Google Scholar]
- Correa-Duarte M. A.; Wagner N.; Rojas-Chapana J.; Morsczeck C.; Thie M.; Giersig M. Fabrication and Biocompatibility of Carbon Nanotube-Based 3D Networks as Scaffolds for Cell Seeding and Growth. Nano Lett. 2004, 4, 2233–2236. 10.1021/nl048574f. [DOI] [Google Scholar]
- Rezwan K.; Chen Q. Z. Z.; Blaker J. J. J.; Boccaccini A. R. Biodegradable and Bioactive Porous Polymer/Inorganic Composite Scaffolds for Bone Tissue Engineering. Biomaterials 2006, 27, 3413–3431. 10.1016/j.biomaterials.2006.01.039. [DOI] [PubMed] [Google Scholar]
- Wu Z. Y.; Hill R. G.; Yue S.; Nightingale D.; Lee P. D.; Jones J. R. Melt-Derived Bioactive Glass Scaffolds Produced by a Gel-Cast Foaming Technique. Acta Biomater. 2011, 7, 1807–1816. 10.1016/j.actbio.2010.11.041. [DOI] [PubMed] [Google Scholar]
- Kankala R. K.; Xu X.-M.; Liu C.-G.; Chen A.-Z.; Wang S.-B. 3D-Printing of Microfibrous Porous Scaffolds Based on Hybrid Approaches for Bone Tissue Engineering. Polymers 2018, 10, 807 10.3390/polym10070807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helguero C. G.; Mustahsan V. M.; Parmar S.; Pentyala S.; Pfail J. P.; Kao I.; Komatsu D. E.; Pentyala S. Biomechanical Properties of 3D-Printed Bone Scaffolds Are Improved by Treatment with CRFP. J. Orthop. Surg. Res. 2017, 12, 195 10.1186/s13018-017-0700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho J. Y.; Kuhn-Spearing L.; Zioupos P. Mechanical Properties and the Hierarchical Structure of Bone. Med. Eng. Phys. 1998, 20, 92–102. 10.1016/S1350-4533(98)00007-1. [DOI] [PubMed] [Google Scholar]
- Livingston T.; Ducheyne P.; Garino J. In Vivo Evaluation of a Bioactive Scaffold for Bone Tissue Engineering. J. Biomed. Mater. Res. 2002, 62, 1–13. 10.1002/jbm.10157. [DOI] [PubMed] [Google Scholar]
- Huebsch N.; Lippens E.; Lee K.; Mehta M.; Koshy S. T.; Darnell M. C.; Desai R. M.; Madl C. M.; Xu M.; Zhao X.; Chaudhuri O.; Verbeke C.; Kim W.; Alim K.; Mammoto A.; Ingber D.; Duda G. N.; Mooney D. J. Matrix Elasticity of Void-Forming Hydrogels Controls Transplanted-Stem-Cell-Mediated Bone Formation. Nat. Mater. 2015, 14, 1269–1277. 10.1038/nmat4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathi M. H.; Hanifi A. Evaluation and Characterization of Nanostructure Hydroxyapatite Powder Prepared by Simple Sol-Gel Method. Mater. Lett. 2007, 61, 3978–3983. 10.1016/j.matlet.2007.01.028. [DOI] [Google Scholar]
- Chen Q. Z.; Thompson I. D.; Boccaccini A. R. 45S5 Bioglass-Derived Glass-Ceramic Scaffolds for Bone Tissue Engineering. Biomaterials 2006, 27, 2414–2425. 10.1016/j.biomaterials.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Zheng K.; Taccardi N.; Beltrán A. M.; Sui B.; Zhou T.; Marthala V. R. R.; Hartmann M.; Boccaccini A. R. Timing of Calcium Nitrate Addition Affects Morphology, Dispersity and Composition of Bioactive Glass Nanoparticles. RSC Adv. 2016, 6, 95101–95111. 10.1039/C6RA05548F. [DOI] [Google Scholar]
- Drisko G. L.; Carretero-Genevrier A.; Perrot A.; Gich M.; Gàzquez J.; Rodriguez-Carvajal J.; Favre L.; Grosso D.; Boissière C.; Sanchez C. Crystallization of Hollow Mesoporous Silica Nanoparticles. Chem. Commun. 2015, 51, 4164–4167. 10.1039/C4CC07828D. [DOI] [PubMed] [Google Scholar]
- Liao C. J.; Lin F. H.; Chen K. S.; Sun J. S. Thermal Decomposition and Reconstruction of Hydroxyapatite in Air Atmosphere. Biomed. Sci. Instrum. 1999, 35, 99–104. 10.1016/S0142-9612(99)00076-9. [DOI] [PubMed] [Google Scholar]
- Giovanardi R.; Montorsi M.; Ori G.; Cho J.; Subhani T.; Boccaccini A. R.; Siligardi C. Microstructural Characterisation and Electrical Properties of Multiwalled Carbon Nanotubes/Glass-Ceramic Nanocomposites. J. Mater. Chem. 2010, 20, 308–313. 10.1039/B914924D. [DOI] [Google Scholar]
- Wang K.; Zhou C.; Hong Y.; Zhang X. A Review of Protein Adsorption on Bioceramics. Interface Focus 2012, 2, 259–277. 10.1098/rsfs.2012.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster T. J.; Ergun C.; Doremus R. H.; Siegel R. W.; Bizios R. Specific Proteins Mediate Enhanced Osteoblast Adhesion on Nanophase Ceramics. J. Biomed. Mater. Res. 2000, 51, 475–483. . [DOI] [PubMed] [Google Scholar]
- Lamprecht C.; Huzil J. T.; Ivanova M. V.; Foldvari M. Non-Covalent Functionalization of Carbon Nanotubes with Surfactants for Pharmaceutical Applications - A Critical Mini-Review. Drug Delivery Lett. 2011, 1, 45–57. 10.2174/2210304x11101010045. [DOI] [Google Scholar]
- Tso S. T.; Pask J. A. Reaction of Fused Silica With Hydrogen Gas. J. Am. Ceram. Soc. 1982, 65, 457–460. 10.1111/j.1151-2916.1982.tb10514.x. [DOI] [Google Scholar]
- El-Ghannam A.; Hamazawy E.; Yehia A. Effect of Thermal Treatment on Bioactive Glass Microstructure, Corrosion Behavior ζ Potential, and Protein Adsorption. J. Biomed. Mater. Res. 2001, 55, 387–395. . [DOI] [PubMed] [Google Scholar]
- El-Ghannam A.; Ducheyne P.; Shapiro I. M. Effect of Serum Proteins on Osteoblast Adhesion to Surface-Modified Bioactive Glass and Hydroxyapatite. J. Orthop. Res. 1999, 17, 340–345. 10.1002/jor.1100170307. [DOI] [PubMed] [Google Scholar]
- Lamprecht C.; Taale M.; Paulowicz I.; Westerhaus H.; Grabosch C.; Schuchardt A.; Mecklenburg M.; Böttner M.; Lucius R.; Schulte K.; Adelung R.; Selhuber-Unkel C. A Tunable Scaffold of Microtubular Graphite for 3D Cell Growth. ACS Appl. Mater. Interfaces 2016, 8, 14980–14985. 10.1021/acsami.6b00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K.; Wu J.; Li W.; Dippold D.; Wan Y.; Boccaccini A. R. Incorporation of Cu-Containing Bioactive Glass Nanoparticles in Gelatin-Coated Scaffolds Enhances Bioactivity and Osteogenic Activity. ACS Biomater. Sci. Eng. 2018, 4, 1546–1557. 10.1021/acsbiomaterials.8b00051. [DOI] [PubMed] [Google Scholar]
- Roohani-Esfahani S. I.; Nouri-Khorasani S.; Lu Z. F.; Appleyard R. C.; Zreiqat H. Effects of Bioactive Glass Nanoparticles on the Mechanical and Biological Behavior of Composite Coated Scaffolds. Acta Biomater. 2011, 7, 1307–1318. 10.1016/j.actbio.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Bellucci D.; Desogus L.; Montinaro S.; Orrù R.; Cao G.; Cannillo V. Innovative Hydroxyapatite/Bioactive Glass Composites Processed by Spark Plasma Sintering for Bone Tissue Repair. J. Eur. Ceram. Soc. 2017, 37, 1723–1733. 10.1016/j.jeurceramsoc.2016.11.012. [DOI] [Google Scholar]
- Filho O. P.; Latorre G. P.; Hench L. L. Effect of Crystallization on Apatite-Layer Formation of Bioactive Glass 45S5. J. Biomed. Mater. Res. 1996, 30, 509–514. . [DOI] [PubMed] [Google Scholar]
- Clupper D. C.; Mecholsky J. J.; LaTorre G. P.; Greenspan D. C. Bioactivity of Tape Cast and Sintered Bioactive Glass-Ceramic in Simulated Body Fluid. Biomaterials 2002, 23, 2599–2606. 10.1016/S0142-9612(01)00398-2. [DOI] [PubMed] [Google Scholar]
- Popov V. N. Carbon Nanotubes: Properties and Application. Mater. Sci. Eng., R 2004, 43, 61–102. 10.1016/j.mser.2003.10.001. [DOI] [Google Scholar]
- Selhuber-Unkel C.; Erdmann T.; López-García M.; Kessler H.; Schwarz U. S.; Spatz J. P. Cell Adhesion Strength Is Controlled by Intermolecular Spacing of Adhesion Receptors. Biophys. J. 2010, 98, 543–551. 10.1016/j.bpj.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban N. Q.; Schwarz U. S.; Riveline D.; Goichberg P.; Tzur G.; Sabanay I.; Mahalu D.; Safran S.; Bershadsky A.; Addadi L.; Geiger B. Force and Focal Adhesion Assembly: A Close Relationship Studied Using Elastic Micropatterned Substrates. Nat. Cell Biol. 2001, 3, 466–472. 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- Fraley S. I.; Feng Y.; Wirtz D.; Longmore G. D. Reply: Reducing Background Fluorescence Reveals Adhesions in 3D Matrices. Nat. Cell Biol. 2011, 13, 5–7. 10.1038/ncb0111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S.; Akiyama S. K.; Yamada K. M. Synergistic Roles for Receptor Occupancy and Aggregation in Integrin Transmembrane Function. Science 1995, 267, 883–885. 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- Baker B. M.; Chen C. S. Deconstructing the Third Dimension – How 3D Culture Microenvironments Alter Cellular Cues. J. Cell Sci. 2012, 125, 3015–3024. 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansukhani A.; Bellosta P.; Sahni M.; Basilico C. Signaling by Fibroblast Growth Factors (FGF) and Fibroblast Growth Factor Receptor 2 (FGFR2)-Activating Mutations Blocks Mineralization and Induces Apoptosis in Osteoblasts. J. Cell Biol. 2000, 149, 1297–1308. 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie P. J. Fibroblast Growth Factor Signaling Controlling Osteoblast Differentiation. Gene 2003, 316, 23–32. 10.1016/S0378-1119(03)00748-0. [DOI] [PubMed] [Google Scholar]
- Debiais F.; Hott M.; Graulet A. M.; Marie P. J. The Effects of Fibroblast Growth Factor-2 on Human Neonatal Calvaria Osteoblastic Cells Are Differentiation Stage Specific. J. Bone Miner. Res. 1998, 13, 645–654. 10.1359/jbmr.1998.13.4.645. [DOI] [PubMed] [Google Scholar]
- Ogiso B.; Hughes F. J.; Melcher A. H.; McCulloch C. A. G. Fibroblasts Inhibit Mineralised Bone Nodule Formation by Rat Bone Marrow Stromal Cells in Vitro. J. Cell. Physiol. 1991, 146, 442–450. 10.1002/jcp.1041460315. [DOI] [PubMed] [Google Scholar]
- Mishra Y. K.; Modi G.; Cretu V.; Postica V.; Lupan O.; Reimer T.; Paulowicz I.; Hrkac V.; Benecke W.; Kienle L.; Adelung R. Direct Growth of Freestanding ZnO Tetrapod Networks for Multifunctional Applications in Photocatalysis, UV Photodetection, and Gas Sensing. ACS Appl. Mater. Interfaces 2015, 7, 14303–14316. 10.1021/acsami.5b02816. [DOI] [PubMed] [Google Scholar]
- Schaber P. M.; Colson J.; Higgins S.; Thielen D.; Anspach B.; Brauer J. Thermal Decomposition (Pyrolysis) of Urea in an Open Reaction Vessel. Thermochim. Acta 2004, 424, 131–142. 10.1016/j.tca.2004.05.018. [DOI] [Google Scholar]
- Kokubo T.; Takadama H. How Useful Is SBF in Predicting in Vivo Bone Bioactivity?. Biomaterials 2006, 27, 2907–2915. 10.1016/j.biomaterials.2006.01.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.