

Abstract

Biosensors and materials for biomedical applications generally require chemical functionalization to bestow their surfaces with desired properties, such as specific molecular recognition and antifouling properties. The use of modified poly(l-lysine) (PLL) polymers with appended oligo(ethylene glycol) (OEG) and thiol-reactive maleimide (Mal) moieties (PLL-OEG-Mal) offers control over the presentation of functional groups. These reactive groups can readily be conjugated to, for example, probes for DNA detection. Here we demonstrate the reliable conjugation of thiol-functionalized peptide nucleic acid (PNA) probes onto predeposited layers of PLL-OEG-Mal and the control over their surface density in the preceding synthetic step of the PLL modification with Mal groups. By monitoring the quartz crystal microbalance (QCM) frequency shifts of the binding of complementary DNA versus the density of Mal moieties grafted to the PLL, a linear relationship between probe density and PLL grafting density was found. Cyclic voltammetry experiments using Methylene Blue-functionalized DNA were performed to establish the absolute probe density values at the biosensor surfaces. These data provided a density of 1.2 × 1012 probes per cm2 per % of grafted Mal, thus confirming the validity of the density control in the synthetic PLL modification step without the need of further surface characterization.
Introduction
DNA biosensors have gained in importance over the last few decades.1 Their applicability ranges from biotechnology and biomedical applications2,3 to food safety and forensics.4−6 Owing to their selectivity and sensitivity, DNA biosensors have become the gold standard for the diagnosis and monitoring of several types of gene-related diseases, such as cystic fibrosis7 and cancer.8
Among all types of DNA biosensors, surface-based ones have several advantages, such as high sensitivity and reduced interference.9,10 The integration of a biorecognition element at the transducing substrate is the most critical step in the creation of a high-performance sensor surface, while at the same time preventing nonspecific adsorption.1 Controlling the chemical and physical nature of the interface determines the performance of the whole biosensing device.11
Several strategies for probe immobilization on the sensor surface have been used depending on the substrate composition (mostly Au, SiO2, or metal oxides): adsorption of DNA probes on positively charged surfaces,12,13 attachment of thiolated DNA probes as self-assembled monolayers (SAMs) on gold substrates14 or modified probes on chemically functionalized surfaces,15,16 silanization on SiO2,17 adsorption by noncovalent interactions (e.g., biotin–streptavidin),18 and layer-by-layer (LbL) polyelectrolyte assembly.19 Although many procedures have been standardized, these approaches can present problems, such as reduced stability or affinity during hybridization, long functionalization times, and solvent incompatibility.
Control over the probe density is an important challenge for optimal performance of a biosensing device.20,21 The probe density (Γ) is a crucial parameter in biosensing because it affects the probe’s orientation and the uniformity of the sensing layer. Moreover, the probe density not only provides possible binding sites for the analyte, but also affects the accessibility of the analyte toward the probe surface by the occurrence of steric and electrostatic effects. In particular, in DNA sensing, the density of probes, which usually consist of complementary single strand DNA (ssDNA), is affected by electrostatic repulsion leading to decreased hybridization efficiency at increased probe densities.20,22,23
The type of immobilization technique (for example, by SAM formation of thiolated probes on gold or by silanization) affects the density and distribution of the probes on a sensing surface. In the case of direct probe adsorption on both unfunctionalized and pretreated surfaces, the following two are the most used approaches to vary the probe density. First, by increasing the deposition time (while keeping the concentration constant), Herne et al. were able to vary the probe density by over an order of magnitude by using HS-ssDNA on gold,24 and the same order of magnitude was calculated for the probes on an impedance-based silicon transducer by Macanovic and co-workers.25 Second, by increasing the concentration of the probe solution for a fixed amount of time, Ricci et al. varied Γ in their E-DNA sensor between 1010 and 1012 molecules/cm2.26 A conceptually different strategy is to employ variation of the molar ratio of two components in the depositing solution: Kjallman and co-workers varied the amounts of thiolated poly(ethylene glycol) (PEG) and thiol-modified hairpin probes during the self-assembly process on a gold surface.27 Yao et al. chose a 1:9 ratio for a biotinylated and an OH-terminated thiol in a mixed self-assembled monolayer, again on a gold substrate.28 Peterson and co-workers varied the ionic strength and applied an electric field to assist the immobilization of negatively charged ssDNA-SH, thus obtaining a high surface density.23
All the methods presented above aim to control the probe density at the surface modification step, requiring characterization of the probe density and the hybridization efficiency after sample preparation using difficult surface-analytical techniques. A method is therefore desired that allows control over Γ in a preceding synthetic step, upon which analysis is conveniently achieved using solution-analytical techniques and which is followed by a straightforward surface immobilization step of a single compound that provides a reproducible density upon adsorption.
The chemi/physisorption of modified polyelectrolytes provides advantages for the immobilization of biomolecules and for biosensing applications.29 At physiological pH, poly(l-lysine) (PLL) polymers readily and strongly adsorb onto a variety of metal oxide surfaces through multivalent electrostatic interactions between the positively charged lysine side-chains and a negatively charged surface.30,31 Consequently, modified PLL polymers easily allow the accommodation of the grafted functional groups over the substrate, maintaining their adsorption properties. This approach has been used, for example, in bestowing antifouling properties on surfaces. Owing to the simple functionalization and easy characterization in solution, PLL-g-PEG polymers were made with different grafting densities to study the influence of interfacial architecture on the resistance to protein adsorption.30,32,33 VandeVondele and Hubbel varied the grafting density of RGD peptide to form PLL-g-PEG-RGD polymers that promote cell adhesion, blocking the nonspecific protein interactions at the same time,34 while Huang et al. formed a bioaffinity sensor to detect rabbit immunoglobulin (RIgG) changing the graft ratio of biotin in PLL-g-PEG-biotin polymers.35 The long PEG type (MWPEG > 2000 Da) used in the previous examples can affect the final output of the detector by positioning the receptors away from the interface and the polydispersity of the PEG chain can prevent the uniform formation of the biorecognition layer. Therefore, Duan et al. grafted short oligo(ethylene glycol) (OEG4)36 in combination with OEG4-biotin to PLL backbones on silicon nano-BioFETs, forming a biosensing surface with uniform layers, maintaining the strong surface stability and antifouling properties.37
Here we show the formation of DNA biorecognition surfaces prepared by the deposition of modified poly(l-lysine) polymers with various ratios of OEG and maleimide (Mal) moieties (PLL-OEG-Mal) on surfaces, so that the probe density control is achieved during a preceding and simple synthetic step, where the degree of functionalization is readily analyzed and quantified by 1H NMR. Afterward, by means of a predictable and straightforward, single-step surface assembly process over gold and silicon oxide, the PLL-OEG-Mal polymers are adsorbed on the sensing surface and coupled subsequently with thiol-modified peptide nucleic acid (PNA) probes, able to detect KRAS sequences of DNA (involved in the early formation of several types of cancer).38,39 We used PNA sequences as probes to achieve a better affinity for cDNA sequences, compared to DNA probes.40 It has been shown that PNA can distinguish single point mutations in nucleotide sequences,41 a property that was used in PCR protocols for detection of cancer-related mutations,42 and their properties can be used for increasing the sensitivity and the selectivity of the biosensing devices.43 The detection of cDNA is evaluated as a function of the degree of Mal grafted to the different PLL-OEG-Mal polymers by quartz crystal microbalance with dissipation (QCM-D), in order to establish the relationship between the probe density and the PLL grafting density with Mal groups. Cyclic voltammetry (CV) experiments have been used to support these findings and to provide absolute density values.
Results and Discussion
Probe Density Control by Grafting Density of Maleimide Groups at PLL
Figure 1a shows the concept of the control over probe density during the preceding synthesis phase of modified PLL, after which the PLL with the desired degree of functionalization is placed on the sensor surface in one step. Increasing amounts of maleimide (Mal) reactive moieties in the synthetic step result in a higher content of Mal grafted to the PLL backbone and to a concomitantly higher density at the substrate upon immobilization of the modified PLL. The density of PNA probes that is displayed on the surface is set during the thiol–ene reaction, as has been demonstrated in earlier studies on other maleimide-functionalized recognition surfaces carrying antifouling PEG moieties.44,45 Consequently, the cDNA hybridization step can, in the envisaged application, be carried out without previous quantification of the total probe density. Aim of this work was to observe the relationship between the Mal grafting density on the PLL and the response of the sensing layer to cDNA after its modification with PNA, and thus to establish the relationship between polymer grafting density and surface probe density.
Figure 1.

(a) Scheme showing the control of the probe density, during the synthesis of PLL-OEG-Mal polymers, which provides the targeted probe density and cDNA binding at the substrate upon immobilization of the modified PLL. x and y indicate the percentages of oligo(ethylene glycol) and maleimide grafted to the PLL side chains, respectively. (b) Synthesis of PLL-OEG-Mal. PLL is reacted with desired relative ratios of Mal-OEG4-NHS (y = 0–9.1%) and methyl-OEG4-NHS ester (x = 15.9–29.1%) to give the final modified PLL with the desired degrees of functionalization.
In order to investigate the surface density control, PLL polymers with various fractions of oligomeric ethylene glycol and maleimide moieties were synthesized (Figure 1b). Specifically, the structure of the functionalized polymers in this work was based on PLL (MW 15–30 kDa) functionalized with side groups with an OEG spacer with four units. The end of the spacer was either a methoxy group (OEG) or a maleimide group (Mal) to have mono or bifunctionalized polymers (PLL-OEG or PLL-OEG-Mal). The name PLL-OEG(x) is used here to refer to PLL-g-OEG4(x%) polymers where x corresponds to the experimentally assessed mole fraction of OEG per lysine monomer unit grafted to the PLL backbone. In the case of maleimide derivatives, PLL-OEG(x)-Mal(y) stands for PLL-g-OEG4(x%)-g-OEG4-Mal(y%) where y is the percentage of maleimide grafted to the PLL, as shown in Figure 1b.
Following a reported procedure,37 OEG and Mal groups were covalently grafted to the PLL backbone in a one-step, one-pot synthesis by NHS ester coupling, as depicted in Figure 1b, using a mixture of NHS-OEG4 and NHS-OEG4-Mal. The grafted OEG/Mal ratio attached to PLL was calculated taking the proper 1H NMR integrals, after normalization by the peak at 4.29 ppm (1H, lysine backbone, NH–CH–C(O)−) (see Figure S1). The fraction of maleimide was varied between 0% and 9%, while the OEG fraction was kept at 15–30% (see Table S1). This degree of OEG was found to be sufficient to prevent nonspecific interactions from both uncharged molecules as PNA and between positively charged PLL and negatively charged DNA, as will be explained below. As suggested previously by Duan and co-workers, the total functionalization of PLL polymer was intentionally kept below 35–40% to have a sufficiently strong surface adhesion as well as enough OEG moieties to avoid nonspecific interactions.37,46
The results presented in Table S1 indicate a small discrepancy between the maximum theoretical amounts, based on full conversion of the supplied NHS esters of OEG and Mal, and the measured grafting densities observed for both the OEG and Mal groups. Typically about 80–90% of the Mal-NHS derivative was coupled successfully to the PLL. The small discrepancy is probably due to the limited stability of the NHS ester in PBS at pH 7.0 leading to competing hydrolysis.47
PLL-Based DNA Sensing Layer Formation
Figure 2a shows a QCM-D time trace of the full process of PLL adsorption, PNA probe binding and cDNA binding (frequency shifts Δf, in blue) as conceptually outlined in Figure 1. The gold QCM substrate was activated by oxygen plasma, mounted in the QCM chamber, and flushed with 0.3 mg/mL PLL-OEG-Mal solutions in PBS buffer at pH 7.2. The initial decrease in Δf reflects the self-assembly of the PLL onto the substrate, which was homogeneously distributed with a thickness of 0.52 nm (±0.14 nm) as confirmed by ellipsometry. The chip was then washed with PBS followed by injection of a 1 μM solution of PNA-thiol (KRAS-WT, protected sequence SPDP-dPEG4-CTA CGC CAC CAG CT-Gly-NH2, 14 nt, synthesized according to a reported procedure,39 deprotected from the 3-(2-pyridyldithio)propionyl (SPDP) protecting group using TCEP gel directly before use) in the same buffer, which provided the final sensing layer, as confirmed by the decrease in frequency. The same response was obtained in the last step, where a 1 μM solution of cDNA fully complementary to the PNA KRAS-WT (also with 14 nt) in PBS buffer (pH 7.2) was used. The same conditions were used for all the surface density experiments (Figure 2 and Figure S2), varying the type of modified PLL and consequently the density of maleimide groups presented on the biorecognition layer.
Figure 2.

(a) Typical QCM-D measurement of the full process of PLL-OEG(30.3)-Mal(1.8) adsorption, PNA coupling and cDNA binding on a gold substrate. Both the main frequency (Δf, blue) and the dissipation (ΔD, red) are displayed. Example of five normalized Δf for the (b) PNA-thiol probe and (c) cDNA absorption steps using different PLL polymers with increasing maleimide densities (Mal = 0.0–9.1%). The dashed lines refer to the positions used to calculate the Δf of the corresponding step. In all the multistep adsorption experiments the concentrations were 0.3 mg/mL for the modified PLL solutions, and 1 μM for both the PNA-thiol and cDNA solutions. PBS washing steps at pH 7.2 (gray bars) were placed between adsorption steps. The fifth overtone was used for both the frequency and the dissipation.
In Figure 2b, an example of five normalized QCM experiments of the PNA probe binding step onto gold substrates is presented, where several PLLs with different maleimide fractions (Mal = 0.0–9.1%, OEG = 15.9–24.8%) were used. Apparently, the change in frequency of the PNA coupling step is proportional to the fraction of maleimide in the PLL that was preadsorbed at the substrate. A similar trend was observed for the following cDNA binding step (Figure 2c), where a maximum Δf of 5.4 Hz was obtained for the substrate functionalized with the PLL with the higher Mal molfraction (green), while no frequency shift was observed for the PLL without Mal (black).
In Figure 2 (and Figure S2), several aspects can be noted. First of all, all steps associated with PLL, PNA, and cDNA attachment were generally clearly visible. Moreover, larger degrees of Mal functionalization of the PLL lead, qualitatively, to larger amounts of both coupled PNA and adsorbed cDNA as indicated by the increasing frequency shifts. The selectivity of the biorecognition layer was tested by flushing 1 μM of fully noncomplementary DNA (ncDNA) over a substrate functionalized with modified PLL and PNA-thiol (Figure S3a). No frequency shift was observed for the ncDNA. Moreover, the biorecognition layer was still active after the passage of ncDNA as shown by the Δf when 1 μM cDNA solution was subsequently flushed over the substrate. In addition, Figure S2a and S3b show that, for samples with only OEG functionalization, the insertion of PNA-thiol through the modified PLL monolayer is blocked, preventing cDNA binding. Figure S3c shows a control experiment on a substrate with only PLL, so in the absence of PNA-thiol, performed to evaluate the influence of DNA interaction with a PLL-coated substrate. The absence of a frequency shift in Figures S3c confirms that the cDNA adsorbs only when complementary PNA is present, and the antifouling behavior is maintained regardless the presence of PNA as well as maleimide moieties (Figure S3b).
The selectivity of the modified PLL polymers was further tested flushing 35 mg/mL (common concentration of human serum albumin, HSA, in blood)48 of bovine serum albumin (BSA) solution in PBS (pH 7.4) over a substrate coated with PLL-OEG(29.1)-Mal(5.5). A swift frequency change of ∼30 Hz was observed, accompanied by a large dissipation change (Figure S4a). Yet, the baseline recovered almost completely (<1 Hz) upon flushing with PBS and in an equally fast manner. Therefore, we attribute the observed frequency change to a change of the solution viscosity, and conclude that no physisorption of BSA occurred. Likewise, the selectivity was tested for a PNA-modified chip (Figure S4b), where the gold substrate was functionalized by PLL-OEG(19.4)-Mal(9.1) and PNA-thiol, followed by flow of BSA solution (35 mg/mL) at PBS 7.2. Subsequently, solutions of 1 μM of ncDNA and cDNA, in the presence of the same BSA concentration, were flushed over the PNA-modified chip. A frequency shift (of 5.5 Hz) was visible (Figure S4b) only for the cDNA, which confirms the intrinsic DNA binding selectivity of the PNA interface.
Other QCM-D experiments were done using SiO2 chips (instead of Au) to demonstrate the versatility of the system with different substrate materials, and different DNA lengths were also used (Figure S5). After the self-assembly of the modified PLL and the anchoring of PNA-thiol probes, 1 μM of longer ncDNA and cDNA were flushed over the QCM substrate. Even with the longer ncDNA (44 nt), which has possibly stronger electrostatic interactions, no frequency shift was recorded. In contrast, as in the case of the Au substrates, the Δf was increasing linearly for cDNA (43 nt) with respect to the density of maleimide groups grafted to the PLL backbone. Notably, the frequency shifts of the cDNA steps shown in Figure S5 were roughly three time higher than the ones observed for the QCM experiments on the Au substrates, when the same modified PLL was used. This difference is caused by the longer cDNA (from 43 nt compared to 14 nt).
Assessment of Surface Probe Density
Relationships between the degree of functionalization and the adsorbed masses of PLL-OEG-Mal, thiol-PNA, and cDNA were quantified gravimetrically by means of QCM-D. In order to establish the response between adsorbed mass from QCM and the degree of PLL polymer functionalization, the frequency shifts, Δf, of adsorption steps for PLL, PNA, and cDNA were plotted either versus the total degree of functionalization (OEG+Mal) or the Mal content, both previously quantified by 1H NMR. In detail, Figure 3a shows the graph obtained plotting Δf of the PLL-OEG-Mal steps versus the total degree of functionalization for each modified PLL polymer used. The linear relationship, with an intercept close to the origin, indicates that the frequency shift has a strong correlation with the grafting density of functionalized side chains, but the main PLL chain has little contribution. This effect is probably primarily governed by the different degrees of hydration of the polymer segments, mostly due to the content of ethylene glycol groups in the side chains, which are typically well hydrated.
Figure 3.

(a) QCM-D frequency shift (fifth overtone) of the PLL-OEG-Mal deposition step versus the total degree of functionalization of the modified PLL, quantified by 1H NMR. (b) QCM-D frequency shift of the cDNA binding step versus the fraction of Mal grafted to the PLL polymer, quantified by 1H NMR. All experiments were performed using 0.3 mg/mL of modified PLL, 1 μM PNA thiol solution (activated by TCEP), and 1 μM cDNA solution in PBS at pH 7.2. PLL-OEG-Mal polymers with different degrees of functionalization were used (Mal = 0.0–9.1%, OEG 15.9–29.1%). Data points represent individual measurements.
Subsequently, to prove the variation of Mal coverage at the interface, the PNA (Figure S6) and cDNA frequency shifts (Figure 3b) were plotted versus the amount of maleimide grafted to the modified PLL self-assembled on the gold substrates. Although the frequency shifts of both the PNA and cDNA steps show a linear dependence on the Mal fraction, the lower values for the PNA step gave a somewhat poorer correlation, so that we decided to use the cDNA dependence primarily. As shown in Figure 3b, the Δf for the cDNA step, which is related to the adsorbed mass of the cDNA, increases linearly with the fraction of Mal groups and passes through the origin, regardless the content of OEG. At low Mal coverage, the efficiency of PNA/DNA hybridization is assumed to be 100%. The PNA/DNA hybridization efficiency is higher than that of DNA/DNA duplex formation because of a higher affinity and the absence of repulsion between the probe molecules.40,49,50 Consequently, due to the linear relationship, we estimated that this efficiency is maintained at higher Mal densities. This trend confirms the possibility to fine-tune the relative probe density at the surface by controlling the fraction of maleimide groups in the preadsorbed PLL. Therefore, using the linear response shown in Figure 3b as a calibration curve allows us to predict the detected amount of cDNA by assessing the Mal fraction by NMR in the PLL synthesis step. In addition, the linearity of the fit, as well as the use of new substrates for each experiment, confirms indirectly the reproducibility of the method and the surface modification.
All in all, these data confirm that the probe density at biosensor surfaces (here of PNA)—and consequently the amount of cDNA detected—can be controlled by simply varying the fraction of grafted Mal during the synthetic step. However, despite the possibility of relative predictions of the cDNA amount over the modified PLL substrate, the absolute density of analyte (here cDNA), and thus of the PNA probe, adsorbed over QCM substrates cannot be accurately calculated, especially because of the high associated water content, which influences the observed frequency shifts and usually leads to an overestimation of the mass of the adsorbed target.51,52
In order to confirm the predictability of the surface density control and to assess the absolute density of DNA detected, cDNA modified with the redox probe Methylene Blue (cDNA-MB, 5′-MB-AG CTG GTG GCG TAG-3′) was used in cyclic voltammetry (CV) experiments. The electroactive cDNA-MB is used to correlate the peak current generated by the MB group to the density of hybridized cDNA at the detection layer. As a proof of concept, four modified PLLs were used: PLL-OEG(24.9), PLL-OEG(28.1)-Mal(3.1), PLL-OEG(29.1)-Mal(5.5), and PLL-OEG(19.4)-Mal(9.1). Gold chips, preincubated with PLL and PNA-thiol, were covered by a 1 μM cDNA-MB solution in PBS for 1 h, and 0.1 M NaClO4 was used as electrolyte. The linear dependence of ip versus the scan rate in the CV experiments (Figure S7) proved that the redox process occurred at the interface (Figure S8).
Figure 4 shows the trend of the calculated surface charge density, translated into molecular surface density, obtained from the CV data as a function of the Mal grafting density. Using differently modified PLLs, four maleimide densities were used. The data shows DNA densities of (0–11) × 1012 molecules of cDNA-MB—corresponding to equal numbers of PNA probes—per cm2, in agreement with the range of probe density values presented in the literature with other types of systems.24,25 These data confirm the quantitative assessment of probe densities displayed at the surface by varying the modified PLL. Importantly, the linear relationship, with a slope of (1.24 ± 0.02) × 1012 probes per cm2 per % of grafted Mal, provides a practical, empirical, but at the same time quantitative, prediction of the probe density that is obtained for a specific degree of Mal functionalization in the first, synthetic step.
Figure 4.
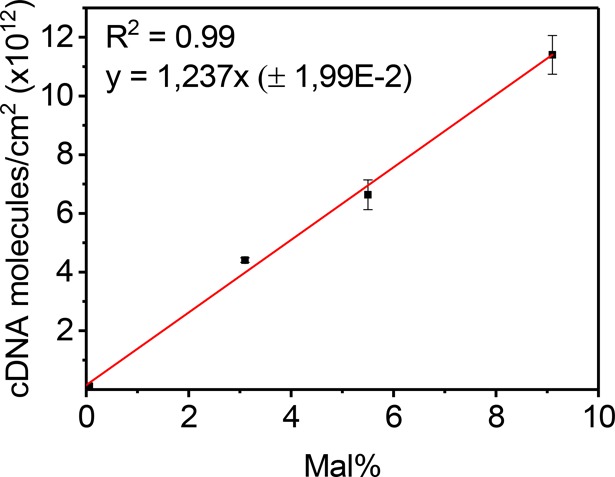
Surface density of cDNA-MB detected from CV experiments using PLL-OEG(24.9) PLL-OEG(28.1)-Mal(3.1), PLL-OEG(29.1)-Mal(5.5), and PLL-OEG(19.4)-Mal(9.1) polymers. Calculations were done by means of eq 2 (Experimental Section). All gold substrates were prepared using 0.3 mg/mL of modified PLL, 1 μM PNA thiol solution (activated by TCEP), and 1 μM MB-modified cDNA solution in PBS at pH 7.2. Fresh solutions of 0.1 M NaClO4 were used as electrolyte. Data points and given single standard deviations are based on 3–4 measurements each.
Conclusions
Summarizing, we have demonstrated control over the probe density at model biosensor surfaces, by the application of poly(l-lysine) polymers with various ratios of OEG and Mal moieties (quantified by 1H NMR) and the conjugation of thiol-functionalized PNA probes to the Mal groups. The QCM-D technique was used to monitor the self-assembly of different PLL-OEG-Mal polymers over negatively charged substrates and the thiol-PNA binding step, forming the biorecognition substrate. The selectivity was tested using both short and long cDNA/ncDNA sequences, as well as BSA solutions, showing that adsorption occurred only for cDNA and that the OEG content grafted to the PLL backbone prevents physisorption. The frequency shifts of the adsorption steps recorded for different PLL-OEG-Mal were plotted against the total density of grafted groups (OEG+Mal) to establish a (relative) linear relationship between the frequency shift from QCM and the degree of Mal functionalization. Quantification using CV allowed to derive absolute values for the surface densities. The PLL-OEG-Mal surface modification presented here led to displayed probe densities in the range of (0–11) × 1012 molecules/cm2, which is in agreement with the surface density values reported in the literature.23,25 All in all, these results underline the concept density control at the preceding synthetic step: with this relationship a desired probe density can now be translated into a target Mal density, which is engineered at the very first synthetic step of the PLL functionalization.
The strategy to equip surfaces with suitably modified PLL is a promising approach because the material surface properties can be tailored by customizing first the PLL with desired functional groups, potentially with reactive, imaging, binding, and/or release properties, thus offering broad applicability in, among others, biosensing and responsive materials. Moreover, the direct solution coating from aqueous media presents a simple and reliable approach to form biorecognition surfaces or, for example, to anchor biomolecules (e.g., protein, enzymes) selectively at a substrate for biomedical applications. Owing to the electrostatic interactions, modified PLL polymers can be applied as passivation/functionalization layer at virtually any surface such as metals, metal oxides, or polymers. This aspect underlines the versatility of the methodology and may assist in the development of applications in lab-on-a-chip, disposable ready-to-use biomedical sensors, wearable devices, or water treatment.
Experimental Section
Materials
Poly(l-lysine) hydrobromide (MW = 15–30 kDa by viscosity), EDC, NHS, and PBS powder (BioPerformance Certified pH 7.4), NaCl, NaClO4, were purchased from Sigma-Aldrich. Methyl-OEG4-NHS ester and Mal-OEG4-NHS ester, the Zeba spin desalting columns (7 kDa MWCO, 5 mL) and the Immobilized TCEP disulfide reducing gel (Tris[2-carboxyethyl] phospine hydrochloride immobilized onto 4% cross-linked beaded agarose) were purchased from ThermoFischer Scientific. cDNA (complementary to KRAS WT: 14 nt, 5′-AGCTGGTGGCGTAG-3′; 43 nt, 5′-ATGACTGAATATAAACTTGTGGTAGTTGGAGCTGGTGGCGTAG-3′) and ncDNA (short 14 nt, 5′-CTACGCCACCAGCT-3′; long 44 nt, 5′-TTGCCCTTCCTTCCCTCCTTCGTCCCCTCCTCACACCCCACCCC-3′) sequences were purchased from Eurofins Genomic. Methylene Blue (MB)-functionalized DNA (cDNA-MB 15 nt, 5′-MB-TAGCTGGTGGCGTAG-3′) was obtained from Biosearch Technologies, USA. Chips for cyclic voltammetry (200 nm gold on glass) were obtained from Ssens bv (The Netherlands), while Au and SiO2 QCM chips (with fundamental frequency of 5 MHz) were purchased from Biolin Scientific.
PNA Deprotection
PNA bearing a disulfide-protected thiol moiety was used in order to avoid dimerization and side-reactions during storage. A glycine residue was used as spacer during synthesis at the C-terminus to facilitate loading of the PNA on the resin, considering that the resulting glycinamide (GlyNH2) residue has no substantial effect on DNA binding. Directly before use in the coupling reactions to Mal, PNA KRAS-WT (SPDP-dPEG4-CTA CGC CAC CAG CT-Gly-NH2, synthesized according to a previously published procedure,39 see also Figure S9, protecting group SPDP = 3-(2-pyridyldithio)propionyl) was deprotected following the procedure of TCEP disulfide reducing gel (ThermoFischer Scientific). 50 μL of TCEP gel slurry were added in a vial and washed with PBS 7.2, centrifuging the vial at 1000g for 1 min. Then, an equal amount of PNA disulfide solution (50 μL) was added to the equilibrated gel solution and gently stirred for at least 1 h. After the incubation, the vial was centrifuged again for 1 min and the supernatant containing the PNA-thiol was recovered and used directly after dilution to the appropriate working concentration.
PLL-OEG-Mal Synthesis
All the PLL-OEG-Mal polymers with different degrees of functionalization of OEG and Mal were synthesized adapting the procedure of Duan et al.37 PLL HBr was dissolved in PBS buffer (pH 7.0) at a concentration of 10 mg/mL. The desired stoichiometric ratios (vs lysine monomer) of methyl-OEG4-NHS ester and Mal-OEG4-NHS ester were added simultaneously to the mixture, under vigorous stirring (argon atmosphere), and reacted for 4 h at room temperature. Thereafter, the crude mixture was dialyzed by Zeba spin desalting columns (7 kDa MWCO, 5 mL, ThermoFischer Scientific). The filtered solution was immediately freeze-dried overnight. Final compounds were stored at −20 °C as powder or stock solutions of 1 mg/mL in PBS 7.2.
1H NMR (400 MHz D2O, pH 6.5) δ [ppm] = 1.26–1.55 ((lysine γ-CH2), 1.63–1.83 (lysine β,δ-CH2), 2.50 (ethylene glycol CH2 from both OEG and Mal coupled, −CH2–C(=O)–NH), 3.00 (free lysine, H2N–CH2), 3.16 (ethylene glycol CH2 of coupled lysine from both OEG and Mal, C(=O)–NH–CH2−), 3.31 (OEG methoxy, −O–CH3), 3.65 (oligo ethylene glycol from both OEG and Mal, CH2–O−), 4.29 (lysine backbone, NH–CH–C(O)−), 6.86 (maleimide from coupled Mal −C(=O)–CH–CH–C(=O)−).
Quartz Crystal Microbalance (QCM)
Silica-coated (50 nm, QSX303) and gold-coated (50 nm, QSX301) QCM-D sensors from LOT-Quantum were washed with water and EtOH, sonicated in EtOH for 5 min, dried in a stream of nitrogen and finally oxidized in oxygen plasma (Plasma Prep II, SPI Supplies; 200–230 mTorr, 40 mA) for 5 or 1 min, respectively. QCM-D measurements were performed using a Q-Sense E4 4-channel quartz crystal microbalance with a peristaltic pump (Biolin Scientific). All experiments were performed in PBS buffer (10 mM, pH 7.2) with a flow rate of 80 μL/min at 22 °C.
The Sauerbrey eq (eq 1)53 establishes the relationship between the measured frequency change (Δf) and the adsorbed mass per unit area (Δm)53
| 1 |
where Δf is the frequency shift, Δm is the mass change, and C is the Sauerbrey constant (17.7 Hz/ng at f = 5 MHz).54 Frequency shifts of the n th harmonic Δf(n) are normalized to yield Δf = Δf(n)/n where n is the number of the overtone (n = 1, 3, 5, etc.).55
The frequency shift for each step was calculated by subtracting the plateau value of the frequency prior to the injection of the molecule (modified PLL, PNA probe, cDNA) to the plateau value after the following PBS washing step. Although eq 1 generally overestimates the adsorbed mass for viscoelastic layers measured in liquid environment,56 the Sauerbrey equation is still applicable when the dissipation change is below 2.0 × 10–6, because the film can then be assumed to be rigid.57 This limit was respected in all the experiments shown here.
Cyclic Voltammetry
Gold substrates were rinsed with Milli-Q water and ethanol and sonicated for 5 min in a mixture 1:1 of the same solvents. They were then dried and activated by oxygen plasma (Plasma Prep II, SPI Supplies; 200–230 mTorr, 40 mA, 5 min) and immersed in 0.3 mg/mL of the respective PLL-OEG-Mal solution for 1 h in PBS buffer (pH 7.0–7.2). Upon rinsing with Milli-Q water and drying, chips were immersed in 1 μM PNA-thiol (KRAS-WT) solution in PBS, pH 7.2 for 1 h. Afterward, substrates were again rinsed with Milli-Q water and dried, and were covered by a 1 μM cDNA-MB solution (5′-MB-AG CTG GTG GCG TAG-3′) in PBS at pH 7.2 for 1 h.
Electrochemical measurements were performed using PLL-modified Au substrates in a three-electrode setup (custom-built glass electrochemical cell) with a platinum disk as counter electrode, a red rod reference electrode (Ag/AgCl, saturated KCl solution, Radiometer Analytical), and the functionalized gold substrates as working electrode (area = 0.44 cm2) vs normal hydrogen electrode (NHE). Data analysis was done using CHI760D software (CH Instruments, Inc. Austin, USA) using eq 2 to quantify the surface coverage Γ:
| 2 |
where ip is the measured peak current (A), F is the Faraday constant (96 485.34 C/mol), n is the number of electrons involved in the redox process (n = 2 for methylene blue), R is the gas constant (8.3145 J/(K·mol)), T the absolute temperature (K), v the scan rate (V/s), A the area of the working electrode (here 0.44 cm2), and Γ is the surface coverage (mol/cm2). CV experiments were recorded at several scan rates between 25 and 500 mV/s and the respective current peaks ip were determined by Gaussian fitting (using the linear baseline correction) in the CHI760D software (detailed description in SI, electrochemical analysis with Figure S8). Afterward, ip was plotted against the scan rate and a linear regression was performed to calculate Γ (forced to pass to the origin). Results are expressed in molecules/cm2, which means that Γ has been multiplied by Avogadro’s number (NA). All measurements were carried out using aqueous 0.1 M NaClO4 as the electrolyte, upon degassing the electrolyte solution for 5 min.
Ellipsometry
Silicon oxide substrates were activated in oxygen plasma for 1 min and functionalized by dipping them in a PLL-OEG(23.0)-Mal(4.1) solution (0.3 mg/mL in PBS 7.2) for 30 min. After rinsing with Milli-Q water, the layer thickness was measured by ellipsometry (Woollam M-2000UI) in the range of 245–1690 nm, with a spatial resolution of 1.6 nm (245–100 nm) and 3.2 (1000–1690 nm) and the beam diameter of 300 μm. The ellipsometry data (values given are averages over 25 spots on the surface) were obtained at an incident angle of 75° and fitted with a Cauchy layer with a refractive index of 1.46.
Acknowledgments
The Horizon 2020 Health project “ULTRAPLACAD” (nr. 633937) is acknowledged for financial support. R.R. acknowledges financial support from the MESA+ Institute for Nanotechnology, University of Twente, Early Diagnostics project.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.8b00733.
1H NMR spectra of PLL-OEG-Mal and PLL-OEG; QCM experiments of biointerface formation and selectivity tests; CV data and data treatment; chemical structure of PNA linker (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- North S. H.; Lock E. H.; Taitt C. R.; Walton S. G. (2010) Critical aspects of biointerface design and their impact on biosensor development. Anal. Bioanal. Chem. 397, 925–933. 10.1007/s00216-010-3637-4. [DOI] [PubMed] [Google Scholar]
- Seeman C. N.; Sleiman F. H. (2017) DNA nanotechnology. Nat. Rev. Mater. 3, 17068. 10.1038/natrevmats.2017.68. [DOI] [Google Scholar]
- Huang Z.; Huang D.; Ni S.; Peng Z.; Sheng W.; Du X. (2010) Plasma microRNAs are promising novel biomarkers for early detection of colorectal cancer. Int. J. Cancer 127, 118–126. 10.1002/ijc.25007. [DOI] [PubMed] [Google Scholar]
- Piliarik M.; Párová L.; Homola J. (2009) High-throughput spr sensor for food safety. Biosens. Bioelectron. 24, 1399–1404. 10.1016/j.bios.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Levin B. C.; Cheng H.; Reeder D. J. (1999) A human mitochondrial DNA standard reference material for quality control in forensic identification, medical diagnosis, and mutation detection. Genomics 55, 135–146. 10.1006/geno.1998.5513. [DOI] [PubMed] [Google Scholar]
- Marchelli R., Tedeschi T., and Tonelli A. (2012) DNA anlyses in Food Safety and quality: current status and expectations, In Detection of Non-Amplified Genomic DNA (Spoto G., and Corradini R., Eds) pp 25–63, Chapter 2, Soft and Biological Matter, Springer, Dordrecht. [Google Scholar]
- Grody W. W.; Cutting G. R.; Klinger K. W.; Richards C. S.; Watson M. S.; Desnick R. J. (2001) Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet. Med. 3, 149–154. 10.1097/00125817-200103000-00010. [DOI] [PubMed] [Google Scholar]
- Bellassai N.; Spoto G. (2016) Biosensors for liquid biopsy: circulating nucleic acids to diagnose and treat cancer. Anal. Bioanal. Chem. 408, 7255–7264. 10.1007/s00216-016-9806-3. [DOI] [PubMed] [Google Scholar]
- Drummond T. G.; Hill M. G.; Barton J. K. (2003) Electrochemical DNA sensors. Nat. Biotechnol. 21, 1192–1199. 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- Wang J. (2002) Electrochemical nucleic acid biosensors. Anal. Chim. Acta 469, 63–71. 10.1016/S0003-2670(01)01399-X. [DOI] [Google Scholar]
- Bizzotto D.; Burgess I. J.; Doneux T.; Sagara T.; Yu H.-Z. (2018) Beyond simple cartoons: challenges in characterizing electrochemical biosensor interfaces. ACS Sensors. 3, 5–12. 10.1021/acssensors.7b00840. [DOI] [PubMed] [Google Scholar]
- Xu C.; Cai H.; Xu Q.; He P.; Fang Y. (2001) Characterization of single-stranded DNA on chitosan-modified electrode and its application to the sequence-specific DNA detection. Fresenius' J. Anal. Chem. 369, 428–432. 10.1007/s002160000673. [DOI] [PubMed] [Google Scholar]
- Belosludtsev Y.; Iverson B.; Lemeshko S.; Eggers R.; Wiese R.; Lee S.; Powdrill T.; Hogan M. (2001) DNA microarrays based on noncovalent oligonucleotide attachment and hybridization in two dimensions. Anal. Biochem. 292, 250–256. 10.1006/abio.2001.5088. [DOI] [PubMed] [Google Scholar]
- Pavlov V.; Xiao Y.; Gill R.; Dishon A.; Kotler M.; Willner I. (2004) Amplified chemiluminescence surface detection of DNA and telomerase activity using catalytic nucleic acid labels. Anal. Chem. 76, 2152–2156. 10.1021/ac035219l. [DOI] [PubMed] [Google Scholar]
- Walsh M. K.; Wang X.; Weimer B. C. (2001) Optimizing the immobilization of single-stranded DNA onto glass beads optimizing the immobilization of single-stranded DNA onto glass beads. J. Biochem. Biophys. Methods 47, 221–231. 10.1016/S0165-022X(00)00146-9. [DOI] [PubMed] [Google Scholar]
- Jung D.-H.; Kim B. H.; Ko Y. K.; Jung M. S.; Jung S.; Lee S. Y.; Jung H.-T. (2004) Covalent attachment and hybridization of DNA oligonucleotides on patterned single-walled carbon nanotube films. Langmuir 20, 8886–8891. 10.1021/la0485778. [DOI] [PubMed] [Google Scholar]
- Howarter J. A.; Youngblood J. P. (2006) Optimization of silica silanization by 3-aminopropyltriethoxysilane. Langmuir 22, 11142–11147. 10.1021/la061240g. [DOI] [PubMed] [Google Scholar]
- Gunnarsson A.; Jönsson P.; Marie R.; Tegenfeldt J. O.; Höök F. (2008) Single-molecule detection and mismatch discrimination of unlabeled DNA targets. Nano Lett. 8, 183–188. 10.1021/nl072401j. [DOI] [PubMed] [Google Scholar]
- Evtugyn G. A.; Hianik T. (2011) Layer-by-layer polyelectrolyte assembles involving DNA as a platform for DNA sensors. Curr. Anal. Chem. 7, 8–34. 10.2174/157341111793797626. [DOI] [Google Scholar]
- Biagiotti V.; Porchetta A.; Desiderati S.; Plaxco K. W.; Palleschi G.; Ricci F. (2012) Probe accessibility effects on the performance of electrochemical biosensors employing DNA monolayers. Anal. Bioanal. Chem. 402, 413–421. 10.1007/s00216-011-5361-0. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Song S.; Zhang L.; Wang L.; Wu H.; Pan D.; Fan C. (2006) Sequence-specific detection of femtomolar DNA via a chronocoulometric DNA sensor (cds): effects of nanoparticle-mediated amplification and nanoscale control of DNA assembly at electrodes. J. Am. Chem. Soc. 128, 8575–8580. 10.1021/ja061521a. [DOI] [PubMed] [Google Scholar]
- Idili A.; Amodio A.; Vidonis M.; Feinberg-Somerson J.; Castronovo M.; Ricci F. (2014) Folding-upon-binding and signal-on electrochemical DNA sensor with high affinity and specificity. Anal. Chem. 86, 9013–9019. 10.1021/ac501418g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson A. W.; Heaton R. J.; Georgiadis R. M. (2001) The effect of surface probe density on DNA hybridization. Nucleic Acids Res. 29, 5163–5168. 10.1093/nar/29.24.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herne T. M.; Tarlov M. J. (1997) Characterization of DNA probes immobilized on gold surfaces. J. Am. Chem. Soc. 119, 8916–8920. 10.1021/ja9719586. [DOI] [Google Scholar]
- Macanovic A.; Marquette C.; Polychronakos C.; Lawrence M. F. (2004) Impedance-based detection of DNA sequences using a silicon transducer with PNA as the probe layer. Nucleic Acids Res. 32, 20. 10.1093/nar/gnh003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci F.; Lai R. Y.; Heeger A. J.; Plaxco K. W.; Sumner J. J. (2007) Effect of molecular crowding on the response of an electrochemical DNA sensor. Langmuir 23, 6827–6834. 10.1021/la700328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjällman T. H. M.; Peng H.; Soeller C.; Travas-Sejdic J. (2008) Effect of probe density and hybridization temperature on the response of an electrochemical hairpin-DNA sensor. Anal. Chem. 80, 9460–9466. 10.1021/ac801567d. [DOI] [PubMed] [Google Scholar]
- Yao D.; Kim J.; Yu F.; Nielsen P. E.; Sinner E.-K.; Knoll W. (2005) Surface density dependence of PCR amplicon hybridization on PNA/DNA probe layers. Biophys. J. 88, 2745–2751. 10.1529/biophysj.104.051656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M.; Shalon D.; Davis R. W.; Brown P. O. (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470. 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Pasche S.; De Paul S. M.; Vörös J.; Spencer N. D.; Textor M. (2003) Poly(l-lysine)-graft-poly(ethylene glycol) assembled monolayers on niobium oxide surfaces: a quantitative study of the influence of polymer interfacial architecture on resistance to protein adsorption by ToF-SIMS and in situ OWLS. Langmuir 19, 9216–9225. 10.1021/la034111y. [DOI] [Google Scholar]
- Huang N. P.; Vörös J.; De Paul S. M.; Textor M.; Spencer N. D. (2002) Biotin-derivatized poly(l-lysine)-g-poly(ethylene glycol): a novel polymeric interface for bioaffinity sensing. Langmuir 18, 220–230. 10.1021/la010913m. [DOI] [Google Scholar]
- Elbert D. L.; Hubbell J. A. (1998) Reduction of fibrous adhesion formation by a copolymer possessing an affinity for anionic surfaces. J. Biomed. Mater. Res. 42, 55–65. . [DOI] [PubMed] [Google Scholar]
- Kenausis G. L.; Vörös J.; Elbert D. L.; Huang N.; Hofer R.; Ruiz-Taylor L.; Textor M.; Hubbell J. A.; Spencer N. D. (2000) Poly(l-lysine)-g-poly(ethylene glycol) layers on metal oxide surfaces: attachment mechanism and effects of polymer architecture on resistance to protein adsorption. J. Phys. Chem. B 104, 3298–3309. 10.1021/jp993359m. [DOI] [Google Scholar]
- VandeVondele S.; Vörös J.; Hubbell J. A. (2003) RGD-grafted poly-l-lysine-graft-(polyethylene glycol) copolymers block non-specific protein adsorption while promoting cell adhesion. Biotechnol. Bioeng. 82, 784–790. 10.1002/bit.10625. [DOI] [PubMed] [Google Scholar]
- Huang N.; Vörös J.; De Paul S. M.; Textor M.; Spencer N. D. (2002) Biotin-derivatized poly(l-lysine)-g-poly(ethylene glycol): a novel polymeric interface for bioaffinity sensing. Langmuir 18, 220–230. 10.1021/la010913m. [DOI] [Google Scholar]
- Cao L.; Chang M.; Lee C. Y.; Castner D. G.; Sukavaneshvar S.; Ratner B. D.; Horbett T. A. (2007) Plasma-deposited tetraglyme surfaces greatly reduce total blood protein adsorption, contact activation, platelet adhesion, platelet procoagulant activity, and in vitro thrombus deposition. J. Biomed. Mater. Res., Part A 81, 827–837. 10.1002/jbm.a.31091. [DOI] [PubMed] [Google Scholar]
- Duan X.; Mu L.; Sawtelle S. D.; Rajan N. K.; Han Z.; Wang Y.; Qu H.; Reed M. a. (2015) Functionalized polyelectrolytes assembling on nano-biofets for biosensing applications. Adv. Funct. Mater. 25, 2279–2286. 10.1002/adfm.201500002. [DOI] [Google Scholar]
- Thierry A. R.; Mouliere F.; El Messaoudi S.; Mollevi C.; Lopez-Crapez E.; Rolet F.; Gillet B.; Gongora C.; Dechelotte P.; Robert B.; et al. (2014) Clinical validation of the detection of kras and braf mutations from circulating tumor DNA. Nat. Med. 20, 430–435. 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- Veerbeek J.; Steen R.; Vijselaar W.; Rurup W. F.; Korom S.; Rozzi A.; Corradini R.; Segerink L.; Huskens J. (2018) Selective functionalization with PNA of silicon nanowires on silicon oxide substrates. Langmuir 34, 11395–11404. 10.1021/acs.langmuir.8b02401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egholm M.; Buchardt O.; Christensen L.; Behrens C.; Freier S. M.; Driver D. A.; Berg R. H.; Kim S. K.; Norden B.; Nielsen P. E. (1993) PNA hybridizes to complementary oligonucleotides obeying the watson-crick hydrogen-bonding rules. Nature 365, 566–568. 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- Corradini R.; Feriotto G.; Sforza S.; Marchelli R.; Gambari R. (2004) Enhanced recognition of cystic fibrosis W1282X DNA point mutation by chiral peptide nucleic acid probes by a surface plasmon resonance biosensor. J. Mol. Recognit. 17, 76–84. 10.1002/jmr.646. [DOI] [PubMed] [Google Scholar]
- Kwon M. J.; Lee S. E.; Kang S. Y.; La Choi Y. (2011) Frequency of kras, braf, and pik3ca mutations in advanced colorectal cancers: comparison of peptide nucleic acid-mediated PCR clamping and direct sequencing in formalin-fixed, paraffin-embedded tissue. Pathol., Res. Pract. 207, 762–768. 10.1016/j.prp.2011.10.002. [DOI] [PubMed] [Google Scholar]
- D’Agata R.; Giuffrida M. C.; Spoto G. (2017) Peptide nucleic acid-based biosensors for cancer diagnosis. Molecules 22, 1951. 10.3390/molecules22111951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman B. T.; Gawalt E. S.; Mrksich M. (2003) Maleimide-functionalized self-assembled monolayers for the preparation of peptide and carbohydrate biochips. Langmuir 19, 1522–1531. 10.1021/la0262304. [DOI] [Google Scholar]
- Lee C. Y.; Nguyen P. C. T.; Grainger D. W.; Gamble L. J.; Castner D. G. (2007) Structure and DNA hybridization properties of mixed nucleic acid/maleimide-ethylene glycol monolayers. Anal. Chem. 79, 4390–4400. 10.1021/ac0703395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z.; Wang Y.; Duan X. (2017) Biofunctional polyelectrolytes assembling on biosensors – a versatile surface coating method for protein detections. Anal. Chim. Acta 964, 170–177. 10.1016/j.aca.2017.01.051. [DOI] [PubMed] [Google Scholar]
- Frey B. L.; Corn R. M. (1996) Covalent attachment and derivatization of poly(l-lysine) monolayers on gold surfaces as characterized by polarization-modulation ft-ir spectroscopy. Anal. Chem. 68, 3187–3193. 10.1021/ac9605861. [DOI] [Google Scholar]
- Peters T. (1996) All about Albumin. Biochemistry, Genetics, and Medical Applications, Academic Press, San Diego, CA. [Google Scholar]
- Nielsen P. E.; Egholm M.; Buchardt O. (1994) Peptide nucleic acid (PNA). a DNA mimic with a peptide backbone. Bioconjugate Chem. 5, 3–7. 10.1021/bc00025a001. [DOI] [PubMed] [Google Scholar]
- Wittung P.; Nielsen P. E.; Buchardt O.; Egholm M.; Nordén B. (1994) DNA-like double helix formed by peptide nucleic acid. Nature 368, 561. 10.1038/368561a0. [DOI] [PubMed] [Google Scholar]
- Larsson C.; Rodahl M.; Höök F. (2003) Characterization of DNA immobilization and subsequent hybridization on a 2D arrangement of streptavidin on a biotin-modified lipid bilayer supported on SiO2. Anal. Chem. 75, 5080–5087. 10.1021/ac034269n. [DOI] [PubMed] [Google Scholar]
- Höök F.; Ray A.; Nordén B.; Kasemo B. (2001) Characterization of PNA and DNA immobilization and subsequent hybridization with DNA using acoustic-shear-wave attenuation measurements. Langmuir 17, 8305–8312. 10.1021/la0107704. [DOI] [Google Scholar]
- Sauerbrey G. (1959) Verwendung von schwingquarzen zur wägung dünner schichten und zur mikrowägung. Eur. Phys. J. A 155, 206–222. 10.1007/BF01337937. [DOI] [Google Scholar]
- Bračič M.; Mohan T.; Kargl R.; Griesser T.; Hribernik S.; Köstler S.; Stana-Kleinschek K.; Fras-Zemljič L. (2014) Preparation of pdms ultrathin films and patterned surface modification with cellulose. RSC Adv. 4, 11955. 10.1039/C3RA47380E. [DOI] [Google Scholar]
- Butergerds D.; Cramer C.; Schonhoff M. (2017) PH-dependent growth laws and viscoelastic parameters of poly-l-lysine/hyaluronic acid multilayers. Adv. Mater. Interfaces 4, 1600592. 10.1002/admi.201600592. [DOI] [Google Scholar]
- Vörös J. (2004) The density and refractive index of adsorbing protein layers. Biophys. J. 87, 553–561. 10.1529/biophysj.103.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt B. D.; Soles C. L.; Lee H. J.; Lin E. K.; Wu W. L. (2004) Moisture absorption and absorption kinetics in polyelectrolyte films: influence of film thickness. Langmuir 20, 1453–1458. 10.1021/la035239i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.