

Abstract
Foveal hypoplasia is a retinal disorder in which there is a lack of full development of the morphology of the fovea. The optical coherence tomography (OCT) and functional findings are presented in relation to the underlying genetic and developmental conditions. Recent advancements of high-resolution OCT imaging have unveiled characteristics of foveal hypoplasia that were not detected by conventional imaging methods. An absence of a foveal pit does not necessarily imply poor visual acuity, and the maturation of the cone photoreceptors is important for the visual acuity. Regardless of the degree of the development of the inner retinal layers, the visual acuity can be preserved as in diseases such as Stickler syndrome that is a newly identified retinal disorder associated with foveal hypoplasia.
Keywords: Achromatopsia, albinism, aniridia, familial exudative vitreoretinopathy, foveal avascular zone, foveal hypoplasia, incontinentia pigmenti, optic nerve hypoplasia, optical coherent tomography, retinopathy of prematurity, Stickler syndrome
Introduction
In humans and some primates, the central retina has a specialized structure called the foveal pit which is referred to as the fovea centralis. The pit is formed by the migration of the inner retinal neurons peripherally, and the underlying cone photoreceptors elongate and are densely packed. Foveal hypoplasia is a condition in which the foveal pit does not fully develop.[1] It is manifested by a reduction or absence of a foveal pit and a foveal avascular zone (FAZ). Foveal hypoplasia has been associated with poor vision and nystagmus,[1] and various diseases are known to be associated with foveal hypoplasia including albinism and aniridia.[2] However, the mechanism causing the lack of development of the foveal pit has not been definitively determined, and the genetic and nongenetic factors, such as prematurity of birth, can affect the development of the fovea. Foveal hypoplasia has been identified by ophthalmoscopy, fluorescein angiography, and histology before the advent of optical coherence tomography (OCT).[3,4]
Advances of high-resolution spectral-domain OCT and swept-source OCT have allowed ophthalmologists to obtain detailed views of the morphology of the retina and to investigate the morphology of normal and pathological maculas.[5,6] Recently, a new imaging method called OCT angiography (OCTA) has allowed clinicians and researchers to obtain detailed information on the retinal microvasculature in en-face images of different layers of the retina.[7] Owing to the progress of OCT imaging, foveal hypoplasia has been identified as a novel feature that is associated with known retinal diseases.
The aim of this review is to present the characteristics of diseases associated with foveal hypoplasia and to present the OCT images and functional assessments in relation to the underlying genetics or development conditions. The diseases can be characterized by their frequency of foveal hypoplasia, the underlining genetic and nongenetic conditions, and the presence or absence of vision impairments [Table 1].
Table 1.
Diseases associated with foveal hypoplasia
| Diseases | Factors (genes; roles) | Type of foveal hypoplasia | Frequency of foveal hypoplasia | Nystagmus and poor vision | Reduced macular pigment |
|---|---|---|---|---|---|
| Albinism | Genetic (albinism genes;[8] melanin synthesis) | I + O | Frequent | Common | Found |
| Aniridia | Genetic (PAX6; pan-ocular genesis) | I + O | Frequent | Common | Found |
| Idiopathic foveal hypoplasia | Genetic (PAX6, GPR143, SLC38A8; special forms of albinism or aniridia) | I + O | Frequent | Common | Found |
| Retinopathy of prematurity | Nongenetic (retinal vascularization) | I + O | Frequent in severe cases | Uncommon for isolated foveal hypoplasia | Not evident |
| Incontinentia pigmenti | Genetic (IKBKG; genesis of ectoderm) | I | Infrequent | Uncommon for isolated foveal hypoplasia | Not evident |
| Achromatopsia | Genetic (achromatopsia genes;[9] cone genesis) | I + O | 50%-80%[10,11,12] | Common due to cone dysfunction | Found (complicated as hyper-FAF with progressive retinal degeneration |
| Optic nerve hypoplasia | Genetic (PAX6, PAX2, and others;[13] ganglion cell genesis) | I | 24%-82%[14,15] | Common due to dysgenesis of retinal ganglion cells | Not evident |
| Familial exudative vitreoretinopathy | Genetic (Wnt signaling genes:[16] Retinal vascularization) | I | 20%[17] | Uncommon for isolated foveal hypoplasia | Not evident |
| Stickler syndrome | Genetic (procollagen genes[18]) | I | 82%[19] | Uncommon | Not evident |
FAF=Fundus autofluorescence, I=Persistence of the inner retinal layer, O=Failed cone specialization in the outer retinal layer
Development of Normal Fovea
Foveal avascular zone
In humans, the normal development of the fovea begins at fetal week 25 and is completed by 15–45 months after birth.[20] The location of the future fovea is determined by fetal week 11.[21] The formation of the pit does not take place until the FAZ is formed.[22,23] Therefore, the morphology of the foveal pit is correlated with the FAZ, i.e. larger FAZs are associated with deeper and broader foveal pits.[24] Dubis et al.[24] analyzed the relationship between the FAZ and size of the foveal pit by OCT and other devices, and they reported that the size of the FAZ in normal participants is highly variable with the area ranging from 0.05 to 1.05 mm2 and the diameter ranging from 0.2 to 1.08 mm.
The macular pigment appears at about 17 weeks of gestational age and is believed to drive the formation of the FAZ.[25] Its spatial distribution is correlated with the size of the FAZ.[26] The fovea is made up of the cone photoreceptors and Müller cells, and the rod photoreceptors are absent from the central fovea.[27] The Müller cells inhibit the migration of astrocytes into the central fovea so that an astrocyte-free zone is formed.[26] The astrocyte-free zone plays an important role in forming the FAZ because astrocytes induce the migration of retinal vessel cells across the retina.[28]
Development of the foveal pit
The development of the foveal pit involves the bidirectional movement of the neurons in the inner and outer retina [Figure 1].[22] The cells of the inner retina are displaced centrifugally to form the foveal pit, and the cone cells are displaced centripetally so that the density of cones in the foveal pit increases.[22]
Figure 1.
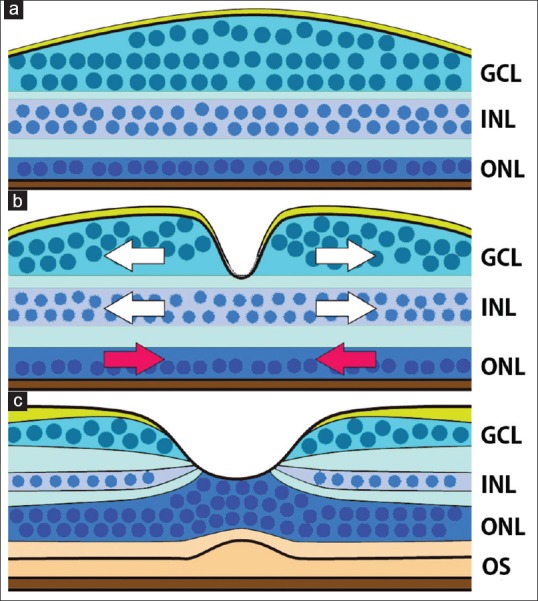
Schematic diagram showing the development of the foveal structure. (a) At the end of the second trimester, a thickened ganglion cell layer is found, however, a foveal pit is not present. The outer nuclear layer is a single-cell layer. (b) A foveal pit forms in the ganglion cell layer and inner nuclear layer. The cells in the ganglion cell layer and inner nuclear layer are displaced centrifugally to form the foveal pit (white arrows), and cone cells in the outer nuclear layer are displace centripetally (red arrows). (c) In the adult retina, the cone cells form a multicellular layer, and the inner and outer segments become prominent. The schema is based on references Springer and Hendrickson [23] and Hendrickson[29]
During the third trimester and after birth, the ganglion cells and cells in the inner nuclear layer continue to be displaced away from the foveal pit and the FAZ.[29] The pit depth is reported to be 137.56 ± 4.3 μm, and the foveal pit diameter is 1.12–2.57 mm according to the analysis using spectral-domain OCT images.[24] Postnatally, the outer retinal layers mature as cone cells migrate centripetally and appear as a multicellular layer.[27] This phenomenon is referred to as cone specialization that includes a thinning of the individual cones and an increase in the density of foveal cones. The inner and outer segments (OSs) elongate, and the axonal processes lengthen and bend to form the “fibers of Henle.”[22,29] The lengthening of the OSs and development of the rod-free zones are believed to be associated with good visual acuity. The rod-free zone is >1000 μm in diameter before birth and becomes reduced to 650–700 μm by 45 months of age.[27] At 14 fetal weeks, the cone density at the fovea is <15,000/mm,2 and it reaches 108,000/mm2 by 4 years of age.[29]
Thomas et al.[30] proposed a structural grading system of foveal hypoplasia based on the cross-sectional OCT images: Grade 1, a shallow foveal pit and presence of a thick outer nuclear layer (ONL) and presence of OSs in the foveal pit; Grade 2, absence of foveal pit; Grade 3, absence of OS lengthening; and Grade 4, absence of ONL widening. Grades 1 and 2 represent deficient foveal pit formation with intact cone specialization, while Grades 3 and 4 represent a failure of cone specialization [Figure 2].
Figure 2.
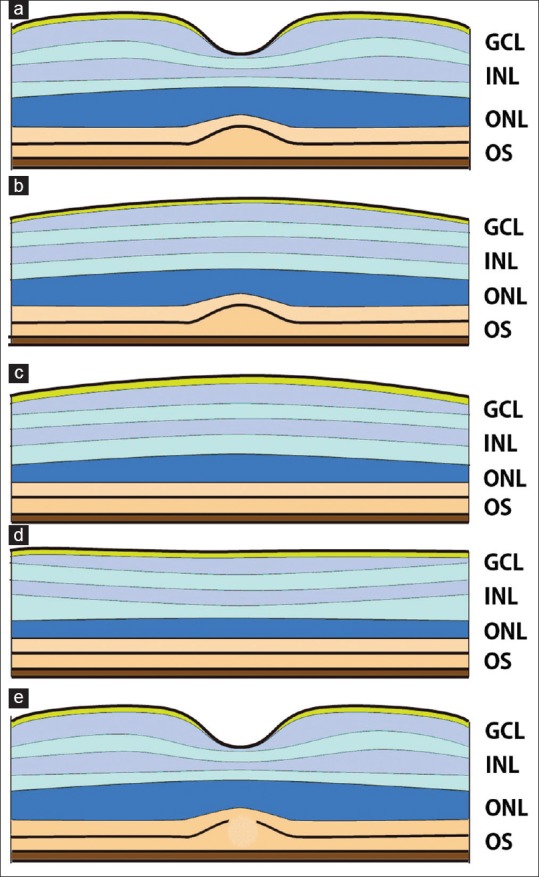
Structural grading system of foveal hypoplasia using cross-sectional optical coherence tomography images after Thomas et al.[30] (a) Grade 1, a shallow foveal pit and presence of outer nuclear layer and outer segment in the fovea. (b) Grade 2, absence of foveal pit. (c) Grade 3, absence of outer segments lengthening. (d) Grade 4, absence of outer nuclear layer widening. (e) Atypical form of foveal hypoplasia with disruption of the inner and outer segments of photoreceptor, a sign of photoreceptor degeneration. Grades 1 and 2 represent deficient foveal pit formation without affecting the cone specialization while Grades 3 and 4 represent a failure in the cone specialization
Diseases Known to be Associated with Foveal Hypoplasia Before Optical Coherence Tomography Imaging
Before the advent of OCT imaging, several diseases were known to be associated with foveal hypoplasia. These include albinism, aniridia, isolated foveal hypoplasia, retinopathy of prematurity, and incontinentia pigmenti (see below). In addition, foveal hypoplasia was also reported to be associated with microcornea and familial and presenile cataract; however, the details have not been published.[2]
Albinism
Oculocutaneous albinism (OCA) is a group of autosomal recessive disorders that is characterized by hypopigmentation of eyes, skin, and hair due to an impairment of melanin synthesis.[8] Ocular albinism (OA) is an X-linked recessive disorder that only affects the eyes. Ocular symptoms in OCA and OA include poor visual acuity, nystagmus, strabismus, hypopigmentation of the uvea, and foveal hypoplasia. Optic nerve fibers misrouting is also a characteristic of OCA and OA.[31] Seven types of human OCA, OCA 1–7, have been identified and six genes have been found to be the cause of these OCAs.[8] In addition, there are several syndromic forms of albinism including the Hermansky–Pudlak and Chediak–Higashi syndromes.[8]
Despite the genetic heterogeneity of albinism, patients with albinism are believed to have foveal hypoplasia consistently. The relationship between the visual function and abnormal pit formation in albinism has been investigated by time-domain and spectral-domain OCT images.[32,33,34,35,36] It has been debated whether the degree of foveal pit formation is correlated with the visual acuity.[33,34,35] Mohammad et al.[36] showed that the visual acuity was not correlated with the presence of the inner retinal layers, and the length of the photoreceptor layer was the best predictor of the visual acuity. This led to a conclusion that the pit formation is not required for cone specialization.[34,35]
It is not known whether the foveal hypoplasia in eyes with OCA and OA is related to the abnormal melanin synthesis.[31] Patients with albinism lack a rod-free zone that is essential for cone specialization.[37,38] Macular pigmentation is also deficient, and the phenomenon is believed to be associated with the poor vision.[39,40]
Aniridia
Aniridia is caused by mutations in the PAX6 gene.[41] Aside from the characteristic iris dysgenesis, the findings in eyes with aniridia include poor vision and nystagmus. The PAX6 gene is a transcription factor that encodes a key regulator of ocular and neural development. As seen in eyes with albinism, a lack of macular pigment is also a characteristic in eyes with aniridia.[42]
Patients with aniridia are consistently associated with foveal hypoplasia with the exception of hypomorphic mutations in the PAX6 gene. Yokoi et al.[43] analyzed 28 patients who had mutations in the PAX6 gene or chromosomal abnormality including the PAX6 gene region, and all but one case had foveal hypoplasia. Hingorani et al.[44] analyzed 43 patients with a PAX6 mutation and found that 34 of 35 patients with foveal hypoplasia had gene truncation mutations, i.e., nonsense, frameshift, splicing, or C-terminal extension mutations, or gene deletions. However, foveal hypoplasia was not found in 6 of 9 patients who had missense mutations. The visual acuities in the better eye ranged from light perception to 20/80 in the patients with truncation mutations while the visual acuities in the better eyes in the patients with missense mutations ranged from 20/30 to 20/250 with a median of 20/60. Two patients lacked nystagmus.[44] Katagiri et al.[45] reported that different OCT images of Henle's fiber layer reflected different grades of foveal maturity with regard to the visual acuity in eyes with aniridia and isolated foveal hypoplasia.
The macular pigment is also deficient in eyes with aniridia and is believed to be associated with poor vision.[40,42]
Isolated foveal hypoplasia
Isolated foveal hypoplasia is a condition that lacks any other ocular manifestations. Hypomorphic mutations of the PAX6 gene are known to cause isolated foveal hypoplasia which has an autosomal dominant inheritance.[46,47] Patients with OA with a GPR143 mutation can have isolated foveal hypoplasia.[48] Recently, mutations in the SLC38A8 gene were identified to cause autosomal recessive, isolated foveal hypoplasia.[49,50] The gene encodes a putative sodium-dependent amino-acid/proton transporter. So far, 11 mutations in the SLC38A8 gene from 11 families have been reported, and all cases showed typical foveal hypoplasia.[49,50,51]
Retinopathy of prematurity
Retinopathy of prematurity is an environmental disorder that is caused by hypoxia of the retina due to arrested development of the retinal vessels.[52] Secondary fibrovascular proliferation results in tractional retinal detachment that can lead to blindness. A pathological expression of vascular endothelial growth factor in the retinal tissues is associated with the progression of the disease process.
A disruption of foveal development is associated with retinopathy of prematurity.[53,54] Thus, morphological abnormalities in the macular region including retention of inner retinal layers and absence of a foveal pit as well as macular edema have been detected in the OCT images.[55,56,57] Perifoveal capillaries are formed during late gestation, and foveal development can be affected by the prematurity.[28] The photoreceptor layer is thin in the fovea of eyes of premature neonates.[55] The diameter of the FAZ is reduced in eyes with retinopathy of prematurity.[54] Villegas et al.[58] reported that an abnormal foveal pit was present in children between 2-and 18-years-of-age with a history of retinopathy of prematurity. Twenty-four of these eyes (64%) had a visual acuity of 20/40 or better. The reduction of the visual acuity was correlated with the presence of myopia but not with the structural changes seen in the OCT images.[58] The macular pigment is deficient in premature babies; however, its relevance to the visual acuity is unknown.[59]
Falavarjani et al.[60] showed that the FAZ area in 43 eyes with a history of prematurity was smaller than that of controls by OCTA. Twelve of these eyes totally lacked the FAZ. Nonobe et al.[61] found that the median FAZ area in 10 patients with a history of retinal coagulation therapy was 0.103 mm2 by OCTA which was significantly smaller than that of normal participants.
Incontinentia pigmenti
Incontinentia pigmenti is an X-linked dominant retinal disorder characterized by pathognomonic skin rashes on the trunk and limbs.[62] The disease affects various tissues of ectodermal origin including the retina and the central nervous system. Mutations in the IKBKG gene cause incontinentia pigmenti.[62]
The disease is associated with occlusive changes throughout the retinal vasculature. The macular abnormalities include an abnormal vascular pattern in the parafovea and foveal hypoplasia.[63,64] However, more than 90% of patients with incontinentia pigmenti have normal vision, and the consequences of the foveal hypoplasia remain unknown.[65] A small case series study (n = 4) using spectral-domain OCT showed that the foveal hypoplasia was associated with eyes with incontinentia pigmenti.[63,66] The eyes with reduced vision were associated with irregularities of the FAZ detected by fluorescein angiography and a persistence of inner retinal layer by OCT.[66] The frequency, severity, and visual impairments of the foveal hypoplasia are yet to be determined in patients with incontinentia pigmenti.
Diseases Newly Identified to be Associated with Foveal Hypoplasia by Optical Coherence Tomography Imaging
With the advent of high-resolution OCT imaging, signs of foveal hypoplasia have been identified in eyes with achromatopsia, optic nerve hypoplasia, familial exudative vitreoretinopathy (FEVR), and Stickler syndrome. The signs are the persistence of the inner retinal layers as detected in the cross-sectional OCT images and reduced size of the FAZ by OCT angiography. In contrast to eyes with achromatopsia or optic nerve hypoplasia in which poor vision is essentially due to cone dysfunction or dysgenesis of retinal ganglion cells, eyes with FEVR and Stickler syndrome tend to have good visual acuities regardless of the severity of foveal pit malformation [Figure 3].
Figure 3.
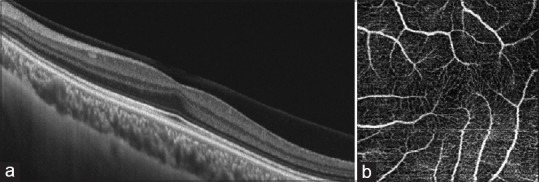
Eye in patient with Stickler syndrome showing foveal hypoplasia. The left eye of a 22-year-old male patient diagnosed with Stickler syndrome with a best-corrected visual acuity of 1.0. (a) Cross-sectional optical coherence tomography image showing persistence of inner retinal layers, and the presence of a widening of the outer nuclear layer and lengthening of the outer segments leading to the “foveal bulge.”(b) Optical coherence tomography -angiographic image showing a lack of foveal avascular zone suggesting foveal hypoplasia
Achromatopsia
Achromatopsia is an autosomal recessive disorder with deficits of cone function.[9] The disease is characterized by low vision, photophobia, and nystagmus. The conventional photopic ERGs are essentially absent whereas the scotopic ERGs remain intact. Mutations in six genes, CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, and ATF6, are known to cause achromatopsia.[9] These genes encode proteins in the cone photoreceptors involved in phototransduction except for the ATF6 gene which is a transcription factor that regulates the unfolded protein response during endoplasmic reticulum maintenance.[9] Foveal hypoplasia in eyes with achromatopsia was first reported by Thiadens et al. as a sign of impaired foveal pit formation using OCT imaging.[10] Foveal hypoplasia was found in 50%–80% of patients with achromatopsia.[10,11,12] Sundaram et al.[11] found no significant differences in the age, contrast sensitivity, retinal sensitivity, and fixation stability between patients with and without foveal hypoplasia. A thickening of the outer nuclear layer was not observed.[10] Notably, the photoreceptor layer in the fovea was disrupted including the absence of the inner segment ellipsoids, hyporeflective zone, and atrophy as seen in the OCT images [Figure 2].[11,12] Adaptive optics scanning light ophthalmoscopy revealed that the foveal cone density is significantly lower than that of normal eyes.[67] These signs in the outer retinal layers seem to progress while the morphology of foveal pit did not progress.[10] Abnormalities of the near-infrared fundus autofluorescence images were also reported in eyes with achromatopsia.[68] According to Greenberg et al.,[69] the degree and distribution of the fundus autofluorescence vary considerably among patients, and 53% (9/17) had reduced or absent autofluorescence. Hyperautofluorescence was also observed as a sign of progressive retinal degeneration.
Optic nerve hypoplasia
Optic nerve hypoplasia is a congenital anomaly of the optic nerve caused by dysgenesis of retinal ganglion cells.[70] Optic nerve hypoplasia is manifested by a small-sized optic nerve head with a peripapillary hypopigmentation referred to as “double-ring sign.”[70] The etiology is highly diverse, and mutations in genes, involved in transcription regulation (e.g., PAX6 and PAX2), chromatin remodeling, a-dystroglycan glycosylation, cytoskeleton protein, RNA splicing, and MAP kinase signaling pathway, have been associated with optic nerve hypoplasia.[13,71] Optic nerve hypoplasia can be manifested by a thinner retinal nerve fiber layer and ganglion cell layer.[70] Foveal hypoplasia was found in a case with mutation in the PAX2 gene.[72] Pilat et al.[14] reported that 82% (18/22) of eyes with optic nerve hypoplasia had a persistence of the inner retinal layers in the fovea in the spectral-domain OCT images. Katagiri et al.[15] reported that 24% (7/29) of eyes with optic nerve hypoplasia had foveal hypoplasia.
Familial exudative vitreoretinopathy
FEVR is a hereditary vitreoretinal disorder characterized by insufficient vascular development in the peripheral retina.[16] Secondary pathological processes lead to various forms of retinal detachments with fibrovascular proliferation. The genes causing FEVR are highly heterogeneous, and aside from very rare causative genes, four genes, FZD4, LRP5, TSPAN12, and NDP, are known to cause FEVR.[16,73] The main causative genes are related to Wnt signaling, a key regulator of cellular proliferation and differentiation, and mutations in these genes account for ~50% of the FEVR cases.[16,73]
Eyes with FEVR have different degrees of severities of the retina, and the vascular system ranging from mild vascular abnormalities in the peripheral retina to total retinal detachment. Secondary proliferative changes in the periphery can affect the macular structure which is seen as an ectopic macula and falciform retinal folds. The deformed macular structures have hindered the detection of microstructural changes in eyes with FEVR. Yonekawa et al.[17] found various abnormal microstructural findings by spectral-domain OCT. Twenty-two (20%) of 52 eyes with Stage 1 and Stage 2 FEVR, i. e. milder forms not accompanied by a retinal detachment, had a persistence of the inner retinal layers in the fovea which is analogous to mild foveal hypoplasia. The persistence of the inner retinal layer did not appear to cause the reduced visual acuity when the underlying outer retinal structures remain intact.[17] As the majority of mild cases of FEVR are asymptomatic, it is difficult to estimate the exact frequency of foveal hypoplasia in FEVR patients.
Stickler syndrome
Stickler syndrome is an inherited connective tissue disease that affects the eyes, ears, joints, and midline facial structures.[18] The ocular features are high myopia, vitreous degeneration, cataracts, perivascular retinal degeneration, and a high incidence of retinal detachments.[74] The retinal detachments occur during childhood and can lead to blindness.[75] Stickler syndrome is caused by mutant procollagen genes in which mutations in the COL2A1 gene account for more than 80% of the patients.[75]
Matsushita et al.[19] showed that 82% of eyes with Stickler syndrome had a persistence of the inner retinal layers with a reduced or absent foveal pit in the OCT images. In addition, the OCT angiography showed smaller FAZ areas and disarrangements of the capillary network around the fovea. All of the eyes in their cohort had fairly good visual acuity, and the foveae were limited to Grades 1 or 2 based on the criteria by Thomas et al.[30] There was not a significant correlation between the visual acuity and degree of foveal hypoplasia. Foveal hypoplasia had not been reported in patients with Stickler syndrome probably because these patients have fairly good visual acuity.[19]
Conclusions
Recent advancements of high-resolution OCT imaging have challenged the earlier concepts of the visual consequences of foveal hypoplasia. A lack of foveal pit does not simply indicate poor visual acuity as clinicians have observed cases with foveal hypoplasia with no visual impairments. On the other hand, the maturation of the cone photoreceptors is important for visual prognosis. The causes of foveal hypoplasia are highly heterogeneous, and other retinal disorders have been found unexpectedly to be associated with foveal hypoplasia as determined by OCT imaging. The en-face images of the FAZ could be useful in the identification of eyes with foveal hypoplasia. Future studies in vivo imaging by OCT will provide more evidence that will be helpful in understanding the physiology and pathology of the fovea.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This work was financially supported by Grants-in-Aid 17K11441 for Scientific Research (C) from the Japan Society for the Promotion of Science.
Conflicts of interest
The authors declare that there are no conflicts of interests of this paper.
Acknowledgments
The author would like to thank Duco Hamasaki, Professor Emeritus, Bascom Palmer Eye Institute, University of Miami, Miami, Florida, for his critical comments and valuable assistance.
References
- 1.Duke-Elder S. Vol. 3. St Louis: CV Mosby; 1963. System of Ophthalmology. [Google Scholar]
- 2.Bringmann A, Syrbe S, Görner K, Kacza J, Francke M, Wiedemann P, et al. The primate fovea: Structure, function and development. Prog Retin Eye Res. 2018 doi: 10.1016/j.preteyeres.2018.03.006. pii: S1350-9462(17) 30116-7. [DOI] [PubMed] [Google Scholar]
- 3.Curran RE, Robb RM. Isolated foveal hypoplasia. Arch Ophthalmol. 1976;94:48–50. doi: 10.1001/archopht.1976.03910030014005. [DOI] [PubMed] [Google Scholar]
- 4.Mietz H, Green WR, Wolff SM, Abundo GP. Foveal hypoplasia in complete oculocutaneous albinism. A histopathologic study. Retina. 1992;12:254–60. doi: 10.1097/00006982-199212030-00011. [DOI] [PubMed] [Google Scholar]
- 5.Hendrickson A, Possin D, Vajzovic L, Toth CA. Histologic development of the human fovea from midgestation to maturity. Am J Ophthalmol. 2012;154:767–78. doi: 10.1016/j.ajo.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vajzovic L, Hendrickson AE, O’Connell RV, Clark LA, Tran-Viet D, Possin D, et al. Maturation of the human fovea: Correlation of spectral-domain optical coherence tomography findings with histology. Am J Ophthalmol. 2012;154:779–89 e2. doi: 10.1016/j.ajo.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Carlo TE, Romano A, Waheed NK, Duker JS. A review of optical coherence tomography angiography (OCTA) Int J Retina Vitreous. 2015;1:5. doi: 10.1186/s40942-015-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Montoliu L, Grønskov K, Wei AH, Martínez-García M, Fernández A, Arveiler B, et al. Increasing the complexity: New genes and new types of albinism. Pigment Cell Melanoma Res. 2014;27:11–8. doi: 10.1111/pcmr.12167. [DOI] [PubMed] [Google Scholar]
- 9.Hirji N, Aboshiha J, Georgiou M, Bainbridge J, Michaelides M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018;39:149–57. doi: 10.1080/13816810.2017.1418389. [DOI] [PubMed] [Google Scholar]
- 10.Thiadens AA, Somervuo V, van den Born LI, Roosing S, van Schooneveld MJ, Kuijpers RW, et al. Progressive loss of cones in achromatopsia: An imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2010;51:5952–7. doi: 10.1167/iovs.10-5680. [DOI] [PubMed] [Google Scholar]
- 11.Sundaram V, Wilde C, Aboshiha J, Cowing J, Han C, Langlo CS, et al. Retinal structure and function in achromatopsia: Implications for gene therapy. Ophthalmology. 2014;121:234–45. doi: 10.1016/j.ophtha.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas MG, Kumar A, Kohl S, Proudlock FA, Gottlob I. High-resolution in vivo imaging in achromatopsia. Ophthalmology. 2011;118:882–7. doi: 10.1016/j.ophtha.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 13.Chen CA, Yin J, Lewis RA, Schaaf CP. Genetic causes of optic nerve hypoplasia. J Med Genet. 2017;54:441–9. doi: 10.1136/jmedgenet-2017-104626. [DOI] [PubMed] [Google Scholar]
- 14.Pilat A, Sibley D, McLean RJ, Proudlock FA, Gottlob I. High-resolution imaging of the optic nerve and retina in optic nerve hypoplasia. Ophthalmology. 2015;122:1330–9. doi: 10.1016/j.ophtha.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katagiri S, Nishina S, Yokoi T, Mikami M, Nakayama Y, Tanaka M, et al. Retinal structure and function in eyes with optic nerve hypoplasia. Sci Rep. 2017;7:42480. doi: 10.1038/srep42480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo H. Complex genetics of familial exudative vitreoretinopathy and related pediatric retinal detachments. Taiwan J Ophthalmol. 2015;5:56–62. doi: 10.1016/j.tjo.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yonekawa Y, Thomas BJ, Drenser KA, Trese MT, Capone A., Jr Familial exudative vitreoretinopathy: Spectral-domain optical coherence tomography of the Vitreoretinal interface, retina, and choroid. Ophthalmology. 2015;122:2270–7. doi: 10.1016/j.ophtha.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 18.Richards AJ, Baguley DM, Yates JR, Lane C, Nicol M, Harper PS, et al. Variation in the vitreous phenotype of stickler syndrome can be caused by different amino acid substitutions in the X position of the type II collagen gly-X-Y triple helix. Am J Hum Genet. 2000;67:1083–94. doi: 10.1016/s0002-9297(07)62938-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsushita I, Nagata T, Hayashi T, Kimoto K, Kubota T, Ohji M, et al. Foveal hypoplasia in patients with stickler syndrome. Ophthalmology. 2017;124:896–902. doi: 10.1016/j.ophtha.2017.01.046. [DOI] [PubMed] [Google Scholar]
- 20.Hendrickson AE, Yuodelis C. The morphological development of the human fovea. Ophthalmology. 1984;91:603–12. doi: 10.1016/s0161-6420(84)34247-6. [DOI] [PubMed] [Google Scholar]
- 21.Linberg KA, Fisher SK. A burst of differentiation in the outer posterior retina of the eleven-week human fetus: An ultrastructural study. Vis Neurosci. 1990;5:43–60. doi: 10.1017/s0952523800000067. [DOI] [PubMed] [Google Scholar]
- 22.Provis JM, Dubis AM, Maddess T, Carroll J. Adaptation of the central retina for high acuity vision: Cones, the fovea and the avascular zone. Prog Retin Eye Res. 2013;35:63–81. doi: 10.1016/j.preteyeres.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Springer AD, Hendrickson AE. Development of the primate area of high acuity 1.Use of finite element analysis models to identify mechanical variables affecting pit formation. Vis Neurosci. 2004;21:53–62. doi: 10.1017/s0952523804041057. [DOI] [PubMed] [Google Scholar]
- 24.Dubis AM, Hansen BR, Cooper RF, Beringer J, Dubra A, Carroll J, et al. Relationship between the foveal avascular zone and foveal pit morphology. Invest Ophthalmol Vis Sci. 2012;53:1628–36. doi: 10.1167/iovs.11-8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gariano RF. Special features of human retinal angiogenesis. Eye (Lond) 2010;24:401–7. doi: 10.1038/eye.2009.324. [DOI] [PubMed] [Google Scholar]
- 26.Balaratnasingam C, Chae B, Remmer MH, Gomez E, Suzuki M, Engelbert M, et al. The spatial profile of macular pigments is related to the topological characteristics of the foveal avascular zone. Invest Ophthalmol Vis Sci. 2015;56:7859–65. doi: 10.1167/iovs.15-17532. [DOI] [PubMed] [Google Scholar]
- 27.Yuodelis C, Hendrickson A. A qualitative and quantitative analysis of the human fovea during development. Vision Res. 1986;26:847–55. doi: 10.1016/0042-6989(86)90143-4. [DOI] [PubMed] [Google Scholar]
- 28.Provis JM, Sandercoe T, Hendrickson AE. Astrocytes and blood vessels define the foveal rim during primate retinal development. Invest Ophthalmol Vis Sci. 2000;41:2827–36. [PubMed] [Google Scholar]
- 29.Hendrickson AE. Primate foveal development: A microcosm of current questions in neurobiology. Invest Ophthalmol Vis Sci. 1994;35:3129–33. [PubMed] [Google Scholar]
- 30.Thomas MG, Kumar A, Mohammad S, Proudlock FA, Engle EC, Andrews C, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011;118:1653–60. doi: 10.1016/j.ophtha.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Creel DJ, Summers CG, King RA. Visual anomalies associated with albinism. Ophthalmic Paediatr Genet. 1990;11:193–200. doi: 10.3109/13816819009020979. [DOI] [PubMed] [Google Scholar]
- 32.Chong GT, Farsiu S, Freedman SF, Sarin N, Koreishi AF, Izatt JA, et al. Abnormal foveal morphology in ocular albinism imaged with spectral-domain optical coherence tomography. Arch Ophthalmol. 2009;127:37–44. doi: 10.1001/archophthalmol.2008.550. [DOI] [PubMed] [Google Scholar]
- 33.Harvey PS, King RA, Summers CG. Spectrum of foveal development in albinism detected with optical coherence tomography. J AAPOS. 2006;10:237–42. doi: 10.1016/j.jaapos.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Marmor MF, Choi SS, Zawadzki RJ, Werner JS. Visual insignificance of the foveal pit: Reassessment of foveal hypoplasia as fovea plana. Arch Ophthalmol. 2008;126:907–13. doi: 10.1001/archopht.126.7.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McAllister JT, Dubis AM, Tait DM, Ostler S, Rha J, Stepien KE, et al. Arrested development: High-resolution imaging of foveal morphology in albinism. Vision Res. 2010;50:810–7. doi: 10.1016/j.visres.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohammad S, Gottlob I, Kumar A, Thomas M, Degg C, Sheth V, et al. The functional significance of foveal abnormalities in albinism measured using spectral-domain optical coherence tomography. Ophthalmology. 2011;118:1645–52. doi: 10.1016/j.ophtha.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 37.Fulton AB, Albert DM, Craft JL. Human albinism. Light and electron microscopy study. Arch Ophthalmol. 1978;96:305–10. doi: 10.1001/archopht.1978.03910050173014. [DOI] [PubMed] [Google Scholar]
- 38.Provis JM, Penfold PL, Cornish EE, Sandercoe TM, Madigan MC. Anatomy and development of the macula: Specialisation and the vulnerability to macular degeneration. Clin Exp Optom. 2005;88:269–81. doi: 10.1111/j.1444-0938.2005.tb06711.x. [DOI] [PubMed] [Google Scholar]
- 39.Rodanant N, Bartsch DU, Bessho K, Freeman WR. Autofluorescence image in ocular albinism. Retina. 2003;23:265–6. doi: 10.1097/00006982-200304000-00031. [DOI] [PubMed] [Google Scholar]
- 40.Seo JH, Yu YS, Kim JH, Choung HK, Heo JW, Kim SJ, et al. Correlation of visual acuity with foveal hypoplasia grading by optical coherence tomography in albinism. Ophthalmology. 2007;114:1547–51. doi: 10.1016/j.ophtha.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 41.Gehring WJ. The master control gene for morphogenesis and evolution of the eye. Genes Cells. 1996;1:11–5. doi: 10.1046/j.1365-2443.1996.11011.x. [DOI] [PubMed] [Google Scholar]
- 42.Abadi RV, Dickinson CM. Blue light hazard and aniridia. Br J Ophthalmol. 1985;69:233–5. doi: 10.1136/bjo.69.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yokoi T, Nishina S, Fukami M, Ogata T, Hosono K, Hotta Y, et al. Genotype-phenotype correlation of PAX6 gene mutations in aniridia. Hum Genome Var. 2016;3:15052. doi: 10.1038/hgv.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hingorani M, Williamson KA, Moore AT, van Heyningen V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci. 2009;50:2581–90. doi: 10.1167/iovs.08-2827. [DOI] [PubMed] [Google Scholar]
- 45.Katagiri S, Yokoi T, Mikami M, Nishina S, Azuma N. Outer retinal deformity detected by optical coherence tomography in eyes with foveal hypoplasia. Graefes Arch Clin Exp Ophthalmol. 2016;254:2197–201. doi: 10.1007/s00417-016-3385-z. [DOI] [PubMed] [Google Scholar]
- 46.Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M. PAX6 missense mutation in isolated foveal hypoplasia. Nat Genet. 1996;13:141–2. doi: 10.1038/ng0696-141. [DOI] [PubMed] [Google Scholar]
- 47.Chauhan BK, Yang Y, Cveklová K, Cvekl A. Functional properties of natural human PAX6 and PAX6(5a) mutants. Invest Ophthalmol Vis Sci. 2004;45:385–92. doi: 10.1167/iovs.03-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu JY, Ren X, Yang X, Guo T, Yao Q, Li L, et al. Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J Hum Genet. 2007;52:565–70. doi: 10.1007/s10038-007-0152-3. [DOI] [PubMed] [Google Scholar]
- 49.Poulter JA, Al-Araimi M, Conte I, van Genderen MM, Sheridan E, Carr IM, et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013;93:1143–50. doi: 10.1016/j.ajhg.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez Y, Gradstein L, Flusser H, Markus B, Cohen I, Langer Y, et al. Isolated foveal hypoplasia with secondary nystagmus and low vision is associated with a homozygous SLC38A8 mutation. Eur J Hum Genet. 2014;22:703–6. doi: 10.1038/ejhg.2013.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toral MA, Velez G, Boudreault K, Schaefer KA, Xu Y, Saffra N, et al. Structural modeling of a novel SLC38A8 mutation that causes foveal hypoplasia. Mol Genet Genomic Med. 2017;5:202–9. doi: 10.1002/mgg3.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lutty GA, Chan-Ling T, Phelps DL, Adamis AP, Berns KI, Chan CK, et al. Proceedings of the third international symposium on retinopathy of prematurity: An update on ROP from the lab to the nursery (November 2003, anaheim, california) Mol Vis. 2006;12:532–80. [PubMed] [Google Scholar]
- 53.Isenberg SJ. Macular development in the premature infant. Am J Ophthalmol. 1986;101:74–80. doi: 10.1016/0002-9394(86)90467-8. [DOI] [PubMed] [Google Scholar]
- 54.Mintz-Hittner HA, Knight-Nanan DM, Satriano DR, Kretzer FL. A small foveal avascular zone may be an historic mark of prematurity. Ophthalmology. 1999;106:1409–13. doi: 10.1016/S0161-6420(99)00732-0. [DOI] [PubMed] [Google Scholar]
- 55.Maldonado RS, O’Connell RV, Sarin N, Freedman SF, Wallace DK, Cotten CM, et al. Dynamics of human foveal development after premature birth. Ophthalmology. 2011;118:2315–25. doi: 10.1016/j.ophtha.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Recchia FM, Recchia CC. Foveal dysplasia evident by optical coherence tomography in patients with a history of retinopathy of prematurity. Retina. 2007;27:1221–6. doi: 10.1097/IAE.0b013e318068de2e. [DOI] [PubMed] [Google Scholar]
- 57.Wu WC, Lin RI, Shih CP, Wang NK, Chen YP, Chao AN, et al. Visual acuity, optical components, and macular abnormalities in patients with a history of retinopathy of prematurity. Ophthalmology. 2012;119:1907–16. doi: 10.1016/j.ophtha.2012.02.040. [DOI] [PubMed] [Google Scholar]
- 58.Villegas VM, Capó H, Cavuoto K, McKeown CA, Berrocal AM. Foveal structure-function correlation in children with history of retinopathy of prematurity. Am J Ophthalmol. 2014;158:508–12. doi: 10.1016/j.ajo.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 59.Bernstein PS, Sharifzadeh M, Liu A, Ermakov I, Nelson K, Sheng X, et al. Blue-light reflectance imaging of macular pigment in infants and children. Invest Ophthalmol Vis Sci. 2013;54:4034–40. doi: 10.1167/iovs.13-11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Falavarjani KG, Iafe NA, Velez FG, Schwartz SD, Sadda SR, Sarraf D, et al. Optical coherence tomography angiography of the fovea in children born preterm. Retina. 2017;37:2289–94. doi: 10.1097/IAE.0000000000001471. [DOI] [PubMed] [Google Scholar]
- 61.Nonobe N, Kaneko H, Ito Y, Takayama K, Kataoka K, Tsunekawa T, et al. Optical coherence tomography angiography of the foveal avascular zone in children with a history of treatment-requiring retinopathy of prematurity. Retina. 2017 doi: 10.1097/IAE.0000000000001937. [Ahead of print]. doi: 10.1097/IAE.0000000000001937. [DOI] [PubMed] [Google Scholar]
- 62.Aradhya S, Woffendin H, Jakins T, Bardaro T, Esposito T, Smahi A, et al. A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum Mol Genet. 2001;10:2171–9. doi: 10.1093/hmg/10.19.2171. [DOI] [PubMed] [Google Scholar]
- 63.Goldberg MF. The blinding mechanisms of incontinentia pigmenti. Ophthalmic Genet. 1994;15:69–76. [PubMed] [Google Scholar]
- 64.Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: A Comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650–7. doi: 10.3928/23258160-20150610-09. [DOI] [PubMed] [Google Scholar]
- 65.Minić S, Trpinac D, Obradović M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85:536–42. doi: 10.1111/cge.12223. [DOI] [PubMed] [Google Scholar]
- 66.Basilius J, Young MP, Michaelis TC, Hobbs R, Jenkins G, Hartnett ME, et al. Structural abnormalities of the inner macula in incontinentia pigmenti. JAMA Ophthalmol. 2015;133:1067–72. doi: 10.1001/jamaophthalmol.2015.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langlo CS, Patterson EJ, Higgins BP, Summerfelt P, Razeen MM, Erker LR, et al. Residual foveal cone structure in CNGB3-associated achromatopsia. Invest Ophthalmol Vis Sci. 2016;57:3984–95. doi: 10.1167/iovs.16-19313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matet A, Kohl S, Baumann B, Antonio A, Mohand-Said S, Sahel JA, et al. Multimodal imaging including semiquantitative short-wavelength and near-infrared autofluorescence in achromatopsia. Sci Rep. 2018;8:5665. doi: 10.1038/s41598-018-23919-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Greenberg JP, Sherman J, Zweifel SA, Chen RW, Duncker T, Kohl S, et al. Spectral-domain optical coherence tomography staging and autofluorescence imaging in achromatopsia. JAMA Ophthalmol. 2014;132:437–45. doi: 10.1001/jamaophthalmol.2013.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mosier MA, Lieberman MF, Green WR, Knox DL. Hypoplasia of the optic nerve. Arch Ophthalmol. 1978;96:1437–42. doi: 10.1001/archopht.1978.03910060185017. [DOI] [PubMed] [Google Scholar]
- 71.Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E, et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003;72:1565–70. doi: 10.1086/375555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Higashide T, Wada T, Sakurai M, Yokoyama H, Sugiyama K. Macular abnormalities and optic disk anomaly associated with a new PAX2 missense mutation. Am J Ophthalmol. 2005;139:203–5. doi: 10.1016/j.ajo.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 73.Nikopoulos K, Venselaar H, Collin RW, Riveiro-Alvarez R, Boonstra FN, Hooymans JM, et al. Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum Mutat. 2010;31:656–66. doi: 10.1002/humu.21250. [DOI] [PubMed] [Google Scholar]
- 74.Donoso LA, Edwards AO, Frost AT, Ritter R, 3rd, Ahmad N, Vrabec T, et al. Clinical variability of stickler syndrome: Role of exon 2 of the collagen COL2A1 gene. Surv Ophthalmol. 2003;48:191–203. doi: 10.1016/s0039-6257(02)00460-5. [DOI] [PubMed] [Google Scholar]
- 75.Fincham GS, Pasea L, Carroll C, McNinch AM, Poulson AV, Richards AJ, et al. Prevention of retinal detachment in stickler syndrome: The cambridge prophylactic cryotherapy protocol. Ophthalmology. 2014;121:1588–97. doi: 10.1016/j.ophtha.2014.02.022. [DOI] [PubMed] [Google Scholar]