

Abstract
Aims
Demonstrate the presence of cytochrome P450 (CYP) and UDP‐glucuronosyltransferase (UGT) proteins and mRNAs in isolated human plasma exosomes and evaluate the capacity for exosome‐derived biomarkers to characterize variability in CYP3A4 activity.
Methods
The presence of CYP and UGT protein and mRNA in exosomes isolated from human plasma and HepaRG cell culture medium was determined by mass spectrometry and reverse transcription–polymerase chain reaction, respectively. The concordance between exosome‐derived CYP3A4 biomarkers and midazolam apparent oral clearance (CL/F) was evaluated in a small proof‐of‐concept study involving six genotyped (CYP3A4 *1/*1 and CYP3A5 *3/*3) Caucasian males.
Results
Exosomes isolated from human plasma contained peptides and mRNA originating from CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 2 J2, 3A4 and 3A5, UGT 1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, 2B10 and 2B15, and NADPH‐cytochrome P450 reductase. Mean (95% confidence interval) exosome‐derived CYP3A4 protein expression pre‐ and post‐rifampicin dosing was 0.24 (0.2–0.28) and 0.42 (0.21–0.65) ng ml–1 exosome concentrate. Mean (95% confidence interval) exosome CYP3A4 mRNA expression pre‐ and post‐rifampicin dosing was 6.0 (1.1–32.7) and 48.3 (11.3–104) × 10–11 2‐ΔΔCt, respectively. R2 values for correlations of exosome‐derived CYP3A4 protein expression, CYP3A4 mRNA expression, and ex vivo CYP3A4 activity with midazolam CL/F were 0.905, 0.787 and 0.832, respectively.
Conclusions
Consistent strong concordance was observed between exosome‐derived CYP3A4 biomarkers and midazolam CL/F. The significance of these results is that CYP3A4 is the drug‐metabolizing enzyme of greatest clinical importance and variability in CYP3A4 activity is poorly described by existing precision dosing strategies.
Keywords: ADME, biomarkers, cytochrome P450, exosomes
What is Already Known about this Subject
Cytochromes P450 (CYP) and UDP‐glucuronosyltransferases (UGT) account for the metabolic clearance of >90% of drugs.
CYP3A4 is the drug‐metabolizing enzyme of greatest clinical importance as it is responsible for the metabolism of >30% of all drugs.
Variability in CYP3A4 activity is primarily driven by differences in protein expression, poorly described by current genotype‐ and biomarker‐based approaches, which has forced some investigators to resort to tissue biopsy.
What this Study Adds
Exosomes isolated from human plasma contain functional protein and mRNA for multiple cytochrome P450 and UDP‐glucuronosyltransferase enzymes.
Exosome‐derived mRNA and protein biomarkers (ADMExosomes) characterize between subject variability in basal CYP3A4 activity and track induction of CYP3A4 expression by rifampicin, a well‐known inducer.
Exosome‐derived CYP3A and UGT proteins are metabolically active under ex vivo conditions and exhibit comparable kinetics to subcellular fractions (microsomes) prepared from human liver tissue.
Human plasma‐derived exosomes present as liquid biopsy and may complement existing drug probe‐based approaches, while possibly circumventing the need for tissue biopsy.
Introduction
Characterization of the physiological and molecular characteristics underpinning variability in drug absorption, distribution, metabolism and excretion (ADME) pathways provides the capacity to account for variability in drug exposure 1. Together the cytochrome P450 (CYP) and UDP‐glucuronosyltransferase (UGT) families of drug‐metabolizing enzymes account for the metabolic clearance of >90% of drugs that are subject to metabolism 2, 3, 4, as such the activity of these enzymes is an important determinant of drug exposure. The activity of CYP and UGT enzymes is determined by their intrinsic clearance (i.e. function) and abundance (i.e. expression). Accordingly, variability in CYP and UGT activity can be caused by differences in either function or expression. To date, attempts to explain variability in CYP and UGT activities have largely focused on the assessment of genotype differences via a pharmacogenomics (PGx) approach. While there are several examples where genetic variants account for a large proportion of observed variability in activity, including CYP2C19, CYP2D6 and UGT1A1, there are many cases in which genotype alone is insufficient to predict patient exposure to a drug 5. Notably, CYP3A4 is the drug‐metabolizing enzyme of greatest clinical importance as it is responsible for the metabolism of >30% of all drugs 6 and variability is primarily driven by differences in protein expression that are poorly described by a PGx approach 7, 8, 9. It is acknowledged that the CYP3A4*22 genotype is associated with a small but significant reduction in CYP3A4 activity 10, and that CYP3A5 is only expressed (*1) in approximately 15% of Caucasians. As such, while CYP3A4*22 and CYP3A5*1 genotypes occur at a low frequency, they should be considered as they can markedly alter clearance of CYP3A substrates in affected individuals 11. Additionally, PGx provides minimal insight regarding the impact of changes in CYP or UGT activity caused by environmental factors such as drug interactions.
It has been proposed that assessment of hepatic mRNA expression may be a robust biomarker to account for variability in CYP3A activity 12. The limitation is that, in generating the evidence to support this claim, Yamaori et al. 12 utilized surgically resected liver tissue to assess expression of hepatic CYP3A mRNAs, an approach is preclusively invasive for routine clinical applications. Attempts to correlate CYP3A4 mRNA expression in more diagnostically amenable samples such as peripheral blood mononuclear cells (PBMCs) with activity or induction have demonstrated that PBMC mRNA expression does not correlate with CYP3A4‐mediated drug clearance 13, 14.
In recent years, exosome‐derived biomarkers have revolutionized the diagnosis of multiple pathologies including cancer, cardiovascular disease and liver injury 15, 16, 17, 18, 19. Exosomes are small membranous vesicles that are secreted into the blood by many organs, including the liver 20. As exosomes contain functional proteins and nucleic acids (double stranded DNA, mRNA and miRNA) derived from the organ from which they originate 21, 22, 23, they reflect the functional state of that organ and are a potential rich source of biomarkers in peripheral blood. Here, we demonstrate for the first time the capacity of exosome‐derived biomarkers for ADME pathways (ADMExosomes) to characterize variability in drug exposure. Specifically, we demonstrate the capacity of ADMExosome biomarkers to characterize between subject variability in basal CYP3A4 expression and to track induction of CYP3A4 expression in a cohort of healthy males. We further provide data demonstrating: (i) protein and the ability to detect mRNA expression for multiple additional CYP and UGTs in circulating exosomes; (ii) ex vivo activity for exosome‐derived CYP3A and UGT proteins; and (iii) in vitro data supporting the capacity of exosome‐derived biomarkers to track induction of CYP3A4.
Methods
Human biological samples
Plasma samples for this proof‐of‐concept study were obtained from healthy Caucasian males aged 21–35 years enrolled in the EPOK‐15 metabolic phenotyping study before and after a 7‐day course of rifampicin (300 mg daily) 7. The cohort (n = 6) was genotype selected for CYP3A4*1/*1 and CYP3A5*3/*3. The study was approved by the Southern Adelaide Clinical Human Research Ethics Committee (SAHREC 11.15), and informed written consent was obtained from each participant. The study was conducted according to the principles stated in the Declaration of Helsinki and is compliant with CPMP/ICH/135/95 GCP standards. The study protocol has been reported previously 7. Exposure to a 1‐mg oral dose of midazolam was assessed before (Day 1) and after (Day 8) a 7‐day course of rifampicin. Timed blood samples were collected prior to and for up to 6 h post midazolam dosing. Between Day 1 and 8 participants self‐administered oral rifampicin (300 mg daily). Rifampicin was dosed daily commencing 2 h after the final sample collection on Day 1, with the final dose administered 16 h prior to the cocktail administration on Day 8.
Liver tissue (H07, H10, H12, H13 and H40) was obtained from the human liver bank of the Department of Clinical Pharmacology of Flinders University. Approval for the use of human liver tissue in xenobiotic metabolism studies was obtained from both the Clinical Investigation Committee of Flinders Medical Centre (Approval ID 59–056), and from the donors' next‐of‐kin. Pooled human liver microsomes (HLM) were prepared by mixing equal protein amounts from the five human livers 24. Microsomes were prepared by differential centrifugation as described previously 25. Exosome and HLM protein concentrations were determined by Lowry protein assay.
Cell culture model
HepaRG cells (passage 14) were seeded at density of 28 000 cells cm–2 in growth medium composed of Williams' medium E with GlutaMAX‐I, supplemented with 10% fetal bovine serum, 100 IU ml–1 penicillin, 100 μg ml–1 streptomycin, 5 μg ml–1 bovine insulin and 50 μmol l–1 hydrocortisone hemisuccinate. After 14 days of cell culture, the medium was supplemented with 2% dimethyl sulfoxide (differentiation medium). The medium was renewed every 48–72 h. Cells were cultured in differentiation medium for 3 weeks prior to experiments, with the medium renewed every 48–72 h. Cells were cultured in growth medium supplemented with 10% exosome depleted fetal bovine serum (Gibco Cat #A2720801) and 2% dimethyl sulfoxide with or without 15 μmol l–1 rifampicin for 96 h prior to harvesting. For mRNA analyses, the cells were harvested in TRIzol reagent (0.5 ml per well in 24‐well plates). For protein analyses, whole cell lysates were prepared in Tris‐EDTA (TE) Buffer and protein concentrations were determined by Lowry assay. At the time of cell harvesting, culture medium containing HepaRG exosomes were collected and filtered through a 0.45 μm filter. All samples were stored at –80°C until use.
Isolation of exosomes from plasma and cell culture medium
Plasma samples (5 or 10 ml) or cell culture medium (12 ml) were filtered using 0.45 μm syringe filters. Up to 10 ml of the resulting filtrate was mixed with 1.25 volumes of binding buffer (Qiagen XBP) and the resulting mixture was added to the reservoir compartment of a preconditioned ExoEasy membrane affinity spin columns (Qiagen). The column was centrifuged at room temperature for 90 s at 400 g and the resulting flow‐through discarded. Wash buffer (10 μl; Qiagen XWP) was added to the reservoir compartment and the column was centrifuged at room temperature for 5 min at 4000 g. Elution buffer (400 μl; Qiagen XE) was added to the reservoir compartment and the spin column was transferred to a fresh collection tube and incubated at room temperature for 90 s. The column was centrifuged at room temperature for 90 s at 400 g and the resulting flow‐through reapplied to the reservoir compartment and incubated at room temperature for a further 90 s. The column was centrifuged at room temperature for 5 min at 4000 g and the resulting flow‐through collected and stored at –80°C until used.
Transmission electron microscopy
To validate the exosomes purity, electron microscope imaging of exosomes was performed. Briefly, 1 μg of exosomes were spotted onto a carbon‐coated 200 mesh grid and dried at room temperature. The grids were washed twice with water for 5 min before staining with 2% w/v uranyl acetate for 5 min. Grids were examined using a Philips CM100 transmission electron microscope (Thermo‐Scientific, Waltham, MA, USA) operated at 60 kV.
Nanoparticle tracking analysis
Nanoparticles in isolated exosome suspensions were analysed using a NanoSight LM10 (NanoSight Ltd, Amesbury, UK). Analysis settings were optimized to ensure a minimum of 4000 tracks per video, and each video was analysed to determine the median particle size and range, and the estimated concentration for each particle size. Experiments were performed at a 1:1000 dilution, yielding particle concentrations in the region of 1 × 109 particles ml–1. All samples were analysed in triplicate.
Immunoblotting of Tsg101
Proteins contained within exosomes isolated from human plasma, exosomes isolated from HepaRG cell culture and HepaRG cell lysate (20 μg total protein) were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proteins were rectilinearly transferred onto nitrocellulose and probed with rabbit anti‐Tsg101 monoclonal antibody (1:1000 dilution; Invitrogen, Sydney, Australia). Membrane‐bound peptides conjugated with horseradish peroxidase were detected by chemiluminescence (Roche Diagnostics, Mannheim, Germany) and subsequently exposed to Omat autoradiographic film (Eastman Kodak, Rochester, NY, USA). Autoradiographs were processed manually with AGFA developer, fixer and replenisher reagents.
Preparation of exosomes for mRNA analysis
Total RNA was isolated from exosomal pellets using TRIzol (Invitrogen). Exosome‐derived RNA concentration and purity determined using a GeneQuant II spectrophotometer. RNA integrity was further evaluated by visualization of a gel smear using an Agilent 2100 Bioanalyzer. RNA was reverse transcribed to cDNA using SuperScript VILO cDNA Synthesis kits (Invitrogen, Cat no: 11754050). Reverse‐transcribed cDNA was preamplified by polymerase chain reaction (PCR) using TaqMan Gene Expression Assays (Life Technologies, for CYP3A4 Assay ID:Hs00604506_m1, Cat no: 4453320) and TaqMan Pre‐Amp Master Mix (Life Technologies, Cat no: 4391128). Briefly, the 50‐μl reaction mixture contained 1–250 ng cDNA, 25 μl TaqMan Pre‐Amp Mater mix (2×), TaqMan gene expression assays consisting of primers and probe (20×). The PCR reaction was initially incubated at 95°C for 10 min, followed by 14 cycles of 95°C for 15 s and 60°C for 4 min.
Preamplified PCR products were diluted in 1:5 ratio with 1× TE buffer, and quantified using quantitative reverse transcription PCR (qRT‐PCR) with the TaqMan Gene Expression Master Mix (Life Technologies, Cat no: 4369016) and the same TaqMan gene expression assay primers and probes used for pre‐amplification. Typically a 20‐μl reaction mixture contained 5 μl preamplified cDNA product, 1 μl TaqMan Gene expression Assay (20×), 10 μl TaqMan Gene Expression Master mix (2×). The PCR reaction was initially incubated at 50°C for 2 min and then 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The expression level of mRNA was analysed using the Rotor‐Gene 6 software (Corbett Life Science). 18S and GAPDH were used as normalizing controls (TaqMan Gene Expression Assay IDs Hs99999901_s1 and Hs02786624_g1 respectively).
CYP3A genotyping
CYP3A genotyping was performed as described previously 7. Briefly, genomic DNA was isolated from whole blood using a QIAsymphony (QIAGEN) automated platform running a DSP DNA Mini Kit, quantified by NanoDrop and normalized to 20 ng μl. DNA was genotyped for CYP3A4 and CYP3A5 using a custom designed TaqMan OpenArray that was analysed on a QuantStudio 12 K Flex Real‐Time PCR System (Applied Biosystems). Specific accession (RS) numbers analysed were: CYP3A4*22 (rs35599367), CYP3A5*3 (rs776746), CYP3A5*5 (rs55965422), CYP3A5*6 (rs10264272) and CYP3A5*7 (rs41303343).
Proteomic screening of exosomes
In‐gel trypsin digestion was performed on 40–70 kDa bands excised from 1D‐sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels. Gel fragments were destained then dehydrated at room temperature using 100 μl of acetonitrile. Protein bands were reduced using 10 μmol l–1 dithiothreitol then alkylated using 50 μmol l–1 iodoacetamide by incubating gel pieces at 60°C for 30 min. Gels were dehydrated using 100 μl acetonitrile and digested with trypsin at an enzyme‐to‐protein ratio of 1:20 for 16 h at 37°C. Peptides were extracted by adding acetonitrile at a final concentration of 50%.
Peptides were separated by liquid chromatography (LC) performed on a Waters XBridge BEH C18 column (150 mm × 0.3 mm, 3.5 μm) with a 45 min acetonitrile gradient using an AB Sciex Ekspert 400 nanoHPLC. Column elutant was monitored using an AB Sciex 5600+ triple time of flight mass spectrometer (MS) operating in positive ion mode, with rolling collision energy and dynamic accumulation enabled. Three technical replicates were performed for each sample. De novo sequencing was performed on raw MS data using Peaks Studio v7.0 software.
Quantification of ExoCYP3A4 expression by ELISA
Exosomes were seeded in 96‐well plates and incubated overnight at 37°C in an atmosphere of 5% CO2. Wells were fixed by addition of fixing solution (100 μl) then quenching buffer (100 μl) with incubation at room temperature for 20 min following addition of each solution. Wells were then blocked by addition of blocking buffer (200 μl) with incubation at room temperature for 1 h and prepared for analysis by addition of CYP3A4 primary antibody (50 μl) with overnight incubation at 4°C then addition of HRP‐conjugated secondary antibody (50 μl) with incubation at room temperature for 90 min. Reactions were terminated by addition of stop solution (50 μl) and the optical density (OD) was quantified at 450 nm. OD450 values were normalized using OD values obtained for GAPDH.
Preparation of plasma for analysis of midazolam concentration
The sample preparation and analysis for the determination of plasma midazolam concentrations has been reported 7. Briefly, 100 μl of plasma sample was diluted in 300 μl of methanol containing 0.1% formic acid and 7.5 ng ml–1 d6‐midazolam (internal standard) then vortexed for 3 min, then centrifuged at 16 000 g for 5 min. A 2.5‐μl aliquot of the resulting supernatant was analysed by LC–MS.
Ex vivo determination of exosome‐derived CYP3A and UGT enzyme kinetics
The ex vivo activity of exosomal CYP and UGT proteins were examined using metabolite formation assays developed for the assessment of human liver microsomal enzyme kinetics 25, 26. Exosomes were activated by incubating on ice for 30 min in the presence of the pore forming peptide alamethicin (50 μg mg–1 protein). Incubations performed to determine microsomal enzyme kinetics contained 50 pg of HLM in place of exosomes.
4‐methylumbeliferone glucuronidation assay
Incubations in 200 μl contained 0.1 mol l–1 phosphate buffer (pH 7.4), 4 μmol l–1 MgCl2, exosomes (0.1 mg protein) and 4‐methylumbeliferone (4‐MU; 5–300 μmol l–1). Following a 5 min preincubation, reactions were initiated by the addition of 5 μmol l–1 UDPGA. Incubations were performed at 37°C in a shaking water bath for 120 min. Reactions were terminated by the addition of 2 μl of perchloric acid (70%). Samples were centrifuged at 4000 g for 10 min. A 2.5 μl aliquot of the supernatant fraction was analysed by LC–MS.
Midazolam 1‐hydroxylation assay
Incubations in 200 μl contained 0.1 mol l–1 phosphate buffer (pH 7.4), exosomes (0.1 mg protein) and midazolam (1–50 μmol l–1). Following a 5 min preincubation, reactions were initiated by the addition of 1 μmol l–1 NADPH generating system (1 μmol l–1 NADP, 10 μmol l–1 glucose‐6‐phosphate, 2 IU ml–1 glucose‐6‐phosphate dehydrogenase and 5 μmol l–1 MgCl2). Incubations were performed at 37°C in a shaking water bath for 45 min. Reactions were terminated by the addition of 400 μl of ice‐cold methanol containing 0.1% formic acid. Samples were centrifuged at 4000 g for 10 min. A 7.5 μl aliquot of the supernatant fraction was analysed by LC–MS.
For experiments performed to assess the impact of exosome‐derived CYP3A4 inhibition, CYP3Cide (0.05, 0.3 or 1.5 μmol l–1) was included in the incubations. Initially developed by Pfizer for ADME/Toxicology research to characterize in vitro and ex vivo CYP3A4 phenotype 27, CYP3Cide is commercially available through Sigma–Aldrich (cat # PZ0195). CYP3Cide is a highly selective inhibitor of CYP3A4 that can be used to distinguish the contributions of CYP3A4 vs. all other CYP, including CYP3A5 and 3A7, on drug metabolism. The IC50 values for inhibition of midazolam 1'‐hydroxylase activity of CYP3A4 (0.3 μmol l–1) is 20‐fold lower than the IC50 value for any other CYP, including CYP3A5 (IC50 = 17 μmol l–1) 11.
Quantification of analyte concentrations
Analytes were separated from their respective sample matrices by UPLC performed on a Waters ACQUITY BEH C18 column (100 mm × 2.1 mm, 1.7 μm) using a Waters ACQUITY UPLC system. Column elutant was monitored by MS, performed on a Waters Q‐ToF Premier MS operating in positive electron spray ionization mode. Selected ion data was extracted at the analyte [M + H]+ precursor m/z. Resulting pseudo‐MRM spectra were analysed using Waters TargetLynx software. Plasma analyte concentrations were determined by comparison of normalized peak areas in participant or ex vivo incubation samples to those of calibrators.
Data analysis
Noncompartmental methods (PK Functions for Microsoft Excel; Department of Pharmacokinetics and Drug Metabolism, Irvine, CA, USA) were used to estimate the area under the plasma–concentration time curve from the time of dosing to the last measured sample (AUC) and maximal concentration for midazolam at baseline (Day 1) and following a 7‐day course of rifampicin (Day 8). Midazolam apparent oral clearance (CL/F) was calculated as the midazolam dose (1 mg) divided by the AUC. Univariate linear regression was performed to evaluate associations between the exosome‐derived CYP3A4 biomarkers and midazolam CL/F. Analyses were also performed to evaluate the concordance between each of the biomarkers.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 28, and are permanently archived in the Concise Guide to PHARMACOLOGY2017/18 29.
Results
Characterization of exosomes
Exosomes were selectively and reproducibly (CV < 10%) isolated from the extracellular medium of cultured HepaRG cells and human plasma. Visualized in Figure 1A the round appearance of the isolated microvesicles is consistent with established morphological criteria of exosomes 30. Nanoparticle tracking analysis (NTA) of the particle size distribution of exosomes isolated from human plasma demonstrated a peak (± standard deviation, SD) at 109.0 ± 3.3 nm. The geometric mean ± SD and 90% confidence interval (CI; ± SD) diameter of isolated microvesicles was 127.0 ± 2.0 nm (89.2 ± 2.4 to 229.9 ± 8.9 nm; Figure 1B). The mean (± SD) yield of exosomes isolated from human plasma was 1.82 x 1012 ± 1.24 × 1011 particles ml–1 of plasma. The mean (± SD) total exosome‐derived protein yield was 0.321 ± 0.017 mg ml–1 of plasma.
Figure 1.
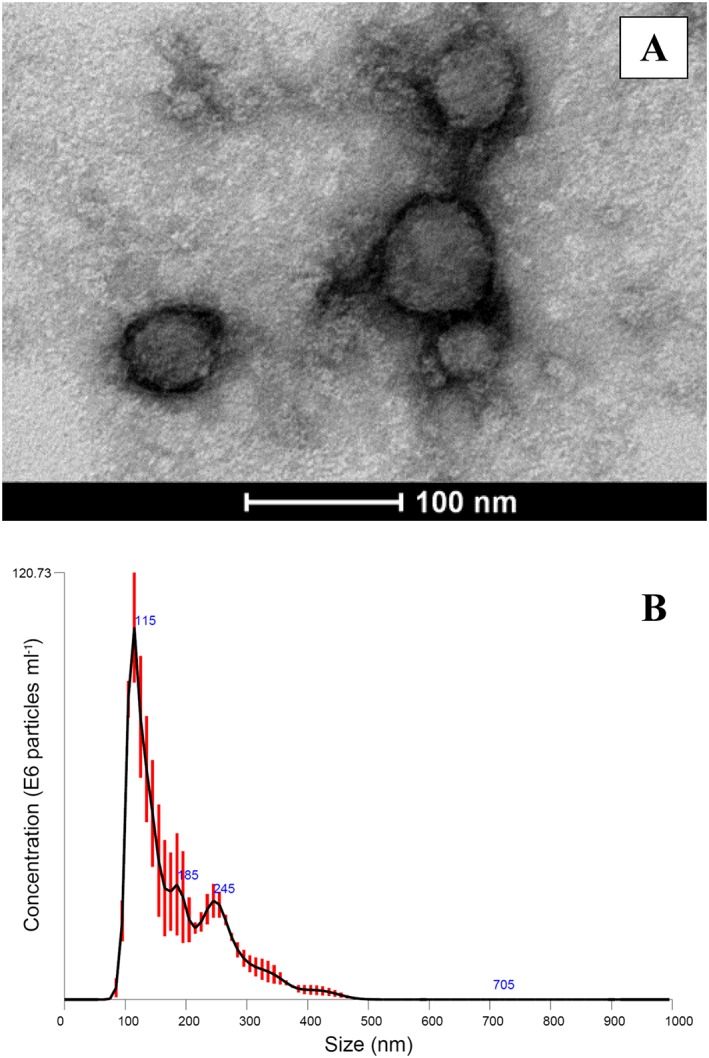
Characterization of exosomes. (A) Transmission electron microscopy image of exosomes isolated from HepaRG cell culture medium. (B) Nanoparticle tracking analysis of exosomes isolated from plasma. Black line represents mean of triplicate analyses, red error bars indicate ±1 standard error of the mean
The identity of the exosomes was confirmed by analysis of tumour susceptibility 101 (Tsg101) expression. Tsg101 is a 44 kDa ubiquitin‐conjugating enzyme present in exosomes secreted by all tissues and is a validated exosomal marker 31. Immunoblotting with an anti‐Tsg101 antibody (Figure S1) demonstrated expression of Tsg101 in exosomes isolated from human plasma, exosomes isolated from HepaRG cell culture medium and HepaRG cell lysates. Enrichment of Tsg101 expression was observed in the exosomes isolated from HepaRG cell culture medium compared to an equivalent quantity of HepaRG cell lysate (10 μg per well).
Detection of CYP and UGT mRNA in exosomes isolated from plasma
The presence of CYP 1A2, 2C8, 2C9, 2D6, 2E1 and 3A4, and UGT 1A1, 1A9, 2B4, 2B7 and 2B10 mRNAs in exosomes isolated from human plasma was confirmed by qRT‐PCR. The relative expression of individual CYP and UGT mRNAs in exosomes are reported in Table S1, along with a comparison to published HLM data.
Exosome‐derived CYP3A4 mRNA abundance increases following rifampicin treatment
When treated with rifampicin (15 μmol l–1) for 96 h, CYP3A4 mRNA expression in HepaRG cell lysate and exosomes isolated from HepaRG cell culture medium increased by 22‐ and 15‐fold, respectively (Figure 2). Similarly, the abundance of CYP3A4 mRNA in exosomes isolated from human plasma (n = 6) post‐rifampicin dosing was on average 8‐fold (90% CI, 2–14‐fold) higher compared to expression in matched pre‐rifampicin dosing samples (Table 1).
Figure 2.
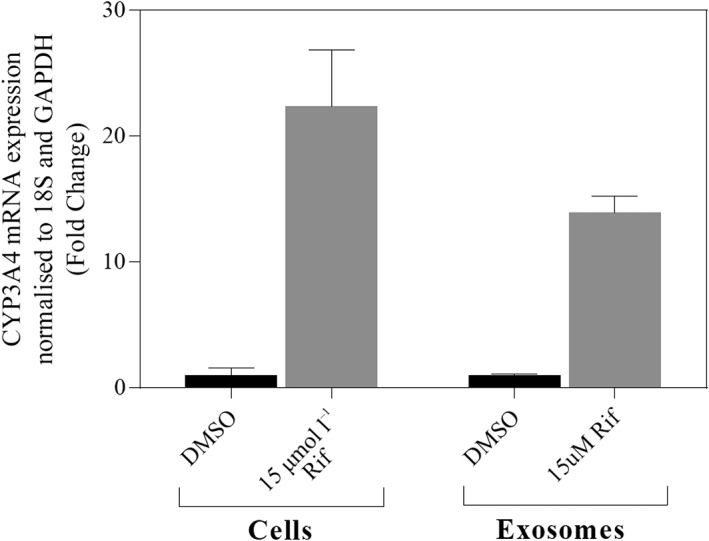
Expression of CYP3A4 mRNA in control and rifampicin (Rif)‐treated HepaRG cells and exosomes isolated from HepaRG culture medium. Error bars represent S. D of independent triplicate experiments. DMSO, dimethyl sulfoxide
Table 1.
Parameters describing in vivo midazolam exposure and exosome CYP3A4 protein and mRNA expression
| Parameter | Study phase | Geometric mean | 95% confidence interval | |
|---|---|---|---|---|
| Lower | Upper | |||
| In vivo Midazolam AUC | Baseline | 8.2 | 4.9 | 11.5 |
| Induced | 2.2 | 1.5 | 2.9 | |
| In vivo Midazolam C max | Baseline | 4.1 | 3.2 | 5.0 |
| Induced | 0.9 | 0.6 | 1.2 | |
| Exosome CYP3A4 protein expression | Baseline | 0.24 | 0.20 | 0.28 |
| Induced | 0.42 | 0.21 | 0.65 | |
| Exosome CYP3A4 mRNA expression | Baseline | 6.0 | 1.1 | 32.7 |
| Induced | 48.3 | 11.3 | 104 | |
| Ex Vivo CYP3A4 activity | Baseline | 25.8 | 22.9 | 30.5 |
| Induced | 38.9 | 27.7 | 68.1 | |
Area under the plasma–concentration time curve (AUC): μg l–1 h; maximal concentration (Cmax): μg l–1; Exosome CYP3A4 protein expression; ng ml–1 concentrated exosome sample, exosome CYP3A4 mRNA expression (x10–11); 2‐ΔΔCt, Ex vivo CYP3A4 activity (rate of 1‐hydroxy midazolam formation at 50 μmol l–1 midazolam); pmol min–1 mg–1
CYP and UGT proteins detected in exosomes isolated from plasma
Mass spectrometry based proteomic profiling of exosomes isolated from human plasma detected 188 unique peptides originating from CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 2 J2, 3A4 and 3A5, and UGT 1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, 2B10 and 2B15. The unique peptides attributed to each protein are summarized in Table S2. The number of unique peptides detected for each protein ranged between 2 and 19, with a mean of 9.65. In addition, 5 unique peptides originating from NADPH‐cytochrome P450 reductase (the redox partner required for CYP activity) were also detected. The capacity to detect peptides originating from CYP and UGT proteins was confirmed in plasma from 3 individuals. It is worth further noting that while cytochrome b5 (34.5 kDa) was not detected in the current analysis as it was not contained within the window of protein bands analysed (40 to 70 kDa), the presence of this protein in human‐derived exosomes has already been established 32, 33. Figure S2 demonstrates the robustness of exosome‐derived protein isolation for a representative enzyme (UGT2B7).
Ex vivo CYP3A and UGT metabolism kinetics in exosomes isolated from plasma
The ex vivo kinetics of UGT and CYP3A catalysed metabolism by exosomes isolated from plasma were assessed using 4‐MU and midazolam as probe substrates, respectively. Parallel experiments were performed using pooled HLM as the enzyme source for comparison of exosome‐ and liver‐derived enzyme kinetics. Kinetic data for the UDPGA‐dependent conversion of 4‐MU to 4‐MU glucuronide and the NADPH‐dependent oxidation of midazolam to 1‐hydroxy midazolam by exosomes and HLMs were well described by the Michaelis–Menten equation (Figure S3). Mean kinetic parameters (Km, Vmax and CLint) describing 4‐MU glucuronide and 1‐hydroxy midazolam formation by exosomes and HLMs are reported in in Table 2.
Table 2.
Parameters describing the ex vivo kinetics of 4‐methylumbilliferone and midazolam metabolism by human liver microsomes and human plasma‐derived exosomes
| Reaction | Enzyme Source | K m | V max | CL int |
|---|---|---|---|---|
| 4‐MU glucuronidation | Exosomes | 181 | 151 | 0.84 |
| HLM | 105 | 10 683 | 101 | |
| Midazolam hydroxylation | Exosomes | 6.5 | 31.1 | 4.8 |
| HLM | 4.4 | 2895 | 658 |
Units: Km; μmol l–1, Vmax; pmol min–1 mg–1, CLint; μl min–1 mg–1.
4‐MU: 4‐methylumbilliferone; HLM, human liver microsomes
No metabolite formation was observed when incubations were performed in the absence of added cofactors. The rate of metabolite formation was diminished by >90% when incubations were performed using exosomes that were not activated by prior incubation with alamethicin on ice; the mean (±SD) maximal rates of 4‐MU glucuronide formation by activated and native exosomes were 150 ± 7.6 and 6.5 ± 0.4 pmol min–1 mg–1, respectively, while the mean (±SD) maximal rates of 1‐hydroxy midazolam formation by activated and native exosomes was 14.3 ± 0.7 pmol min–1; mg–1 and 0.35 ± 0.07 pmol min–1 mg–1. As discussed below, the conversion of midazolam (50 μmol l–1) to 1‐hydroxy midazolam by exosomes was further assessed as an ex vivo activity biomarker for CYP3A4 activity and induction (Table 1, Figure 3 and S4). Furthermore, the highly selective CYP3A4 inhibitor CYP3Cide inhibited exosome catalysed midazolam hydroxylation (measured at 5 μmol l–1 midazolam) in a dose‐dependent manner; formation of 1‐hydroxy midazolam was inhibited by 32 ± 4, 61 ± 6 and 95 ± 3% at CYP3Cide concentrations of 0.05, 0.3 and 1.5 μmol l–1. This inhibition profile is consistent with the reported in vitro IC50 value for 1‐hydroxy midazolam formation by recombinant and human liver microsome‐derived CYP3A4 (viz. 0.3 μmol l–1) 27.
Figure 3.
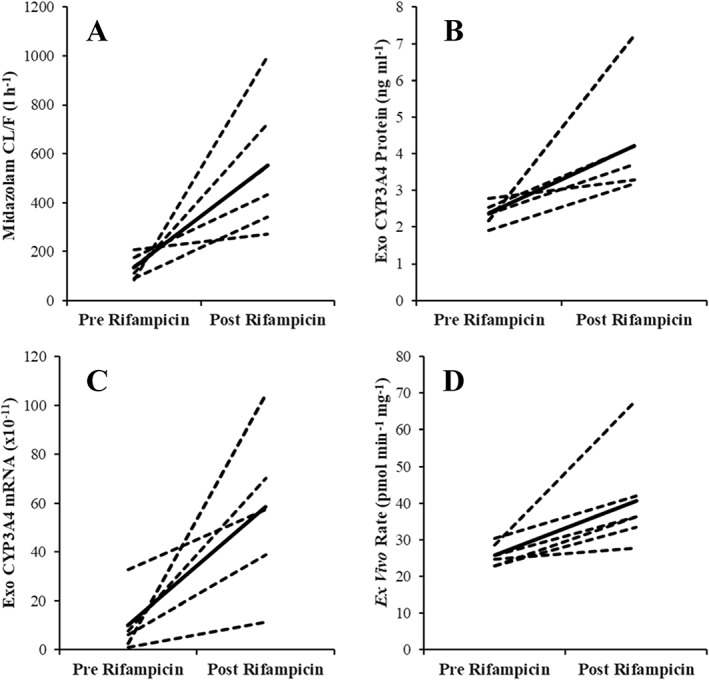
Spaghetti plots showing change in midazolam apparent oral clearance (CL/F; A) and exosome‐derived CYP3A4 biomarkers. (B) Exosome‐derived CYP3A4 mRNA expression (C) exosome‐derived CYP3A4 mRNA expression. (D) Ex vivo CYP3A4 activity (rate of 1‐hydroxymidazolam formation)
Plasma exosome‐derived CYP3A4 protein abundance correlates with midazolam exposure
Parameters (geometric mean and 90% CI) describing midazolam exposure (AUC and maximal concentration), exosome‐derived CYP3A4 mRNA and protein expression, and ex vivo CYP3A4 activity, before and after rifampicin dosing (300 mg daily for 7 days) are reported in Table 1. The change in each parameter post‐/pre‐rifampicin is visualized in Figure 3.
Figure 4 shows the concordance between exosome‐derived CYP3A4 biomarkers (mRNA expression, protein expression and ex vivo activity) and midazolam CL/F. R2 values for the correlations of exosome‐derived CYP3A4 protein expression (Figure 4A), exosome‐derived CYP3A4 mRNA expression (Figure 4B) and ex vivo CYP3A4 activity (Figure 4C) with midazolam CL/F were 0.905, 0.787 and 0.832, respectively. The concordance between the different exosome‐derived CYP3A4 biomarkers was also assessed; R2 values for the correlation of ex vivo CYP3A4 activity (Figure 4D) and exosome derive CYP3A4 mRNA expression (Figure 4E) with exosome‐derived CYP3A4 protein expression were 0.928 and 0.794, respectively, while the R2 value for the correlation of ex vivo CYP3A4 activity with exosome‐derived CYP3A4 mRNA expression (Figure 4F) was 0.638.
Figure 4.
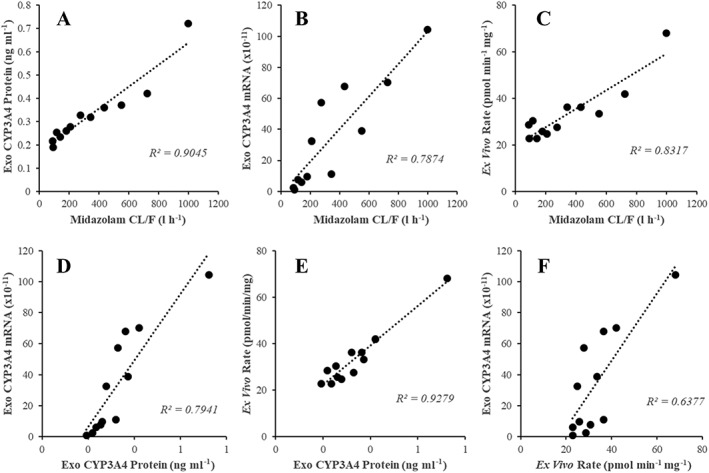
Concordance of exosome‐derived CYP3A4 biomarkers and midazolam CL/F in a cohort of healthy males (n = 6). (A) Exosome‐derived CYP3A4 protein expression vs. midazolam CL/F. (B) Exosome‐derived CYP3A4 mRNA expression vs. midazolam CL/F. (C) Ex vivo CYP3A4 activity (rate of 1‐hydroxymidazolam formation) vs. midazolam CL/F. (D) Exosome‐derived CYP3A4 mRNA expression vs. exosome‐derived CYP3A4 protein expression. (E) Ex vivo CYP3A4 activity vs. exosome‐derived CYP3A4 protein expression. (F) Ex vivo CYP3A4 activity vs. exosome‐derived CYP3A4 protein expression
Figure S4(A–C) demonstrates the concordance between the change in (∆) exosome‐derived CYP3A4 biomarkers and the change in midazolam CL/F post‐/pre‐rifampicin dosing. R2 for the correlation of the change in exosome‐derived CYP3A4 biomarkers and the change in midazolam CL/F was invariably >0.828. The R2 value for the correlation of the change in exosome‐derived CYP3A4 biomarkers post‐/pre‐rifampicin dosing (Figure S4D–F) was invariably >0.959.
Discussion
To the best of our knowledge this is the first report describing the clinical use of human plasma‐derived exosomes to characterize variability in drug exposure and support extensive ADME profiling. Data presented in this manuscript demonstrate consistent strong concordance between exosome‐derived CYP3A4 biomarkers and midazolam CL/F. Of note is the concordance between exosome‐derived CYP3A4 protein expression and midazolam CL/F, and between the change in exosome‐derived CYP3A4 protein expression and the change in midazolam CL/F post‐/pre‐rifampicin dosing. The significance of these results is that CYP3A4 is the drug‐metabolizing enzyme of greatest clinical importance 6 and variability in CYP3A4 activity is poorly described by existing precision dosing strategies 34.
To place the data presented in Figure 4 in context, commonly used endogenous probes (e.g. 4β‐ hydroxycholesterol) are only able to explain a small proportion (R2 0.1–0.4) of variability in CYP3A4 activity 35. The R2 (0.904) for data presented in Figure 4A indicate that assessment of exosome‐derived CYP3A4 protein expression can define >90% of variability in CYP3A4 activity. Furthermore, currently the best endogenous probes explain at best only half the variability in CYP3A4 induction and with this level of prediction the value of such markers to detect CYP3A4 induction is being considered 35, 36. Similarly, the concordance (R2 = 0.917) between the change in exosome‐derived CYP3A4 protein expression and the change in midazolam CL/F post‐/pre‐rifampicin dosing (Figure S4A) support the ability of exosome‐derived CYP3A4 protein expression to track the >90% of variability in CYP3A4 induction following treatment with rifampicin.
When considering alternate approaches to account for variability in CYP3A activity, the CYP3A4*22 genotype is associated with a marked reduction in CYP3A4 activity but occurs at a low frequency; i.e. 5–7% in Caucasian populations 10. Likewise, CYP3A5 is highly polymorphic, but is only expressed (*1) in approximately 15% of Caucasians. As such CYP3A4*22 and CYP3A5*1 genotypes should be considered for as they can markedly alter clearance of CYP3A substrates in affected individuals 11. However, due to their respective low frequencies, these genotypes do not explain a meaningful proportion of the observed variability in CYP3A activity 37. In the current study, participants were genotype selected for CYP3A4*1/*1 and CYP3A5*3/*3 to mitigate the contribution of CYP3A5 to midazolam clearance 11 and to minimize genetic sources of variability in midazolam exposure.
Historically, direct assessment of CYP3A4 induction has necessitated liver biopsy or elaborate deconvolution (intravenous vs. oral) of probe drug pharmacokinetics 38, 39, 40. For example, following rifampicin dosing (600 mg daily) for 4–7 days, liver biopsy demonstrates that hepatic CYP3A4 protein and mRNA is induced ~4‐fold. The results of this study present exosome‐derived CYP3A4 biomarker tracking as a viable alternative to tissue biopsy. This strategy could facilitate the assessment of time‐ and dose‐dependent variability in induction of CYP3A4.
Proteomic screening and RT‐PCR analyses demonstrated the presence of multiple CYP and UGT proteins and mRNAs in exosomes isolated from plasma (Tables S1 and Figure 3). These data are supported by prior untargeted profiling of exosomes isolated from human urine showing detectable levels of protein from all subcellular compartments including the endoplasmic reticulum, which explicitly identified CYP2D6 41. A recent study 42 proposed the specific packaging and circulation of CYP2E1 in exosomes on the basis of mRNA expression. Here, we similarly demonstrate that of the isoforms screened, CYP2E1 accounted for 63.5% relative CYP mRNA expression in exosomes. Notably, CYP2E1 is also the predominant mRNA detected in human liver, accounting for 34.5% of total hepatic CYP mRNA expression, and 67.9% of relative expression for the isoforms screened. Kumar et al. 42 also demonstrated protein expression for multiple CYP in exosomes, although their conclusions in regard to protein quantification must be considered in the context that protein expression was quantified by immunoblot band intensity using isoform specific antibodies. As band intensity depends on the efficiency of antibody binding, the lack of external standardization precluded meaningful comparison of protein abundances.
We further demonstrate the ex vivo kinetics of CYP3A and UGT substrates by exosome‐derived proteins. As described in Table 2, the kinetic behaviour of exosome and HLM‐derived enzymes were comparable. Consistent with previous reports for HLM 43, 44, the kinetics of 4‐MU glucuronide and 1‐hydroxy midazolam formation by exosome‐ and HLM‐derived CYP and UGT proteins were best described by single‐Km Michaelis–Menten kinetics. In this regard, the Km values for exosomal and microsomal reactions were comparable for both substrates. Further confirming the identity of CYP3A4 catalysed midazolam hydroxylation in exosomes, the highly selective CYP3A4 inhibitor CYP3Cide inhibited exosome catalysed 1‐hydroxy midazolam formation by >95% at a CYP3Cide concentration of 1.5 μmol l–1. This concentration of CYP3Cide is five times the IC50 for recombinant CYP3A4, and 10‐fold lower than the IC50 value for any other CYP isoform 27. Absolute CYP and UGT activity per mg of total protein was approximately 100‐times lower for exosomes compared to HLM. The lower apparent CYP and UGT activity in exosomes vs. HLM is probably due to the more crude composition of exosome preparations compared to HLM. Specifically, HLM exclusively contain endoplasmic reticulum proteins whereas exosomes contain proteins from all subcellular compartments 41, and HLM contain only hepatic proteins whereas plasma exosomes are derived from all organs and hepatic proteins account for only a small proportion of the total exosome protein yield.
When assessing CYP and UGT activities using plasma‐derived exosomes it is necessary to disrupt the external vesicle membrane to allow efficient access of the substrate and cofactor to the internally localized enzyme. This was achieved by incubating exosomes on ice in the presence of the pore forming peptide alamethicin 45 for 30 min prior to activity assays. No metabolite formation was observed for incubations performed in the absence of added cofactor for either CYP or UGT. This observation indicates that while active in an ex vivo system, exosome‐derived CYP and UGT proteins are unlikely to possess endogenous drug‐metabolizing activity in the blood.
In conclusion, data presented here support the potential for exosome‐derived biomarkers for ADME pathways (ADMExosomes) to provide new capacity to characterize variability in drug exposure. While the current analysis focusses on drug metabolizing enzymes, specifically CYP and UGTs, it is likely that this strategy is equally applicable to other ADME proteins such as drug transporters. The enhanced capacity to characterize variability in the expression and activity of key drug‐metabolizing enzymes using a diagnostically amenable sample type is a major advance for the assessment of variability in drug exposure with potential applications throughout drug development and in precision dosing. ADMExosomes represent an intriguing novel strategy with the potential to markedly advance ongoing efforts to develop biomarkers for ADME proteins driven by the increasing need to understand variability in drug exposure due to genotype, phenotype, and drug interactions. More broadly, there is great potential for plasma‐derived exosomes (ADMExosomes) to serve as liquid biopsy in support of ADME phenotyping of diseased, organ‐impaired, nonadult, pregnant and elderly subjects.
Competing Interests
L.S.W., A.D.R. and J.‐C.M. are employees and stock holders of Pfizer, World Wide Research and Development. All authors declare that there are no competing interests.
This study was supported by a project grant [1100179] from the National Health and Medical Research Council of Australia.
The authors thank Dr Christine Guguen‐Guillouzo, Dr Philippe Gripon and Dr Christian Trepo of INSERM for providing undifferentiated HepaRG cells.
Contributors
A.R., P.I.M. and M.J.S. designed the research. W.R., M.V.D., D.W. and L.S.W. performed the research. A.R., A.D.R., R.M., J.‐C.M. and M.J.S. analysed the data. A.R., P.I.M., A.D.R., J.‐C.M. and M.J.S. wrote the manuscript. A.R. and M.J.S. contributed new methods or reagents.
Supporting information
Figure S1 Immunoblot demonstrating Tsg101 (37 kDa) expression in exosomes isolated plasma, exosomes isolated from HepaRG culture medium and HepaRG cell lysate
Figure S2 Immunoblot demonstrating UGT2B7 (50 kDa) expression in exosomes isolated from the plasma of six healthy individuals (P1 to P6). Expression of UGT2B7 for each participant was assessed in independent duplicate samples
Figure S3 Kinetics of 4‐methylumbeliferone (4‐MU) glucuronidation and midazolam 1‐hydroxylation by exosome‐ and human liver microsomes (HLM)‐derived proteins. (A) 4‐MU glucuronidation by exosomes; (B) 4‐MU glucuronidation by HLM; (C) midazolam hydroxylation by exosomes; (D) midazolam hydroxylation by HLM
Figure S4 Concordance of the change in (∆) exosome‐derived cytochrome P450 (CYP)3A4 biomarkers and ∆ midazolam apparent oral clearance (CL/F) in a cohort of healthy males (n = 6) post‐/pre‐rifampicin dosing. (A) ∆ Exosome‐derived CYP3A4 protein expression vs. ∆ midazolam CL/F, (B) ∆ Exosome‐derived CYP3A4 mRNA expression vs. ∆ midazolam CL/F, (C) ∆ Ex vivo CYP3A4 activity (rate of 1‐hydroxymidazolam formation) vs. ∆ midazolam CL/F, (D) ∆ Exosome‐derived CYP3A4 mRNA expression vs. ∆ exosome‐CYP3A4 protein expression, (E) ∆ Ex vivo CYP3A4 activity vs. ∆ exosome‐derived CYP3A4 protein expression, (F) ∆ Ex vivo CYP3A4 activity vs. ∆ exosome‐derived CYP3A4 protein expression
Table S1 Relative percent mean cytochrome P450 and UDP‐glucuronosyltransferase mRNA expression in human liver and exosomes isolated from plasma
Table S2 Summary of unique cytochrome P450 and UDP‐glucuronosyltransferase peptides detected in exosomes isolated from plasma.
Rowland, A. , Ruanglertboon, W. , van Dyk, M. , Wijayakumara, D. , Wood, L. S. , Meech, R. , Mackenzie, P. I. , Rodrigues, A. D. , Marshall, J.‐C. , and Sorich, M. J. (2019) Plasma extracellular nanovesicle (exosome)‐derived biomarkers for drug metabolism pathways: a novel approach to characterize variability in drug exposure. Br J Clin Pharmacol, 85: 216–226. 10.1111/bcp.13793.
References
- 1. Rowland A, van Dyk M, Hopkins A, Mounzer R, Polasek TM, Rostami‐Hodjegan A, et al Physiologically based pharmacokinetic modeling to identify physiological and molecular characteristics driving variability in drug exposure. Clin Pharmacol Ther 2018. 10.1002/cpt.1076. [DOI] [PubMed] [Google Scholar]
- 2. Nebert DW, Russell DW. Clinical importance of cytochromes P450. Lancet 2002; 360: 1155–1162. [DOI] [PubMed] [Google Scholar]
- 3. Rowland A, Miners JO, Mackenzie PI. The UDP‐glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 2013; 45: 1121–1132. [DOI] [PubMed] [Google Scholar]
- 4. Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, et al Drug‐drug interactions for UDP‐glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos 2004; 32: 1201–1208. [DOI] [PubMed] [Google Scholar]
- 5. Dias MM, Sorich MJ, Rowland A, Wiese MD, McKinnon RA. The routine clinical use of Pharmacogenetic tests: what it will require? Pharm Res 2017; 34: 1544–1550. [DOI] [PubMed] [Google Scholar]
- 6. Zanger U, Schwab M. Cytochrome P450 enzymes in drug metabolism. Pharmacol Ther 2013; 138: 103–141. [DOI] [PubMed] [Google Scholar]
- 7. van Dyk M, Marshall J, Sorich M, Wood LS, Rowland A. Assessment of inter‐racial variability in CYP3A4 activity and inducibility among healthy adult males. Eur J Clin Pharmacol 2018; 74: 913–920. [DOI] [PubMed] [Google Scholar]
- 8. Klein K, Zanger UM. Pharmacogenomics of cytochrome P450 3A4: recent rrogress toward the "missing heritability" problem. Front Genet 2013; 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 2013; 138: 103–141. [DOI] [PubMed] [Google Scholar]
- 10. Elens L, van Gelder T, Hesselink DA, Haufroid V, van Schaik RHN. CYP3A4*22: promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics 2013; 14: 47–62. [DOI] [PubMed] [Google Scholar]
- 11. Tseng E, Walsky RL, Luzietti RA Jr, Harris JJ, Kosa RE, Goosen TC, et al Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A‐cleared drugs assessed in vitro using a CYP3A4‐selective inactivator (CYP3cide). Drug Metab Dispos 2014; 42: 1163–1173. [DOI] [PubMed] [Google Scholar]
- 12. Yamaori S, Yamazaki H, Iwano S, Kiyotani K, Matsumura K, Saito T, et al Ethnic differences between Japanese and Caucasians in the expression levels of mRNAs for CYP3A4, CYP3A5 and CYP3A7. Xenobiotica 2005; 35: 69–83. [DOI] [PubMed] [Google Scholar]
- 13. Gashaw I, Kirchheiner J, Goldammer M, Bauer S, Seidemann J, Zoller K, et al Cytochrome p450 3A4 ribonucleic acid induction by rifampin in peripheral blood mononuclear cells. Clin Pharmacol Ther 2003; 74: 448–457. [DOI] [PubMed] [Google Scholar]
- 14. Haas CE, Brazeau D, Cloen D, Booker BM, Frerichs V, Zaranek C, et al Cytochrome P450 mRNA expression in peripheral blood lymphocytes as a predictor of enzyme induction. Eur J Clin Pharmacol 2005; 61: 583–593. [DOI] [PubMed] [Google Scholar]
- 15. Li Y, Elashoff D, Oh M, Sinha U, St John MAR, Zhou X, et al Serum circulating human mRNA profiling and its utility for oral cancer detection. J Clin Oncol 2006; 24: 1754–1760. [DOI] [PubMed] [Google Scholar]
- 16. Patino WD, Mian OY, Kang JG, Matoba S, Bartlett LD, Holbrook B, et al. Circulating transcriptome reveals markers of atherosclerosis. Proc Natl Acad Sci USA 2005; 102: 3423–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heravi FM, Bala S, Kodys K, Szabo G. Exosomes derived from alcohol‐treated hepatocytes horizontally transfer liver specific miRNA‐122 and sensitize monocytes. Sci Report 2015; 5: 9991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mohr S, Liew CC. The peripheral‐blood transcriptome: new insights into disease and risk assessment. Trend Mol Med 2007; 13: 422–432. [DOI] [PubMed] [Google Scholar]
- 19. King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012; 12: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Imani Fooladi AA, Mahmoodzadeh Hosseini H. Biological functions of exosomes in the liver in health and disease. Hepat Mon 2014; 14: e13514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valadi H, Ekstrom K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007; 9: 654–659. [DOI] [PubMed] [Google Scholar]
- 22. Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa‐Silva B, et al Double‐stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014; 24: 766–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013; 200: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Korprasertthaworn P, Polasek TM, Sorich MJ, McLachlan AJ, Miners JO, Tucker GT, et al In vitro characterization of the human liver microsomal kinetics and reaction phenotyping of olanzapine metabolism. Drug Metab Dispos 2015; 43: 1806–1814. [DOI] [PubMed] [Google Scholar]
- 25. Rowland A, Elliot DJ, Knights KM, Mackenzie PI, Miners JO. The "albumin effect" and in vitro‐in vivo extrapolation: sequestration of long‐chain unsaturated fatty acids enhances phenytoin hydroxylation by human liver microsomal and recombinant cytochrome P450 2C9. Drug Metab Dispos 2008; 36: 870–877. [DOI] [PubMed] [Google Scholar]
- 26. Rowland A, Knights KM, Mackenzie PI, Miners JO. The "albumin effect" and drug glucuronidation: bovine serum albumin and fatty acid‐free human serum albumin enhance the glucuronidation of UDP‐glucuronosyltransferase (UGT) 1A9 substrates but not UGT1A1 and UGT1A6 activities. Drug Metab Dispos 2008; 36: 1056–1062. [DOI] [PubMed] [Google Scholar]
- 27. Walsky RL, Obach RS, Hyland R, Kang P, Zhou S, West M, et al Selective mechanism‐based inactivation of CYP3A4 by CYP3cide (PF‐04981517) and its utility as an in vitro tool for delineating the relative roles of CYP3A4 versus CYP3A5 in the metabolism of drugs. Drug Metab Dispos 2012; 40: 1686–1697. [DOI] [PubMed] [Google Scholar]
- 28. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2002; 2: 569–579. [DOI] [PubMed] [Google Scholar]
- 31. Conde‐Vancells J, Rodriguez‐Suarez E, Embade N, Gil D, Matthiesen R, Valle M, et al Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J Proteome Res 2008; 7: 5157–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He M, Qin H, Poon TC, Sze SC, Ding X, Co NN, et al Hepatocellular carcinoma‐derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 2015; 36: 1008–1018. [DOI] [PubMed] [Google Scholar]
- 33. Skogberg G, Gudmundsdottir J, van der Post S, Sandström K, Bruhn S, Benson M, et al Characterization of human thymic exosomes. PLoS One 2013; 8: e67554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang D, Sadee W. The making of a CYP3A biomarker panel for guiding drug therapy. J Personal Med 2012; 2: 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Diczfalusy U, Nylén H, Elander P, Bertilsson L. 4β‐hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br J Clin Pharmacol 2011; 71: 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanebratt KP, Diczfalusy U, Bäckström T, Sparve E, Bredberg E, Böttiger Y, et al. Cytochrome P450 induction by rifampicin in healthy subjects: determination using the Karolinska cocktail and the endogenous CYP3A4 marker 4beta‐hydroxycholesterol. Clin Pharmacol Ther 2008; 84: 589–594. [DOI] [PubMed] [Google Scholar]
- 37. Hohmann N, Haefeli WE, Mikus G. CYP3A activity: towards dose adaptation to the individual. Expert Opin Drug Metab Toxicol 2016; 12: 479–497. [DOI] [PubMed] [Google Scholar]
- 38. Ged C, Rouillon J, Pichard L, Combalbert J, Bressot N, Bories P, et al The increase in urinary excretion of 6 beta‐hydroxycortisol as a marker of human hepatic cytochrome P450IIIA induction. Br J Clin Pharmacol 1989; 28: 373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marschall HU, Wagner M, Zollner G, Fickert P, Diczfalusy U, Gumhold J, et al Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology 2005; 129: 476–485. [DOI] [PubMed] [Google Scholar]
- 40. Lemahieu WPD, Maes BD, Ghoos Y, Rutgeerts P, Verbeke K, Vanrenterghem Y. Measurement of hepatic and intestinal CYP3A4 and PGP activity by combined po + iv [14C] erythromycin breath and urine test. Am J Physiol Gastrointes Liver Physiol 2003; 285: G470–G482. [DOI] [PubMed] [Google Scholar]
- 41. Moon PG, Lee JE, You S, Kim TK, Cho JH, Kim IS, et al Proteomic analysis of urinary exosomes from patients of early IgA nephropathy and thin basement membrane nephropathy. Proteomics 2011; 11: 2459–2475. [DOI] [PubMed] [Google Scholar]
- 42. Kumar S, Sinha N, Gerth KA, Rahman MA, Yallapu MM, Midde NM. Specific packaging and circulation of cytochromes P450, especially 2E1 isozyme, in human plasma exosomes and their implications in cellular communications. Biochem Biophys Res Commun 2017; 491: 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miners JO, Lillywhite KJ, Matthews AP, Jones ME, Birkett DJ. Kinetic and inhibitor studies of 4‐methylumbelliferone and 1‐naphthol glucuronidation in human liver microsomes. Biochem Pharmacol 1988; 37: 665–671. [DOI] [PubMed] [Google Scholar]
- 44. von Moltke LL, Greenblatt DJ, Schmider J, Duan SX, Wright CE, Harmatz JS, et al Midazolam hydroxylation by human liver microsomes in vitro: inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents. J Clin Pharmacol 1996; 36: 783–791. [DOI] [PubMed] [Google Scholar]
- 45. Boase S, Miners JO. In vitro‐in vivo correlations for drugs eliminated by glucuronidation: investigations with the model substrate zidovudine. Br J Clin Pharmacol 2002; 54: 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Immunoblot demonstrating Tsg101 (37 kDa) expression in exosomes isolated plasma, exosomes isolated from HepaRG culture medium and HepaRG cell lysate
Figure S2 Immunoblot demonstrating UGT2B7 (50 kDa) expression in exosomes isolated from the plasma of six healthy individuals (P1 to P6). Expression of UGT2B7 for each participant was assessed in independent duplicate samples
Figure S3 Kinetics of 4‐methylumbeliferone (4‐MU) glucuronidation and midazolam 1‐hydroxylation by exosome‐ and human liver microsomes (HLM)‐derived proteins. (A) 4‐MU glucuronidation by exosomes; (B) 4‐MU glucuronidation by HLM; (C) midazolam hydroxylation by exosomes; (D) midazolam hydroxylation by HLM
Figure S4 Concordance of the change in (∆) exosome‐derived cytochrome P450 (CYP)3A4 biomarkers and ∆ midazolam apparent oral clearance (CL/F) in a cohort of healthy males (n = 6) post‐/pre‐rifampicin dosing. (A) ∆ Exosome‐derived CYP3A4 protein expression vs. ∆ midazolam CL/F, (B) ∆ Exosome‐derived CYP3A4 mRNA expression vs. ∆ midazolam CL/F, (C) ∆ Ex vivo CYP3A4 activity (rate of 1‐hydroxymidazolam formation) vs. ∆ midazolam CL/F, (D) ∆ Exosome‐derived CYP3A4 mRNA expression vs. ∆ exosome‐CYP3A4 protein expression, (E) ∆ Ex vivo CYP3A4 activity vs. ∆ exosome‐derived CYP3A4 protein expression, (F) ∆ Ex vivo CYP3A4 activity vs. ∆ exosome‐derived CYP3A4 protein expression
Table S1 Relative percent mean cytochrome P450 and UDP‐glucuronosyltransferase mRNA expression in human liver and exosomes isolated from plasma
Table S2 Summary of unique cytochrome P450 and UDP‐glucuronosyltransferase peptides detected in exosomes isolated from plasma.