

Abstract
Aims
Infection‐induced inflammation is associated with adverse long‐term outcomes in preterm infants. Pentoxifylline (PTX) is a candidate for adjunct immunomodulatory therapy in preterm infants with late‐onset sepsis (LOS) and necrotizing enterocolitis (NEC), but pharmacokinetic data in this population are extremely limited. This study aims to characterize the pharmacokinetic properties of intravenous PTX and its metabolites in preterm infants.
Method
An open label pilot clinical study of intravenous PTX as an adjunct therapy in preterm infants (gestation <32 weeks) with suspected LOS or NEC was undertaken. PTX was infused for 12 h for two days (60 mg kg−1 per 12 h), and in infants with confirmed diagnosis of LOS or NEC, for 6 h for another 4 days (30 mg kg−1 per 6 h). Plasma concentrations of PTX and its principal metabolites from collected blood samples were measured using a validated LCMS assay. NONMEM was used to analyse the data using population pharmacokinetic modelling.
Results
The preterm infants (n = 26) had a median (range) gestation of 24.8 weeks (23.3–30.4) and birthweight of 689 g (370–1285). PTX was well tolerated and without treatment‐limiting adverse effects. Changes in size (weight) and maturation were successfully modelled for PTX and metabolites. After allometric scaling, clearance increased with postmenstrual age, increasing by approximately 30% per week for PTX and M1 (lisofylline) and simulations of current dosing demonstrated a six‐fold difference in exposure between 24 and 35 weeks postmenstrual age.
Conclusions
The developed model can be used to explore dosing strategies based on size and maturation for preterm infants.
Keywords: late‐onset sepsis, lisofylline, necrotizing enterocolitis, pentoxifylline, population pharmacokinetics, preterm infant
What is Already Known about this Subject
There is growing evidence that pentoxifylline is of clinical benefit in preterm infants with sepsis.
Currently there are limited reports, consisting of point concentrations only, of the pharmacokinetics of pentoxifylline and its metabolites in infants.
The influences of size and maturation are key in determining appropriate dosing regimens in infants and children.
What this Study Adds
This is the first reported population model of PTX and its metabolites.
The effect of maturation after allometric scaling, based on postmenstrual age, on clearance is clearly defined for the population of interest.
The model developed here can be used for future studies to employ less demanding, potentially opportunistic, sampling to explore pharmacokinetic/pharmacodynamic (PKPD) relationships which will allow for precision medicine.
Introduction
Despite the advances in neonatal medicine and dramatically improved survival, the incidence of impaired developmental outcomes has not changed. Postnatal inflammation, most commonly initiated by late‐onset sepsis (LOS) or necrotizing enterocolitis (NEC), affects up to 50% of extremely preterm infants, can disturb white matter integrity and is associated with increased risk of long‐term disability 1. Systemic inflammation in preterm infants often is poorly controlled and pervasive due to immature regulation of the host response 2, 3. Current management includes early initiation of empiric antibiotic therapy as well as supportive care; however, there is no intervention available which specifically targets the systemic inflammation.
Pentoxifylline (PTX), a methylxanthine derivative and phosphodiesterase inhibitor, is an attractive candidate for adjunct immunomodulatory therapy in this high‐risk population, but population‐specific pharmacokinetic information is limited. In previous neonatal pilot studies, PTX improved endothelial cell function, reduced coagulopathy in sepsis and NEC and diminished intestinal permeability by reducing myeloperoxidase activity and oxygen free radical generation 4. PTX also significantly reduces inflammatory host responses to various stimuli in neonatal blood in vitro and in vivo 5, 6, 7. A recent Cochrane review concluded that PTX increases survival and shortens length of hospital stay in neonates with sepsis, and recommended appropriately powered clinical trials be undertaken to confirm these findings 8. Despite PTX being used in neonates for many years in some European neonatal intensive care units (NICUs), the pharmacokinetics of PTX in this vulnerable population remains incompletely defined. There are limited data on concentrations in children, infants or neonates 9, 10 and no pharmacometric analyses in these populations. In adults, PTX and the main active metabolite in adults (M1) are found in higher concentrations, whilst other metabolites with activity (M4 and M5) are found at lower concentrations 11. PTX and M1 primarily undergo hepatic elimination, while M5 is the main product found in urine 11. Accordingly, adults with hepatic impairment have significantly raised concentrations of PTX and M1, while those with renal impairment have increased M5 concentrations but no change in PTX and M1 12. Hence, significant effects of infant hepatic and renal maturation and weight on PTX metabolism are anticipated 13. PTX and metabolite concentrations in blood achieved by standard dosing regimen need to be determined 9, 10.
A method to determine the concentrations of PTX and its principal metabolites M1, M4 and M5 in the small peripheral blood volumes, like those typically available from extremely preterm infants, has recently been reported 14. The dosing schedule of PTX used in this study was based on that of previous clinical trials in newborn infants that suggested clinical benefits in a Cochrane review 8. The present study aimed to utilize this assay to develop a population pharmacokinetic model of PTX and its metabolites in extremely preterm infants with suspected LOS or NEC to assist with determining optimal dosing in this vulnerable and hitherto poorly studied population.
Methods
An open‐label pilot clinical study was conducted in the NICU at King Edward Memorial Hospital, Perth, the only perinatal referral centre in Western Australia. Study recruitment occurred between April and November 2016. The primary outcome of the study was to characterize the pharmacokinetics of intravenous PTX in extremely preterm infants. Secondary outcomes were neonatal morbidity and mortality until discharge from the NICU, including: mortality, need for surgery for NEC, intraventricular haemorrhage, periventricular leukomalacia, duration of mechanical ventilation, chronic lung disease, retinopathy of prematurity and length of hospital stay.
The study was approved by the institutional human research ethics committees (King Edward Memorial Hospital: 2015218EW; Curtin University: HR60/2016). Infants were enrolled once written informed consent was obtained from parents or guardians. The Australian National Health and Medical Research Council guidelines were followed for protecting confidentiality and data storage. Study data were collected and managed using REDCap (Research Electronic Data Capture) electronic data capture tools 15.
Infants were eligible for the study if they met the following criteria: (1) gestation <32 weeks, (2) postnatal age > 72 h, (3) < 6 h from the time a blood culture was taken for suspected LOS or NEC, and (4) written informed consent. Infants with major congenital malformations and chromosomal aberrations were excluded from the study. All adverse events that may lead to cessation of the study intervention were monitored.
Once infants were enrolled into the study, a baseline peripheral blood sample (0.2 ml) was obtained either by venepuncture or heel prick prior to starting therapy, to assess for potential interfering substances and allow the quantification of caffeine level prior to starting PTX (to be reported separately). PTX was infused for 12 h for the first 2 days (60 mg kg−1 d−1), and in infants with confirmed diagnosis of LOS or NEC, for 6 h for up to an additional 4 days (30 mg kg−1 d−1) unless (i) the infant died or (ii) the intravenous access was discontinued earlier due to clinical improvement. Confirmed LOS was defined as antibiotic therapy ≥ 5 days with positive blood culture and CRP (C‐reactive protein) >20 mg l−1. Clinical LOS was defined as antibiotic therapy ≥ 5 days with positive blood culture or CRP >20 mg l−1. For NEC, only Bell's stage 2 (definite NEC; abdominal x‐ray changes seen) or greater were considered 16.
Further planned peripheral blood samples were taken at 6–12 h, 12–18 h and > 18 h of infusion, timed with routine blood draws where possible and collected by venepuncture or heel prick. In infants with confirmed LOS or NEC in whom PTX treatment was continued until Day 6, one further blood sample was obtained. Additionally, blood sample surpluses from the routine clinical pathology laboratory around the time of therapy were opportunistically collected. These were obtained after tests performed for clinical reasons were complete. This allowed an increased number of samples available per infant, and without the need for additional dedicated study blood draws. Stability of PTX and its metabolites in such samples had been ascertained previously 14.
Analytical method
Analyte concentrations in plasma (10 μl) were measured simultaneously using a validated liquid chromatography mass‐spectroscopy (LCMS) assay 14. The precision (relative standard deviation, RSD ≤ 8%) and accuracy (RSD ≤ 13%) were within acceptable limits across the concentration range of 0.1–50 μg ml−1 for PTX and its metabolites. The lower limit of quantification (LLOQ) was 0.01 μg ml−1 for PTX, M1 (lisofylline), M4 and M5. The limit of detection for PTX, M1, M4 and M5 in plasma were 0.001 μg ml−1, 0.001 μg ml−1, 0.003 μg ml−1 and 0.0025 μg ml−1, respectively. The intra‐day and inter‐day accuracy and precision were within ±20% of the nominal value at the LLOQ for all analytes in plasma.
Pharmacokinetic modelling
Loge plasma concentration–time datasets for PTX, M1, M4 and M5 up to 48 h after the final dose were analysed by nonlinear mixed effects modelling using NONMEM (v 7.2.0, ICON Development Solutions, Ellicott City, MD, USA) with an Intel Visual FORTRAN 10.0 compiler. First order conditional estimation (FOCE) with interaction estimation method was used. The minimum value of the objective function (OFV), conditional weighted residuals (CWRES) plots and visual diagnostic plots were used to choose suitable models during the model‐building process. A significance level of P < 0.01 was set for comparison of nested models. Residual variability (RV) was estimated as additive error for the log‐transformed data. Different RV estimates for opportunistic vs. planned samples were tested. Inter‐individual variability (IIV) and correlation between IIV terms was added to parameters for which it could reasonably be estimated from the data. For one infant who had received nebulized PTX prior to entry to the study, these doses of PTX were included in the model with a parameter estimating the absolute bioavailability of nebulized PTX (Fneb) to account for concentration prior to intravenous (IV) dosing.
Initial models of PTX alone were trialled with one, two and three compartments with first‐order elimination. Once a satisfactory structure for PTX was obtained, additional compartments for disposition of M1 were added. Models with absolute and complete conversion to M1 as well as a more physiological model with reversible production of M1 were tested. Next M4 and M5 data were added with additional compartments incorporated for each metabolite and modelled with first‐order elimination. The relative molecular weight of metabolite to parent compounds were included in the model. To allow identifiability in the pharmacokinetic model, the model assumed complete conversion to each metabolite. Therefore, all of the parameters for each of the metabolites were relative to their metabolic conversion rather than representing a physiologically meaningful parameter (FM1, FM4 and FM5 for M1, M4 and M5, respectively). The parameters for each metabolite are equivalent to those if modelled alone with PTX. Although this resulted in a physiologically implausible model, the structure allows the modelling of all four analytes to be performed simultaneously, while maintaining identifiability within the model. Alternative assumptions of metabolism of PTX, while changing the numerical estimates of parameters, would not affect the subsequent steps such as covariate selection, evaluation or simulation results.
In exploring covariate relations, the effect of size on clearance and volume was first considered followed by the maturation of clearance 13. Allometric scaling using body weight (WT, in grams) was incorporated a priori with fixed exponents of ¾ for clearance and 1 for volume using a reference weight of 1000 g. Therefore, clearance terms were multiplied by and volume terms by . Given that WT changed over the course of the study, daily weight data were incorporated into the dataset to allow for these changes.
Maturation of clearance was included as an additional factor Fmat. First, the effect of postmenstrual age in weeks (PMA, gestational age at birth + postnatal age) was tested with different structures. PMA was chosen over gestational age and/or postnatal age as it presented the most parsimonious way to account for both antenatal and postnatal maturation. This included (i) a linear model:
where θlin is the parameter describing the linear change of clearance with PMA and is the median of PMA in the population; (ii) an exponential model:
where θexp is the parameter defining the exponential change in clearance with PMA; (iii) an asymptotic exponential model:
where βcl is the fractional estimate of clearance at the median PMA while Tcl is the maturation half‐life; and finally (iv) a sigmoid Emax model:
where Hill is the Hill coefficient and MAT50 is the PMA where 50% maturation occurs. After the effect of PMA was explored, plots of clearance against postnatal age and gestational age at birth were examined to investigate possible unaccounted effects on maturation of clearance.
After the effect of size and maturation was assessed, the potential influence of a limited number (given the small numbers of infants) of other covariates including gender and confirmed LOS/NEC were first assessed by examining plots of covariate vs. individual parameter. These were then assessed within NONMEM using a stepwise forwards (P < 0.05) and backwards (P < 0.01) approach.
Model evaluation
Initially, plots of observed vs. individual and population predicted values, and time vs. CWRES, were assessed. A bootstrap procedure using Perl speaks NONMEM (PSN) with 1000 samples was performed, and the parameters derived from this analysis summarized as median and 2.5th and 97.5th percentiles (95% empirical CI) to facilitate evaluation of final model parameter estimates. In addition, prediction corrected visual predictive checks (pcVPCs) and numerical predictive checks (NPCs) were performed with 1000 datasets simulated from the final models. These were performed for each analyte and stratified according to weight, age and type of sample (opportunistic vs. planned). The observed 10th, 50th and 90th percentiles were plotted with their respective simulated 95% CIs to assess the predictive performance of the model.
Simulations
Once a final population pharmacokinetic model was established, simulations were performed to assess the current (as described above) and possible alternative dose regimens. Given the optimal concentration and profile for PTX and its metabolites for sepsis in preterm infants is not known, alternative dose regimens included adjustments to dose according to age with the goal of achieving a more consistent overall exposure (area under the plasma concentration–time curve, AUC) for all preterm infants. The differences in secondary parameters after a single 12 h infusion including AUC over 24 h (AUC0–24), concentration at 12 h (end of infusion, C12), concentration at 24 h (C24) as well as ratio between concentration at these time points over the age range of the infant in the study population were examined. Using weight‐for‐age statistical data for preterm infants, 1000 male and 1000 female infants for each gestational age from 24 to 35 weeks postmenstrual age were used for simulations 17. Results for each gender were combined for each week of gestational age and summarized as median and 95% simulation intervals. These simulations focused primarily on PTX and M1, the primary active compounds, while changes in M4 and M5 were also assessed in case of significant disparity in exposure of these other metabolites.
Statistical considerations
Continuous clinical data were summarized using medians and interquartile ranges (IQR), and categorical data were summarized using frequency distributions. Univariate pre‐ and post‐PTX comparisons were conducted using a signed ranks paired Wilcoxon test. Clinical data analysis was performed using SPSS version 20 (IBM, Armonk, New York). All hypothesis tests were two‐sided, and P‐values <0.05 were considered significant.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 18, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 19.
Results
During the study, 27 preterm infants developed clinical signs consistent with suspected LOS or NEC and received intravenous PTX (Figure 1). Post‐PTX infusion blood samples were available from 26 infants; all analyses and results presented are based on these infants.
Figure 1.
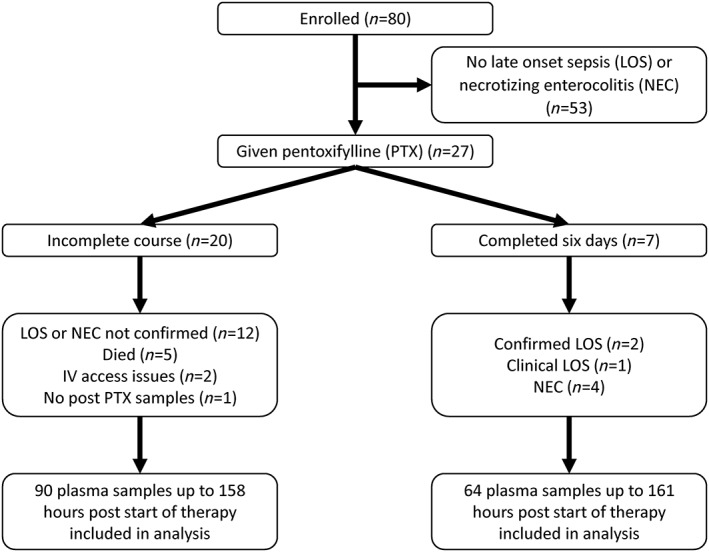
Disposition of infants in the study
The basic clinical characteristics of the participants are described in Table 1 while Table 2 describes the neonatal morbidities of the study cohort. Further, during the episodes of suspected LOS or NEC leading to administration of PTX, all infants were mechanically ventilated and three (11%) also received inhaled nitric oxide. As per current best clinical practice, all infants received full intensive care support and medications administered included antibiotics (gentamicin, vancomycin, metronidazole, meropenem), inotropes (dopamine, dobutamine), sedative (morphine), caffeine, total parenteral nutrition and blood products as required.
Table 1.
Basic clinical characteristics of the study cohort
| Parameter | Preterm infants (n = 26) |
|---|---|
| Birth gestational age (weeks) | 24.8 (23.9–27.2) [23.3–30.4] |
| Age at start of PTX infusion (days) | 12.5 (7–30) [3–60] |
| Birth weight (grams) | 689 (615–922) [370–1285] |
| Birth weight z‐score | −0.07 (−0.49–0.52) [(−2.7)–1.3] |
| Post menstrual age at initiation of therapy (weeks) | 28.5 (25.5–30.6) [24.2–35.2] |
| Weight at initiation of therapy (grams) | 974 (706–1108) [470–1936] |
| APGAR <7 at 5 min | 8 (30.8) |
| Male | 15 (57.7) |
| Caesarean section | 12 (46.2) |
| Multiple births | 2 (7.7) |
| PPROM >24 h | 5 (19.2) |
| Antenatal glucocorticoid exposure | |
| Complete | 15 (57.7) |
| Incomplete | 9 (34.6) |
| None | 2 (7.7) |
| CRIB II score | 14 (9–16) [2–18] |
Data are described as median (IQR) [min‐max] or n (%), as appropriate. PPROM: preterm prelabour rupture of membranes
Table 2.
Neonatal outcomes
| Parameter | Preterm infants (n = 26) |
|---|---|
| LOS or NEC outcome during PTX therapy | |
| LOS or NEC not confirmed | 14 (53.4) |
| Clinical LOS | 3 (11.5) |
| Confirmed LOS | 4 (15.4) |
| NEC | 5 (19.2) |
| Peak CRP during PTX infusion | 25 (12–106) [5–200] |
| Respiratory support during admission | |
| CPAP | 23 (88.5) |
| Duration (h) | 956 (466–1324) [29–2507] |
| Endotracheal ventilation | 24 (92.3) |
| Duration (h) | 588 (207–834) [17–2388] |
| Surfactant therapy | 24 (92.3) |
| Mechanical ventilation at any time during PTX infusion | 17 (65.4%) |
| Nitric oxide at any time during infusion | 3 (11.5%) |
| Chronic lung disease | 16 (61.5%) |
| Patent ductus arteriosus | 22 (84.6) |
| Intraventricular haemorrhage (grade III/IV) | 4 (15.4) |
| Periventricular leukomalacia | 0 (0%) |
| Retinopathy of prematurity | |
| Any | 14 (53.9) |
| Requiring intervention | 5 (19.2) |
| Mortality | 6 (23.1%) |
| Age at discharge home | 116 (90–151) [58–265] |
Data are described as median (IQR) [min‐max] or n (%), as appropriate
Five infants died during the intervention period; two had LOS, two had NEC and one died from complications of spontaneous intestinal perforation. One further infant died after the intervention period from severe chronic lung disease with respiratory failure. Due to the limited sample size and the open‐label design of this pilot study, no statistical hypothesis tests could be performed on clinical characteristics. Table 3 describes all relevant clinical and laboratory parameters prior to and following PTX administration. No adverse events were assessed to be related to PTX administration, separate from the expected clinical outcomes seen in these critically unwell preterm infants 20, 21, 22. Specifically, the mortality was similar to local experience as well as a large prospective registry of LOS in very low birth weight neonates 21.
Table 3.
Laboratory and clinical outcomes
| Parameter | Prior to starting PTX treatment | Post PTX treatment start a | Post PTX – Pre PTX pairwise differences | p‐value e |
|---|---|---|---|---|
| Heart rate (bpm) b | ||||
| Day 1 60 mg kg −1 day −1 | 154 (145–163)[130–180]n = 26 | 154 (149–175)[136–195]n = 26 | 5 (−2–12)[−9–23] | 0.010f |
| Day 2 60 mg kg −1 day −1 | 160 (153–170)[139–190]n = 25 | 164 (157–170)[138–188]n = 25 | 1 (−3–7)[−21–20] | 0.263 |
| Blood pressure | 38 (32–44)[18–59]n = 23 | 38 (30–47)[16–60]n = 26 | −2 (−7–5)[−14–14] | 0.701 |
| White blood cell (× 10 3 /μl) | 12.3 (9.3–18.7)[5.5–29.9]n = 26 | 16.5 (10.5–23.9)[5.5–36.4]n = 19 | 2.4 (−1.3–12.0)[−8.4–19.3] | 0.026g |
| Platelet count (× 10 3 /μl) | 212 (137–432)[72.0–623.0]n = 26 | 192 (98–‐244)[41.0–638.0]n = 19 | 8 (−51–26)[−198–273] | 0.470 |
| Neutrophils absolute count (× 10 3 mm −3 ) | 5.8 (3.4–11.1)[1.6–19.4]n = 26 | 9.8 (5.1–15.8)[2.2–24.0]n = 19 | 2.7 (−0.3–10.3)[−7.8–19.9] | 0.036h |
| Hematocrit (%) | 34 (29–37)[23–54]n = 26 | 31 (29–32)[23–43]n = 20 | −4.5 (−6.8–3.8)[−12–7] | 0.076 |
| Metabolic acidosis c (pH <7.25 and base excess > −10 mEq l −1 ) | 2 (7.7) | 3 (11.5) | ||
| IVH Grade III/IV | 4 (15.4) | 4 (15.4)d |
Data are described as median (IQR [min‐max] or n (%), as appropriate.
Within 7 days after starting PTX
Mean 4 h before starting PTX treatment and mean 4 h after starting treatment;
Arterial blood when available, capillary blood otherwise
No new incidence or worsening during PTX treatment
P‐values for signed ranks paired Wilcoxon test
19/26 (73.1%) infants had an increase in their HR following PTX infusion, by median of 10 bpm (IQR: 3–14) [range: 1–23]
14/19 (73.7%) neonates had an increase in the white cell counts (median of 8.0 (IQR: 1.4–14.2) [range: 0.2–19.3]
14/19 (73.7%) had an increase in the neutrophil counts (median of 5.7 (IQR: 1.4–11.0) [range: 0.2–19.9]
Pharmacokinetic analysis
From the 26 infants there were 154 concentrations for each analyte with no concentration below the limit of quantification (BLQ) for PTX or M1, 10% for M4 and 2% for M5. Given these proportions were low, BLQ data were not included in the analysis. As RV estimates for opportunistic vs. planned samples were not different, only a single RV estimate was used for each analyte.
A single compartment for PTX with first‐order elimination was sufficient with no significant improvement in OFV or diagnostic plots with the addition of further compartments. Similarly, a single compartment for each of the metabolites was appropriate with the assumption there was complete conversion for PTX to each of the metabolites (see Methods section). More complex models including those with reversible conversion to M1 were also tested. With these models, however, there was either no further improvement in the fit of the data, poor estimation of parameters or a model that had parameters that were unidentifiable. Therefore, the structural parameters in the final model were VPTX, V/FM1, V/FM4, V/FM5, CLPTX, CL/FM1, CL/FM4 and CL/FM5 – the central volume and clearance terms for PTX, M1, M4 and M5, respectively. These are equivalent to parameters if each metabolite was modelled alone with PTX, with a single compartment for each analyte.
Fneb was also used to estimate the absolute bioavailability of PTX in the one infant who received nebulized PTX prior to IV dosing. This was determined to be 4.8% in the base model and was fixed to this value during further modelling.
IIV for VPTX, V/FM4, V/FM5, CLPTX, CL/FM1, CL/FM4 and CL/FM5 was able to be estimated. CLPTX with CL/FM1, VPTX with V/FM1 and V/FM4 with V/FM5 were found to be highly correlated (r 2 > 0.95) and therefore the correlation between these parameters was fixed to 1. This high correlation likely results from similar chemical properties and biological processes. Correlation of CLPTX with CL/FM4, CL/FM4 with V/FM4 and VPTX with V/FM4 was also included in the final model.
For maturation of clearance, an exponential model performed better than a linear model for all analytes. Although the asymptotic exponential and sigmoid Emax models are preferred for extrapolation outside the observed age range, when tested, the result was poor precision of estimates of clearance (relative standard error > 100%). This likely relates to the age range in the present study only representing the very early phase of maturation and not including older infants, children or adults. Therefore, an exponential model of maturation was selected to describe these changes with a single additional parameter for each analyte, θexp,PTX, θexp,M1, θexp,M4 and θexp,M5. There was an estimated 31% and 29% increase each week in clearance for PTX and M1, respectively (weekly increased calculated based on θexp for each exponential equation). M4 and M5 had a slower rate of maturation over the age range, 6.4% and 14%, respectively. Once maturation due to postmenstrual age was accounted for, there was no additional effect of postnatal age or gestational age at birth noted on clearance parameters. Appendix A outlines the changes in the OFV with the steps taken in model building for size and maturation. Plots of each individual's parameter estimate along the population distribution (ETA), after inclusion of the above covariates, against PMA, WT, gestational age and postnatal age are presented in Appendix B. No other significant covariate relationships were identified.
Final model parameter estimates, including shrinkage for variable model parameters (which were low), and bootstrap results are presented in Table 4. Figures 2 and 3 present goodness‐of‐fit plots and pcVPCs for each analyte. Stratified pcVPCs and NPC demonstrated similar good predictive performance of the model (data not shown). For all analytes there is a small bias at lower, likely clinically insignificant concentrations, in the plots of observed vs. population predicted concentrations. No other bias was noted during model evaluation. Table 5 presents the relative changes in clearance over the age and weight range of the study cohort. The spread of samples relative to infusion times is also demonstrated within the pcVPC plots.
Table 4.
Final population pharmacokinetic estimates and bootstrap results for pentoxifylline, M1, M4 and M5 metabolites in preterm infants
| Parameter | Mean | RSE% | Bootstrap median [95% CI] |
|---|---|---|---|
| Objective Function Value | −260.778 | −316.306 [−563.415–123.104] | |
| Structural model parameters | |||
| CL PTX (l h −1 kg −1 ) | 0.305 | 10 | 0.303 [0.246–0.371] |
| V PTX (l kg −1 ) | 3.15 | 9 | 3.15 [2.68–3.71] |
| CL/F M1 (l h −1 kg −1 ) | 0.122 | 12 | 0.121 [0.095–0.155] |
| V/F M1 (l kg −1 ) | 0.0628 | 80 | 0.069 [0.011–0.206] |
| CL/F M4 (l h −1 kg −1 ) | 4.51 | 24 | 4.33 [3.09–6.98] |
| V/F M4 (l kg −1 ) | 29.6 | 40 | 27.2 [15.9–50.5] |
| CL/F M5 (l h −1 kg −1 ) | 0.624 | 21 | 0.605 [0.449–0.874] |
| V/F M5 (l kg −1 ) | 3.91 | 38 | 3.78 [2.26–6.67] |
| F neb (%) | 4.77 | 6 | 4.77 [4.68–5.26] |
| Covariate relationships (%) | |||
| θ exp, PTX | 0.267 | 14 | 0.263 [0.207–0.406] |
| θ exp, M1 | 0.255 | 14 | 0.254 [0.192–0.379] |
| θ exp, M4 | 0.0616 | 54 | 0.066 [0.005–0.129] |
| θ exp, M5 | 0.133 | 25 | 0.137 [0.074–0.22] |
| Variable model parameters [shrinkage%] | |||
| IIV on CL PTX | 46 [3] | 15 | 46 [31–68] |
| IIV on V PTX | 25 [21] | 18 | 25 [14–33] |
| IIV on CL/F M1 | 51[3] | 10 | 50 [37–73] |
| IIV on CL/F M4 | 78 [1] | 29 | 73 [29–109] |
| IIV on V/F M4 | 143 [4] | 18 | 132 [94–187] |
| IIV on CL/F M5 | 79 [1] | 21 | 74 [41–104] |
| IIV on V/F M5 | 142 [4] | 17 | 135 [96–177] |
| r (CL PTX , CL/F M1 ) | 1 | FIXED | |
| r (CL PTX , CL/F M4 ) | −0.187 | 65 | −0.201 [−0.563–0.032] |
| r (V PTX , V/F M4 ) | −0.422 | 42 | −0.463 [−0.803–0.079] |
| r (CL/F M4 , CL/F M5 ) | 1 | FIXED | |
| r (CL/F M4 , V/F M4 ) | 0.786 | 37 | 0.777 [0.457–0.943] |
| r (V/F M4 , V/F M5 ) | 1 | FIXED | |
| RV for pentoxifylline (%) | 35 [6] | 11 | 34 [27–41] |
| RV for M1 (%) | 36 [6] | 13 | 35 [27–44] |
| RV for M4 (%) | 52 [5] | 21 | 50 [32–70] |
| RV for M5 (%) | 40 [9] | 14 | 38 [27–49] |
Estimate fixed in the final model, relative standard error and bootstrap results obtained from base model.
terms are: CLPTX (clearance of pentoxifylline), VPTX (volume of distribution of pentoxifylline), CL/FM1 (relative clearance of M1), V/FM1 (relative volume of distribution of M1), CL/FM4 (relative clearance of M4), V/FM4 (relative volume of distribution of M4), CL/FM5 (relative clearance of M5), V/FM5 (relative volume of distribution of M5), Fneb (absolute bioavailability of nebulized pentoxifylline), θexpt (exponent of exponential maturation of clearance), r (correlation coefficient), IIV (inter‐individual variability) and RV (residual variability). IIV and RV are presented as
Figure 2.
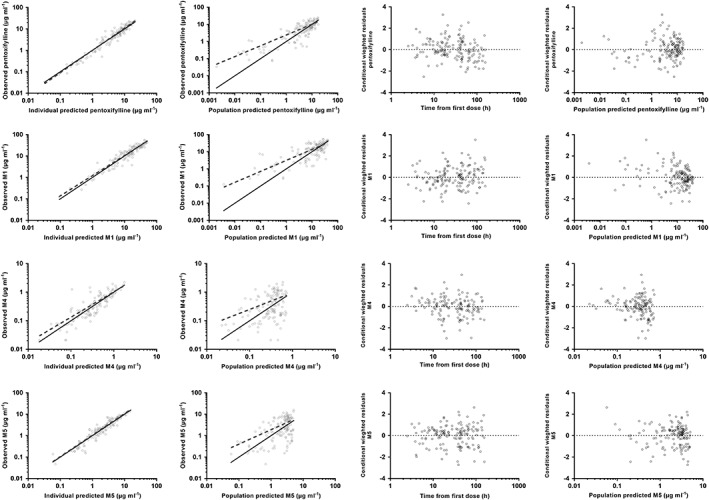
Goodness‐of‐fit plots of the final population pharmacokinetic model. From left to right: observed vs. individual predicted plasma concentration; observed vs. population predicted plasma concentrations; conditional weighted residuals (CWRES) vs. time, and conditional weighted residuals (CWRES) vs. population predicted concentrations for pentoxifylline, M1, M4 and M5 from top to bottom
Figure 3.
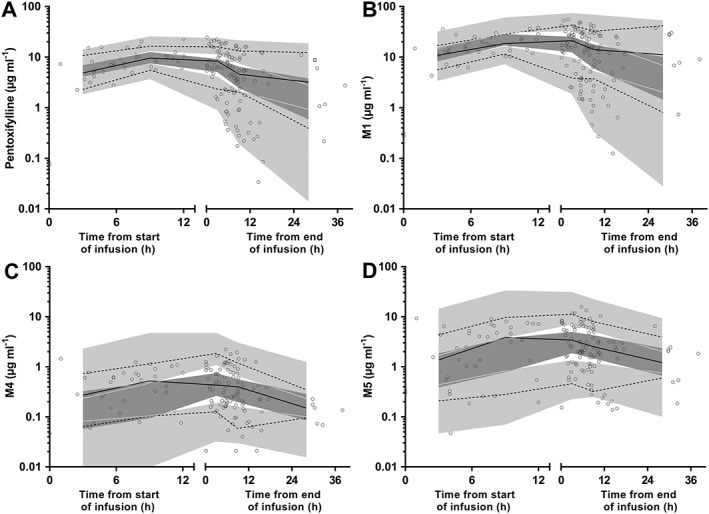
Prediction corrected visual predictive check for plasma pentoxifylline (A), M1 (B), M4 (C) and M5 (D) concentrations (μg ml−1 on log10 scale) for preterm infants. Observed 50th (solid line), 10th and 90th (dotted lines) percentiles within their simulated 95% CI (grey shaded areas) with overlying data points (○)
Table 5.
Relative median population clearance of pentoxifylline over the postmenstrual age and weight ranges of the studies infants. The values are relative to clearance at 25 weeks with a weight of 500 g
| Postmenstrual age (weeks) | Weight (g) | ||||||
|---|---|---|---|---|---|---|---|
| 500 | 750 | 1000 | 1250 | 1500 | 1750 | 2000 | |
| 25 | 1 | 1.36 | 1.68 | 1.99 | 2.28 | 2.56 | 2.83 |
| 26 | 1.31 | 1.77 | 2.20 | 2.60 | 2.98 | 3.34 | 3.69 |
| 27 | 1.71 | 2.31 | 2.87 | 3.39 | 3.89 | 4.36 | 4.82 |
| 28 | 2.23 | 3.02 | 3.75 | 4.43 | 5.08 | 5.70 | 6.30 |
| 29 | 2.91 | 3.94 | 4.89 | 5.78 | 6.63 | 7.45 | 8.23 |
| 30 | 3.80 | 5.15 | 6.39 | 7.56 | 8.66 | 9.72 | 10.75 |
| 31 | 4.96 | 6.73 | 8.35 | 9.87 | 11.31 | 12.70 | 14.04 |
Simulations
Simulations for PTX and M1 after a single 12‐h infusion of the current dose regimen of 5 mg kg−1 h−1 demonstrated higher exposure (AUC0–24, C12 and C24) with a lower C24:C12 ratio in younger infants (Figure 4). For both PTX and M1, when compared to a 30‐week old infant, the median AUC0–24 was approximately double in a 24‐week old infant and around one third in a 35‐week old infant. The range of exposure over 24 h for the range of simulated ages was therefore six‐fold in the age range of this study. For C12 (representing the peak concentration for PTX), the pattern was similar with the eldest simulated infants having 4.3 and 3.4 times lower median concentrations of PTX and M1, respectively, when compared to the youngest simulated infants. Median C24 concentrations were 33% and 21% lower than median C12 concentrations for the simulated 24‐week‐old infants, due to slow clearance in this population. By contrast median concentrations at 24 h were negligible in the simulated 35‐week‐old infants. An alternative dose regimen was simulated with dose rate adjusted for postmenstrual age. This was 3 mg kg−1 d−1 for 24–28 weeks, 5 mg kg−1 h−1 for 29–31 weeks, 7.5 mg kg−1 h−1 for 32–33 weeks and 10 mg kg−1 h−1 for 34–35 weeks. Simulations from this regimen resulting in less variability in AUC0–24 of PTX and M1 over the simulated age range (Figure 5). Simulations of the current dose regimen for M4 and M5 revealed less significant changes over the age range with wider simulation intervals. For the alternative regimen there was increasing concentrations of both metabolites, however, the combined concentration of M4 and M5 remained below that of PTX and M1.
Figure 4.
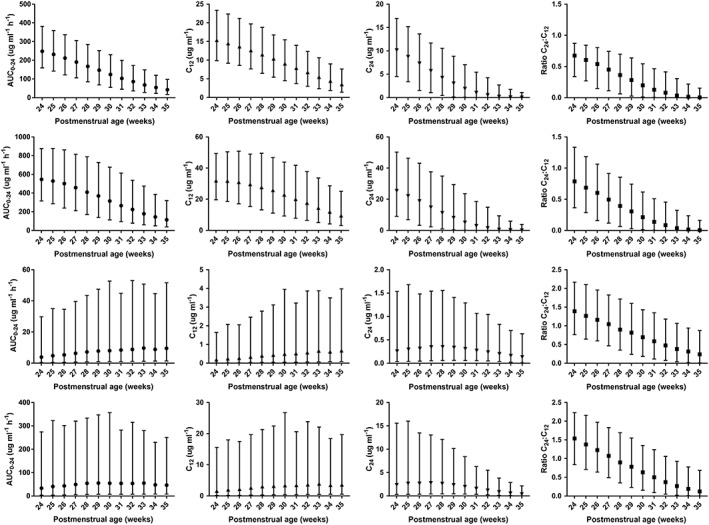
Summary of simulation results of a single 12 h infusion of 5 mg kg−1 h−1 pentoxifylline for 2000 infants (equal male:female) for each week of postmenstrual age from 24 to 35 weeks summarized as median and 95% simulation interval. From left to right are the results for area under the plasma concentration vs. time curve to 24 h (AUC0–24 in μg ml−1 h−1), concentration at end of infusion (12 h, C12 in μg ml−1), concentration at 24 h (C24 in μg ml−1) and ratio of C24:C12 for pentoxifylline, M1, M4 and M5 from top to bottom
Figure 5.
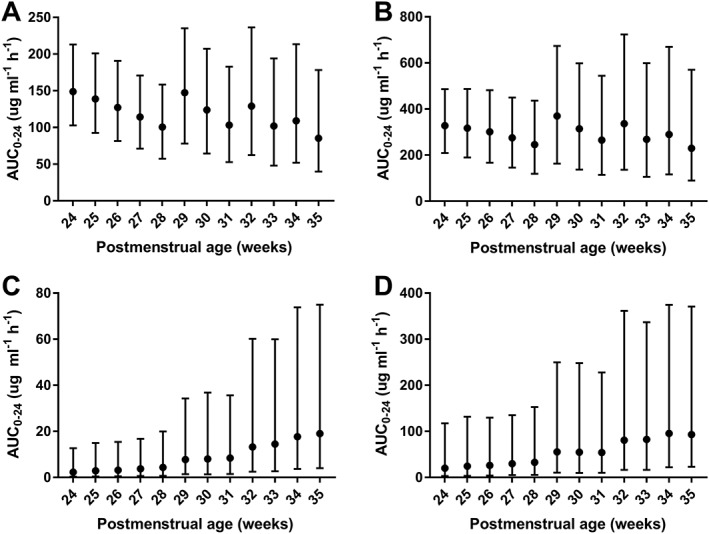
Summary of area under the plasma concentration vs. time curve to 24 h (AUC0–24 in μg ml−1 h−1) for pentoxifylline (A), M1 (B), M4 (C) and M5 (D) from simulation results of a single 12 h infusion of potential alternative dose regimen of pentoxifylline for 2000 infants (equal male:female) for each week of postmenstrual age from 24 to 35 weeks summarized as median and 95% simulation interval
Discussion
Here we report the results of a pilot study of intravenous PTX in very preterm infants with suspected LOS or NEC and to our knowledge provide the first pharmacokinetic modelling for this population. Based on recently reported, validated LCMS‐based methodology, analysis of PTX and metabolite concentrations was achievable in the limited blood volumes typically available from preterm infants 14. The population model developed here simultaneously accounts for the pharmacokinetics of PTX and its three principal metabolites. Further, this model allows for integration of the effects of postmenstrual age and the rapid changes in bodyweight occurring during the first weeks of life.
Utilization of a population approach to pharmacokinetics had several benefits in this case.
This included allowing for the analysis of variable times of collection from the different infants, which fits better into clinical practice. In addition, there were unplanned opportunistic plasma samples available from surplus blood taken as a part of routine clinical care. These samples were able to be incorporated into the final model without the need for a separate residual variability term, despite their different handling, and did not show any bias in simulation‐based diagnostics (i.e. pcVPC).
The flexible nature of population modelling also allowed for a dynamic method to account for residual PTX in the baseline sample from nebulized doses. By allowing the nebulized doses to be included in the model, an additional benefit included an estimate of the absolute bioavailability of nebulized PTX (around 5%). Although only from a single infant, this estimate is close to a point estimate from a previous publication where concentrations were around 20 times less when comparing nebulized to IV doses 10.
Finally, by developing a population model this can be supplemented with more limited sampling strategies in future studies to further refine the effects of size and maturation and allow estimation of pharmacokinetic parameters from fewer samples using Bayesian forecasting.
Importantly from the clinical perspective, intravenous administration of PTX was found to be feasible, well tolerated and without adverse effects, even in the smallest and most unwell infants. The dosing schedule of PTX used in this study was based on that of previous clinical trials in newborn infants that suggested clinical benefits, such as improved survival and reduced length of stay, and represent the currently available best evidence 8. Using this schedule, the concentrations of PTX and its metabolites achieved in our study of very preterm infants are in the range of those previously reported in infants and adults 9, 11, 23, 24, 25, 26. Currently there are no data to support particular pharmacokinetic targets associated with outcomes in this population. The model can be utilized to explore alternative dosing regimens in a flexible way, including but not limited to different dose, duration of infusion and frequency of administration.
As was anticipated, given slower clearance in adults with hepatic failure 12 and maturation of hepatic elimination in infants 13, we observed significant changes in the concentrations of PTX and M1 with a faster clearance in older infants, after allometric scaling. To maintain similar exposure across gestational ages, dose adjustments may therefore be advisable that account for changing maturity and bodyweight. The findings from this study are planned to be expanded in a larger cohort of preterm infants with a comprehensive range of postmenstrual ages and bodyweights from an ongoing large randomized controlled trial of PTX as adjunct therapy in extremely preterm infants with suspected LOS and/or NEC. This larger dataset also plans to investigate for potential clinically efficacious pharmacokinetic targets. It is conceivable, given in vitro differences in the effects of PTX between preterm and term infants, that these targets will be different through the age range of these preterm infants 6, 7. For example, potentially, a more pronounced hyperinflammatory state in younger infants may require higher concentrations of PTX for equivalent effect.
Our study also has some unavoidable limitations. This includes the lack of a comparator group and limited sample size that did not allow characterization of PTX effects on clinical outcomes. Currently there is an ongoing multinational randomized clinical trial which is designed to investigate this further. Despite the limited sample size, the primary objective was still met with the developed model having good predictive performance. The use of an exponential maturation model limits the ability to use the model for prediction outside of the age range of the study population. Given that currently the evidence is for the use of PTX in a preterm population, the age range of dosing incorporated here, namely 24–35 weeks postmenstrual age, is sufficiently wide to include most infants who will received PTX in this setting. Additionally, the present data can be pooled with future data sets in older infants, children and even adults, if the need to build a model of maturation to adulthood is required.
In summary, this report presents the first population pharmacokinetic model of PTX and its principal metabolites and indicates that preterm infants' elimination capacity matures rapidly with increasing postmenstrual age. There are plans for future studies to use this model to examine pharmacokinetic‐pharmacodynamic relationships which may warrant changes to the current dose regimes.
Competing Interests
There are no competing interests to declare.
This study was supported by a grant of the Channel 7 Telethon Trust and Women and Infants Research Foundation (Principal Investigator: T.S.) and the Telethon‐Perth Children's Hospital Research Fund, a joint initiative of the Channel 7 Telethon Trust and the Western Australian Department of Health (Principal Investigator: K.T.B.). We thank the families, participants and all staff that contributed to this study and made it possible.
Appendix A.
Changes in objective function value (OFV) with steps taken in model building for pentoxifylline and metabolites
| Step taken | Change in OFV |
|---|---|
| Inclusion of allometric scaling | −50.614 |
| Linear maturation model | −74.779 |
| Exponential maturation model | −102.145 |
| Asymptotic exponential maturation model | −102.145 |
| Sigmoid E max maturation model | −100.927 |
Appendix B.
Plots of ETA terms within NONMEM against key covariates
ETA1 corresponds to CLPTX and CL/FM1, ETA2 corresponds to CL/FM3 and CL/FM4, ETA3 corresponds to V/FM4 and V/FM5 and ETA4 corresponds to VPTX. Covariates included are gestational age (GEST), postnatal age (PNA), postmenstrual age (PMA) and weight (WT).
Salman, S. , Hibbert, J. , Page‐Sharp, M. , Manning, L. , Simmer, K. , Doherty, D. A. , Patole, S. , Batty, K. T. , and Strunk, T. (2019) Effects of maturation and size on population pharmacokinetics of pentoxifylline and its metabolites in very preterm infants with suspected late‐onset sepsis or necrotizing enterocolitis: a pilot study incorporating clinical outcomes. Br J Clin Pharmacol, 85: 147–159. 10.1111/bcp.13775.
References
- 1. Strunk T, Inder T, Wang X, Burgner D, Mallard C, Levy O. Infection‐induced inflammation and cerebral injury in preterm infants. Lancet Infect Dis 2014; 14: 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Volpe JJ. Postnatal sepsis, necrotizing entercolitis, and the critical role of systemic inflammation in white matter injury in premature infants. J Pediatr 2008; 153: 160–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoon BH, Romero R, Kim CJ, Koo JN, Choe G, Syn HC, et al High expression of tumor necrosis factor‐alpha and interleukin‐6 in periventricular leukomalacia. Am J Obstet Gynecol 1997; 177: 406–411. [DOI] [PubMed] [Google Scholar]
- 4. Harris E, Schulzke SM, Patole SK. Pentoxifylline in preterm neonates: a systematic review. Paediatr Drugs 2010; 12: 301–311. [DOI] [PubMed] [Google Scholar]
- 5. Shabaan AE, Nasef N, Shouman B, Nour I, Mesbah A, Abdel‐Hady H. Pentoxifylline therapy for late‐onset sepsis in preterm infants: a randomized controlled trial. Pediatr Infect Dis J 2015; 34: e143–e148. [DOI] [PubMed] [Google Scholar]
- 6. Schuller SS, Wisgrill L, Herndl E, Spittler A, Forster‐Waldl E, Sadeghi K, et al Pentoxifylline modulates LPS‐induced hyperinflammation in monocytes of preterm infants in vitro . Pediatr Res 2017; 82: 215–225. [DOI] [PubMed] [Google Scholar]
- 7. Speer EM, Dowling DJ, Ozog LS, Xu J, Yang J, Kennady G, et al Pentoxifylline inhibits TLR‐ and inflammasome‐mediated in vitro inflammatory cytokine production in human blood with greater efficacy and potency in newborns. Pediatr Res 2017; 81: 806–816. [DOI] [PubMed] [Google Scholar]
- 8. Haque KN, Pammi M. Pentoxifylline for treatment of sepsis and necrotizing enterocolitis in neonates. Cochrane Database Syst Rev 2011; CD004205. [DOI] [PubMed] [Google Scholar]
- 9. Szymura‐Oleksiak J, Bury J, Lauterbach R, Pawlowski M. Serum concentrations of pentoxifylline and its metabolites in premature infants with sepsis when administered by continuous intravenous infusion. Pharmaceut Sci 1997; 367–371. [Google Scholar]
- 10. Lauterbach R, Szymura‐Oleksiak J. Nebulized pentoxifylline in successful treatment of five premature neonates with bronchopulmonary dysplasia. Eur J Pediatr 1999; 158: 607. [DOI] [PubMed] [Google Scholar]
- 11. Smith RV, Waller ES, Doluisio JT, Bauza MT, Puri SK, Ho I, et al Pharmacokinetics of orally administered pentoxifylline in humans. J Pharm Sci 1986; 75: 47–52. [DOI] [PubMed] [Google Scholar]
- 12. Sanofi‐aventis . FDA Product Label: Trental® (pentoxifylline) extended‐release tablets, 400 mg, 2015.
- 13. Anderson BJ, Holford NH. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 2009; 24: 25–36. [DOI] [PubMed] [Google Scholar]
- 14. Page‐Sharp M, Strunk T, Salman S, Hibbert J, Patole SK, Manning L, et al Simultaneous determination of pentoxifylline, metabolites M1 (lisofylline), M4 and M5, and caffeine in plasma and dried blood spots for pharmacokinetic studies in preterm infants and neonates. J Pharm Biomed Anal 2017; 146: 302–313. [DOI] [PubMed] [Google Scholar]
- 15. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap) – a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009; 42: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee JS, Polin RA. Treatment and prevention of necrotizing enterocolitis. Semin Neonatol 2003; 8: 449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fenton TR, Kim JH. A systematic review and meta‐analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr 2013; 13: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174 (Suppl. 1): S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Costeloe KL, Hennessy EM, Haider S, Stacey F, Marlow N, Draper ES. Short term outcomes after extreme preterm birth in England: comparison of two birth cohorts in 1995 and 2006 (the EPICure studies). BMJ 2012; 345: e7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, et al Late‐onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics 2002; 110: 285–291. [DOI] [PubMed] [Google Scholar]
- 22. Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, et al Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 2010; 126: 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beermann B, Ings R, Mansby J, Chamberlain J, McDonald A. Kinetics of intravenous and oral pentoxifylline in healthy subjects. Clin Pharmacol Ther 1985; 37: 25–28. [DOI] [PubMed] [Google Scholar]
- 24. Mauro VF, Mauro LS, Hageman JH. Alteration of pentoxifylline pharmacokinetics by cimetidine. J Clin Pharmacol 1988; 28: 649–654. [DOI] [PubMed] [Google Scholar]
- 25. Mauro VF, Mauro LS, Hageman JH. Comparison of pentoxifylline pharmacokinetics between smokers and nonsmokers. J Clin Pharmacol 1992; 32: 1054–1058. [DOI] [PubMed] [Google Scholar]
- 26. Nisi A, Panfili M, De Rosa G, Boffa G, Groppa F, Gusella M, et al Pharmacokinetics of pentoxifylline and its main metabolites in patients with different degrees of heart failure following a single dose of a modified‐release formulation. J Clin Pharmacol 2013; 53: 51–57. [DOI] [PubMed] [Google Scholar]