

Abstract
Periodontitis is one of the most common inflammatory diseases, with a prevalence of 11% worldwide for the severe forms and an estimated heritability of 50%. It is classified into the widespread moderate form chronic periodontitis (CP) and the rare early-onset and severe phenotype aggressive periodontitis (AgP). These different disease manifestations are thought to share risk alleles and predisposing environmental factors. To obtain novel insights into the shared genetic etiology and the underlying molecular mechanisms of both forms, we performed a two step-wise meta-analysis approach using genome-wide association studies of both phenotypes. Genotypes from imputed genome-wide association studies (GWAS) of AgP and CP comprising 5,095 cases and 9,908 controls of North-West European genetic background were included. Two loci were associated with periodontitis at a genome-wide significance level. They located within the pseudogene MTND1P5 on chromosome 8 (rs16870060-G, P = 3.69 × 10−9, OR = 1.36, 95% CI = [1.23–1.51]) and intronic of the long intergenic non-coding RNA LOC107984137 on chromosome 16, downstream of the gene SHISA9 (rs729876-T, P = 9.77 × 10−9, OR = 1.24, 95% CI = [1.15–1.34]). This study identified novel risk loci of periodontitis, adding to the genetic basis of AgP and CP.
Subject terms: Genome-wide association studies, Sequence annotation, Genetic databases
Introduction
Periodontitis (PD) is a common inflammatory disease of the oral cavity that leads to the resorption of alveolar bone, making it the major cause of tooth loss in adults above 40 years [1]. The inflammation is often chronic and can affect large areas of the gingival tissues. Since periodontal disease is painless, the inflammation is usually long-term persisting and it is common for PD to have reached advanced degrees of severity before it is diagnosed and treatment is started. Recurrent and persistent inflammations caused by bacteria are also recognized as continually renewing reservoirs for the systemic spread of bacterial antigens, cytokines, and other proinflammatory mediators and may bring a burden onto the rest of the body [2]. Accordingly, researchers have hypothesized the etiologic role of PD in the pathogenesis of various systemic illnesses like diabetes mellitus [3], cardiovascular disease [4], and osteoporosis [5], bridging the once-wide gap between medicine and dentistry.
Severe forms of PD are prevalent with > 9% in adults with an age of 30 years and older [6]. PD is classified into the widespread forms chronic periodontitis (CP) and the rare, early-onset and much more severe disease phenotype of aggressive periodontitis (AgP), which has a prevalence of <0.1% globally [7]. Both forms have an estimated heritability of 50%, with AgP having a stronger and better-established heritable component compared to CP [8–10]. CP and AgP have a similar etiology and histopathology and can be considered as parts of the same disease spectrum. The different disease manifestations develop as a consequence of individual combinations of genetic risk loci that determine the individual immune response in conjunction with external factors like smoking. In this view, the different disease manifestations of a complex disease such as PD are not considered as confined entities [11].
To date, several genome-wide association studies (GWAS) for CP have been published [9, 12–17] but in these studies no common allele reached genome-wide significance, likely related to an insufficiently large number of available case-control samples of this late-onset disease phenotype. Previously, we performed a GWAS of AgP and identified a single nucleotide polymorphism (SNP) within the gene GLT6D1 to be associated with AgP at a genome-wide significance level [18]. This association was replicated in a Sudanese-African case-control sample of AgP, but could not be validated for the more moderate late-onset form CP [19]. This suggests a role in the etiology of early-onset very severe disease phenotypes of AgP alone. In an expanded AgP case-control sample, we have recently performed a second GWAS and validated the most significant associations with CP in a German CP case-control sample. In this study, two common variants at SIGLEC5 and DEFA1A3 reached genome-wide significance in the combined sample [20]. Currently, these variants are the only shared risk variants of CP and AgP identified by GWAS, together with two haplotype blocks at PLG (plasminogen) and at the gene cluster PF4 (platelet factor 4)/PPBP (pro-platelet basic protein)/CXCL5 (C-X-C motif chemokine ligand 5), which were identified by different approaches [21–23]. In the current study, we aimed to identify additional common susceptibility variants for AgP and CP by conducting a meta-analysis with our North-West European GWAS of AgP and a European-American GWAS of CP. Suggestive associations were validated in imputed genotypes from our previously used German CP case-control sample. We identified two novel loci, which showed genome-wide significant associations with PD.
Materials and methods
Participating studies
The meta-analysis samples consisted of case-control GWA studies of German and Dutch AgP [20] and of European American [9] and German CP [17] patients (Supplementary Table 1).
The German AgP sample (AgP-Ger) included 680 cases and 3,973 controls. Cases were recruited across Germany by the biobank Popgen [24], University-Hospital Schleswig-Holstein, Germany. Controls originate from North-Germany and West-Germany and were recruited from the Competence Network “FoCus–Food Chain Plus” [25], the Dortmunder Gesundheitsstudie–DOGS [26] and the Heinz Nixdorf Recall Studies 1–3 [27]. The Dutch AgP sample (AgP-NL) consisted of 171 cases and 2607 controls. The Dutch cases were recruited from the ACTA (Academisch Centrum Tandheelkunde Amsterdam) and the Dutch controls were recruited from Rotterdam and Wageningen by the B-Proof Study [28]. Inclusion criteria for AgP were ≥2 affected teeth with ≥ 30% bone loss in patients <36 years of age; disease phenotype was diagnosed by full mouth dental radiographs. Genotyping was performed on Illumina Omni Bead Chips for German and Dutch AgP cases and subsequent genotype imputation was performed on the 1000 Genomes Phase 3 reference.
The European American CP (CP-EA) sample included 958 severe (sev) CP cases, 2293 moderate (mod) CP cases and 1909 controls from the Atherosclerosis Risk in Communities (ARIC) Study and were described before [29]. In brief, the patients were classified by the Centers for Disease Control/American Academy of Periodontology (CDC/AAP) consensus three-level classification system [30]. The CDC/AAP taxonomy uses clinical attachment loss (CAL) and PD criteria to define three CP categories as healthy-mild, moderate, and severe CP cases, the first being the control. Genotyping was carried out using the Affymetrix Genome-Wide Human SNP Array 6.0 and the subsequent genotype imputation was performed on the HapMap Phase II reference with individuals of Northern and Western European (CEU) ancestry.
The German CP (CP-Ger) sample consisted of 993 cases and 1419 controls from a meta-analysis of SHIP and SHIP-TREND cohorts [31–33]. In brief, subjects within the first and the third tertile of proportion of proximal sites with attachment loss (AL) ≥ 4 mm were contrasted after stratification by sex and 10-year age groups. Age-specific tertiles were defined to include severely diseased cases within each age stratum. Thus, also young and severely diseased subjects were captured and included in the third tertile. Otherwise, those in the third tertile would have been the older ones and those in the first tertile would have included the younger ones only. Individuals aged > 60 years were excluded. The identification of these subjects is important as we assume that genetic predispositions might manifest especially in younger age, while in older subjects these effects are overlaid with risk factor associated disease progression. This case-control sample was previously described in detail [17]. Cases and controls were genotyped either with the Affymetrix Genome-Wide Human SNP Array 6.0 or the Illumina Human Omni 2.5 array and imputed on the 1000 Genomes Phase 1 reference.
Data availability
Genotype data for aggressive periodontitis samples and the chronic periodontitis sample with German descent are available upon request from the biobanks PopGen (https://www.epidemiologie.uni-kiel.de/biobanking) and ShIP (https://www.fvcm.med.uni-greifswald.de/index.html). Summary statistics for CP-EU can also be downloaded (see ref. [9]).
Filtering
In the post imputation QC processing, variants were excluded, which passed the following cut-off criteria: AgP: Hardy-Weinberg-Equilibrium P-value (PHWE) < 10−4, imputation quality (INFO) of < 0.8 [20]; CP (Germany): PHWE ≤ 0.001, imputation quality (r2HAT) ≤ 0.3 [17]; CP (European-US): PHWE < 10−5 (ARIC) and PHWE < 10−6 (Health ABC), imputation quality < 0.8 [9];
Moreover, we filtered out variants with a minor allele frequency (MAF) < 0.05 because our study lacked statistical power to analyze rare variants. The variant sets did not completely overlap between the different genotype data sets which was mainly due to the different human genome references that were used to impute the different data sets. Only variants with genotype data available in each study were analyzed in the meta-analysis.
Meta-analysis
Firstly, hypotheses on candidate gene regions were generated by meta-analyzing the genome-wide association scans of AgP-Ger, AgP-NL, CP-EA-sev, and CP-EA-mod based on the additive model using logistic regression. In this stage, we did not include CP-Ger, because the complete genome-wide summary statistics were not available to us. Subsequently, all variants with Pmeta < 10−5 were selected and clustered into genomic loci. Our clustering algorithm compared pairs of variants and assigned pairs with a maximum distance of 200 kilo base pairs (kb) to the same locus. For each locus, we then interrogated for CP-GER the genotypes of all variants in the range of the variant with lowest P-value +/− 500 kb. However, of 35 loci that we meta-analyzed with the CP-GER sample, from this sample no genotype data were available for four loci.
Secondly, we ran a validation meta-analysis for the prioritized candidate risk loci by pooling CP-Ger with the other samples and filtered them again. We considered variants to be significantly associated with PD, if the pooled P-value in this meta-analysis was genome-wide significant (i.e., P < 5 × 10−8). Additionally, we considered variants as suggestively associated with PD if the P-value was <10−6 in the final meta-analysis. By default, we applied a fixed effects model. However, for variants with a high degree of heterogeneity, i.e., a P-value of Cochran’s Q P(Q) < 0.05 and a heterogeneity index I2 > 0.5, we applied a random effects model instead. We used the P-value correction method for shared controls by Zaykin and Kozbur to account for inflation when pooling summary statistics of CP-EA-mod and CP-EA-sev [34].
Functional annotation
Variants were annotated using the Genehopper database (DB) [35]. Genehopper DB integrates data of many public sources by applying periodically executed extraction, transformation and loading (ETL) processes. Specifically, we used integrated datasets of linkage equilibrium (LD), expression quantitative trait loci (eQTL) mappings, topologically associated domain (TAD) boundaries and GWAS studies to annotate our findings. Identified loci were annotated using LD information (correlation measures r2 and D’) from the European reference population (EUR) of 1000 Genomes Phase 3. EQTL mapping information was gathered from Genotype-Tissue Expression project (GTEx) [36], Haploreg v4.1 [37], GRASP v2.0 [38], SCAN [39], seeQTL [40], and Blood eQTL Browser [41]. EQTLs describe genetic variants that contribute to the variation of gene expression levels. Information about TAD boundaries derived by Hi-C experiments was taken from Dixon et al. [42]. A TAD is a genomic region defined by interactome boundaries that represent a spatial compartment in the genome. TADs are essentially cell-type independent and physical interactions occur more frequently inside a TAD. We utilized TAD boundaries to separate genes, which reside in the same TAD as the associated LD block, i.e., genes in cis, from genes that reside outside the TAD of the associated LD block, i.e., genes in trans. The dataset used contained TADs with a length of ~ 853 kb in average (maximum length = 4.44 mega base pairs, minimum length = 0.8 kb). Variant consequence information was taken from Ensembl Variation DB and additionally, we annotated variants using combined annotation dependent depletion (CADD) score. The CADD score combines several annotations into a single Phred-scaled pathogenicity score between 1 to 99 by applying the formula −10 × log10(variant_rank/number_of_variants) to the list of variants sorted by their pathogenicity. Accordingly, a score ≥10 indicates that the variant belongs to the 10% most deleterious substitutions in the human genome, a score ≥ 20 indicates that the variant belongs to the 1% most deleterious substitutions. To illuminate the relationship to other traits and diseases, we took information of phenotype associations and links from the NHGRI-EBI Catalogue of published GWAS [43].
Results
Meta-analysis
The genetic datasets of the four samples AgP-Ger (6,416,838 variants), AgP-NL (6,319,518 variants), CP-EA-mod (2,135,233 variants), CP-EA-sev (1,809,893 variants) contained a total of 1,722,107 variants being present in all datasets. In the first meta-analysis comprising 4102 cases and 8489 controls (due to the shared controls between CP-EA-sev and CP-EA-med, this control group was only counted once), 35 distinct loci met the pre-specified criteria of Pmeta < 10−5 (Fig. 1a, Supplementary Table 2).
Fig. 1.

Description of the analysis workflow. a A two-step meta-analysis was performed because for the German CP case-control sample (SHiP cohort), the complete GWAS dataset was not available to be directly included into the meta-analysis. Thus, in the first step, hypotheses on candidate gene regions were generated with the genome-wide summary statistics of the N.-W.-EU AgP-GWAS and the EU-USA CP-GWAS data alone. Next, genotypes of the priorized candidate risk loci were obtained from the SHiP cohorts and included into the dataset of the first meta-analysis. This gave the validation meta-analysis dataset, which provided the highest statistical power. This increase in statistical power resulted in genome-wide significance of two loci. (N.W. EU North-West Europeans, EU-USA European-US Americans, NL The Netherlands, PD Periodontitis, AgP Aggressive Periodontitis). b In small sample sizes such as the Dutch AgP case sample, chance effects are immanent and can skew the results, e.g., by random depletion of risk alleles in a particular sample. The misrepresentation of the allele frequency in a small case sample can have strong effects on the inter sample heterogeneity. However, a low inter sample heterogeneity is the prerequisite for applying the fixed effects model in which the effects of each study sample are weighted by the sample size. Otherwise the random effects model must be applied in which all study samples are weighted equally, irrespective of the sample size. Therefore, the two-step meta-analysis of a was repeated without the Dutch AgP case-control sample. In this analysis, we re-discovered the association of SNP rs729876 with an equal association P-value. Additionally, a second association of SNP rs16870060 passed the genome-wide significance threshold (P = 5 × 10E-08)
Subsequently, these 35 loci were meta-analyzed including the additional sample of CP-Ger, which raised the overall sample size to 5,095 cases and 9,908 controls. In this meta-analysis, SNP rs729876, located downstream of SHISA9 on chromosome (chr) 16, intronic to the long intergenic noncoding RNA (lincRNA) LOC107984137, reached genome-wide significance (P = 1.21 × 10−8, OR = 1.23, 95% CI = [1.15–1.32], Table 1, Fig. 2). Three more variants had a P < 10−6. SNP rs11084095, located at SIGLEC5 (sialic acid binding Ig-like lectin 5), reached the second highest significance level in this study with P = 5.09 × 10−8 (OR = 1.17, 95% CI = 1.11–1.24). SIGLEC5 was earlier identified by us as a susceptibility locus for PD with genome-wide significance, tagged by the GWAS SNP rs4284742 [20]. SNP rs4284742 was not included in the variant set of CP-EA. The other variants were SNP rs9982623, intronic to MCM3AP (minichromosome maintenance complex component 3 associated protein; chr21), with P = 8.65 × 10−7 (OR = 1.23, 95% CI = 1.13–1.33) and SNP rs9984417, located between the processed pseudogene MAPK6PS2 and lincRNA AP000959.2 on chr21, with P = 9.31 × 10−7 (OR = 1.16, 95% CI = 1.09–1.23).
Table 1.
Six variants were identified to be associated with AgP and CP at the significance level P < 10−6 in the meta-analyses using either all samples (all) or after excluding the small sample AgP-NL (no AgP-NL)
| Lead variant (HGVS) | Locus | Nearest gene(s) | A1 | A2 | EAF Cas | EAF Con | Stage | OR [95% CI] | Mod | P | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
rs16870060a (chr8:g.104098515 G > T) |
8q22.3 | MTND1P5 | G | T | 0.94 | 0.90 | AgP-Ger | 1.46 [1.20–1.79] | 2.14E-04 | |
| 0.88 | 0.91 | AgP-NL | 0.70 [0.48–1.03] | 7.20E-02 | |||||||
| 0.92 | 0.89 | CP-EA-sev | 1.29 [1.09–1.51] | 2.37E-03 | |||||||
| 0.91 | 0.89 | CP-EA-mod | 1.39 [1.10–1.76] | 6.21E-03 | |||||||
| 0.91 | 0.90 | CP-Ger | 1.35 [1.05–1.74] | 2.06E-02 | |||||||
| Pooled (all) | 1.26 [1.05–1.50] | RE | 1.14E-02 | ||||||||
| Pooled (no NL) | 1.36 [1.23–1.51] | FE | 3.69E-09 | ||||||||
| 2 |
rs729876 (chr16:g.13388778 T > C) |
16p13.12 | LOC107984137 | T | C | 0.84 | 0.80 | AgP-Ger | 1.26 [1.08–1.46] | 2.63E-03 | |
| 0.81 | 0.79 | AgP-NL | 1.09 [0.83–1.44] | 5.38E-01 | |||||||
| 0.84 | 0.80 | CP-EA-sev | 1.19 [1.05–1.35] | 6.28E-03 | |||||||
| 0.82 | 0.80 | CP-EA-mod | 1.40 [1.17–1.68] | 2.74E-04 | |||||||
| CP-Ger | 1.20 [1.03–1.40] | 1.97E-02 | |||||||||
| Pooled (all) | 1.23 [1.15–1.32] | FE | 1.21E-08 | ||||||||
| Pooled (no NL) | 1.24 [1.15–1.34] | FE | 9.77E-09 | ||||||||
| 3 |
rs11084095 (chr19:g.52127030 G > A) |
19q13.41 | SIGLEC5 - AC018755.18 | A | G | 0.46 | 0.41 | AgP-Ger | 1.29 [1.14–1.45] | 2.61E-05 | |
| 0.47 | 0.42 | AgP-NL | 1.24 [0.99–1.56] | 6.37E-02 | |||||||
| 0.41 | 0.39 | CP-EA-sev | 1.13 [1.03–1.24] | 1.32E-02 | |||||||
| 0.42 | 0.39 | CP-EA-mod | 1.10 [0.96–1.26] | 1.63E-01 | |||||||
| 0.83 | 0.80 | CP-Ger | 1.15 [1.01–1.31] | 3.18E-02 | |||||||
| Pooled (all) | 1.17 [1.11–1.24] | FE | 5.09E-08 | ||||||||
| Pooled (no NL) | 1.16 [1.10–1.23] | FE | 2.60E-07 | ||||||||
| 4 |
rs2064712 (chr6:g.161216608 G > A) |
6q26 | AL109933.3 - AL391361.2 | A | G | 0.19 | 0.15 | AgP-Ger | 1.39 [1.18–1.64] | 6.29E-05 | |
| 0.09 | 0.13 | AgP-NL | 0.69 [0.48–0.97] | 3.56E-02 | |||||||
| CP-EA-sev | 1.12 [0.97–1.30] | 1.14E-01 | |||||||||
| CP-EA-mod | 1.31 [1.08–1.60] | 7.16E-03 | |||||||||
| CP-Ger | 1.19 [1.00–1.41] | 5.49E-02 | |||||||||
| Pooled (all) | 1.16 [0.99–1.36] | RE | 7.49E-02 | ||||||||
| Pooled (no NL) | 1.24 [1.14–1.35] | FE | 5.29E-07 | ||||||||
| 5 |
rs9982623 (chr21:g.47691216 C > T) |
21q22.3 | MCM3AP | C | T | 0.88 | 0.86 | AgP-Ger | 1.25 [1.06–1.48] | 9.41E-03 | |
| 0.88 | 0.87 | AgP-NL | 1.05 [0.76–1.46] | 7.61E-01 | |||||||
| 0.89 | 0.86 | CP-EA-sev | 1.35 [1.17–1.55] | 3.93E-05 | |||||||
| 0.89 | 0.86 | CP-EA-mod | 1.26 [1.03–1.54] | 2.40E-02 | |||||||
| 0.86 | 0.86 | CP-Ger | 1.07 [0.90–1.27] | 4.66E-01 | |||||||
| Pooled (all) | 1.23 [1.13–1.33] | FE | 8.65E-07 | ||||||||
| Pooled (no NL) | 1.24 [1.14–1.35] | FE | 5.67E-07 | ||||||||
| 6 |
rs9984417 (chr21:g.23847110 A > T) |
21q21.1 | MAPK6PS2 - AP000959.2 | T | A | 0.65 | 0.61 | AgP-Ger | 1.20 [1.06–1.36] | 3.03E-03 | |
| 0.68 | 0.65 | AgP-NL | 1.21 [0.96–1.53] | 1.12E-01 | |||||||
| CP-EA-sev | 1.16 [1.05–1.29] | 3.59E-03 | |||||||||
| CP-EA-mod | 1.10 [0.96–1.27] | 1.81E-01 | |||||||||
| CP-Ger | 1.13 [1.00–1.29] | 5.91E-02 | |||||||||
| Pooled (all) | 1.16 [1.09–1.23] | FE | 9.31E-07 | ||||||||
| Pooled (no NL) | 1.15 [1.09–1.23] | FE | 3.21E-06 |
Note: Genomic positions are based on hg19/GRCh37
avariants that came up only after exclusion of AgP-NL, bold rows indicate P-values of association <10E-6
A1 effect allele, A2 non-effect allele, EAF effect allele frequency, Cas cases, Con controls, OR odds ratio, CI confidence interval, Mod model, RE random effects, FE fixed effects, P P-value
Fig. 2.

Regional association plots of the identified loci with P < 10−6 for the lead variants. Due to the low number of variants that were existent in all samples, this plot is based on the results of the meta-analysis in the first stage including AgP-GER, AgP-NL, CP-EA-sev and CP-EA-mod and excluding CP-Ger. a SNP rs729876 at 16p13.12, b SNP rs11084095 at 19p13.41, c SNP rs9982623 at 21q22.3, and d SNP rs9984417 at 21q22.1
Meta-analysis without smallest study sample
In addition, we performed a meta-analysis without the smallest study sample of AgP-NL for 1,749,472 variants (Fig. 1b). In the first step of this analysis (3931 cases and 5882 controls), 39 loci surpassed our preassigned selection criteria and were selected for follow-up in the second step (4924 cases and 7301 controls, Supplementary Table 3). By using this approach we were able to detect two variants with genome-wide significance: SNP rs729876 (P = 9.77 × 10−9, OR = 1.24, 95%, CI = 1.15–1.34) had been identified in the first meta-analysis with the Dutch sample included. Additionally, we detected the association of SNP rs16870060 with P = 3.69 × 10−9 (OR = 1.36, 95% CI = 1.23–1.51, Table 1, Fig. 3). SNP rs16870060 is intronic to the pseudogene MTND1P5 (mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 1 pseudogene 5, chr8). Three other variants were associated with P < 10−6, two (SNPs rs4801882 and rs9982623) of which were concordant with the top variants or with their high LD (r2 > 0.8) variants of the above-mentioned analysis based on all samples, and P-values differed only slightly. The third variant, SNP rs2064712, was associated with P = 5.29 × 10−7 (OR = 1.24, 95% CI = [1.14–1.35]) and located on chromosome 6 between unprocessed pseudogene AL109933.3 and lincRNA AL391361.2 and 42 kb downstream of PLG (Plasminogen). The PLG locus had earlier been identified by us as a susceptibility locus for PD in the same analyses samples in a candidate-gene association study, where it was tagged by the GWAS SNP rs1247559 (r2 < 0.2 with rs2064712) [22]. Rs1247559 had a P-value of 2.25 × 10−5 in the meta-analysis of AgP-Ger, CP-EA-mod and CP-EA-sev and was not pursued further in the current study. For the six variants described in this section, we compared the pooled P-value for CP-EA-mod and CP-EA-sev with and without correction for shared controls. A maximum difference of less than one potency indicated a reasonable inflation (Supplementary Table 4).
Fig. 3.
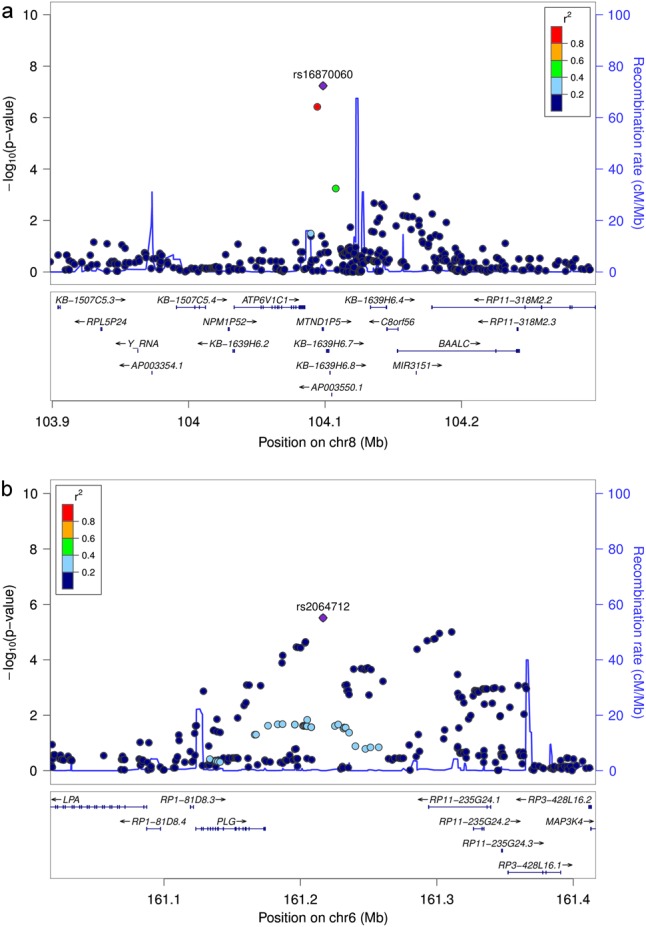
Regional association plots of the loci with P < 10−6 for the lead variants identified after removing the smallest sample of AgP-NL. Due to the low number of variants that were existent in all samples, this plot is based on the results of the meta-analysis in the first stage including AgP-GER, CP-EA-sev, and CP-EA-mod and excluding CP-Ger. a SNP rs16870060 at 8q22.3 and b SNP rs2064712 at 6q26
In-silico characterization of selected variant effects
For the lead variants in the six distinct loci, we defined LD blocks by incorporating their high LD variants (r2 > 0.8) resulting in 52 variants, including the lead variants (Supplementary Table 5). These variants were examined for known associations with other traits, putative effects on genes and their impact on the pathogenesis of PD.
eQTL. Examination of the 52 variants indicated cis-regulatory and trans-regulatory effects on multiple genes for all loci (Supplementary Table 8). The subset of eQTLs reported in gastrointestinal tissue, bone tissue and blood are summarized in Table 2. The LD block tagged by the genome-wide significant lead SNP rs729876 at 16p13.12 indicated a trans-effect on gene expression of HOXC10 (homeobox C10, chr12), with P = 7 × 10−6 in peripheral blood monocytes. For the LD block of the second SNP with genome-wide significant association with PD, rs16870060 at 8q22.3, trans eQTL effects were reported to the genes ORM1 (orosomucoid 1; chr9) with P = 2 × 10−6 in peripheral blood monocytes.
Table 2.
For each locus, cis and trans eQTLs of the lead variant and the high LD variants were compiled from public resources using the web application at http://www.genehopper.de/qtlizer
| Lead variant | Type | Gene | Samples (best P-value) |
|---|---|---|---|
| rs16870060 | trans | ORM1 | Cells-Monocytes (0.0000022) |
| rs729876 | trans | HOXC10 | Cells-Monocytes (0.0000071) |
| rs11084095 | cis | SIGLEC5 | Cells-Monocytes (6.4e-23)a, Cells-Macrophages (1.2e-10), Artery-Tibial (5.9e-9)a, Colon-Sigmoid (1.7e-8)a, Stomach (9.6e-8)a+, Esophagus-Muscularis (0.0000024)a, Peripheral blood (0.00033)a, Cells-Lymphoblastoid cell lines (0.07) |
| cis | SIGLEC14 | Whole blood (0.0000077)a | |
| rs9982623 | cis | DIP2A | Blood (3.6e-7), Pancreas (0.0000022)a |
| cis | PRMT2 | Cells-Monocytes (1.1e-9) | |
| cis | PCNT | Cells-Monocytes (6.7e-12), Blood (1.3e-10), Cells-Macrophages (1e-8), Artery-Aorta (5.1e-7)a, Artery-Tibial (9.4e-7)a, Esophagus-Muscularis (0.0000066)a, Artery-Coronary (0.0000087)a, Whole blood (0.000014)a, Colon-Sigmoid (0.000027)a, Cells-Lymphoblastoid cell lines (0.13) | |
| cis | FTCD | Esophagus-Muscularis (0.0000053)a, Muscle skeletal (0.000012)a | |
| cis | AP001469.9 | Esophagus-Muscularis (3.3e-19)a, Artery-Tibial (2e-14)a, Muscle skeletal (1.1e-11)a+, Colon-Sigmoid (1.7e-11)a+, Colon-Transverse (6.5e-11)a, Esophagus-Mucosa (1.3e-9)a, Esophagus-Gastroesophageal junction (3.6e-9)a+, Artery-Aorta (3.9e-9)a+, Spleen (4.4e-9)a, Stomach (8.9e-9)a+, Whole blood (1.7e-7)a, Pancreas (0.0000011)a, Cells-Transformed fibroblasts (0.0000012)a, Heart-Atrial appendage (0.0000066)a, Heart-Left ventricle (0.000012)a+ | |
| cis | C21orf58 | Peripheral blood (0.000081)a | |
| cis | LSS | Peripheral blood (2.3e-132)a+, Cells-Monocytes (7.1e-34)a+, Blood (8.1e-32)a+, Whole blood (1.6e-19)a, Atherosclerotic aortic root (2.3e-13)a+, Osteoblasts (3.2e-13)a+, Muscle skeletal (3.2e-13)a, Pancreas (2.7e-11)a+, Cells-Lymphoblastoid cell lines (3.6e-11), Esophagus-Mucosa (6.6e-11)a+, Spleen (6.7e-9)a+, Esophagus-Muscularis (2.5e-8)a, Ileum - Pre-pouch (5.1e-8), Small intestine-Terminal ileum (3.3e-7)a, Cells-T cells (0.0000012)+, Artery-Aorta (0.0000036)a, Cells-Transformed fibroblasts (0.0000081)a, Blood cells in celiac disease (0.000013)+, Cells-CD4 + lymphocytes (0.000014) | |
| cis | SPATC1L | Peripheral blood (2.9e-29)a, Artery-Tibial (6e-10)a, Blood (1.7e-8), Cells-Lymphoblastoid cell lines (2.2e-8)a, Muscle skeletal (8.3e-8)a, Esophagus-Muscularis (1.7e-7)a, Cells-Transformed fibroblasts (1.8e-7)a, Pancreas (2.2e-7)a, Esophagus-Gastroesophageal junction (0.0000015)a, Stomach (0.0000027)a, Colon-Transverse (0.0000028)a, Esophagus-Mucosa (0.0000065)a, Heart-Left ventricle (0.000015)a, Heart-Atrial appendage (0.00002)a | |
| cis | MCM3AP | Peripheral blood (3.3e-45)a, Blood (7.2e-32)a, Cells-CD4 + lymphocytes (7.1e-24)a + , Osteoblasts (5.1e-14)a+, Cells-Lymphoblastoid cell lines (3.2e-11), Sputum (0.000022), Blood cells in celiac disease (0.000029) | |
| cis | MCM3AP-AS1 | Peripheral blood (3.3e-45)a, Esophagus-Muscularis (8.5e-8)a, Esophagus-Mucosa (2.8e-7)a, Muscle skeletal (5.4e-7)a, Blood (0.0000037), Esophagus-Gastroesophageal junction (0.000005)a, Artery-Tibial (0.0000059)a, Artery-Aorta (0.00004)a | |
| cis | YBEY | Peripheral blood (9.8e-198)a, Muscle skeletal (1.2e-43)a, Artery-Tibial (6.1e-39)a, Esophagus-Mucosa (3.8e-34)a, Cells-Transformed fibroblasts (2.5e-32)a, Heart-Atrial appendage (8.4e-32)a, Esophagus-Muscularis (1.5e-28)a, Blood (1.2e-26)a, Heart-Left ventricle (2.6e-26)a, Artery-Aorta (5.9e-25)a, Stomach (2.3e-24)a, Colon-Transverse (2e-23)a, Esophagus-Gastroesophageal junction (3.6e-22)a, Whole blood (1.6e-21)a, Pancreas (2.7e-21)a, Spleen (2.4e-20)a, Colon-Sigmoid (1.2e-19)a, Osteoblasts BMP2 (4.2e-18)a, Cells-Lymphoblastoid cell lines (7.9e-17)a, Artery-Coronary (5.5e-16)*, Osteoblasts PGE2 (1.2e-15)a, Osteoblasts DEX (1.1e-13)a, Osteoblasts (1.4e-13)a, Cells-EBV-transformed lymphocytes (5.7e-11)a, Testis (5.8e-11)a, Minor salivary gland (8.8e-10)a, Small intestine-Terminal ileum (7.2e-7)a | |
| trans | WDR4 | Cells-Monocytes (0.0000092) | |
| rs9984417 | trans | ATP8B4 | Cells-Monocytes (0.0000095) |
To condense information, only eQTLs of gastrointestinal tissue, bone tissue and blood are shown (the complete list of eQTL is given in Supplementary Table 8)
ais study-wide significant; (+) is best, i.e., high LD block of lead variant is best associated (regarding P-value) with the gene among all associations
GWAS Catalog
To assess the relationships of the identified loci with other traits and diseases, we screened the LD blocks for entries in the NHGRI-EBI GWAS Catalog with P < 10−5. None of the LD blocks was associated with other phenotypes. However, the regions defined by the lead variant +/− 500 kb are associated with several other phenotypes with genome-wide significance, including lipoprotein levels (SNP rs118039278, P = 1 × 10−396, 231 kb distance to lead variant, r2 < 0.2) and coronary artery disease (SNP rs55730499, P = 3 × 10−154, 211 kb distance to lead variant, r2 < 0.2) at 6q.26, blood protein levels (SNP rs12459419, P = 3 × 10−165, 399 kb distance to lead variant, r2 < 0.2), plasma plasminogen levels (SNP rs10412972, P = 8 × 10−9, 28 kb distance to lead variant, r2 < 0.2) and high density lipoprotein (HDL) cholesterol levels (SNP rs17695224, P = 7 × 10−16, 197 kb distance to lead SNP, r2 < 0.2) at 19q13.41. Moreover, at 19q13.41 the previously reported association of GWAS SNP rs4284742 (4.7 kb distance to lead variant, r2 = 0.2) with AgP is located. The complete list of associations is shown in Supplementary Table 6.
TADs
We could identify 20 genes at 5q31.1, 29 genes at 8q22.3, 7 genes at 13q21.1, 19 genes at 16p13.12, 65 genes at 19q13.41, and 31 genes at 21q22.3 to be located inside the same TAD as the associated LD block at each locus (Supplementary Table 7).
CADD
We annotated the LD blocks with the CADD score (Supplementary Table 9) and compared the pathogenicity of the variants in the high LD blocks as shown in Supplementary Fig. 1.
Discussion
This study identified two novel susceptibility loci of PD with genome-wide significance.
SNP rs729876 showed genome-wide significant association with PD in both meta-analyses samples and the association was consistent with the same risk allele in all samples. The variant is located within the intronic region of the lincRNA LOC107984137, the function of which is unknown. Currently, it is not clear if this SNP does affect the function of this lincRNA and/or of other genes. Regarding expression effects, eQTL data indicated tissue specific effects on the expression of the genes HOXC10 in blood monocytes and ZC3H7A (zinc finger CCCH-type containing 7A) and MYH11 (myosin, heavy chain 11) in the brain. Experimental work suggests that the chimeric protein β/MYH11 inhibits the function of RUNX1 (runt-related transcript factor 1) [44]. RUNX1 plays a role in hematopoiesis and bone formation [45, 46]. These eQTL effects did not pass the study-wide significance thresholds and we point out the suggestive nature of these observations and emphasize the need for more extensive experimental follow-up before a mechanism that links the effect of the associated SNPs with the cis-regulation or trans-regulation of these genes can be proposed.
We were aware that the very small size of the Dutch AgP case sample (N = 171 cases) in conjunction with the comparably large Dutch control sample (N = 2607 controls) might have a potentially large impact on the inter-sample heterogeneity. Small samples a generally impaired by random depletion, as well as enrichment of risk alleles due to chance effects. This random misrepresentation of the allele frequency in the case sample can have strong effects on the results, especially if the control sample is large like in the Dutch AgP case-control sample, and might skew the results of the meta-analysis. Therefore, we performed the meta-analysis again without the AgP-NL GWAS sample. In this analysis, SNP rs16870060 showed the strongest of association in our meta-analysis, with P = 3.7 × 10−9. In the AgP-NL case sample, the frequency of the effect allele was lower than that of the controls, which was reverse to the allele frequencies of the other AgP and CP samples. This, together with the large size of the Dutch AgP-controls neutralized the association results. This may indicate a false positive association finding, but we consider it more likely that either the small number of cases diluted the association, or rs16870060, with high likelihood not being the causative variant of the association, shows a different recombination pattern in the Dutch cases sample. SNP rs16870060 is located within an intron of the processed pseudogene MTND1P5, 13 kb downstream of the protein coding gene ATP6V1C1 (ATPase H + transporting V1 subunit C1). eQTL data indicated no cis-effect of variants of this haplotype block on these genes, but suggest tissue specific trans-effects on ARHGEF28 (rho guanine nucleotide exchange factor 28) in the liver and on ORM1 in monocytes. The biological function of ARHGEF28 is yet not clearly described. However, ORM1 is an interesting novel candidate gene for PD because it encodes a key acute phase plasma protein, which is increased due to acute inflammation. The specific function of ORM1 has not yet been determined; however, it is probably involved in aspects of immunosuppression [47, 48]. In addition, ORM1 was experimentally shown to interact with PAI-1 (plasminogen activator inhibitor-1) and the binding of PAI-1 to ORM1 results in significant stabilization of its inhibitory activity toward plasminogen activators [49]. Likewise, ORM1 may play a significant role in the regulation of fibrinolysis. We consider this as relevant, because variants with putative cis-effects on the expression of PLG were previously found to be associated with both AgP and CP [21, 22] and an association with the PLG locus was among the top 6 associations in the current study. SNP rs16870060 showed an increased CADD pathogenicity score ( > 8), indicating some biological effect. However, the regulatory effect of rs16870060 or the associated putative causative variants on the expression of ORM1 in blood monocytes requires further experimental validation.
We note that among the suggestive associations with P < 10−6, the chromosomal region at 21q22.3 (tagged by lead variant rs9982623), spans a particularly broad area, which covers the genes LSS (Lanosterol synthase), YBEY (ybeY metallopeptidase), and PCNT (pericentrin). Because this locus encompasses a cluster of SNPs with strong associations, we suspect it to be less likely a false positive association compared to the other suggestive loci tagged by rs10491294 and rs11084095. eQTL data indicated strong cis-effect of variants of this LD block on these genes, as well as on the genes proximal and distal to this associated region, SPATC1L (spermatogenesis and centriole associated 1-like) and PRMT2 (protein arginine N-methyltransferase 2), respectively.
The limitation of this study was that not all case-control samples were employed in the initial meta-analysis and that the samples were imputed to different genome assemblies. Future studies are required to address these shortcomings and to increase the number of investigated variants and the statistical power, which may allow the identification of additional susceptibility loci. Although we could have identified the genome-wide significant loci by excluding smallest sample of AgP-NL completely from the analysis, we decided to make use of the Dutch case-sample because it can provide some additional statistical power. Thus, we included the Dutch case-control sample in the first meta-analysis, because this approach gave the highest statistical power.
In conclusion, the current study identified two novel susceptibility variants for PD increasing the number of genome-wide significant associations of common susceptibility variants to five (previously identified variants are located at DEFA1A3, SIGLECS [20], and GLT6D1 [18]). These novel variants are associated at a genome-wide significance level, providing statistical evidence for the relevance of these loci in the disease etiology. The putative causative variants underlying the associations, as well as their target genes need to be identified. For the latter, the lincRNA LOC107984137 and ORM1 could provide the first targets. In addition, we could replicate the previously reported association of SIGLEC5 and PLG with PD, using a set of different genetic variants, adding evidence to these previously reported risk genes of PD. We further suggest the chromosomal regions at 21q21.1 (tagged by rs9984417) and 21q22.3, spanning the genes LSS, YBEY, and PCNT to harbor putative susceptibility variants for PD.
Electronic supplementary material
Acknowledgements
This work was supported by a research grant of the German Research Foundation DFG (Deutsche Forschungsgemeinschaft; GZ: SCHA 1582/3-1). Collection of the AgP cases was additionally supported by the German Ministry of Education and Research through the POPGEN biobank project (01GR0468). The Dortmund Health Study (DHS) is supported by the German Migraine & Headache Society (DMKG) and by unrestricted grants of equal share from Almirall, Astra Zeneca, Berlin Chemie, Boehringer, Boots Health Care, Glaxo-Smith-Kline, Janssen Cilag, McNeil Pharma, MSD Sharp and Dohme and Pfizer to the University of Muenster (collection of sociodemographic and clinical data). Blood collection in the Dortmund Health Study was done through funds from the Institute of Epidemiology and Social Medicine University of Muenster and genotyping supported by the German Ministry of Research and Education (BMBF, grant no. 01ER0816). FOCUS was supported by the Federal Ministry of Education and Research BMBF (FKZ 0315540 A). The HNR study is supported by the Heinz Nixdorf Foundation (Germany). Additionally, the HNR study was funded by the German Ministry of Education and Science and the German Research Council (DFG; Project SI 236/8-1, SI236/9-1, ER 155/6-1). The genotyping of the Illumina HumanOmni-1 Quad BeadChips of the HNR subjects was financed by the German Centre for Neurodegenerative Diseases (DZNE), Bonn. SHIP is part of the Community Medicine Research net (http://www.community-medicine.de) of the University of Greifswald, Germany, which is funded by the Federal Ministry of Education and Research (grants no. 01ZZ9603, 01ZZ0103, and 01ZZ0403), the Ministry of Cultural Affairs, as well as the Social Ministry of the Federal State of Mecklenburg-West Pomerania. Generation of genome-wide SNP data has been supported by the Federal Ministry of Education and Research (grant no. 03ZIK012) and a joint grant from Siemens Healthcare, Erlangen, Germany and the Federal State of Mecklenburg, West Pomerania. The University of Greifswald is a member of the Caché Campus program of the InterSystems GmbH.
Authors contribution
MM, SO, TK, AU, LdG, JE, and AS where involved in the study design. GR, BL, KD, SO, AT, BH, TK, CB, YJS, CG, IS, NV, AU, LdG, JW, KB, BK, PH, ML, WL, AF, HD, JE, and AS managed the studies. BL, SJ, SO, BH, TK, CB, YJS, CG, IS, NV, AU, LdG, JW, KB, BK, PH, HD, JE, and AS were involved in the subject recruitment. SO, AT, CG, ML, WL, AF, and AS were involved in the genotyping. MM applied the methods and did the analysis. MM, GR, and AS did the interpretation of the results. MM did the drafting of the manuscript. GR, BL, KD, AT, IA, BH, WL, JE, and AS were involved in the critical review of the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41431-018-0265-5) contains supplementary material, which is available to authorized users.
References
- 1.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. CDC Periodontal Disease Surveillance workgroup: James Beck (University of North Carolina, Chapel Hill, USA), Gordon Douglass (Past President, American Academy of Periodontology) RP (University of W. Prevalence of Periodontitis in Adults in the United States: 2009 and 2010. J Dent Res. 2012;91:914–20. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, Amar S. Periodontal disease and systemic conditions: a bidirectional relationship. Odontology. 2006;94:10–21. doi: 10.1007/s10266-006-0060-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Preshaw PM, Alba AL, Herrera D, et al. Periodontitis and diabetes: a two-way relationship. Diabetologia. 2012;55:21–31. doi: 10.1007/s00125-011-2342-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck JD, Eke P, Heiss G, et al. Periodontal disease and coronary heart disease. Circulation. 2005;112:19 LP–24. doi: 10.1161/CIRCULATIONAHA.104.511998. [DOI] [PubMed] [Google Scholar]
- 5.Wang CWJ, McCauley LK. Osteoporosis and periodontitis. Curr Osteoporos Rep. 2016;14:284–91. doi: 10.1007/s11914-016-0330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcenes W, Kassebaum NJ, Bernabé E, et al. Global burden of oral conditions in 1990-2010: a systematic analysis. J Dent Res. 2013;92:592–7. doi: 10.1177/0022034513490168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Susin C, Haas AN, Albandar JM. Epidemiology and demographics of aggressive periodontitis. Periodontol 2000. 2014;65:27–45. doi: 10.1111/prd.12019. [DOI] [PubMed] [Google Scholar]
- 8.Michalowicz BS, Diehl SR, Gunsolley JC, et al. Evidence of a substantial genetic basis for risk of adult periodontitis. J Periodontol. 2000;71:1699–707. doi: 10.1902/jop.2000.71.11.1699. [DOI] [PubMed] [Google Scholar]
- 9.Divaris K, Monda KL, North KE, et al. Exploring the genetic basis of chronic periodontitis: a genome-wide association study. Hum Mol Genet. 2013;22:2312–24. doi: 10.1093/hmg/ddt065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Offenbacher S, Divaris K, Barros SP, et al. Genome-wide association study of biologically informed periodontal complex traits offers novel insights into the genetic basis of periodontal disease. Hum Mol Genet. 2016;25:2113–29. doi: 10.1093/hmg/ddw069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13:135–45. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng P, Wang X, Casado PL, et al. Genome wide association scan for chronic periodontitis implicates novel locus. BMC Oral Health. 2014;14:84. doi: 10.1186/1472-6831-14-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong KW, Shin MS, Ahn YB, Lee HJ, Kim HD. Genomewide association study on chronic periodontitis in Korean population: results from the Yangpyeong health cohort. J Clin Periodontol. 2015;42:703–10. doi: 10.1111/jcpe.12437. [DOI] [PubMed] [Google Scholar]
- 14.Sanders AE, Sofer T, Wong Q, et al. Chronic periodontitis genome-wide association study in the hispanic community health study/study of Latinos. J Dent Res. 2017;96:64–72. doi: 10.1177/0022034516664509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaffer JR, Polk DE, Wang X, et al. Genome-wide association study of periodontal health measured by probing depth in adults ages 18-49 years. G3. 2014;4:307–14. doi: 10.1534/g3.113.008755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu S, Momozawa Y, Takahashi A, et al. A genome-wide association study of periodontitis in a Japanese population. J Dent Res. 2015;94:555–61. doi: 10.1177/0022034515570315. [DOI] [PubMed] [Google Scholar]
- 17.Teumer A, Holtfreter B, Völker U, et al. Genome-wide association study of chronic periodontitis in a general German population. J Clin Periodontol. 2013;40:977–85. doi: 10.1111/jcpe.12154. [DOI] [PubMed] [Google Scholar]
- 18.Schaefer AS, Richter GM, Nothnagel M, et al. A genome-wide association study identifies GLT6D1 as a susceptibility locus for periodontitis. Hum Mol Genet. 2010;19:553–62. doi: 10.1093/hmg/ddp508. [DOI] [PubMed] [Google Scholar]
- 19.Hashim NT, Linden GJ, Ibrahim ME, et al. Replication of the association of GLT6D1 with aggressive periodontitis in a Sudanese population. J Clin Periodontol. 2015;42:319–24. doi: 10.1111/jcpe.12375. [DOI] [PubMed] [Google Scholar]
- 20.Munz M, Willenborg C, Richter GM, et al. A genome-wide association study identifies nucleotide variants at SIGLEC5 and DEFA1A3 as risk loci for periodontitis. Hum Mol Genet. 2017;33:272–9. doi: 10.1093/hmg/ddx151. [DOI] [PubMed] [Google Scholar]
- 21.Schaefer AS, Bochenek G, Jochens A, et al. Genetic evidence for PLASMINOGEN as a shared genetic risk factor of coronary artery disease and periodontitis. Circ Cardiovasc Genet. 2015;8:159–67. doi: 10.1161/CIRCGENETICS.114.000554. [DOI] [PubMed] [Google Scholar]
- 22.Munz Matthias, Chen Hong, Jockel-Schneider Yvonne, Adam Knut, Hoffman Per, Berger Klaus, Kocher Thomas, Meyle Jörg, Eickholz Peter, Doerfer Christof, Laudes Matthias, Uitterlinden André, Lieb Wolfgang, Franke Andre, Schreiber Stefan, Offenbacher Steven, Divaris Kimon, Bruckmann Corinna, Loos Bruno G., Jepsen Søeren, Dommisch Henrik, Schäefer Arne S. A haplotype block downstream of plasminogen is associated with chronic and aggressive periodontitis. Journal of Clinical Periodontology. 2017;44(10):962–970. doi: 10.1111/jcpe.12749. [DOI] [PubMed] [Google Scholar]
- 23.Shusterman A, Munz M, Richter G, et al. The PF4/PPBP/CXCL5 gene cluster is associated with periodontitis. J Dent Res. 2017;96:945–52. doi: 10.1177/0022034517706311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krawczak M, Nikolaus S, von Eberstein H, Croucher PJP, El Mokhtari NE, Schreiber S. PopGen: Population-Based Recruitment of Patients and Controls for the Analysis of Complex Genotype-Phenotype Relationships. Public Health Genom. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 25.Müller N, Schulte DM, Türk K, et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J Lipid Res. 2015;56:1034–42. doi: 10.1194/jlr.P052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berger K. DOGS. Bundesgesundheitsblatt-Gesundh-Gesundh. 2012;55:816–21. doi: 10.1007/s00103-012-1492-5. [DOI] [PubMed] [Google Scholar]
- 27.Schmermund A, Möhlenkamp S, Stang A, et al. Assessment of clinically silent atherosclerotic disease and established and novel risk factors for predicting myocardial infarction and cardiac death in healthy middle-aged subjects: Rationale and design of the Heinz Nixdorf RECALL study. Am Heart J. 2002;144:212–8. doi: 10.1067/mhj.2002.123579. [DOI] [PubMed] [Google Scholar]
- 28.van Wijngaarden JP, Dhonukshe-Rutten RAM, van Schoor NM, et al. Rationale and design of the B-PROOF study, a randomized controlled trial on the effect of supplemental intake of vitamin B12 and folic acid on fracture incidence. BMC Geriatr. 2011;11:80. doi: 10.1186/1471-2318-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hill C, Gerardo D, James F, et al. The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. the ARIC investigators. Am J Epidemiol. 1989;129:687–702. doi: 10.1093/oxfordjournals.aje.a115184. [DOI] [PubMed] [Google Scholar]
- 30.Page RC, Eke PI. Case definitions for use in population-based surveillance of periodontitis. J Periodontol. 2007;78:1387–99. doi: 10.1902/jop.2007.060264. [DOI] [PubMed] [Google Scholar]
- 31.John U, Greiner B, Hensel E, et al. Study of Health In Pomerania (SHIP): a health examination survey in an east German region: objectives and design. Soz Prav. 2001;46:186–94. doi: 10.1007/BF01324255. [DOI] [PubMed] [Google Scholar]
- 32.Hensel E, Gesch D, Biffar R, et al. Study of Health in Pomerania (SHIP): a health survey in an East German region. Objectives and design of the oral health section. Quintessence Int. 2003;34:370–8. [PubMed] [Google Scholar]
- 33.Volzke H, Alte D, Schmidt CO, et al. Cohort Profile: The Study of Health in Pomerania. Int J Epidemiol. 2011;40:294–307. doi: 10.1093/ije/dyp394. [DOI] [PubMed] [Google Scholar]
- 34.Zaykin DV, Kozbur DO. P-value based analysis for shared controls design in genome-wide association studies. Genet Epidemiol. 2010;34:725–38. doi: 10.1002/gepi.20536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munz M, Tönnies S, Balke WT, Simon E. Multidimensional gene search with Genehopper. Nucleic Acids Res. 2015;43:98–103. doi: 10.1093/nar/gkv511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.GTEx Consortium Gte. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward LD, Kellis M. HaploRegv4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877–D881. doi: 10.1093/nar/gkv1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eicher JD, Landowski C, Stackhouse B, et al. GRASPv2.0: an update on the Genome-Wide Repository of Associations between SNPs and phenotypes. Nucleic Acids Res. 2015;43:D799–804. doi: 10.1093/nar/gku1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gamazon ER, Zhang W, Konkashbaev A, et al. SCAN: SNP and copy number annotation. Bioinformatics. 2010;26:259–62. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia K, Shabalin AA, Huang S, et al. seeQTL: a searchable database for human eQTLs. Bioinformatics. 2012;28:451–2. doi: 10.1093/bioinformatics/btr678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–43. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang G. Molecular basis for a dominant inactivation of RUNX1/AML1 by the leukemogenic inversion 16 chimera. Blood. 2004;103(8):3200–3207. doi: 10.1182/blood-2003-07-2188. [DOI] [PubMed] [Google Scholar]
- 45.Ono M, Yaguchi H, Ohkura N, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–9. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 46.Stein GS, Lian JB, Wijnen AJvan, et al. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene. 2004;23:4315–29. doi: 10.1038/sj.onc.1207676. [DOI] [PubMed] [Google Scholar]
- 47.Fey GH, Fuller GM. Regulation of acute phase gene expression by inflammatory mediators. Mol Biol Med. 1987;4:323–38. [PubMed] [Google Scholar]
- 48.Williams JP, Weiser MR, Pechet TT, Kobzik L, Moore FD, Hechtman HB. alpha 1-Acid glycoprotein reduces local and remote injuries after intestinal ischemia in the rat. Am J Physiol. 1997;273:G1031–5. doi: 10.1152/ajpgi.1997.273.5.G1031. [DOI] [PubMed] [Google Scholar]
- 49.Boncela J, Papiewska I, Fijalkowska I, Walkowiak B, Cierniewski CS. Acute Phase Protein α 1 -Acid Glycoprotein Interacts with Plasminogen Activator Inhibitor Type 1 and Stabilizes Its Inhibitory Activity. J Biol Chem. 2001;276:35305–35311. doi: 10.1074/jbc.M104028200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genotype data for aggressive periodontitis samples and the chronic periodontitis sample with German descent are available upon request from the biobanks PopGen (https://www.epidemiologie.uni-kiel.de/biobanking) and ShIP (https://www.fvcm.med.uni-greifswald.de/index.html). Summary statistics for CP-EU can also be downloaded (see ref. [9]).