

Abstract
Aims
Risdiplam (RG7916, RO7034067) is an orally administered, centrally and peripherally distributed, survival of motor neuron 2 (SMN2) mRNA splicing modifier for the treatment of spinal muscular atrophy (SMA). The objectives of this entry‐into‐human study were to assess the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics of risdiplam, and the effect of the strong CYP3A inhibitor itraconazole on the PK of risdiplam in healthy male volunteers.
Methods
Part 1 had a randomized, double‐blind, adaptive design with 25 subjects receiving single ascending oral doses of risdiplam (ranging from 0.6–18.0 mg, n = 18) or placebo (n = 7). A Bayesian framework was applied to estimate risdiplam's effect on SMN2 mRNA. The effect of multiple doses of itraconazole on the PK of risdiplam was also assessed using a two‐period cross‐over design (n = 8).
Results
Risdiplam in the fasted or fed state was well tolerated. Risdiplam exhibited linear PK over the dose range with a multi‐phasic decline with a mean terminal half‐life of 40–69 h. Food had no relevant effect, and itraconazole had only a minor effect on plasma PK indicating a low fraction of risdiplam metabolized by CYP3A. The highest tested dose of 18.0 mg risdiplam led to approximately 41% (95% confidence interval 27–55%) of the estimated maximum increase in SMN2 mRNA.
Conclusions
Risdiplam was well tolerated and proof of mechanism was demonstrated by the intended shift in SMN2 splicing towards full‐length SMN2 mRNA. Based on these data, Phase 2/3 studies of risdiplam in patients with SMA are now ongoing.
Keywords: genetic diseases, pharmacokinetics–pharmacodyamics, phase 1, neuroscience
What is Already Known about this Subject
Risdiplam is an orally administered, centrally and peripherally distributed, survival of motor neuron 2 (SMN2) splicing modifier that increased SMN protein levels in mice.
In healthy volunteers, SMN2 mRNA is an appropriate biomarker for the assessment of risdiplam's effect.
Bayesian adaptive designs guide dose decisions and escalations to optimize efficiency.
What this Study Adds
Bayesian statistical methods were successfully applied to characterize the effect of risdiplam on SMN2 mRNA.
An 18.0‐mg dose of risdiplam led to approximately half the estimated maximum increase in SMN2 mRNA.
These data were crucial to initiate risdiplam Phase 2/3 studies in patients with Type 1 and Type 2/3 spinal muscular atrophy.
Introduction
Spinal muscular atrophy (SMA) is a severe, progressive, neuromuscular disease caused by deletions and/or loss‐of‐function mutations in the survival of motor neuron 1 (SMN1) gene 1. SMA is characterized by the degeneration of α‐motor neurons in the ventral horns of the spinal cord. This leads to muscle atrophy and, depending on the type of SMA, loss of physical strength and the ability to walk, eat or breathe 2. A second SMN gene, SMN2, produces only low levels of full‐length (FL) SMN2 mRNA since, in the majority of transcripts, exon 7 is removed by splicing (∆7 mRNA). SMN2 therefore produces low levels of FL functional SMN protein, which are not sufficient to compensate fully for the SMN1 gene‐related deficits 3. SMN protein is ubiquitously expressed throughout the body and thought to have diverse roles within the cell 4. Emerging evidence suggests cells and tissue throughout the body may be selectively vulnerable to reduced levels of SMN 5. Therefore, increasing SMN in both central and peripheral compartments has the potential to have broader therapeutic benefit than targeting motor neurons alone 6. Modifying SMN2 mRNA splicing to increase the inclusion of exon 7, has the potential to increase SMN2FL mRNA production and subsequently increase levels of functional SMN protein in patients with SMA.
A number of small molecule splicing modifiers with high specificity for SMN2 pre‐mRNA, were identified including RO6885247 (RG7800), the predecessor to risdiplam (RG7916, RO7034067) 7, 8. In the Phase 1 MOONFISH trial of the investigational medicine, RO6885247, in people with SMA (Types 1–3) 9, there were no serious adverse events (SAEs) or withdrawals due to SAEs. The dosing was suspended in MOONFISH as a precautionary measure due to unexpected observations of retinal toxicity in a chronic toxicity study in monkeys. As monkeys do not possess the SMN2 gene, this finding was considered to reflect off‐target toxicity of RO6885247. Further studies to understand the causes of this effect were considered necessary before proceeding with RO6885247 clinical development. The related compound, risdiplam, is an orally administered, centrally and peripherally distributed, splicing modifier with high specificity for SMN2 pre‐mRNA 7, 8, and has received Orphan Drug Designation and US Food and Drug Administration Fast Track Designation 10. In preclinical studies, an analogue of risdiplam increased survival in SMN ∆7 mice (an experimental model of SMA), restored synapse numbers in the spinal cord and promoted muscle growth resulting in increased muscle size and function 11. Risdiplam was optimized for pharmacokinetic (PK) characteristics and showed improved specificity at the SMN2 splice target compared with other SMN2 splicing modifiers 12.
Appropriate selection of dosing in entry‐into‐human (EIH) and early clinical trials of an investigational medicine is vital to safeguard subjects 13, 14. In conjunction with conventional approaches, Bayesian adaptive designs can be used to determine dosing decisions and guide dose escalations based on safety, tolerability, pharmacodynamics (PD) and exposure 15, 16. Following careful starting dose selection based on preclinical data, subsequent doses are selected (up to an approximate 3‐fold increase) guided by data from the previous dose level by applying Bayesian adaptive methods 15.
In this EIH study (NCT02633709) 17, a maximum exposure was set to limit exposure in each individual subject and a target exposure was identified to assess the potency of risdiplam. Dose selection was guided by Bayesian adaptive design methods. Emergent safety, PK and PD data, and physiologically based PK (PBPK) modelling, were used to determine appropriate doses of risdiplam in the single ascending dose part and to investigate potential food effects and drug–drug interactions (DDIs).
The primary objective of this EIH study in healthy male subjects was to assess the safety and tolerability of oral single ascending doses of risdiplam. Secondary objectives were to investigate the PK of risdiplam, the PD effect of risdiplam on SMN2 mRNA, the effects of food on PK and the effect of itraconazole (a strong CYP4503A inhibitor) on safety, tolerability and PK of risdiplam 18. These data were crucial to advance the clinical development of risdiplam and initiate Phase 2/3 studies in patients with Type 1 and Type 2/3 SMA 19, 20, 21. To our knowledge, this is the first EIH study to utilize Bayesian adaptive design principles in combination with emergent PD data to guide dose escalation.
Methods
Ethics
The study protocol was reviewed and approved by the Stiching Beoordeling Ethiek Biomedisch Onderzoek Review Board prior to initiation of the study and no modifications were made following receipt of ethical approval. The study was conducted in accordance with the Medicines Evaluation Board, the regulatory authority in The Netherlands and with the principles of the Declaration of Helsinki and Good Clinical Practice. Subjects provided written informed consent prior to undergoing any procedure. The study took place between 07 January and 04 August 2016 at Pharmaceutical Research Associates, Health Sciences, Zuidlaren, The Netherlands.
Subjects
To fulfil the key inclusion criteria for this study, subjects had to be healthy adult males, aged 18–45 years with a body mass index of 18–30 kg m–2. Subjects were healthy as defined by prestudy medical and surgical history and a complete physical and ophthalmological examination including vital signs, 12‐lead electrocardiography (ECG), haematology, blood chemistry, serology and urinalysis. Key exclusion criteria included participation in an investigational drug or device study within 90 days prior to screening, as calculated from the day of follow‐up from the previous study. A list of randomized treatment assignments was generated by the sponsor's statistician with an internal validated computerized system (Kit Label Database, version 4.0.1; F. Hoffmann–La Roche, Basel, Switzerland). Subjects were randomized to placebo or active treatment in each cohort. The randomized treatment assignment was allocated from the list sequentially to the subjects. The investigator or designee entered the corresponding subject number for allocation to the cohort in each subject's source data and electronic case report form. SMN1 and SMN2 genotype was determined for all subjects.
Study design
The single‐centre study was designed to have three major parts: Part 1 was a randomized, investigator/subject‐blind, adaptive single ascending dose, placebo‐controlled, parallel study that also studied the effect of food on the PK of risdiplam. Per protocol, Part 2 (food effect) of the study was omitted since an exploratory investigation of the effect of food on the bioavailability of risdiplam was performed in Part 1. Part 3 investigated the PK interaction between itraconazole 18 and risdiplam, using an open‐label, one‐sequence, two‐period crossover design. In both Parts 1 and 3, single doses of risdiplam or placebo were administered orally as a drinking solution together with degassed Sprite (as risdiplam is sensitive to degradation at a higher pH). The subjects, investigator and all individuals in direct contact with the subject at the investigational site (except the on‐site pharmacist) were blinded to treatment allocation.
Part 1: single ascending dose
The starting dose of 0.6 mg risdiplam was selected as it was expected to be well tolerated and it was predicted to be associated with no or only minimal PD effect(s) in humans (≤10% effect of the maximum effect on SMN2 mRNA). This dose is well below the estimated therapeutic dose of 2–22 mg (defined as half of the maximum effect on SMN2 mRNA increase) determined based on PBPK and PK/PD modelling. Further, also based on PBPK modelling, it was predicted that a starting dose of 0.6 mg of risdiplam would be approximately 40‐fold below the individual exposure cap of 1500 ng ml–1 h for the area under the plasma concentration curve over 24 h (AUC0–24h; see below). Subsequent doses were selected in an adaptive manner during the study based on emerging data (Figure 1). The protocol allowed for up to 69 subjects with at least three active and one placebo subjects per dose level; Table 1 shows the number of subjects and doses of risdiplam.
Figure 1.
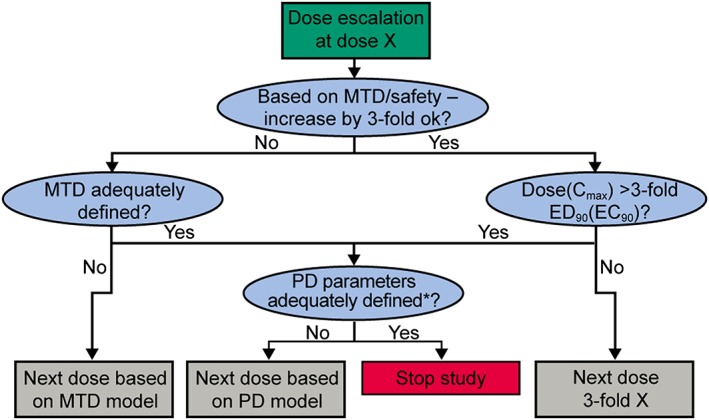
Bayesian decision tree for dose escalation. *Based on the possible range that can be investigated from the assessment of the MTD/safety (cap exposure). PD, pharmacodynamic; MTD, maximum tolerated dose
Table 1.
Doses of risdiplam in Part 1
| Cohort | Dose (mg) | Route | Numberof subjects | Food state | |
|---|---|---|---|---|---|
| Active drug | Placebo | ||||
| 1 | 0.6 | Oral | 3 | 2 | Fasted |
| 2 | 2.0 | Oral | 3 | 1 | Fasted |
| 3 | 6.0 | Oral | 3 | 1 | Fasted |
| 4 | 18.0 | Oral | 3 | 1 | Fasted |
| 5 | 6.0 | Oral | 3 | 1 | Fed |
| 6 | 18.0 | Oral | 3 | 1 | Fasted |
The study consisted of six successive cohorts of 4–5 healthy subjects receiving a single oral dose of 0.6–18.0 mg of risdiplam or placebo. In the first cohort, three subjects were dosed with 0.6 mg under fasted conditions, and two subjects received placebo. To avoid simultaneous exposure of all subjects on the same day, Cohort 1 was split into two groups: a sentinel group of two subjects were dosed on one day (one received active treatment, one placebo) and three subjects were dosed on the following day (two received active treatment, one placebo).
A continual reassessment method (CRM), with control for the probability of over‐dosing, based on occurrence of dose‐limiting events (DLE) and/or based on estimation of PD parameters (e.g. effective dose/effective concentration ED/EC50, ED/EC90) was used to inform dose escalation decisions (Figure 1). After each cohort of subjects completed dosing, the model was updated with the DLE observed occurrence and a new maximum tolerated dose (MTD) estimate was derived together with new estimates of PD parameters (e.g. ED/EC50, ED/EC90). The selection criteria for subsequent doses were based on DLE probability and precision of the estimation for PK/PD relationship (e.g. percentage coefficient of variation [CV%] from ED/EC50, ED/EC90 e.g. ≤75%; based on estimates obtained from the related compound RG7800). In addition, the dose escalation was planned to be stopped once the dose reached the plateau of the dose/PD, PK/PD relationship (dose or maximum observed plasma concentration [Cmax] 3‐fold above the ED90 or EC90, respectively).
The highest dose to be tested within the study was planned to lead to a plasma exposure not greater than 1500 h ng ml–1 for AUC0–24 h in any individual subject. This exposure cap was based on the no observed adverse effect levels for toxicity observed in animal studies in accordance with International Conference on Harmonization guidelines. Planned dose escalation was designed not to exceed a 3.3‐fold increase in risdiplam plasma exposure.
In both Cohorts 2 and 3, three subjects received 2.0 mg or 6.0 mg risdiplam under fasted conditions, respectively, whereas one subject per cohort received placebo. In Cohorts 4 and 6, three subjects received 18.0 mg risdiplam under fasted conditions, and one subject received placebo. Dose escalation was stopped with Cohort 4, when exposure levels approached the exposure cap. Additional subjects were added to the 18.0 mg dose group (Cohort 6) to better characterize the effect of risdiplam on SMN2 mRNA.
In Cohort 5, to study the effect of food, three healthy subjects received a single oral dose of risdiplam in the fed state, whereas one subject received placebo (Table 1). A taste questionnaire was completed by each subject approximately 1 min after administration of the study drug on Day 1.
Part 3: itraconazole interaction
Based on preclinical data including preliminary PBPK simulations and emergent PK data from Part 1, subjects received a single oral dose of risdiplam, ensuring that the pre‐exposure cap defined in Part 1 was not exceeded. In Period 1, subjects were administered a single oral dose of risdiplam (6.0 mg) 30 min after starting a standardized light breakfast (fed state) on the morning of Day 1. Following a wash‐out period of approximately 14 days, subjects entered Period 2: itraconazole 200 mg was administered (as oral capsules, 30 min after food) twice daily (12 h apart) from Day 1–8; on Day 4 a single oral dose of risdiplam (6.0 mg) was concomitantly administered in the fed state. A DDI simulation with itraconazole was performed using PBPK modelling (Simcyp) with different scenarios of fraction metabolized (fm) by CYP3A.
Sample collection
In Part 1 and Part 3 treatment Period 1, venous blood samples were collected from each subject at predose and at regular intervals up to 216 h after dosing with risdiplam to assess the PK of risdiplam. Capillary blood samples were also collected at predose and regular intervals up to 48 h after dosing with risdiplam in Part 1. In Part 3 treatment Period 2, blood samples were collected from each subject regularly for up to 28 days after the last dose of risdiplam for PK analysis of itraconazole and risdiplam. Venous blood (2.0 ml) for itraconazole PK analysis was drawn into K2 EDTA vacutainer tubes (BD, 368841) and maintained on wet ice. Blood samples for risdiplam PK analysis (2.0 ml venous or 0.25 ml capillary) were drawn into vacutainer tubes containing K3 EDTA (venous: Greiner Vacuette, 454 087; capillary: Greiner MiniCollect, 450 476) and maintained on wet ice. All blood samples were centrifuged at 1500× g for 10 min (venous) or 3000× g for 30 min (capillary) at 4°C, within 30 min of collection then decanted into prelabelled sample tubes (itraconazole: Sarstedt, 60.549; risdiplam venous: Elkay, 8545AMX; risdiplam capillary: Sarstedt, 72.730.004 + 65.716.009) and stored frozen (at –70°C or colder) until analysis. Subjects enrolled in Part 1 of the study also had a 3.0 ml whole blood sample taken for DNA extraction to determine the copy numbers of SMN1 and SMN2. Blood samples for SMN protein quantitation and SMN mRNA analysis were taken at predose and regular intervals up to 144 h and 96 h, respectively.
For SMN protein quantification, 3.0 ml of blood were drawn into P700 tubes (BD vacutainer, 366 473) and maintained on wet ice before freezing for storage (at –70°C or colder after 1–2 h at –20°C) until analysis. For in vivo analysis of SMN mRNA, 1.25 ml of blood were drawn into PAXgene tubes (762165), left overnight at –20°C before storage (at –70°C or colder) until analysis. For ex vivo analysis of SMN mRNA, 6.0 ml of blood were drawn into sodium citrate tubes (BD vacutainer, 366 575) and stored at 37°C for a maximum of 30 min before analysis. These predose samples were spiked with stock solutions of risdiplam to achieve test concentrations of 24, 80 and 400 ng ml–1 of risdiplam. In Part 1, urine samples were collected from each subject at predose and regularly up to 48–72 h after dosing. Urine samples (3.5 ml) were stored frozen (at –70°C or colder) in polypropylene tubes (Sarstedt 60.611.011) containing 0.3% Tween 80 until analysis.
Assay description
Concentrations of risdiplam were quantified in human plasma and urine using a validated liquid chromatography‐mass spectrometry (LC–MS/MS) assay (Quantum triple stage quadrupole, Thermo Scientific; calibration range 0.250–250 ng ml–1). The lower limit of quantification was 0.250 ng ml–1 for both venous plasma and urine samples and 1.00 ng ml–1 for capillary plasma samples. The interassay precision (% CV) of quality control (QC) samples was ≤6.6% for plasma and ≤ 6.5% for urine. The interassay accuracy of the QC samples was 100.0–100.8% for plasma and 94.0–97.9% for urine.
Concentrations of itraconazole and its metabolites were quantified in human plasma using a validated LC–MS/MS assay 22. The calibration range was 5.00–2500 ng ml–1 for itraconazole and hydroxy‐itraconazole, and 0.400–200 ng ml–1 for ketoitraconazole and N‐desalkyl‐itraconazole. The interassay precision (% CV) of the QC samples was ≤5.2% and the accuracy was 96.7–101.5% for itraconazole and its metabolites.
Safety assessments
Safety was assessed throughout the study based on adverse events (AEs), clinical laboratory parameters (haematology, serum chemistry, urinalysis), physical examination, vital signs (body temperature, blood pressure and pulse rate), and 12‐lead ECGs. Owing to the retinal toxicity observed in preclinical studies of the related compound, RO6885247 9, a number of ophthalmological assessments were also conducted (ophthalmological examination, fundus photography and auto‐fluorescence, visual acuity tests, visual field test, optical coherence tomography).
PK analysis
PK parameters were calculated using noncompartmental methods (Phoenix WinNonlin Version 6.4; Certara, Princeton, NJ, USA). The Cmax and the time to Cmax were taken directly from the observed plasma concentration vs. time profiles. AUC–time curves were calculated using the linear log trapezoidal method over 24 and 120 h after risdiplam administration (AUC0 – 24h, AUC0 – 120h) and extrapolated to infinity (AUC∞).
PD analysis
Splicing modification of SMN mRNA
In Part 1, the effect of risdiplam on relative changes in SMN1, SMN2FL, and SMNΔ7 mRNA levels were determined in vivo using whole blood samples and multiplex real‐time quantitative polymerase chain reaction (PCR) technology (Roche, Pleasanton, CA, USA) 23. To determine the effects of risdiplam on SMN2 mRNA splicing modification over a wider concentration range (24–400 ng ml–1) than could be tested in vivo due to the exposure cap, predose blood samples were incubated at 37°C for 4 h with different concentrations of risdiplam (plus vehicle only and unmodified blood samples as controls). Individual relative changes in gene expression were calculated by subtracting the cycle threshold of the target gene from the reference gene as described previously 23. The mean of all predose results was used as baseline and data were analysed using Phoenix WinNonlin version 6.4.
SMN protein
In Part 1, the concentration of SMN protein was determined in whole blood samples taken from each subject via immunoassay as described previously 23.
SMN copy number
Genomic DNA from whole blood was extracted and genotyped for gene copy numbers of SMN1 and SMN2 using a droplet digital PCR (ddPCR) SMN1 (#186‐3500) and SMN2 (#186‐3503) Copy Number Determination Kit (Bio‐Rad Laboratories, Hercules, California, USA) on a ddPCR platform using an internal RPP30 gene standard. Copy numbers were quantified using standard controls for zero, one and two copies of SMN1, and two, three and four copies for SMN2 as provided by the kits 23.
Statistical analysis supporting dose escalation decision
The dose escalation was based on the dual assessment of safety/tolerability and the PD effect. The modified CRM (mCRM) with control for the probability of over‐dosing was used for the assessment of safety/tolerability. This part of the Bayesian framework has been described in detail previously 15, 16. In addition, the mCRM was further modified to allow for the estimation of PD parameters by means of the following maximum effect (Emax) model with an additive error:
where ν denotes the Hill parameter, the residual errors ε i are normally distributed around 0 with variance of σ 2 s. Emax was set to 1 based on the ex vivo SMN mRNA assay (at the maximum tested concentration of 400 ng ml–1), which served as a reference value for each individual subject.
The priors chosen for the parameters were:
a uniform distribution in the interval [0.2, 5] for ν
for log (ED50) that ED50 lies with ~90% probability in the interval (1.5–60 mg), centred at ~9 mg. A suitable choice is a normal distribution centred at log(9) and variance 1.12.
The decision to escalate to the next dose was made after reviewing all safety information up to 48 h, PK data over at least 24 h postdose and available and modelled PD data in at least four subjects in each cohort. Doses could have been repeated or adjusted downward on safety, tolerability, PK and/or PD observations at each dose level. Intermediate doses could also have been proposed to be investigated. Dose escalation would have been stopped if severe or clinically significant drug‐related changes in vital signs, ECGs, laboratory abnormalities or AEs of the same type occurred in 50% or more subjects receiving risdiplam.
Statistical analysis for assessing dose proportionality
To test for possible deviations from dose proportionality and the effect of food, a one‐way analysis of variance (ANOVA) model with factor dose group was repeatedly applied to the logarithmically transformed and dose‐normalized PK parameters, Cmax and AUC∞. Least square means and 95% confidence intervals (CI) were derived for AUC∞, and Cmax of each dose level of risdiplam in Part 1.
Statistical analysis for assessing a food effect
One separate dose cohort (6.0 mg risdiplam) was treated after a standardized high‐fat, high‐calorie meal, and compared with the corresponding fasted cohort (3). An ANOVA model with the logarithmically transformed and dose‐normalized PK parameters as dependent variables and food condition as an independent factor was applied. The geometric mean and 95% CIs for both food conditions (fasted and fed) were estimated.
Statistical analysis for assessing DDI with itraconazole
In Part 3, to assess the effect of itraconazole on the PK of risdiplam, a mixed‐effects ANOVA model with independent variable treatment (risdiplam alone vs. risdiplam + itraconazole) and random effect subject was applied to the logarithmically transformed parameters, Cmax and AUC0–∞.
Under this model the geometric means:
where PK stands either for Cmax or for AUC∞, were obtained by exponentiation of the corresponding estimated differences from the ANOVA model.
Results
Subject demographics
In total, 25 subjects were included in the Part 1 PK/PD analysis and safety populations. In Part 3, eight subjects received risdiplam in Period 1. One subject withdrew during Period 2 due to a protocol violation before receiving risdiplam plus itraconazole, therefore the PK analysis and safety populations in Period 2 included seven subjects. Subject demographics are shown in Table 2.
Table 2.
Demographics
| Part 1 | Part 3 | ||||||
|---|---|---|---|---|---|---|---|
| Cohort | All | 1 | 2 | 3 | 4 & 6 | 5 | N/A |
| Food state | Both | Fasted | Fasted | Fasted | Fasted | Fed | Fed |
| Dose | Placebo | 0.6 mg | 2.0 mg | 6.0 mg | 18.0 mg | 6.0 mg | 6.0 mg |
| Male, n (%) | 7 (100) | 3 (100) | 3 (100) | 3 (100) | 6 (100) | 3 (100) | 8 (100) |
| Race, n (%) | |||||||
| White | 3 (43) | 3 (100) | 2 (67) | 2 (67) | 6 (100) | 3 (100) | 5 (63) |
| Black | 2 (29) | – | – | 1 (33) | – | – | 1 (13) |
| Asian | 2 (29) | – | 1 (33) | – | – | – | 1 (13) |
| Multiple | – | – | – | – | – | – | 1 (13) |
| Age, mean years (SD) | 27.0 (6.9) | 23.7 (5.1) | 25.0 (7.0) | 23.0 (3.0) | 22.8 (2.1) | 32.0 (10.1) | 29.4 (8.0) |
| Weight, mean kg (SD) | 80.6 (8.4) | 84.2 (5.9) | 68.5 (17.5) | 85.0 (6.5) | 75.3 (8.5) | 80.7 (14.6) | 80.8 (11.5) |
| BMI mean kg m –2 (SD) | 24.3 (3.5) | 24.5 (1.6) | 21.1 (3.4) | 26.2 (1.6) | 23.2 (1.4) | 24.5 (2.9) | 25.0 (3.5) |
BMI, body mass index; N/A, not applicable; SD, standard deviation
SMN genotype
All subjects had at least two copies of the SMN1 gene, with one subject having three. The copy number of the SMN2 gene was more variable (0–2), with two subjects having no copies, 10 subjects having one copy and 13 subjects having two copies of the SMN2 gene.
Taste questionnaire
Subjects reported similar overall taste intensity across the doses tested, and compared with placebo, however, the risdiplam oral solution was perceived to be more bitter for doses of 2.0–18.0 mg (Table 3).
Table 3.
Mean intensity scores of the taste assessment on Day 1
| Risdiplam dose | ||||||
|---|---|---|---|---|---|---|
| Dose | Placebo | 0.6 mg | 2.0 mg | 6.0 mg | 18.0 mg | 6.0 mg |
| Male, n (%) | 7 (100) | 3 (100) | 3 (100) | 3 (100) | 6 (100) | 3 (100) |
| Overall taste intensity, mean score (SD) | 3.14 (1.07) | 2.33 (0.58) | 3.33 (0.58) | 2.67 (0.58) | 3.33 (0.82) | 2.67 (0.58) |
| Bitter, mean score (SD) | 1.43 (0.79) | 1.33 (0.58) | 2.33 (1.53) | 2.33 (1.53) | 2.50 (1.22) | 2.33 (0.58) |
1 = no taste; 5 = very intense taste; SD, standard deviation
Safety assessments
Risdiplam was well tolerated in the fasted or fed state at single 0.6–18.0 mg doses in Part 1 and at the dose of 6.0 mg when co‐administered with itraconazole in Part 3 of the study (Table 4). No deaths, moderate or severe AEs, withdrawals due to AEs, or SAEs were reported. All AEs resolved within a short period without sequelae. Only two AEs were considered related to the study drug by the investigator: pollakiuria in the placebo cohort and headache in the 18.0 mg risdiplam (fasted) cohort; they were limited in duration and resolved spontaneously without sequelae. The most frequently reported AEs were headache (four subjects), abdominal pain (three subjects), diarrhoea (two subjects) and nasopharyngitis (two subjects). No clinically significant treatment‐ or dose‐dependent changes compared to baseline were observed in physical examination, vital signs, laboratory parameters or ophthalmological assessments. No individual post‐dose QT value >500 ms and QTcF value >450 ms, with a change from baseline that was >30 ms, were reported. An exploratory analysis did not reveal any effect of risdiplam concentration on the ΔQTcF (data not shown).
Table 4.
Summary of AEs following single oral administration of risdiplam and placebo (Part 1) and risdiplam and itraconazole (Part 3)
| Part 1 | Part 3 | |||||||
|---|---|---|---|---|---|---|---|---|
| Cohort | All | 1 | 2 | 3 | 4 and 6 | 5 | N/A | N/A |
| Food state | Both | Fasted | Fasted | Fasted | Fasted | Fed | Fed | Fed |
| Dose | Placebo | 0.6 mg | 2.0 mg | 6.0 mg | 18.0 mg | 6.0 mg | 6.0 mg | 6.0 mg plus itraconazole 200 mg BID |
| Male, n (%) | 7 (100) | 3 (100) | 3 (100) | 3 (100) | 6 (100) | 3 (100) | 8 (100) | 7 (100)a |
| AEs, n | 4 | 6 | 0 | 0 | 9 | 2 | 2 | 4 |
| Subjects with AEs, n (%) | 3 (42.9) | 2 (66.7) | 0 | 0 | 4 (66.7) | 2 (66.7) | 2 (25.0) | 4 (57.1) |
| Subjects with drug‐related AEs, n (%) | 1 (14.3) | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 |
In Part 3, eight subjects received risdiplam in Period 1 but one subject withdrew during Period 2 due to a protocol violation before receiving risdiplam plus itraconazole. Therefore, the pharmacokinetic analysis and safety populations in Period 2 included seven subjects
AE, adverse event; BID, twice a day; N/A, not applicable
Risdiplam pharmacokinetics
Mean risdiplam plasma concentrations vs. time profiles after administration of single ascending doses of risdiplam to healthy male subjects are presented in Figure 2A. Risdiplam exhibited linear PK over the 0.6–18.0 mg dose range, with both Cmax and AUC increasing in an approximate dose‐proportional manner. ANOVA provided no evidence to reject the null hypothesis of dose proportionality for both Cmax (P = 0.23) and AUC∞ (P = 0.271). The apparent terminal elimination half‐life of risdiplam was 40.1–68.7 h for 2.0–18.0 mg doses although the 0.6 mg dose exhibited a steeper decline with an apparent terminal elimination half‐life of 24.8 h (Table 5). Urinary excretion of unchanged risdiplam was low, accounting for 3.6–4.3% of the dose range administered.
Figure 2.
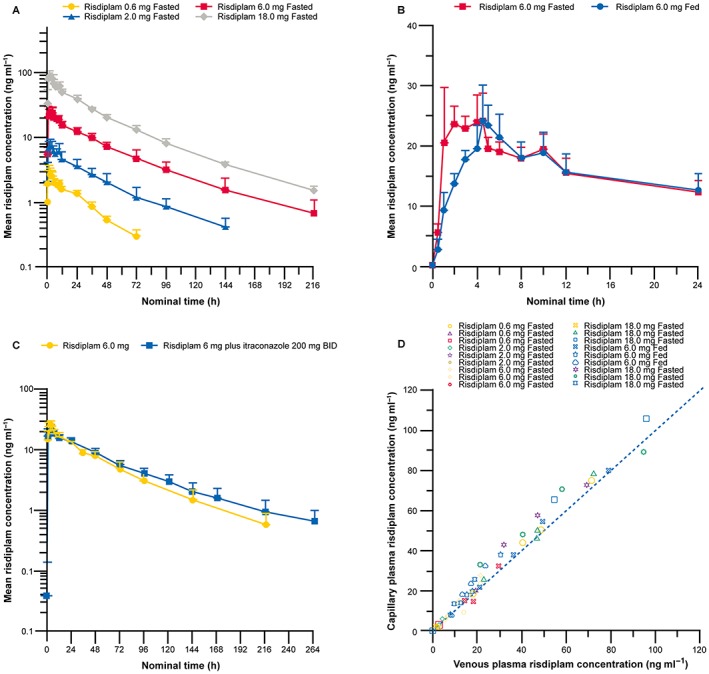
(A) Mean plasma concentration vs. time profiles of risdiplam following single oral doses of 0.6–18.0 mg in the fasted state (semi‐log scale). (B) Plasma concentration vs. time profiles of 6.0 mg risdiplam administered under fasted or fed conditions. (C) Plasma concentration vs. time profiles of risdiplam following single oral doses of 6.0 mg in the fed state alone or in combination with itraconazole (semi‐og scale). (D) Combined individual capillary vs. venous plasma concentrations of risdiplam. (A–C) Error bars represent standard deviation. BID, twice a day
Table 5.
Pharmacokinetic parameters following single oral dose administration of risdiplam (Part 1) and for risdiplam with or without itraconazole (Part 3)
| Part 1 | Part 3 | ||||||
|---|---|---|---|---|---|---|---|
| Cohort | 1 | 2 | 3 | 4 & 6 | 5 | N/A | N/A |
| Food state | Fasted | Fasted | Fasted | Fasted | Fed | Fed | Fed |
| Dose | 0.6 mg | 2 mg | 6 mg | 18 mga | 6 mg | 6 mgb | 6 mg plus itraconazole 200 mg BIDc |
| Male, n (%) | 3 (100) | 3 (100) | 3 (100) | 6 (100) | 3 (100) | 8 (100) | 7 (100) |
| t max mean h ± SD | 3.17 ± 1.26 | 2.67 ± 1.53 | 2.00 ± 1.00 | 2.33 ± 10.3 | 4.67 ± 0.29 | 3.38 ± 0.74 | 3.29 ± 1.11 |
| C max mean ng ml –1 ± SD | 2.88 ± 0.79 | 8.36 ± 0.77 | 24.9 ± 5.15 | 94.0 ± 13.6 | 25.1 ± 4.71 | 27.1 ± 4.46 | 23.6 ± 2.91 |
| AUC 0‐24h mean h ng ml –1 ± SD | 41.9 ± 6.71 | 119 ± 27.8 | 394 ± 56.9 | 1300 ± 107 | 374 ± 64.2 | 410 ± 58.9 | 398 ± 41.1 |
| AUC 0‐120h mean h ng ml –1 ± SD | – | – | – | – | – | 935 ± 148 | 1030 ± 149 |
| AUC ∞ mean h ng ml –1 ± SD | 87.3 ± 11.7 | 303 ± 86.3 | 1100 ± 291 | 3300 ± 241 | 1020 ± 168 | 1090 ± 219 | 1300 ± 269 |
| t ½ mean h ± SD | 25.8 ± 9.06 | 40.1 ± 0.92 | 48.3 ± 9.42 | 68.9 ± 5.85 | 43.3 ± 3.58 | 44.1 ± 6.75 | 60.0 ± 14.6 |
| Cl/F mean l h –1 ± SD | 6.96 ± 1.00 | 7.00 ± 2.06 | 5.70 ± 1.40 | 5.48 ± 0.41 | 6.00 ± 1.09 | 5.71 ± 1.15 | 4.79 ± 0.93 |
| V z /F mean l ± SD | 254 ± 70.2 | 405 ± 120 | 385 ± 25.0 | 256 ± 76.7 | 377 ± 90.0 | 356 ± 46.5 | 400 ± 51.4 |
Values for AUC0‐24h, AUC∞, t1/2, Cl/F, and Vz/F were not determined for one subject who withdrew from the study because of personal reasons on Day 2. bValues for AUC0‐120h, AUC∞, t1/2, Cl/F, and Vz/F were not determined for one subject who withdrew from the study because of personal reasons after completing follow‐up visit 2. cIn Part 3, eight subjects received risdiplam in Period 1 but one subject withdrew during Period 2 due to a protocol violation before receiving risdiplam plus itraconazole. Therefore, the PK analysis and safety populations in Period 2 included seven subjects.
AUC∞, area under the plasma concentration vs. time curve extrapolated to infinity; AUC0 – 24h, area under the plasma concentration vs. time curve over 24 h after risdiplam administration; AUC0 – 120h, area under the plasma concentration vs. time curve over 120 h after risdiplam administration; BID, twice a day; Cl/F, apparent oral plasma clearance (for risdiplam only); N/A, not applicable; tmax, time to maximum observed plasma concentration; t½, apparent terminal elimination half‐life; Vz/F, apparent volume of distribution (for risdiplam only) ; SD, standard deviation
In Part 1, an exploratory comparison of Cohort 3 (fasted) and Cohort 5 (fed), suggested that food had no relevant effect on Cmax and AUC∞ of risdiplam. Due to the limited number of subjects, no P‐values are reported and these results should be interpreted with caution (Figure 2B). In Part 3, itraconazole had a minor effect on the PK of a single oral dose of risdiplam (Figure 2C, Table 5), resulting in a slight increase of the AUC0–120h (11%, 95% CI: 1–21%), and a slight reduction of the Cmax (9%, 95% CI: 1–18%). Risdiplam concentrations in capillary plasma were generally in good agreement with those observed in venous plasma, with slightly higher exposures in capillary plasma (Figure 2D).
Risdiplam PD
The two subjects (one from Cohort 3 and one from Cohort 5) with no SMN2 copies were excluded from risdiplam PD analysis.
SMN mRNA
Administration of single oral doses of 0.6–18.0 mg risdiplam increased SMN2FL mRNA and decreased SMN2Δ7 mRNA levels, which resulted in a dose‐dependent increase of SMN2FL/SMN2Δ7 mRNA ratios (Figure 3A). SMN1 mRNA levels remained unaffected by risdiplam (Figure 3B). The SMN2FL/SMN2Δ7 mRNA ratio appeared to be a sensitive biomarker to monitor the inclusion of SMN2 exon 7 and the effect of risdiplam, as it reduced the confounding effects of SMN2FL and SMN2Δ7 mRNA level fluctuations. The median time to maximum effect was reached at 4–8 h postdose, across all dose cohorts and food had a negligible effect on the splice modification effects of risdiplam (Figure 3A). The highest tested dose of 18.0 mg of risdiplam led to a 64% increase in SMN2FL mRNA as compared with baseline. The results from the ex vivo assay revealed that the mean fold increase in the baseline ratio of SMN2FL mRNA was 1.28, 1.86 and 2.13 at concentrations of 24, 80 and 400 ng ml–1 respectively (Figure 3B). Overall the results of the ex vivo assay were consistent with the in vivo SMN mRNA results.
Figure 3.
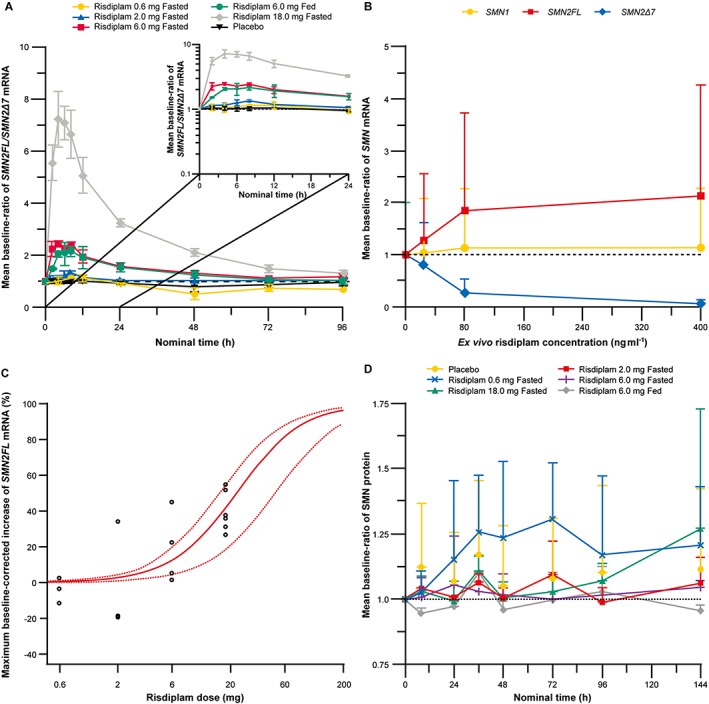
(A) Baseline ratio of SMN2FL/SMNΔ7 mRNA over time following single oral dose administration of 0.6–18.0 mg risdiplam in the fasted or fed state (B) Baseline ratio of ex vivo SMN mRNA vs. ex vivo risdiplam concentrations. (C) Estimated individual maximum increase in SMN2FL mRNA vs. dose of risdiplam. (D) Baseline ratio of SMN protein in blood vs. nominal time. (A, B and D) Error bars represent standard deviation
Bayesian methods
Dose escalation was based on the dual assessment of safety/tolerability and the PD effect applied to a mCRM. As no DLEs were observed and risdiplam was safe and tolerated in all cohorts, the dose was escalated by approximately 3‐fold to 2.0 mg and 6.0 mg. To comply with the prespecified individual subject exposure cap of 1500 h ng ml–1 for AUC0–24h, the dose was escalated to 18.0 mg instead of 20.0 mg. The dose escalation was discontinued after 18.0 mg. The highest observed individual plasma exposure for AUC0‐24h (1470 ng h ml–1) was close to the individual exposure cap (1500 ng h ml–1). The 18.0 mg dose was administered to Cohort 4 and Cohort 6 (Table 1) in the fasted state (n = 6 for risdiplam, and n = 2 for placebo), to adequately estimate PD parameters. The highest tested risdiplam dose of 18.0 mg led to a 46% (95% CI: 30–62%) placebo‐corrected increase in SMN2FL mRNA. This corresponds to 41% (95% CI: 27–55%) of the maximum placebo‐corrected increase in SMN2FL mRNA (where 100% is equivalent to the 113% increase in SMN2FL mRNA observed in the ex vivo experiments at a concentration of 400 ng ml–1 risdiplam, Figure 3B). Statistical analysis revealed an ED50 of approximately 20 mg risdiplam (Figure 3C) for the SMN2FL mRNA increase.
SMN protein
Administration of single oral 0.6–18.0 mg risdiplam doses did not lead to a change of SMN protein concentration in blood and did not reveal any dose‐related changes (Figure 3D).
Discussion
This study demonstrated that single oral, 0.6–18.0 mg doses of risdiplam in the fasted or fed state were well tolerated. This further validates the use of an exposure‐based cap derived from animal toxicity data in this single ascending dose study. PD data indicated proof of mechanism for risdiplam and supported further development of the compound.
In Part 1 (single ascending dose), the first cohort received a starting dose of 0.6 mg risdiplam in the fasted state, selected based on preclinical data and was well below the estimated therapeutic dose of 2–22 mg. The 0.6 mg dose was associated with little or no PD effect on SMN mRNA. For subsequent doses administered, all PK/PD and safety data were analysed in an adaptive manner to inform the decision to proceed to the next cohort. The second and third cohorts received 2.0 mg and 6.0 mg risdiplam in the fasted state, respectively. The fourth cohort was due to receive a dose of 20.0 mg (as predicted by the model algorithm); however, 18.0 mg (fasted) was chosen to ensure that no individual subject would exceed the exposure cap of 1500 h ng ml–1 and to investigate a broad range of concentrations for the assessment of the PD effect. To investigate the effect of food (which was a requirement before investigating the interaction with itraconazole), Cohort 5 received a 6.0 mg dose of risdiplam in the fed state. As the variability of all PK parameters of risdiplam was low (CV% < 30% for Cmax and AUC0–24h) in the preceding dose levels (0.6 mg, 2.0 mg, 6.0 mg and 18.0 mg), a dedicated food effect part was considered unnecessary and a between subject comparison was justified. Finally, Cohort 6 also received an 18.0 mg (fasted) dose to better characterize the effect of risdiplam on SMN2 mRNA. This dose represented the highest exposure that did not exceed the exposure cap, though only around half of the maximum PD effect was achieved. Two subjects had no copies of SMN2 as has been described previously 3, 23 and were excluded from the PD analysis. Statistical analysis revealed an ED50 of approximately 20 mg risdiplam for the SMN2 mRNA increase, assuming that the Emax in vivo would be the same as the observed Emax ex vivo. However, the Emax on SMN2 mRNA has not been investigated in vivo and is fixed to the Emax (+113%) of the ex‐vivo SMN mRNA assay at a maximum tested concentration of 400 ng ml–1. It is unknown if by increasing the dose, an increase in SMN2 mRNA of 113% could be achieved as was observed in the ex vivo assay. Peak PK/PD effect was observed approximately 5 h after the 18.0 mg dose. No obvious difference in the effect on SMN2 mRNA was observed between subjects with one or two copies of the SMN2 gene. However, the sample size was too small to make any conclusions. Consistent with evidence from animal studies 7, the observed dose‐dependent and concentration‐dependent PD effect on SMN2 splicing provided proof of mechanism for the splicing modifier, risdiplam.
However, no change in SMN protein concentration was observed. This was expected because, in healthy subjects, the majority of SMN1 mRNA is FL compared with only ~10% of SMN2 mRNA 24. Therefore, after a single dose of risdiplam, only a minor effect on SMN protein was expected in healthy subjects, and this could even be masked by natural SMN protein level variability. The observed SMN2 mRNA changes suggest that risdiplam may increase SMN protein levels in patients with SMA. As the lack of SMN protein is thought to cause motor neuron degeneration and muscle atrophy, modifying SMN2 mRNA could lead to beneficial effects in patients with SMA 25. Since risdiplam is both centrally and peripherally distributed, it has the potential to increase SMN protein throughout the body and could have beneficial effects that extend beyond the motor neuron 5.
The study revealed favourable PK properties as linear PK were observed over the investigated dose range of 0.6–18.0 mg. Furthermore, there was no meaningful difference in PK in the fasted and fed state, implying that risdiplam can be administered with or without food. Risdiplam concentrations were similar between venous and capillary plasma; this may have important implications for minimizing the volume of blood sampling required, particularly in neonatal patients 26 such as those with Type 1 SMA or older patients in whom venous blood sampling may be a challenge.
Identifying the bitter taste of the risdiplam formulation used in this study was useful for developing an appropriate, artificially flavoured formulation for the ongoing study in paediatric patients 26.
In this study, itraconazole was used to study potential DDIs. It is suggested, based on preclinical data, that risdiplam is mainly metabolized by flavin‐containing monooxygenase (FMO). In contrast to cytochrome P (CYPs), FMO are not readily induced or inhibited by foreign chemicals and produce reaction products that are readily excretable 27. Therefore, drugs that are metabolized predominantly by FMOs are less likely to elicit DDI and by extension, potentially harmful side effects 27. Itraconazole had a minor effect on the intestinal and hepatic metabolism of risdiplam. The PBPK models indicated that the risdiplam fmCYP3A4 was approximately 20%, which would yield a ratio of 1.2 in AUC∞ between risdiplam and itraconazole vs. risdiplam alone. Sensitivity analysis with a fmCYP3A4 of approximately 50% and 95% was predicted to result in a ratio of approximately 1.6 and 3, respectively. Taken together, these data suggest a low likelihood of clinically significant DDI with selective inhibitors and inducers of CYP3A enzymes.
The use of Bayesian adaptive design, while common place in EIH oncology trials, is an emerging area of interest in other therapeutic areas 15. In our study, dose selection was set up to be successfully guided by a Bayesian adaptive design combining safety data (DLE occurrence) with a priori PK and PD data and the estimation of PD parameters with good precision (≤75%). As planned, adaptive dose selection, with a prespecified exposure cap, enabled us to prioritize safety and minimize exposure since dose escalation only occurred after review of safety, tolerability and available PK and PD data for the previous dose(s). Furthermore, Bayesian adaptive design enabled us to assign a greater number of subjects at more informative pharmacological dose levels than traditional methods while reducing the number placed on inactive doses. In the absence of DLEs, the dual Bayesian assessment was driven by the characterization of the PK/PD relationship. Based on data across all cohorts investigated, the dose/PD relationship could be described with sufficient accuracy in line with the predefined criteria PK/PD relationship (e.g. CV% from ED/EC50, ED/EC90 e.g. ≤ 75%). Our experience with this study supports the use of Bayesian adaptive design principles in EIH trials for a wide range of indications as advocated elsewhere 15.
The results and analysis of this study utilizing Bayesian adaptive methods were used for dose selection for subsequent studies with additional model‐based analysis techniques (PBPK modelling, population PK and PK/PD analysis). Anticipated therapeutic risdiplam doses were determined and will be administered across all age ranges of SMA patients in the ongoing Phase 2/3 studies, comprising exploratory dose‐finding and confirmatory parts 19, 20, 21.
In addition to the issues associated with detecting SMN protein changes in healthy volunteers, our study has further limitations including the small sample size and lack of female subjects. Furthermore, this study only enrolled adult volunteers, while many SMA patients are infants or children 28. Safety and tolerability data were limited to single‐dose administration and data from multiple doses and long‐term administration are required to further characterize R07034067. The exposure cap, while necessary, also limited our investigation of the full PK/PD relationship of risdiplam at higher doses. The taste of the risdiplam oral solution and completing the taste questionnaire had the potential to unblind both participants and the investigator. However, this was considered to be of minor concern for a Phase 1 study.
At present, there is only one approved treatment for SMA. Nusinersen is an SMN2‐directed antisense oligonucleotide delivered centrally by intrathecal injection, recently approved by the Food and Drug Administration for the treatment of SMA in paediatric and adult patients, and by the European Medicines Agency for the treatment of 5q SMA 29, 30. Nevertheless, there remain several unmet needs in the treatment of SMA. For instance, not all patients respond to nusinersen 31 and intrathecal administration has been associated with AEs, including headache, back pain, nausea and post‐lumbar puncture syndrome and may be challenging in patients with SMA who frequently experience scoliosis 32. Furthermore, some patients require anaesthesia and hospitalization when receiving drugs intrathecally; therefore, an oral formulation would offer greater convenience and reduced burden for patients and their families, and to the treating physicians and healthcare system 32. In view of research suggesting that beyond the motor neuron, SMN depletion affects cells and tissues in the central nervous system and the periphery 5, 33, 34, 35, there is also a need for a peripherally acting disease‐modifying medicine for patients with SMA.
In conclusion, this study demonstrated that single doses of risdiplam were well tolerated at the doses tested. Bayesian adaptive design principles were successfully applied to enable more subjects to receive pharmacologically active doses, while reducing the number of individuals receiving inactive doses. Proof of mechanism of risdiplam was demonstrated by the intended shift in SMN2 splicing towards SMN2FL mRNA. Based on these data, three Phase 2/3 studies evaluating safety and efficacy of risdiplam in patients with Type 1 and Type 2/3 SMA, are currently ongoing and recruiting patients 19, 20, 21. These studies, in a wide range of participants, will provide critical information regarding the potential therapeutic benefit of risdiplam for the treatment of patients with SMA.
Competing Interests
S.S., A.G., B.J., P.J., N.P., Y.C., N.F., T.B., H.K., A.M., H.R., A.P., L.M., C.C. and O.K. are employees of F. Hoffmann‐La Roche Ltd. N.A.K. is an employee of PRA Health Science.
The authors thank all the subjects who participated in this study. This study was funded by F. Hoffmann‐La Roche Ltd. The authors thank Tom Priddle, DPhil, of Nucleus Global, UK for providing medical writing support, which was funded by F. Hoffmann‐La Roche Basel Ltd, Switzerland in accordance with Good Publication Practice (GGP3) guidelines.
Qualified researchers may request access to individual patient level data through the clinical study data request platform ( www.clinicalstudydatarequest.com ). Further details on Roche's criteria for eligible studies are available here ( https://clinicalstudydatarequest.com/Study‐Sponsors/Study‐Sponsors‐Roche.aspx ). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here ( https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm ).
Sturm, S. , Günther, A. , Jaber, B. , Jordan, P. , Al Kotbi, N. , Parkar, N. , Cleary, Y. , Frances, N. , Bergauer, T. , Heinig, K. , Kletzl, H. , Marquet, A. , Ratni, H. , Poirier, A. , Müller, L. , Czech, C. , and Khwaja, O. (2019) A phase 1 healthy male volunteer single escalating dose study of the pharmacokinetics and pharmacodynamics of risdiplam (RG7916, RO7034067), a SMN2 splicing modifier. Br J Clin Pharmacol, 85: 181–193. 10.1111/bcp.13786.
References
- 1. Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci 2015; 35: 8691–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol 2012; 11: 443–452. [DOI] [PubMed] [Google Scholar]
- 3. Butchbach ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci 2016; 3: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Singh RN, Howell MD, Ottesen EW, Singh NN. Diverse role of survival motor neuron protein. Biochim Biophys Acta 2017; 1860: 299–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 2013; 19: 40–50. [DOI] [PubMed] [Google Scholar]
- 6. Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, et al Peripheral SMN restoration is essential for long‐term rescue of a severe spinal muscular atrophy mouse model. Nature 2011; 478: 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, et al Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014; 345: 688–693. [DOI] [PubMed] [Google Scholar]
- 8. Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, et al Discovery of risdiplam, a selective survival of motor neuron‐2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem 2018; 61: 6501–6517. [DOI] [PubMed] [Google Scholar]
- 9. ClinicalTrials.gov . NCT02240355: A study of RO6885247 in adult and pediatric patients with spinal muscular atrophy (MOONFISH) 2015; 2015.
- 10. PTC Therapeutics Press Release . RG7916 granted orphan drug designation in the U.S. for the treatment of spinal muscular atrophy 2017; 2017.
- 11. Feng Z, Ling KK, Zhao X, Zhou C, Karp G, Welch EM, et al Pharmacologically induced mouse model of adult spinal muscular atrophy to evaluate effectiveness of therapeutics after disease onset. Hum Mol Genet 2016; 25: 964–975. [DOI] [PubMed] [Google Scholar]
- 12. Sivaramakrishnan M, McCarthy KD, Campagne S, Huber S, Meier S, Augustin A, et al Binding to SMN2 pre‐mRNA‐protein complex elicits specificity for small molecule splicing modifiers. Nat Commun 2017; 8: 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. European Medicines Agency . Guideline on strategies to identify and mitigate risks for first‐in‐human and early clinical trials with investigational medicinal products 2017; 2017. [DOI] [PMC free article] [PubMed]
- 14. European Medicines Agency . Guideline on strategies to identify and mitigate risks for first‐in‐human clinical trials with investigational medicinal products 2007; 2017. [DOI] [PMC free article] [PubMed]
- 15. Guede D, Reigner B, Vandenhende F, Derks M, Beyer U, Jordan P, et al Bayesian adaptive designs in single ascending dose trials in healthy volunteers. Br J Clin Pharmacol 2014; 78: 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sturm S, Delporte ML, Hadi S, Schobel S, Lindemann L, Weikert R, et al Results and evaluation of a first‐in‐human study of RG7342, a mGlu5 positive allosteric modulator, utilizing Bayesian adaptive methods. Br J Clin Pharmacol 2018; 84: 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. ClinicalTrials.gov . NCT02633709: A study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of RO7034067 (RG7916) given by mouth in healthy volunteers. Feb 2016; Feb 2016.
- 18. Liu L, Bello A, Dresser MJ, Heald D, Komjathy SF, O'Mara E, et al Best practices for the use of itraconazole as a replacement for ketoconazole in drug‐drug interaction studies. J Clin Pharmacol 2016; 56: 143–151. [DOI] [PubMed] [Google Scholar]
- 19. ClinicalTrials.gov . NCT02913482 a study to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of RO7034067 in infants with Type1 spinal muscular atrophy (FIREFISH) 2017; 2017.
- 20. ClinicalTrials.gov . NCT02908685 A study to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of RO7034067 in Type 2 and 3 spinal muscular atrophy participants (SUNFISH) March 2017, 2017.
- 21. ClinicalTrials.gov . NCT03032172 a study of RO7034067 in adult and pediatric participants with spinal muscular atrophy (JEWELFISH) 2017; 2017.
- 22. Liang X, Van Parys M, Ding X, Zeng N, Bi L, Dorshort D, et al Simultaneous determination of itraconazole, hydroxy itraconazole, keto itraconazole and N‐desalkyl itraconazole concentration in human plasma using liquid chromatography with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2016; 1020: 111–119. [DOI] [PubMed] [Google Scholar]
- 23. Czech C, Tang W, Bugawan T, Mano C, Horn C, Iglesias VA, et al Biomarker for spinal muscular atrophy: expression of smn in peripheral blood of SMA patients and healthy controls. PLoS One 2015; 10: e0139950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, et al The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 1997; 6: 1205–1214. [DOI] [PubMed] [Google Scholar]
- 25. Swoboda KJ. Of SMN in mice and men: a therapeutic opportunity. J Clin Invest 2011; 121: 2978–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ward RM, Benjamin D, Barrett JS, Allegaert K, Portman R, Davis JM, et al Safety, dosing, and pharmaceutical quality for studies that evaluate medicinal products (including biological products) in neonates. Pediatr Res 2017; 81: 692–711. [DOI] [PubMed] [Google Scholar]
- 27. Phillips IR, Shephard EA. Drug metabolism by flavin‐containing monooxygenases of human and mouse. Expert Opin Drug Metab Toxicol 2017; 13: 167–181. [DOI] [PubMed] [Google Scholar]
- 28. Verhaart IEC, Robertson A, Wilson IJ, Aartsma‐Rus A, Cameron S, Jones CC, et al Prevalence, incidence and carrier frequency of 5q‐linked spinal muscular atrophy ‐ a literature review. Orphanet J Rare Dis 2017; 12: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Biogen Inc . SPINRAZA™ (nusinersen) US prescribing information. 2016; 2017.
- 30. Biogen Inc . SPINRAZA™ (nusinersen) EU SmPC. 2017; 2017.
- 31. Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med 2017; 377: 1723–1732. [DOI] [PubMed] [Google Scholar]
- 32. Hache M, Swoboda KJ, Sethna N, Farrow‐Gillespie A, Khandji A, Xia S, et al Intrathecal injections in children with spinal muscular atrophy: nusinersen clinical trial experience. J Child Neurol 2016; 31: 899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wijngaarde CA, Blank AC, Stam M, Wadman RI, van dB, van dP. Cardiac pathology in spinal muscular atrophy: a systematic review. Orphanet J Rare Dis 2017; 12: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowerman M, Michalski JP, Beauvais A, Murray LM, DeRepentigny Y, Kothary R. Defects in pancreatic development and glucose metabolism in SMN‐depleted mice independent of canonical spinal muscular atrophy neuromuscular pathology. Hum Mol Genet 2014; 23: 3432–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bowerman M, Swoboda KJ, Michalski JP, Wang GS, Reeks C, Beauvais A, et al Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol 2012; 72: 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]