

Abstract
Background
Trimethylamine N-oxide (TMAO), a small molecule produced by the metaorganismal metabolism of dietary choline, has been implicated in human disease pathogenesis, including known risk factors for Alzheimer’s disease (AD), such as metabolic, cardiovascular, and cerebrovascular disease.
Methods
In this study, we tested whether TMAO is linked to AD by examining TMAO levels in cerebrospinal fluid (CSF) collected from a large sample (n = 410) of individuals with Alzheimer’s clinical syndrome (n = 40), individuals with mild cognitive impairment (MCI) (n = 35), and cognitively-unimpaired individuals (n = 335). Linear regression analyses were used to determine differences in CSF TMAO between groups (controlling for age, sex, and APOE ε4 genotype), as well as to determine relationships between CSF TMAO and CSF biomarkers of AD (phosphorylated tau and beta-amyloid) and neuronal degeneration (total tau, neurogranin, and neurofilament light chain protein).
Results
CSF TMAO is higher in individuals with MCI and AD dementia compared to cognitively-unimpaired individuals, and elevated CSF TMAO is associated with biomarkers of AD pathology (phosphorylated tau and phosphorylated tau/Aβ42) and neuronal degeneration (total tau and neurofilament light chain protein).
Conclusions
These findings provide additional insight into gut microbial involvement in AD and add to the growing understanding of the gut–brain axis.
Electronic supplementary material
The online version of this article (10.1186/s13195-018-0451-2) contains supplementary material, which is available to authorized users.
Keywords: Alzheimer’s disease, Cerebrospinal fluid, Biomarkers, Trimethylamine N-oxide, Microbiota, Gut bacteria, Amyloid, Tau, Neurofilament light
Background
The human gut is home to trillions of microbes, including bacteria, eukaryotes, and viruses, that participate in a lifelong symbiotic relationship with their human hosts. Resident gut microbes perform essential functions for human health ranging from regulating nutrition and metabolism to influencing immune system development and function [1]. Gut microbes impact human health and disease at least in part by metabolizing dietary and host-derived substrates, and generating biologically active compounds including signaling compounds (e.g., agonists of G-protein coupled receptors), biological precursors, and toxins [2–4]. The microbial-derived metabolite trimethylamine N-oxide (TMAO) has been implicated in metabolic [5], cardiovascular [6, 7], and cerebrovascular [8] disease. The production of TMAO occurs via a two-step process. First, gut microbes enzymatically generate trimethylamine (TMA) from dietary constituents such as choline or l-carnitine [9]. TMA then enters the circulation and is oxidized to TMAO in the liver by flavin-containing monooxygenase 1 and 3 (FMO1 and FMO3) [6]. A recent study [10] demonstrated that TMAO is measurable in cerebrospinal fluid (CSF), suggesting that this microbial-derived metabolite reaches the central nervous system (CNS), and may therefore be relevant to neurological function or disorders. Indeed, mice treated with dietary TMAO show increased brain aging and cognitive impairment, likely due to increased oxidative stress, mitochondrial dysfunction, and inhibition of mammalian target of rapamycin (mTOR) signaling in the brain [11].
Alzheimer’s disease (AD) pathology is characterized by extracellular beta-amyloid (Aβ) plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein [12]. The underlying etiology of AD is highly complex and multifactorial. A variety of genetic and environmental factors have been implicated in AD etiopathogenesis, including contributions from gut microbiota [13–15]. While it has been hypothesized that TMAO could be associated with AD pathology [16], this relationship has not yet been fully investigated in humans with Alzheimer’s clinical syndrome (AD dementia) [17]. In this study, we examined levels of TMAO in a large sample of CSF collected from individuals with AD dementia, individuals with mild cognitive impairment (MCI), and cognitively-unimpaired individuals. We also investigated the relationships between CSF TMAO, AD biomarkers (Aβ and phosphorylated tau), and biomarkers of neuronal and synaptic degeneration (total tau, neurofilament light chain protein, and neurogranin). We found that CSF TMAO levels are elevated in individuals with AD dementia, and that elevated CSF TMAO is associated with elevated AD pathology and neuronal degeneration as measured in CSF.
Methods
Participants
We identified 414 individuals in the Wisconsin Alzheimer’s Disease Research Center (ADRC) clinical core (n = 277) and the Wisconsin Registry for Alzheimer’s Prevention (WRAP) study (n = 137) who had undergone lumbar puncture with CSF collection, as well as TMAO and biomarker quantification. The ADRC clinical core study consists of participants who fall along the clinical continuum of cognitive function, including AD dementia, MCI, and cognitively-unimpaired controls. The WRAP study is a large (> 1500 subjects), ongoing (> 15 years), prospective longitudinal investigation of the genetic, biological, and lifestyle factors that contribute to the development of AD dementia and cognitive decline [18]. Individuals in the WRAP study were recruited as cognitively-unimpaired, asymptomatic middle-aged adults and undergo biannual comprehensive medical and cognitive evaluation. Because both the WRAP study and the ADRC clinical core are enriched for risk of late-onset AD (~ 70% of WRAP subjects have a parental family history of AD, and ~ 50% of participants 45–65 years old in the ADRC study have a parental history of AD), the APOE ε4 genotype is more prevalent. General exclusion criteria for the ADRC and WRAP studies include any significant neurologic disease (other than AD dementia), history of alcohol/substance dependence, major psychiatric disorders (including untreated major depression), or other significant medical illness. APOE ε4 genotyping procedures have been described previously [19], and participants were categorized as noncarriers (zero ε4 alleles) or APOE ε4 carriers (one or two ε4 alleles). The University of Wisconsin Health Science Institutional Review Board approved all study procedures, and all experiments were performed in accordance with relevant guidelines and regulations. All participants provided written informed consent to be involved in this study.
Diagnostic classification
Participants underwent a comprehensive neuropsychological battery to determine their cognitive status. Participants with MCI and AD dementia were diagnosed using available clinical and cognitive information in accordance with the updated 2011 National Institute on Aging–Alzheimer’s Association workgroup diagnostic criteria [20, 21]. All participants in the ADRC clinical core are discussed at a consensus review committee consisting of physicians, neuropsychologists, and nurse practitioners. Biomarker data are not used in determining clinical diagnosis. Participants in the WRAP study are reviewed selectively when flagged after cognitive abnormalities are detected by algorithm on neuropsychological tests, at which point cases are discussed at a consensus review committee meeting [18]. Of the 414 identified participants, four individuals with a diagnosis of nonneurodegenerative cognitive impairment at the time of CSF collection were excluded from the present analyses, resulting in a total of 410 participants: n = 335 cognitively-unimpaired participants (Control group), n = 35 MCI (MCI group), and n = 40 AD dementia (AD group).
Lumbar puncture and CSF collection
Lumbar puncture and CSF collection procedures have been described previously [22]. Briefly, CSF was collected via lumbar puncture in the morning after a 12-h fast with a Sprotte 25 or 24-gauge spinal needle at the L3/4 or L4/5 interspace using gentle extraction into propylene syringes. CSF (~ 22 ml) was then combined, gently mixed, and centrifuged at 2000 × g for 10 min. Supernatants were frozen in 0.5 ml aliquots in polypropylene tubes and stored at − 80 °C.
CSF biomarker quantification
CSF AD biomarkers included the Aβ42/Aβ40 ratio, phosphorylated tau (p-tau), and the p-tau/Aβ42 ratio. CSF Aβ is an indicator of amyloid burden, with greater amyloid deposition in the brain being reflected by lower levels in the CSF. The Aβ42/Aβ40 ratio (which normalizes CSF Aβ42 for the total amount of Aβ peptides that are present in CSF) was used given that it shows better correspondence with brain amyloid deposition as well as superior diagnostic performance compared to CSF Aβ42 alone [23]. p-tau is a marker of tau phosphorylation believed to be associated with neurofibrillary tangle pathology, with higher levels reflecting a more intense tau phosphorylation process; the ratio of p-tau/Aβ42 incorporates both facets of pathology, with higher values indicating greater AD pathology [24]. For the Aβ42/Aβ40 ratio, CSF Aβ42 and CSF Aβ40 were quantified separately by electrochemiluminescence (ECL) using an Aβ triplex assay (MSD Human Aβ peptide Ultra-Sensitive Kit; Meso Scale Discovery, Gaithersburg, MD, USA). For p-tau and the p-tau/Aβ42 ratio, CSF p-tau and Aβ42 were quantified using commercially available sandwich ELISAs (INNOTEST β-amyloid1–42 and Phospho-Tau[181 P], respectively; Fujirebio Europe, Ghent, Belgium).
CSF biomarkers of neuronal degeneration included total tau (t-tau), neurofilament light chain protein (NFL, a marker of axonal degeneration), and neurogranin (a marker of synaptic degeneration). CSF t-tau and NFL were quantified using commercially available sandwich ELISAs: t-tau, INNOTEST hTau Ag (Fujirebio Europe); and NFL, NF-Light ELISA kit (Uman Diagnostics AB, Umeå, Sweden). CSF neurogranin was quantified using a sandwich ELISA as described previously [25]. All CSF assays were performed in two batches (n = 192 samples in batch 1, n = 218 samples in batch 2), and all statistical analyses accounted for batch variation (see Statistical analysis).
CSF TMAO quantification
CSF TMAO was quantified via an untargeted plasma metabolomics analysis performed by Metabolon, Inc. (Durham, NC, USA) using ultrahigh performance liquid chromatography tandem mass spectrometry (UHPLC-MS) as described previously [26] (details presented in Additional file 1: Methods). All samples were sent to Metabolon in one shipment. Raw data were extracted, peak identified, and QC processed using Metabolon’s hardware and software. TMAO levels were expressed as scaled intensity units (SIU) using the QC-processed mass-to-charge ratio (m/z) area-under-the-curve values for TMAO and scaled to a median value of 1.
Statistical analysis
Our analysis approach first examined differences in CSF TMAO levels between clinical diagnostic groups, and then extended these analyses in order to characterize the biological relationships between CSF TMAO and biomarkers of both AD pathology and neurodegeneration. To determine CSF TMAO differences between groups, a multiple linear regression model was conducted in R (v3.5.0) to test the effect of age, sex, APOE ε4 genotype, and clinical diagnosis (Control, MCI, AD dementia) on CSF TMAO levels. CSF TMAO was natural log transformed to account for a nonnormal distribution. Secondarily, linear regression models were used to determine the relationship between CSF TMAO and CSF biomarkers (Aβ42/Aβ40, p-tau, p-tau/Aβ42 ratio, t-tau, NFL, and neurogranin). Separate models were run for each CSF biomarker, and each model included covariates of age, sex, and the nuisance covariate of CSF analysis batch (to account for batch variation). Given that TMAO has been implicated in cardiovascular disease, and that vascular disease risk factors are associated with AD and neurodegeneration, the same linear regression models were run for each CSF biomarker with the addition of peripheral vascular disease measures as covariates (BMI, blood pressure, total cholesterol, HDL cholesterol, and fasting glucose). Nonnormally distributed variables were natural log transformed.
Results
Participant characteristics
Participant characteristics are reported in Table 1. The Control group tended to be younger and had a higher proportion of females compared to the MCI and AD dementia groups. As expected, the APOE ε4 genotype was more prevalent in the MCI and AD dementia groups. There were no differences between groups with respect to cardiovascular disease risk factors including BMI, blood pressure, total cholesterol, HDL cholesterol, and fasting glucose.
Table 1.
Participant characteristics
| Sample characteristic | All | Control | MCI | AD dementia |
|---|---|---|---|---|
| N | 410 | 335 | 35 | 40 |
| Age (years) | 63.8 ± 9.0 | 61.9 ± 7.9 | 73.2 ± 8.5 | 71.9 ± 8.6 |
| Sex (% female) | 63 (258/410) | 69 (231/335) | 31 (11/35) | 40 (16/40) |
| APOE ε4 genotype, % positive (n) | 43 (178/410) | 39 (130/335) | 54 (19/35) | 73 (29/40) |
| AD parental history, % positive (n) | 58 (236/410) | 64 (214/335) | 31 (11/35) | 28 (11/40) |
| Ethnicity, % Caucasian (n) | 97 (396/410) | 96 (321/335) | 100 (35/35) | 100 (40/40) |
| Education (years) | 15.9 ± 2.6 | 16.1 ± 2.5 | 16.3 ± 2.7 | 14.7 ± 2.8 |
| Body mass index (BMI) | 28.3 ± 5.5 | 28.5 ± 5.7 | 28.2 ± 4.4 | 26.6 ± 3.9 |
| Systolic blood pressure (mmHg) | 128 ± 17 | 125 ± 16 | 130 ± 18 | 134 ± 16 |
| Diastolic blood pressure (mmHg) | 74 ± 9 | 75 ± 9 | 76 ± 10 | 75 ± 9 |
| Total cholesterol (mg/dl) | 195 ± 37 | 199 ± 46 | 184 ± 44 | 190 ± 37 |
| HDL cholesterol (mg/dl) | 62 ± 18 | 64 ± 39 | 57 ± 19 | 56 ± 14 |
| Fasting glucose (mg/dl) | 97 ± 20 | 99 ± 51 | 99 ± 15 | 100 ± 18 |
| CSF data | ||||
| TMAO (SIU) | 1.5 ± 1.7 | 1.3 ± 1.5 | 2.1 ± 1.4 | 2.8 ± 2.9 |
| p-tau (pg/ml) | 49.3 ± 22.0 | 45.1 ± 16.4 | 61.3 ± 36.2 | 74.7 ± 26.9 |
| p-tau/Aβ42 | 0.09 ± 0.07 | 0.07 ± 0.04 | 0.14 ± 0.11 | 0.21 + 0.09 |
| Aβ42/Aβ40 | 0.086 ± 0.024 | 0.090 ± 0.022 | 0.076 ± 0.028 | 0.062 ± 0.022 |
| t-tau (pg/ml) | 370 ± 228 | 310 ± 141 | 552 ± 338 | 722 ± 300 |
| NFL (pg/ml) | 841 ± 655 | 705 ± 489 | 1247 ± 863 | 1628 ± 935 |
| Neurogranin (pg/ml) | 352 ± 208 | 328 ± 182 | 430 ± 308 | 488 ± 244 |
All data presented as mean ± standard deviation unless otherwise indicated
MCI mild cognitive impairment, AD Alzheimer’s disease, APOE ε4 apolipoprotein E epsilon 4 allele, HDL high-density lipoprotein, CSF cerebrospinal fluid, TMAO trimethylamine N-oxide, SIU scaled intensity units, p-tau phosphorylated tau, Aβ beta-amyloid, t-tau total tau, NFL neurofilament light chain protein
CSF TMAO is elevated in individuals with MCI and AD dementia
CSF TMAO levels were elevated in individuals with AD dementia (β = 0.50, p < 0.0001) and MCI (β = 0.29, p < 0.05) compared to cognitively-unimpaired individuals (Fig. 1; Table 2), controlling for age, sex, and APOE ε4 genotype. Older age was associated with higher CSF TMAO (β = 0.02, p < 0.0001), but there were no main effects of sex or APOE ε4 genotype, and CSF TMAO levels did not differ between the MCI and AD groups.
Fig. 1.
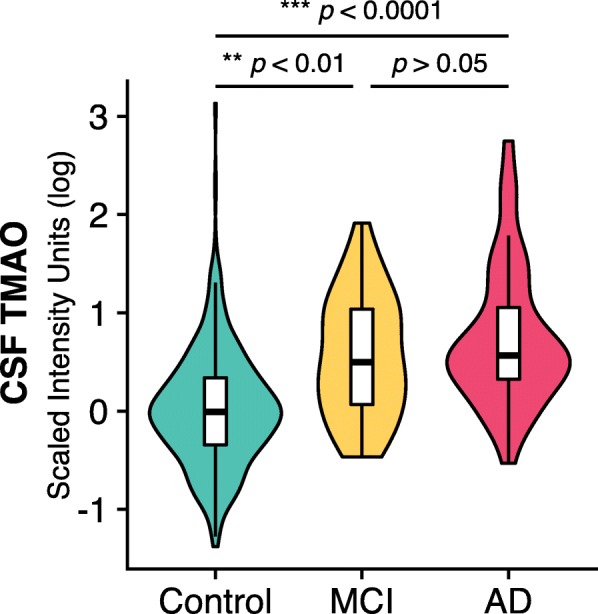
CSF TMAO levels are elevated in individuals with AD dementia and MCI compared to cognitively-unimpaired individuals, after controlling for age, sex, and APOE ε4 genotype. Data presented as violin plots (displaying scaled distribution of data for each group) with inset Tukey boxplots showing median, interquartile range (IQR), and 1.5 × IQR. AD Alzheimer’s disease, CSF cerebrospinal fluid, MCI mild cognitive impairment, TMAO trimethylamine N-oxide
Table 2.
Summary of multiple linear regression of age, sex, APOE ε4 genotype, and diagnosis on CSF TMAO level
| Variable | β (standard deviation) | t | p value |
|---|---|---|---|
| Age | 0.022 (0.004) | 6.1 | 5.6 × 10−8*** |
| Sex | −0.02 (0.06) | −0.03 | 0.75 |
| APOE ε4 genotype | −0.02 (0.06) | −0.3 | 0.77 |
| Diagnosis | |||
| Control vs MCI | 0.29 (0.11) | 2.6 | 0.01* |
| Control vs AD | 0.50 (0.11) | 4.7 | 4.1 × 10− 6*** |
| MCI vs AD | 0.21 (0.13) | 1.5 | 0.12 |
APOE ε4 apolipoprotein E epsilon 4 allele, MCI mild cognitive impairment, AD Alzheimer’s disease
Overall model statistics: F5,404 = 23.1, adjusted R2 = 0.21, p < 2.2 × 10− 16
*p < 0.05
***p < 0.001
CSF TMAO is associated with CSF biomarkers of AD and neuronal degeneration
With respect to CSF AD biomarkers, there was a significant positive relationship between CSF TMAO and p-tau (β = 0.09, p = 0.006; Fig. 2a) and p-tau/Aβ42 (β = 0.11, p = 0.013; Fig. 2b). No significant relationship between CSF TMAO and Aβ42/Aβ40 (β = − 0.003, p = 0.13; Fig. 2c) was observed. Additionally, CSF TMAO was positively associated with both CSF t-tau (β = 0.10, p = 0.01; Fig. 2d) and CSF NFL (β = 0.085, p = 0.007; Fig. 2e), but there was no relationship between CSF TMAO and CSF neurogranin (β = 0.004, p = 0.92; Fig. 2f). Additional file 1: Figure S1 shows the relationships between CSF TMAO and biomarkers colored by diagnostic group. Including peripheral cardiovascular disease risk factors as covariates did not change these associations (see Additional file 1: Table S1).
Fig. 2.

Relationship between CSF TMAO and CSF AD biomarkers (a–c) and biomarkers of neuronal degeneration (d–f). CSF TMAO is significantly positively correlated with phosphorylated tau (p-tau), p-tau/Aβ42, total tau (t-tau), and neurofilament light chain protein (NFL), after controlling for age and sex. Scatterplots show individual data points (n = 410) colored by 2D kernel density estimation. Hotter colors represent higher density; black line represents best linear fit between variables; shading represents 95% confidence interval of fit. CSF TMAO expressed as natural log-transformed scaled intensity units (SIU). Aβ, beta-amyloid CSF cerebrospinal fluid, TMAO trimethylamine N-oxide
Discussion
Understanding the contributions of the gut microbiota to neurological function and disease is an expanding area of research, particularly with respect to neurodegenerative disorders. A recent study [16], which used publicly available databases and a data-driven hypothesis-free computational approach to address the links between gut microbiota and AD, proposed that the gut microbial-derived metabolite TMAO is highly associated with AD. In the present study, we provide biochemical evidence revealing that CSF TMAO is higher in individuals with MCI and AD dementia, and elevated CSF TMAO is associated with both increased AD pathology (as measured by CSF biomarkers) as well as markers of neuronal degeneration.
Specifically, we found that CSF TMAO was associated with CSF p-tau as well as p-tau/Aβ42, but not Aβ42/Aβ40, potentially indicating that TMAO is more closely related to tau pathology than amyloid deposition alone. Additionally, we examined CSF biomarkers of neuronal degeneration, including t-tau, NFL, and neurogranin. CSF t-tau and NFL are thought to reflect axonal integrity [27] (with higher levels indicating greater axonal degeneration), while neurogranin is expressed in dendritic spines and reflects synaptic integrity [24]. We found that CSF TMAO was associated with increased CSF t-tau and NFL, but not neurogranin, suggesting that TMAO is related to axonal injury, but not dendritic degeneration. Taken together, our results suggest that while TMAO may not be a primary driver of amyloid production, it may impact vulnerable neurons and contribute to neurodegeneration.
As a metaorganismal metabolite, the production and accumulation of TMAO is dependent on both bacterial and host metabolism. The gene cluster required for bacterial enzymatic conversion of choline to TMA is distributed widely and discontinuously among gut bacterial taxa [9, 28, 29]. Thus, the presence of TMA-producing bacteria cannot be predicted from bacterial 16S rRNA gene sequencing studies. In the host, oxidation of TMA via FMO3 in the liver can also regulate TMAO levels [30]. Additionally, while both vegetarians and omnivores are able to convert choline to TMA [7, 31], long-term dietary habits can influence TMAO accumulation via changes in gut microbiota composition, which modulates TMA production potential.
TMAO is thought to contribute to disease pathogenesis through a variety of mechanisms including altering lipid and hormonal homeostasis, promoting platelet hyperreactivity [8], modulating cholesterol and sterol metabolism, decreasing reverse cholesterol transport [7], and inducing endothelial dysfunction through activation of the NLRP3 inflammasome [32]. In the brain, TMAO has been shown to induce neuronal senescence, increase oxidative stress, impair mitochondrial function, and inhibit mTOR signaling [11], all of which contribute to brain aging and cognitive impairment. Additionally, TMAO upregulates macrophage scavenger receptors and induces CD68 expression [7, 33], a cellular marker positively associated with dementia [34].
Vascular risk factors are increasingly recognized as important contributors to AD dementia [35], and cerebrovascular pathology commonly coexists with AD pathology at autopsy [36]. TMAO is causally linked with exacerbation of atherosclerosis in a genetically modified mouse model [6, 7], and the presence of intracranial atherosclerosis is an independent risk factor for dementia [37]. Thus, one potential mechanism by which TMAO may play a role in AD pathology is through the promotion of cerebrovascular disease. Of note, in the present study, cognitively-unimpaired, MCI, and AD individuals did not differ with respect to cardiovascular disease risk factors (BMI, blood pressure, cholesterol, and fasting glucose), suggesting that differences observed in TMAO between groups did not reflect underlying differences in cardiovascular disease status. Moreover, controlling for peripheral vascular disease risk factors did not change the associations between CSF TMAO and biomarkers of AD and neurodegeneration, suggesting that TMAO may have an impact independent of vascular effects. However, our study did not examine direct measures of central vascular disease, and future studies are needed to more fully examine the relationship between TMAO and cerebrovascular health.
TMAO is elevated in individuals with diabetes [38] and has been shown to promote insulin resistance in mice fed a high-fat diet [5]. Given that diabetes and insulin resistance are risk factors for developing AD [39, 40], elevated TMAO in the CNS may exacerbate central insulin resistance and AD pathogenesis. Finally, mitochondrial dysfunction and increased oxidative stress are ubiquitous features of AD pathology [41]; mice treated with dietary TMAO show increased brain aging with similar features [11], suggesting elevated TMAO may accelerate neurotoxicity and neurodegeneration in the context of AD pathology. However, additional work is needed to determine the potentially multifactorial pathways by which TMAO impacts the brain. Given that our results indicate that TMAO may be more relevant to neurodegenerative changes rather than initiation of Alzheimer’s-specific amyloid pathology, CSF TMAO levels should be investigated in other neurodegenerative disorders (e.g., Parkinson’s disease).
Conclusions
In this study, we demonstrate that the gut microbiota-derived metabolite TMAO is elevated in the CSF of individuals with MCI and AD dementia, and that levels of CSF TMAO are associated with CSF biomarkers of AD pathology and neuronal degeneration. These results provide additional evidence for an association between TMAO and AD, and further inform the role of gut microbiota in AD. Longitudinal studies are needed to determine whether elevated TMAO during mid-life predicts subsequent development or exacerbation of AD pathology. In this scenario, pharmacological agents designed to inhibit gut microbial TMAO production may be useful in slowing AD pathology [42].
Additional file
Methods. Additional details from Metabolon for methodology of CSF TMAO quantification. Figure S1. Relationship between CSF TMAO and CSF AD biomarkers (A–C) and biomarkers of neuronal degeneration (D–F). Table S1. Summary of multiple linear regressions testing relationships between CSF TMAO and CSF biomarkers of AD pathology and neurodegeneration. (PDF 895 kb)
Acknowledgements
The authors would like to thank Ruocheng Dong, Chuck Illingworth, Nancy Davenport-Sis, and the staff and researchers at the Wisconsin ADRC and the WRAP for their assistance in study organization, participant recruitment, and facilitating data availability. Most importantly, the authors extend thanks to the committed research participants who made this work possible.
Funding
This study was supported by NIH grants F30AG059346 (to NMV), R01AG037639 (to BBB), R01AG027161 (to SCJ), R01AG054047 (to CDE), and P50 AG033514 (to SA); a pilot award made possible by the Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS) (grant UL1TR002373); the Geriatric Research, Education, and Clinical Center of the William S. Middleton Memorial Veterans Hospital; the Swedish Alzheimer Foundation (Nos. AF-553101 and AF-646211); the Torsten Söderberg Foundation (to KB); the Research Council of Sweden (project 14002) (to KB); the Swedish Brain Foundation (project FO2015–0021) (to KB); LUA/ALF Västra Götalandsregionen Sweden (project ALFGBG-139671) (to KB); the European Research Council (No. 681712) (to HZ); Swedish State Support for Clinical Research (No. ALFGBG-441051) (to HZ); the Knut and Alice Wallenberg Foundation (Wallenberg Academy Fellow 2013) (to HZ); and the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison with funding from the Wisconsin Alumni Research Foundation (UW-Madison Microbiome Initiative).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- AD
Alzheimer’s disease
- ADRC
Alzheimer’s Disease Research Center
- APOE ε4
Apolipoprotein E epsilon 4 allele
- Aβ
Beta-amyloid
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- ECL
Electrochemiluminescence
- ELISA
Enzyme-linked immunosorbent assay
- FMO1
Flavin-containing monooxygenase 1
- FMO3
Flavin-containing monooxygenase 3
- MCI
Mild cognitive impairment
- mTOR
Mammalian target of rapamycin
- NFL
Neurofilament light chain protein
- NLRP3
NACHT, LRR and PYD domains-containing protein 3
- p-tau
Phosphorylated tau
- SIU
Scaled intensity units
- TMA
Trimethylamine
- TMAO
Trimethylamine N-oxide
- t-tau
Total tau
- UHPLC-MS
Ultrahigh performance liquid chromatography tandem mass spectrometry
- WRAP
Wisconsin Registry for Alzheimer’s Prevention
Authors’ contributions
NMV, KAR, BBB, FER, CDE, SCJ, CMC, and SA contributed to study concept and experimental design. NMV, KAR, BFD, CDE, SCJ, CMC, KB, HZ, BBB, and FER participated in data acquisition, analysis, or interpretation. NMV, KAR, BBB, and FER drafted the manuscript. BFD, CDE, SCJ, CMC, SA, KB, and HZ provided critical revision of the manuscript for important intellectual content. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The University of Wisconsin Health Science Institutional Review Board approved all study procedures, and all experiments were performed in accordance with relevant guidelines and regulations. All participants provided written informed consent to be involved in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Nicholas M. Vogt, Email: nvogt@wisc.edu
Kymberleigh A. Romano, Email: romanok@ccf.org
Burcu F. Darst, Email: bdarst@wisc.edu
Corinne D. Engelman, Email: corinne.engelman@wisc.edu
Sterling C. Johnson, Email: scj@medicine.wisc.edu
Cynthia M. Carlsson, Email: cmc@medicine.wisc.edu
Sanjay Asthana, Email: sa@medicine.wisc.edu.
Kaj Blennow, Email: kaj.blennow@neuro.gu.se.
Henrik Zetterberg, Email: henrik.zetterberg@clinchem.gu.se.
Barbara B. Bendlin, Email: bbb@medicine.wisc.edu
Federico E. Rey, Email: ferey@wisc.edu
References
- 1.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. 2013;110:4410–4415. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell. 2016;64:982–992. doi: 10.1016/j.molcel.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 5.Gao X, Liu X, Xu J, Xue C, Xue Y, Wang Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J Biosci Bioeng. 2014;118:476–481. doi: 10.1016/j.jbiosc.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Rio D, Zimetti F, Caffarra P, Tassotti M, Bernini F, Brighenti F, et al. The gut microbial metabolite trimethylamine-N-oxide is present in human cerebrospinal fluid. Nutrients. 2017;9:1–7. 10.3390/nu9101053. [DOI] [PMC free article] [PubMed]
- 11.Li D, Ke Y, Zhan R, Liu C, Zhao M, Zeng A, et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell. 2018;49:e12768–e12713. doi: 10.1111/acel.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scheltens P, Blennow K, Breteler MMB, De Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet. 2016;388:505–517. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 13.Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep. 2017;7:41802. doi: 10.1038/srep41802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhan X, Stamova B, Jin L-W, Decarli C, Phinney B, Sharp FR. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology. 2016;87:2324–2332. doi: 10.1212/WNL.0000000000003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer's disease. Sci Rep. 2017;7:13537. doi: 10.1038/s41598-017-13601-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu R, Wang Q. Towards understanding brain-gut-microbiome connections in Alzheimer's disease. BMC Syst Biol. 2016;10(Suppl 3):63. doi: 10.1186/s12918-016-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson SC, Koscik RL, Jonaitis EM, Clark LR, Mueller KD, Berman SE, et al. The Wisconsin Registry for Alzheimer's Prevention: a review of findings and current directions. Alzheimers Dement. 2018;10:130–142. doi: 10.1016/j.dadm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson SC, La Rue A, Hermann BP, Xu G, Koscik RL, Jonaitis EM, et al. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE ε3/ε3 genotype. Alzheimers Dement. 2011;7:456–465. doi: 10.1016/j.jalz.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer‘s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer‘s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Starks EJ, Patrick O’Grady J, Hoscheidt SM, Racine AM, Carlsson CM, Zetterberg H, et al. Insulin resistance is associated with higher cerebrospinal fluid tau levels in asymptomatic APOEɛ4 carriers. J Alzheimers Dis. 2015;46:525–533. doi: 10.3233/JAD-150072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewczuk P, Lelental N, Spitzer P, Maler JM, Kornhuber J. Amyloid-β 42/40 cerebrospinal fluid concentration ratio in the diagnostics of Alzheimer's disease: validation of two novel assays. J Alzheimers Dis. 2015;43:183–191. doi: 10.3233/JAD-140771. [DOI] [PubMed] [Google Scholar]
- 24.Blennow K, Zetterberg H. The past and the future of Alzheimer's disease CSF biomarkers—a journey toward validated biochemical tests covering the whole spectrum of molecular events. Front Neurosci. 2015;9:1–8. doi: 10.3389/fnins.2015.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Öhrfelt A, Andersson K, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement. 2015;11:1180–1190. doi: 10.1016/j.jalz.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 26.Evans AM, Bridgewater BR, Liu Q, Mitchell MW, Robinson RJ, Dai H, et al. High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high-throughput profiling metabolomics. Metabolomics. 2014;04.
- 27.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 28.Romano KA, Vivas EI, Amador-noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability. MBio. 2015;6:1–8. doi: 10.1128/mBio.02481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Del Campo A, Bodea S, Hamer HA, Marks JA, Haiser HJ, Turnbaugh PJ, et al. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. MBio. 2015;6. 10.1128/mBio.00042-15. [DOI] [PMC free article] [PubMed]
- 30.Zhu W, Buffa JA, Wang Z, Warrier M, Schugar R, Shih DM, et al. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J Thromb Haemost. 2018;16:1857–1872. doi: 10.1111/jth.14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obeid R, Awwad HM, Keller M, Geisel J. Trimethylamine-N-oxide and its biological variations in vegetarians. Eur J Nutr. 2017;56:2599–2609. doi: 10.1007/s00394-016-1295-9. [DOI] [PubMed] [Google Scholar]
- 32.Chen ML, Zhu XH, Ran L, Lang HD, Yi L, Mi MT. Trimethylamine-N-oxide induces vascular inflammation by activating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. J Am Heart Assoc. 2017;6:e006347. doi: 10.1161/JAHA.117.006347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc. 2016;5:e002767–e002713. doi: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minett T, Classey J, Matthews FE, Fahrenhold M, Taga M, Brayne C, et al. Microglial immunophenotype in dementia with Alzheimer's pathology. J Neuroinflammation. 2016;13:135. doi: 10.1186/s12974-016-0601-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneider JA, Bennett DA. Where vascular meets neurodegenerative disease. Stroke. 2010;41:S144–S146. doi: 10.1161/STROKEAHA.110.598326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, OBrien RJ. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann Neurol. 2010;68:231–240. doi: 10.1002/ana.22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, et al. Diabetes is associated with higher trimethylamine N-oxide plasma levels. Exp Clin Endocrinol Diabetes. 2016;124:251–256. doi: 10.1055/s-0035-1569330. [DOI] [PubMed] [Google Scholar]
- 39.Rawlings AM, Sharrett AR, Schneider ALC, Coresh J, Albert M, Couper D, et al. Diabetes in midlife and cognitive change over 20 years: a cohort study. Ann Intern Med. 2014;161:785–793. doi: 10.7326/M14-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.la Monte de SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis. 2005;7:45–61. doi: 10.3233/JAD-2005-7106. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Wang W, Li L, Perry G, Lee H-G, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta. 2014;1842:1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, et al. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat Med. 2018;24:1407–1417. doi: 10.1038/s41591-018-0128-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods. Additional details from Metabolon for methodology of CSF TMAO quantification. Figure S1. Relationship between CSF TMAO and CSF AD biomarkers (A–C) and biomarkers of neuronal degeneration (D–F). Table S1. Summary of multiple linear regressions testing relationships between CSF TMAO and CSF biomarkers of AD pathology and neurodegeneration. (PDF 895 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.