

Abstract
Individuals of the basidiomycete fungus Armillaria are well known for their ability to spread from woody substrate to substrate on the forest floor through the growth of rhizomorphs. Here, we made 248 collections of A. gallica in one locality in Michigan's Upper Peninsula. To identify individuals, we genotyped collections with molecular markers and somatic compatibility testing. We found several different individuals in proximity to one another, but one genetic individual stood out as exceptionally large, covering hundreds of tree root systems over approximately 75 hectares of the forest floor. Based on observed growth rates of the fungus, we estimate the minimum age of the large individual as 2500 years. With whole-genome sequencing and variant discovery, we also found that mutation had occurred within the somatic cells of the individual, reflecting its historical pattern of growth from a single point. The overall rate of mutation over the 90 mb genome, however, was extremely low. This same individual was first discovered in the late 1980s, but its full spatial extent and internal mutation dynamic was unknown at that time. The large individual of A. gallica has been remarkably resistant to genomic change as it has persisted in place.
Keywords: Armillaria, mutation, single-nucleotide polymorphism, genome stability
1. Introduction
In the late 1980s, Smith et al. [1] discovered a genetic individual (designated C1) of the fungus Armillaria gallica that extended over at least 37 hectares of forest floor and encompassed hundreds of tree root systems in Michigan's Upper Penninsula. Based on observed growth rates, C1 was estimated at the time to be at least 1500 years old with a mass of more than 105 kg. The conclusion was that C1 was among the largest and oldest organisms on earth, a remarkable claim given that Armillaria is essentially a microorganism existing largely as microscopic hyphae embedded in their substrates. Nearly three decades on, we returned to the site of C1 with the tools of whole-genome sequencing. Here, we show that C1 is still alive on its original site, but it is older and larger than originally estimated, at least 2500 years and 4 × 105 kg, respectively. For these revised estimates, we relied on direct measurements of rhizomorph growth rate and on a sampling of rhizomorphs in forest soil as reported previously [1]. We also show that mutation has occurred within the somatic cells of C1, reflecting its historical pattern of growth from a single point. The overall rate of mutation, however, was extremely low. On the spectrum of mutability in somatic cells, Armillaria occupies the extreme of stability, opposite the extreme of instability as typified by cancer [2].
Since the discovery of the C1 [1,3] individual, much has been learned about genomes, gene content and gene expression in Armillaria. A general picture of how individuals of this fungus become established and persist in woodlands has emerged. Because of its broad host range, capacity for enzyme secretion and prolific production of rhizomorphs, its unique organs of local dispersal, Armillaria appears to be adapted for persistent growth in local habitats [4]. Since its discovery, it has become clear that C1 is not unique in its size and age. Any temporally continuous forest could support large, old Armillaria individuals. Indeed, at least two other individuals of a sibling Armillaria species (A. solidipes) have been reported to occupy larger areas than C1 [5–7].
Despite the general information on colonization and persistence of Armillaria, little is known about the dynamics of recruitment of new individuals into populations and how a resident individual might compete with newcomers. We suspect that the size distributions of Armillaria individuals skews towards the very small, possibly microscopic, with most individuals going extinct quickly after birth and only a very small minority achieving large size. We also speculate that resident individuals do not necessarily compete with newcomers; the residents merely tend to reach newly available food first. These ideas remain to be tested.
Armillaria lives both as a saprophyte on dead wood and as a necrotrophic parasite, killing host plant tissues in advance of its growing mycelia by secreting hydrolytic enzymes and possibly virulence factors [8]. The range of potential host plants is broad, encompassing both angiosperm and conifer trees [9]. The large genome size of Armillaria relative to its sister taxa is due to increased gene content, particularly in plant cell wall degrading enzymes, rather than to the proliferation of transposable elements [8]. Individuals of Armillaria are established when mating occurs between haploid spores or their germlings [10,11]. After mating, a diploid mycelium is established rather than the dikaryotic mycelium typical of other basidiomycetes. From mycelium, rhizomorphs develop and push through the soil in much the same way as a plant root. Rhizomorphs function as foraging organs in locating and infecting new sources of food in dead wood or weakened trees. They can also persist in stasis in soil until new substrates become available. Gene expression in rhizomorph development shares much the same signature of multicellularity found in fruit-body development [8]. Rhizomorphs also share with vegetative mycelium a pattern of gene expression supporting enzyme production [8].
Against this background, the focus of this study was on the mutability of Armillaria—detecting genomic change in the spatial record left from the proliferation of somatic cells in space from a single zygote ancestor. Our motivation for this study goes back nearly three decades [1], but the means (the precision tools of whole-genome sequencing) only a few years. Our early expectation was that C1 would be highly heterogeneous due to mutation accumulation over its long life. Contrary to this expectation, we found here that C1 remains a cohesive and uniform unit due to an extremely low rate of mutation.
2. Methods
(a). Sample
In 2015–2017, we revisited the location of C1 and made 248 collections linked to GPS coordinates (electronic supplementary material, table S1). Collections were mostly pure cultures from rhizomorphs, but in the fall of 2015 and of 2016, samples also included fruit-body tissues which were used directly in DNA analysis. Subsequent methods generally followed Anderson & Catona [12].
(b). Culturing and DNA extraction
Rhizomorphs were cut into 2 cm segments and placed in 2.5% hypochlorite bleach for ten minutes to surface disinfect. The rhizomorphs were then trimmed to less than 1 cm and placed on 2% malt-extract agar medium. Liquid cultures were in 2% malt extract without agar. Mycelium was harvested, flash frozen in liquid nitrogen and then lyophilized. DNA was extracted with a CTAB method as described earlier [13].
(c). Somatic compatibility testing
To determine whether or not a collection belonged to C1, we tested for somatic compatibility (electronic supplementary material, table S1). Somatic compatibility distinguishes mycelia of the same individual which merge seamlessly in culture from mycelia of different individuals which react with a zone of cell death and pigmentation [14]. We noted one large grouping of 110 isolates, which was later confirmed to represent C1, and a smaller grouping of eight isolates, which was later confirmed to represent the C2 individual also described earlier [1,3]. In this study, both C1 and C2 were still centred on their same respective localities as previously reported [1,3].
(d). Initial genotyping
Polymorphic molecular markers were also used to test whether a new collection represented the C1 identity or not (electronic supplementary material, table S1). For example, in one segment of DNA, homozygosity for the absence of a MboI site has a frequency of 0.64 in the general population [15]. In addition, the 3′ end of the 25S rRNA gene is heterozygous in C1 for a length polymorphism, a genotype that has a frequency of 0.21 in the population. The frequency of the combined genotype of the C1 individual over the two DNA regions is 0.13. The combination of the two DNA regions and somatic compatibility drives the probability of a spurious match with a non-C1 individual much lower.
(e). Illumina sequencing
HiSeq Illumina sequencing was by paired-end with 155 bp reads at the Centre for Applied Genomics at the Hospital for Sick Children, Toronto. The Illumina sequences for collections of C1 are deposited as accession PRJNA393342 in the SRA at NCBI. A total of 15 strains were Illumina-sequenced, but only 14 were included in the search for variants listed in electronic supplementary material, table S2. One strain, no. Ar188, had lower than expected coverage, produced a higher than expected number of false calls, and was therefore excluded from this screen. Ar188 genotypes determined from Illumina sequences, however, had sufficient coverage to be included in figure 3.
Figure 3.

Variation at nine selected genomic sites mapped on all isolates of C1. Black dots represent Illumina-sequenced isolates. The sites listed below were identified from the Illumina-sequenced strains in electronic supplementary material, table S3. The sites were then PCR amplified and Sanger-sequenced in other C1 isolates (electronic supplementary material, table S4). Site variation is mapped to branches in the phylogenetic tree and spatial sectors encompassing derived genotypes are delineated with dashed lines. Site no. 1, scaffold 8, position 1 611 588; site no. 2, sc 24, pos 198 035; site no. 3, sc 85, pos 12 187; site no. 4 sc9, pos 1 017 767, site no. 5, sc 1, pos 1 829 026; site no. 6, sc 41, 509 147; site no. 7, sc 23, pos 681 486; site no. 8, sc 1, pos 1 019 568; site no. 9 sc 2, pos 3 135 125 (electronic supplementary material, table S3). The dots that are not encircled have only reference alleles at the nine sites assayed. Note that not all isolates were Sanger-sequenced over all nine sites. For certain sites, we sequenced enough isolates to encircle the variants by isolates carrying only reference alleles.
(f). Bioinformatics
The pipeline mapped the raw fasta files onto the reference genome, produced the pileup files from the resulting .bam files, and then filtered the pileup files to discover the variation among the Illumina-sequenced strains. The pipeline is described in detail in electronic supplementary material, Bioinformatics S1. We restricted our effort to the discovery of single-nucleotide polymorphisms (SNPs), which we were best positioned to detect and assay. We did not attempt to detect indels and rearrangements, although they probably exist. Provisional genome annotation was done with GeneMarkES under the default settings [16] and SNPs were annotated with snpEff [17].
3. Results and discussion
The C1 individual was first identified in the late 1980s by making spatially mapped collections of the fungus on the Michigan site and then genotyping them over multiple loci [1,3]. The C1 designation used here corresponds to Clone 1 in the original publication [1] and to ‘The Humungous Fungus' as named by the news media. All collections of C1 had the same multilocus genotype and shared an identical mitochondrial type. Other nearby individuals had different multilocus genotypes and mitochondrial types [3,15]. Another individual of lesser spatial extent, designated C2 here, was described in addition to C1.
In 2015–2017, we revisited the site of C1 and made 248 collections linked to GPS coordinates (electronic supplementary material, table S1). Figure 1 shows distribution of isolates representing C1, C2 and all other genotypes (which were excluded here from further analysis). Fifteen of these collections of C1 were Illumina-sequenced (electronic supplementary material, table S1) to approximately 100× average coverage (NCBI Accession: PRJNA393342). The sequence reads were initially aligned with a 98 kb mtDNA reference, which was derived from another individual of A. gallica from Ontario, Canada [12] (JGI Armillaria gallica 21-2 v1). C1, C2 and three additional individuals from the Michigan site each had a unique mtDNA genotype based on well-defined SNPs (electronic supplementary material, table S2). The recent Illumina-sequenced collections of C1 have the same mtDNA genotype as a living strain of C1 that was collected in the late 1980s. Similarly, the recent collections of C2 are linked to a C2 strain from the late 1980s by having the identical mtDNA genotype (electronic supplementary material, table S2).
Figure 1.

Map of all collections of Armillaria. Black dots, C1; open circles, C2; grey dots, all other individuals combined. The outline of the pine plantation and Paint Pond Road are included as alignment features. The present sample, which was larger and more broadly distributed than the previous [1], was designed to find the approximate borders of C1. The dashed line encompasses collections of C1 and includes some non-C1 individuals. Other individuals surround C1. The present sample reveals that C1 is larger and older than originally reported [1]. Based on previous growth rate measurements and estimation of fungal biomass, the revised estimates for minimum age and mass are 2500 years and 4 × 105 kg, respectively.
To test for the signature of mutation, we searched for variation in the nuclear genome of the Illumina-sequenced strains. After all filters were applied, 163 variants were found (electronic supplementary material, table S3). In this search, the key requirements for eliminating false calls were to require (a) that candidate sites had at most two alleles among the sequence reads, one reference and one alternate, (b) that one or more strains have 0% or 100% of the reads representing the alternate allele (a ‘purity’ criterion), and (c) that in the heterozygous sites the allele frequencies averaged 50% (30–70%).
Two general kinds of genetic changes were seen. First, for 151 cases, only single sites experienced the change and adjacent sites in the genome were not affected. We interpret all of these changes as point mutations resulting in a gain of heterozygosity from a homozygous ancestral state. The heterozygous strains after mutation had a nearly equal mix of reference and alternate alleles among the Illumina reads, while the homozygous strains had purely one allele. Second, there were regions in strains that were homozygous at a number of adjacent sites that were otherwise heterozygous in other strains; we interpret these events as loss of heterozygosity (LOH) in which a string of adjacent SNP sites that were formerly heterozygous all become homozygous simultaneously, as can happen with mitotic gene conversion. That only six LOH tracts were observed (electronic supplementary material, table S3), and that the preponderance of the original heterozygosity in the individual has been maintained, is remarkable given the potential for mitotic gene conversion and crossing over, which would lead to homozygosity over substantial portions of chromosome arms.
The predominant pattern of both point mutation and LOH was that genetic changes were observed as singletons, present in no more than one Illumina-sequenced strain (130 of 163 sites; electronic supplementary material, table S3). As expected for recent mutations mostly unaffected by selection [18], the majority (73%) of point mutations were due to C to T transitions (electronic supplementary material, table S3). Based on our provisional gene annotation, the distribution of effects for the 151 point mutations was 88 non-coding, 18 coding and synonymous and 45 coding and non-synonymous (electronic supplementary material, table S3), which is consistent with an overall pattern of neutrality, while not ruling out a fitness effect for any particular mutation.
On average, each strain had about 10 singleton changes that were not shared among the other strains. A minority of changes (33 sites), however, were shared among two or more of the strains. These shared changes were particularly informative because they reflect the historical growth pattern of the individual, while the singletons reflect a history of units of the individual persisting in place over time, presumably after the period of general expansion had occurred. This interpretation arises from the fact that individuals of Armillaria cannot exist in stasis over the long term. Existing food sources become exhausted and new sources of food become available, meaning that growth of the fungus is necessary even to remain in the same place.
To examine the spatial relationships of the mutations, we constructed a phylogeny of 14 Illumina-sequenced isolates of C1 using the variants in electronic supplementary material, table S3 as characters and maximum parsimony as the optimality criterion (figure 2). This approach was feasible because a branching filamentous fungus, whether growing in a Petri dish or on the forest floor, represents a microcosm for the bifurcating tree of life. The phylogeny shows a high degree of internal consistency and the nesting of the clades in space can be interpreted as reflecting past growth and colonization patterns.
Figure 2.
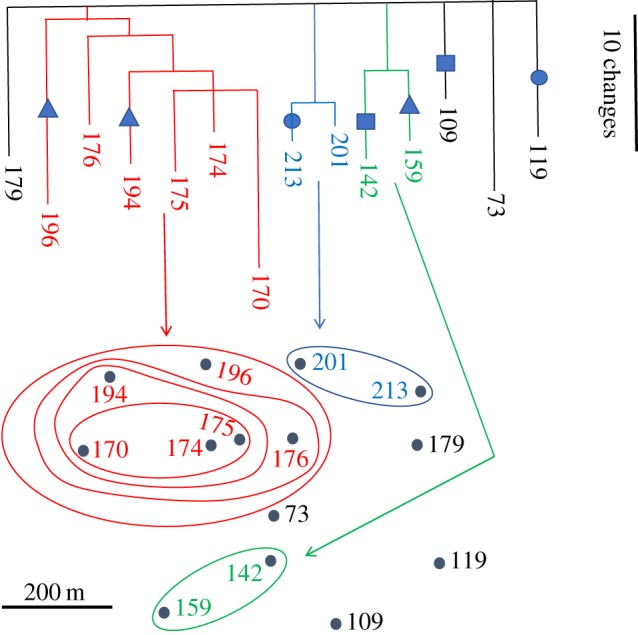
Phylogeny of Illumina-sequenced strains of A. gallica tied to spatial origin. The variants for this geophylogeny are listed in electronic supplementary material, table S3. Symbols represent sites at which changes occur in more than one branch of the tree. Circles, contig 12, position 174 630; squares, contig 61, position 336 513, triangles, contig 33, position 701 686 (electronic supplementary material, table S3).
Three of the 33 sites in the phylogeny, however, showed homoplasy; at these sites, changes occurred two or three times in different branches of the tree. All three site changes occurred in terminal branches of the phylogeny and led to homozygosity. Rather than being point mutations (among the 151 classified as such above), these three changes may actually represent LOH events in which the tract affected is too small to encompass adjacent sites. These three examples of homoplasy do not allow conclusions about the underlying cause, but three equally remote possibilities emerge. The homoplasy could be to recurrent change at the sites in question. With 100 mb in the genome, however, it is improbable that any change would be seen twice, unless there was a large fitness gain associated (and note that two of the three sites in question are non-coding, electronic supplementary material, table S3). A second explanation is that each change happened only once and then somatic recombinants became spatially separated within the individual. The potential for somatic recombination in fungi has ample precedent [19,20]. A third possibility is that each change happened only once, but each derived genotype was somehow maintained at low frequency with subsequent, independent fixation in some (but not all) lineages.
Next, for an independent test of the phylogenetic pattern among Illumina-sequenced strains, we tested nine sites in electronic supplementary material, table S3 among the additional isolates of C1 that were not Illumina-sequenced. Once again, the changes showed a nesting pattern in space, with the spatially discrete sectors reflecting their mutational history (figure 3). A plausible branching pattern for the mycelium would reflect the phylogeny of nuclei over the growth by C1 from its point of origin.
Despite our ability to identify and map mutant alleles in the Armillaria mycelium, estimating the rate of mutation is problematic because we do not know the number of cell divisions intervening between any two isolates. We can estimate the number of genetic differences among the Illumina-sequenced isolates experimentally, but the number of intervening cell divisions is inseparable from the mutation rate (no. differences = no. cell divisions times the mutation rate). Mutation rates, however, have been estimated in other organisms [21] and we can therefore assume an average mutation rate on the low end of the spectrum, 10−10 per base per cell replication (note that mutation rate varies across the genome and among individuals [22]). With a genome of 100 mb, one mutation is expected every 100 cell divisions in a haploid genome and every 50 cell divisions in a diploid genome such as that of A. gallica. The pairs of strains were separated by an average of ca 20 mutations, including those spatially separated by 1 km. In order to produce this number of mutations, the number of cell divisions would be restricted to roughly 1000 cell divisions over 1 km, or only one division for every 1 m of growth. If we assume a higher mutation rate, then the value of one cell division per metre of growth must increase even further. For example, a doubling of mutation rate to 2 × 1010, would restrict the number of cell divisions to 500, or only one division for every 2 m of growth over the 1 km span. Such a dearth of cell division is hard to reconcile with the length of cells within fungal hyphae, on the order of tens to hundreds of micrometres.
How might individuals of Armillaria protect themselves from mutation during cell division and growth? We see four possibilities, which are not mutually exclusive. First, the rhizomorph tip may represent a mixed pool of cells such that the fixation probability of any new mutant allele would be reduced by the effective number of cells in that pool. In the rhizomorph lineage, there would be no bottleneck to a single cell and fixation of the genotype would be necessary for the change to be detected by our methods. We speculate that the fold reduction in mutation rate could range up into the hundreds, perhaps into the thousands. Second, the tips of rhizomorphs, which represent the inoculum potential for colonization, may remain relatively quiescent with respect to cell division, much like the apical meristem of plant roots [12,23] and germline cells in mammals [24]. The rhizomorph tip, however, may be propelled forward by cell division and elongation behind the tip. In this way, the rhizomorph tip may minimize cell division even as it moves through its substrate. There is also precedent for the avoidance of cell division in the shoots of plants in which axial meristems are derived from apical meristems with remarkably few intervening cell divisions [25]. A third possibility is that repair processes may have been driven to higher efficiency by natural selection up to the point where genetic drift negates any additional diminishing fitness benefit [26,27]. The fourth possibility is that the distribution of DNA strands after replication is asymmetric. Cells perpetuating the lineage might tend to receive old DNA while cells committed solely to local development and not to perpetuating the lineage would receive new DNA [28]. In Armillaria, this would mean that cells in the rhizomorph tip would retain the old DNA, whereas the subtending cells (committed to local, dead-end development) would receive the new DNA. In this way, the rhizomorph tips perpetuating the lineage would retain fewer mutations than cells committed to local differentiation. The extent to which each of these mechanisms may contribute to stability remains to be determined.
Another factor lowering overall mutation rate may be due to the nature of the environment in which Armillaria exists. One common mutagenic agent, UV radiation, is low in the forest soils and woody substrates in which Armillaria lives and damage such as pyrimidine dimer formation may be lower than in other environments.
4. Conclusion
Here, we followed clonal evolution within cell lineages of a single fungal individual of A. gallica in a spatial context. This follows an earlier analysis of a much smaller individual of the same species in Ontario [12]. Our picture of clonal evolution in individuals of Armillaria closely parallels that in cancer progression within single individuals [29–32]. Cancer progression, however, is accompanied by extreme genomic instability [2], not necessarily due to loss of function in DNA repair processes, but rather to loss of control over DNA replication. The rate of replication increases to the extent that fidelity suffers and DNA damage accumulates rapidly. Evolution in cancer occurs on a time span shorter than the lifespan of the individual affected. Evolution occurs similarly in Armillaria individuals, but over a span of centuries and millennia, and is characterized by extreme genomic stability. The genomic stability of Armillaria and the underlying mechanisms allowing such stability may provide a useful counterpoint to cancer.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Data accessibility
The Illumina sequences for 15 collections of C1 are deposited as accession PRJNA393342 in the SRA at NCBI. All other data are in electronic supplementary information as tables S1–S4. The gene annotation file is available in the Dryad Digital Repository at: https://doi.org/10.5061/dryad.dv03078 [33].
Authors' contributions
J.N.B., J.B.A. and M.L.S. planned the study and did the field collecting. D.K. did the laboratory culturing and somatic compatibility tests, DNA extractions, PCR, Sanger sequencing and preparation of DNA for Illumina sequencing. N.R. and H.W. did the bioinformatic analysis of the Illumina sequences and subsequent nuclear SNP discovery. J.B.A. did the analysis of mtDNA variation. J.N.B., J.B.A. and M.L.S. wrote the paper with input from D.K., N.R. and H.W.
Competing interests
The authors have no competing financial interests.
Funding
J.B.A, M.L.S. and N.R. were supported by Discovery Grants from the Natural Sciences and Engineering Research Council of Canada.
References
- 1.Smith ML, Bruhn JN, Anderson JB. 1992. The fungus Armillaria bulbosa is among the largest and oldest organisms. Nature 356, 428–431. ( 10.1038/356428a0) [DOI] [Google Scholar]
- 2.Negrini S, Gorgoulis VG, Halazonetis TD. 2010. Genomic instability: an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228. ( 10.1038/nrm2858) [DOI] [PubMed] [Google Scholar]
- 3.Smith ML, Duchesne LC, Bruhn JN, Anderson JB. 1990. Mitochondrial genetics in a natural population of the plant pathogen Armillaria. Genetics 126, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sipos G, Anderson J, Nagy L. 2018. Armillaria. Curr. Biol. 28, R297–R298. ( 10.1016/j.cub.2018.01.026) [DOI] [PubMed] [Google Scholar]
- 5.Shaw CG, Roth LF. 1976. Persistence and distribution of a clone of Armillaria mellea in a Ponderosa Pine forest. Phytopathology 66, 1210–1213. ( 10.1094/Phyto-66-1210) [DOI] [Google Scholar]
- 6.Anderson J, Ullrich R, Roth L, Filip G. 1979. Genetic identification of clones of Armillaria mellea in coniferous forests in Washington. Phytopathology 69, 1109–1111. ( 10.1094/Phyto-69-1109) [DOI] [Google Scholar]
- 7.Ferguson B, Dreisbach T, Parks C, Filip G, Schmitt C. 2003. Coarse-scale population structure of pathogenic Armillaria species in a mixed-conifer forest in the Blue Mountains of northeast Oregon. Can. J. For. Res. 33, 612–623. ( 10.1139/x03-065) [DOI] [Google Scholar]
- 8.Sipos G, et al. 2018. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat. Ecol. Evol. 1, 1931–1941. ( 10.1038/s41559-017-0347-8) [DOI] [PubMed] [Google Scholar]
- 9.Shaw CG, Kile GA. 1991. Armillaria root disease. Washington, DC: United States Department of Agriculture Forest Service. [Google Scholar]
- 10.Korhonen K, Hintikka V. 1974. Cytological evidence for somatic diploidization in dikaryotic cells of Armillariella mellea. Arch. Microbiol. 95, 187–192. ( 10.1007/BF02451760) [DOI] [Google Scholar]
- 11.Ullrich RC, Anderson JB. 1978. Sex and diploidy in Armillaria mellea. Exp. Mycol. 2, 119–129. ( 10.1016/S0147-5975(78)80025-5) [DOI] [Google Scholar]
- 12.Anderson JB, Catona S. 2014. Genomewide mutation dynamic within a long-lived individual of Armillaria gallica. Mycologia 106, 642–648. ( 10.3852/13-367) [DOI] [PubMed] [Google Scholar]
- 13.Smith ML, Bruhn JN, Anderson JB. 1994. Relatedness and spatial distribution of Armillaria genets infecting red pine seedlings. Phytopathology 84, 822–829. ( 10.1094/Phyto-84-822) [DOI] [Google Scholar]
- 14.Worrall JJ. 1997. Somatic incompatibility in basidiomycetes. Mycologia 89, 24–36. ( 10.1080/00275514.1997.12026751) [DOI] [Google Scholar]
- 15.Saville BJ, Yoell H, Anderson JB. 1996. Genetic exchange and recombination in populations of the root-infecting fungus Armillaria gallica. Mol. Ecol. 5, 485–497. ( 10.1111/j.1365-294X.1996.tb00341.x) [DOI] [PubMed] [Google Scholar]
- 16.Borodovsky M, Lomsadze A. 2011. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinform. 35, 4.6.1–4.6.10. ( 10.1002/0471250953.bi0406s35) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cingolani P, Platts A, Wangle L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. ( 10.4161/fly.19695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trivedi J, Lachapelle J, Vanderwolf KJ, Misra V, Willis CKR, Ratcliffe JM, Ness RW, Anderson JB, Kohn LM. 2017. Fungus causing white-nose syndrome in bats accumulates genetic variability in North America with no sign of recombination. mSphere 2, e00271-17 ( 10.1128/mSphereDirect.00271-17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark TA, Anderson JB. 2004. Dikaryons of the basidiomycete fungus Schizophyllum commune: evolution in long-term culture. Genetics 167, 1663–1675. ( 10.1534/genetics.104.027235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho DB, Smith ML, Anderson JB. 1995. Genetic exchange between diploid and haploid mycelia of Armillaria gallica. Mycol. Res. 99, 641–647. ( 10.1016/S0953-7562(09)80520-0) [DOI] [Google Scholar]
- 21.Lynch M, Ackerman MS, Gout JF, Long H, Sung W, Thomas WK, Foster PL. 2016. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 17, 704–714. ( 10.1038/nrg.2016.104) [DOI] [PubMed] [Google Scholar]
- 22.Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD. 2015. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res. 25, 1739–1749. ( 10.1101/gr.191494.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aanen D. 2014. How a long-lived fungus keeps mutations in check. Science 346, 922–923. ( 10.1126/science.1261401) [DOI] [PubMed] [Google Scholar]
- 24.Milholland B, Dong X, Zhang L, Hao XX, Suh Y, Vijg J. 2017. Differences between germline and somatic mutation rates in humans and mice. Nat. Commun. 8, 15183 ( 10.1038/ncomms15183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burian A, de Reuille PB, Kuhlemeier C. 2016. Patterns of stem cell divisions contribute to plant longevity. Curr. Biol. 26, 1385–1394. ( 10.1016/j.cub.2016.03.067) [DOI] [PubMed] [Google Scholar]
- 26.Lynch M. 2010. Evolution of the mutation rate. Trends Genet. 26, 345–352. ( 10.1016/j.tig.2010.05.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch M. 2011. The lower bound to the evolution of mutation rates. Genome Biol. Evol. 3, 1107–1118. ( 10.1093/gbe/evr066) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aanen DK, Debets AJM. 2018. Mutation-rate plasticity and the germline of unicells. BioRxiv. ( 10.1101/307371) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown D, et al. 2017. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat. Commun. 8, 14944 ( 10.1038/ncomms14944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crespi B, Summers K. 2005. Evolutionary biology of cancer. Trends Ecol. Evol. 20, 545–552. ( 10.1016/j.tree.2005.07.007) [DOI] [PubMed] [Google Scholar]
- 31.Navin NE, Hicks J. 2010. Tracing the tumor lineage. Mol. Oncol. 4, 267–283. ( 10.1016/j.molonc.2010.04.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sprouffske K, Merlo LMF, Gerrish PJ, Maley CC, Sniegowski PD. 2012. Cancer in light of experimental evolution. Curr. Biol. 22, R762–R771. ( 10.1016/j.cub.2012.06.065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson JB, Bruhn JN, Kasimer D, Wang H, Rodrigue N, Smith ML. 2018. Data from: Clonal evolution and genome stability in a 2500-year-old fungal individual Dryad Digital Repository. ( 10.5061/dryad.dv03078) [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Anderson JB, Bruhn JN, Kasimer D, Wang H, Rodrigue N, Smith ML. 2018. Data from: Clonal evolution and genome stability in a 2500-year-old fungal individual Dryad Digital Repository. ( 10.5061/dryad.dv03078) [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
The Illumina sequences for 15 collections of C1 are deposited as accession PRJNA393342 in the SRA at NCBI. All other data are in electronic supplementary information as tables S1–S4. The gene annotation file is available in the Dryad Digital Repository at: https://doi.org/10.5061/dryad.dv03078 [33].