

Hexuronate catabolic pathways and their transcriptional networks are highly variable among different bacteria. We identified novel transcriptional regulators that control the hexuronate and aldarate utilization genes in four families of proteobacteria. By regulon reconstruction and genome context analysis we identified several novel components of the common hexuronate/aldarate utilization pathways, including novel uptake transporters and catabolic enzymes. Two novel families of lactonases involved in the oxidative pathway of hexuronate catabolism were characterized. Novel transcriptional regulons were validated via in vitro binding assays and gene expression studies with Polaromonas and Ralstonia species. The reconstructed catabolic pathways are interconnected with each other metabolically and coregulated via the GguR regulons in proteobacteria.
KEYWORDS: proteobacteria, comparative genomics, hexuronate metabolism, lactonase, regulon reconstruction, transcriptional regulation
ABSTRACT
We used comparative genomics to reconstruct d-galacturonic and d-glucuronic acid catabolic pathways and associated transcriptional regulons involving the tripartite ATP-independent periplasmic (TRAP) family transporters that bind hexuronates in proteobacteria. The reconstructed catabolic network involves novel transcription factors, catabolic enzymes, and transporters for utilization of both hexuronates and aldarates (d-glucarate and meso-galactarate). The reconstructed regulons for a novel GntR family transcription factor, GguR, include the majority of hexuronate/aldarate utilization genes in 47 species from the Burkholderiaceae, Comamonadaceae, Halomonadaceae, and Pseudomonadaceae families. GudR, GulR, and UdhR are additional local regulators of some hexuronate/aldarate utilization genes in some of the above-mentioned organisms. The predicted DNA binding motifs of GguR and GudR regulators from Ralstonia pickettii and Polaromonas were validated by in vitro binding assays. Genes from the GulR- and GguR-controlled loci were differentially expressed in R. pickettii grown on hexuronates and aldarates. By a combination of bioinformatics and experimental techniques we identified a novel variant of the oxidative pathway for hexuronate utilization, including two previously uncharacterized subfamilies of lactone hydrolases (UxuL and UxuF). The genomic context of respective genes and reconstruction of associated pathways suggest that both enzymes catalyze the conversion of d-galactaro- and d-glucaro-1,5-lactones to the ring-opened aldarates. The activities of the purified recombinant enzymes, UxuL and UxuF, from four proteobacterial species were directly confirmed and kinetically characterized. The inferred novel aldarate-specific transporter from the tripartite tricarboxylate transporter (TTT) family transporter TctC was confirmed to bind d-glucarate in vitro. This study expands our knowledge of bacterial carbohydrate catabolic pathways by identifying novel families of catabolic enzymes, transcriptional regulators, and transporters.
IMPORTANCE Hexuronate catabolic pathways and their transcriptional networks are highly variable among different bacteria. We identified novel transcriptional regulators that control the hexuronate and aldarate utilization genes in four families of proteobacteria. By regulon reconstruction and genome context analysis we identified several novel components of the common hexuronate/aldarate utilization pathways, including novel uptake transporters and catabolic enzymes. Two novel families of lactonases involved in the oxidative pathway of hexuronate catabolism were characterized. Novel transcriptional regulons were validated via in vitro binding assays and gene expression studies with Polaromonas and Ralstonia species. The reconstructed catabolic pathways are interconnected with each other metabolically and coregulated via the GguR regulons in proteobacteria.
INTRODUCTION
Hexuronic acids d-galacturonic (d-GalA) and d-glucuronic acid (d-GlcA) are commonly found in all three domains of life. d-GalA is the main sugar residue of pectins in the primary cell wall of plants (1, 2). d-GlcA is common in glycosaminoglycan chains of proteoglycans in animals and glucuronans in algae. Both hexuronic acids also can be found in hemicellulose in the cell walls of plants. Many bacteria can assimilate hexuronates using either one of two known catabolic pathways (Fig. 1). In the isomerase pathway of hexuronate utilization that was described for many models, including Escherichia coli and Bacillus subtilis (3–6), the 2-keto-3-deoxy-d-gluconate (2K3DG) intermediate is produced using hexuronate isomerase (UxaC), d-mannonate/d-altronate hydrolases (UxuA/UxaA), and two cognate oxidoreductases (UxuB/UxaB). 2K3DG is converted to d-glyceraldehyde-3-phosphate and pyruvate by the subsequent action of the KdgK kinase and the KdgA aldolase. The isomerase pathway of hexuronate catabolism is conserved in other gammaproteobacteria, including the Enterobacteriales, Vibrionales, and Pasteurellales (3).
FIG 1.

Reconstruction of hexuronate and aldarate utilization metabolic pathways and regulons in proteobacteria. The oxidative pathway for utilization of d-glucoronate and d-galacturonate (hexuronates) described in this work in the Burkholderiaceae, Comamonadaceae, Halomonadaceae, and Pseudomonadaceae feeds the downstream catabolic pathway for d-glucarate and meso-galactarate (aldarates). Two alternative aldarate catabolic pathways were described for E. coli (GarR-GarL) and Acinetobacter baylyi (KdgD and KgsD). The alternative hexuronate catabolic pathway known in E. coli and several other lineages of proteobacteria is shown in the gray box. Four novel TFs identified in this work as cognate regulators of the hexuronate and aldarate utilization pathway genes are shown in colored ovals, and their predicted target genes are marked by circles of corresponding colors. Numbers in circles show the number of genomes where the target gene is preceded by a candidate binding site for GguR (black), GudR (blue), UdhR (purple), and GulR (green) regulators. Known and predicted transporters for hexuronates and aldarates are shown in the pink box. New transporters and enzymes identified in this work are marked with an asterisk.
In the oxidative pathway of hexuronate utilization, which was characterized in Agrobacterium tumefaciens, d-GlcA and d-GalA are first oxidized to produce d-glucaro- or d-galactaro-1,5-lactone by hexuronate dehydrogenase (Udh) (7–9). The galactarolactone is converted to 5-keto-4-deoxy-d-galactarate (KDG) via the subsequent action of δ-lactone isomerase (Gli) and γ-lactone cycloisomerase (Gci) (10, 11); glucarolactone presumably has a similar fate, although Gli is inactive with glucarolactone as a substrate (data not shown). In the last two steps of this pathway, KDG dehydratase (KdgD) and α-ketoglutaric semialdehyde dehydrogenase (KgsD) produce α-ketoglutarate as a final product of the oxidative pathway (12). It is important to note that KDG is a common intermediate of the catabolic pathway for utilization of two aldaric acids (aldarates), namely, d-glucarate (d-GlcAA) and meso-galactarate (meso-GalAA), which are converted to KDG via committed dehydratases, GudD and GarD, respectively (Fig. 1). In E. coli, KDG produced from aldarate degradation is further converted to glycerate-2-phosphate (glycerate-2P) by subsequent action of GarL aldolase and GarR reductase (13). An alternative d-GlcAA catabolic pathway involving KdgD and KgsD enzymes operates in Acinetobacter baylyi (14).
Transcriptional regulators and uptake transporters are important components of carbohydrate utilization networks (15, 16). Sugar-responsive transcription factors (TFs) mediate specific induction of a catabolic pathway. In E. coli and other enterobacteria, hexuronate metabolic genes are controlled by two related TFs from the GntR family, ExuR and UxuR, that respond to d-GalA and d-fructuronate, respectively (3, 17). Hexuronates are taken up by enterobacteria via the ExuT transporter from the major facilitator superfamily (MFS) (18, 19). Similar ExuT-like hexuronate permeases operate in bacteria from other taxonomic groups, including Bacillus subtilis and Ralstonia solanacearum (5, 20). Our previous large-scale screening of 158 solute-binding proteins (SBPs) from the tripartite ATP-independent periplasmic (TRAP) transporter family using differential scanning fluorimetry (DSF) identified a large group of hexuronate-specific TRAP transporters (termed UxuPQM) in various lineages of proteobacteria, including Agrobacterium, Bradyrhizobium, Burkholderia, Polaromonas, and Pseudomonas (21). More recently, through gene deletion and growth assays, the essential role of the UxuPQM-like system, loci Atu3135 to Atu3137, in d-GalA uptake was verified in A. tumefaciens (22). Uptake of aldarates in E. coli is mediated by two paralogous transporters, GudP and GarP (23).
In this work, we performed comparative genomics reconstruction of hexuronate/aldarate regulons and pathways in beta- and gammaproteobacteria that have incomplete versions of the oxidative hexuronate catabolic pathways. The Pseudomonadaceae, Burkholderiaceae, and Comamonadaceae genomes include orthologues of gene for the Udh dehydrogenase but lack orthologues of the genes for the Gli and Gci enzymes required for further catabolism of the δ-lactone products. We combined the bioinformatics reconstructions of four novel transcriptional regulons for hexuronate/aldarate pathway genes with in vitro characterization of two representative regulators from Polaromonas and Ralstonia species and in vivo gene expression studies in the Ralstonia species. Two previously uncharacterized subfamilies of lactone hydrolases predicted via metabolic pathway reconstruction were confirmed to catalyze the direct conversion of the δ-lactone products of the uronate dehydrogenase reaction to the ring-opened diacids (aldarates). The inferred novel variant of the oxidative hexuronate utilization pathway merges with the downstream aldarate catabolic pathway, and the two pathway genes are coregulated via the novel KDG-responsive regulator GguR. Finally, we assigned the role of the tripartite tricarboxylate transporter (TTT) family transporter TctC in aldarate uptake and confirmed its specificity in vitro. The functional diversity of hexuronate/aldarate catabolic pathways in various taxonomic groups of proteobacteria is discussed.
RESULTS
Comparative genomics analysis of metabolic pathways and regulons. (i) Chromosomal colocalization of hexuronate utilization genes.
We analyzed genomic context of four novel TRAP family transporters encoded by the uxuPQM genes in gamma- and betaproteobacteria that were previously shown to bind d-GalA and d-GlcA (21). In Chromohalobacter salexigens, a gammaproteobacterium from the Halomonadaceae family, the uxuPQM-containing operon encodes known enzymes from the oxidative pathway of hexuronate catabolism, Udh, Gci, and Gli, involved in the biochemical conversion of hexuronates to KDG (Fig. 2). In Pseudomonas putida, a representative of the Pseudomonadaceae family of gammaproteobacteria, uxuPQM is likely cotranscribed with genes encoding the uronate dehydrogenase Udh, a putative lactonase from the senescence marker protein 30 (SMP-30)/gluconolactonase/luciferin-regenerating enzyme (LRE)-like region (SGL) family (PF08450 in the Pfam database) and a putative aldose 1-epimerase (AldE) from the PF01263 family. In Polaromonas sp. strain JS666, a betaproteobacterium from the Comamonadaceae family, a large uxuPQM-containing gene cluster encodes orthologues of Udh, AldE, and UxuL, three d-galactarate catabolic enzymes (GarD, GarL, and GarR), as well as the substrate-binding component of a hypothetical TTT family transporter (named TctC). In Burkholderia ambifaria, a member of the Burkholderiaceae family of betaproteobacteria, uxuPQM genes form a candidate operon with a putative enzyme from the 7-bladed beta-propeller lactonase family (PF10282). The reaction products of Udh from the oxidation of d-GalA and d-GlcA are d-galactaro- and d-glucaro-1,5-lactone, respectively (24). We propose that two previously uncharacterized subfamilies of lactonases from the PF08450 and PF10282 families (named UxuL and UxuF) hydrolyze 1,5-lactones formed during the oxidative pathway of hexuronate utilization. Thus, the proposed novel lactonases fill a gap between the upstream hexuronate oxidation process and the downstream pathway for aldaric acid utilization (Fig. 1).
FIG 2.


Genomic contents of the GguR, GudR, GulR, and UdhR regulons in four lineages of beta- and gammaproteobacteria: Comamonadaceae (A), Burkholderiaceae (B), Pseudomonadaceae (C), and Halomonadaceae (D). Genes (shown by rectangles) with the same functional roles are marked in matching colors. The first genes in putative operons are marked with a small arrow inside the gene. Loci separated by a spacer are not clustered on the chromosome. Transcriptional regulators are in black. Candidate regulator binding sites are shown by colored circles corresponding to GguR, GudR, GulR, and UdhR. Sequence logos representing the consensus binding-site motifs are shown at the bottom.
To further confirm this hypothesis, we analyzed the genomic context of the uxuL and uxuF orthologues in related proteobacteria (see Fig. S1 in the supplemental material). In most Pseudomonadaceae and Comamonadaceae genomes, as well as in two Ralstonia spp., uxuL is clustered with the uronate dehydrogenase gene, udh, and hexuronate transporter genes, uxuPQM or exuT. In two Acidovorax species, Burkholderia, Polaromonas, and Ralstonia species, and Variovorax paradoxus, uxuL is colocalized with the meso-GalAA dehydratase gene garD. The uxuF orthologue in Hylemonella gracilis is colocalized with d-GlcAA and meso-GalAA dehydratase genes gudD and garD, whereas in Variovorax and Acidovorax spp., it is clustered with two paralogues of tctC transporter genes. In most Burkholderia spp., uxuF is clustered with a novel MFS family transporter gene (named gguT; see below), whereas in Burkholderia phymatum, it belongs to the garLR-uxuF-gudD-kdgD operon, involved in d-GlcAA catabolism. The gene neighborhood analysis suggests that both novel lactonases are involved in hexuronate/aldarate metabolism. We further complemented this analysis by genomic reconstruction of potential transcriptional regulons involving hexuronate and/or aldarate utilization genes.
(ii) Novel glucarate and galactarate utilization regulator GguR.
The hexuronate/aldarate utilization gene loci in many species from the Burkholderiaceae, Comamonadaceae, Halomonadaceae, and Pseudomonadaceae families encode a novel GntR family transcriptional regulator, which was named GguR (Fig. 2). The identified GguR regulators are distantly related to the UxuR/ExuR regulators of hexuronate metabolism from E. coli (∼30% identity). The presence of the gguR regulator in the genomes of four analyzed families of proteobacteria correlate with the presence of genes encoding aldarate catabolic enzymes, including GarD, GudD, KdgD, and KgsD (Table S1). GguR regulators are present in two copies in several genomes from the Pseudomonas, Burkholderia, and Halomonas lineages. To get insight into a possible evolutionary history of the duplicates, we constructed a phylogenetic tree for all GguR proteins analyzed in this study (Fig. S2). GguR paralogues found in all Pseudomonas spp. except P. mendocina and P. stutzeri are in two diverged branches of the tree, suggesting their early emergence via lineage-specific gene duplication. Closely related GguR paralogues in Halomonas lutea, B. phymatum, and Burkholderia glumae likely appeared via at least two species-specific duplications.
(iii) Identification of GguR-binding sites and regulons.
To identify GguR-binding motifs and reconstructed regulons, we applied a comparative genomics approach to each analyzed taxonomic group of proteobacteria. Briefly, noncoding upstream regions of gguR-containing operons from closely related species were collected and used as inputs for the DNA motif discovery tool. A highly conserved palindromic DNA motif with consensus sequence GTTGT(A/C)N(G/T)ACAAC was identified as a candidate GguR-binding motif in the Burkholderiaceae, Comamonadaceae, and Pseudomonadaceae families, whereas a modified GguR motif consensus sequence was identified in the Halomonadaceae family (Fig. S2). The metabolic and genomic contexts of the reconstructed GguR regulons in the four taxonomic groups of beta- and gammaproteobacteria (47 species) are summarized in Fig. 1 and 2, while the detailed information on predicted binding sites is available in Table S1. The core conserved part of the GguR regulons includes the garD, gudD, kdgD, kgsD, udh, uxuPQM, and gguR genes, which are preceded by candidate GguR-binding sites in all four studied lineages. The first four genes involved in the aldarate catabolic pathway are members of the GguR regulons in the majority of the analyzed genomes. In contrast, the hexuronate utilization genes udh and uxuPQM are less frequent members of the GguR regulons due to their absence in some genomes. The novel lactonase genes, uxuL and uxuF, belong to the reconstructed GguR regulons in all genomes where they are present except those of two Ralstonia spp. (Fig. S1). In the Halomonadaceae, the GguR regulons include genes involved in an alternative hexuronate utilization pathway (Udh-Gli-Gci). Three Halomonas spp. also contain a second GguR-regulated gene locus involved in aldarate utilization. The garL to garR genes, involved in an alternative KDG catabolic pathway, belong to the reconstructed GguR regulons in Polaromonas spp., two Burkholderia spp., and Leptothrix cholodnii. The known MFS family aldarate transporter, GarP, belongs to the GguR regulons in the majority of the Burkholderiaceae and Pseudomonadaceae genomes. Additional lineage-specific members of GguR regulons include several novel transporters and enzymes that are described in the sections below.
(iv) Identification of additional TF regulons.
Some hexuronate/aldarate utilization gene loci in the studied lineages of proteobacteria lack upstream GguR-binding sites but contain genes encoding hypothetical TFs other than GguR. Using gene neighborhood analysis, we identified three novel TFs, named GudR, GulR, and UdhR, presumably controlling these catabolic gene clusters and reconstructed their local regulons (Fig. 2). The LacI family UdhR regulator was identified in 6 out of 10 Burkholderia spp., in which it presumably controls the udh and exuT genes, involved in hexuronate uptake and catabolism. The GudR regulator representing another branch of TFs from the LacI family was identified in some Comamonadaceae genomes, in which it controls the gudD and/or tctC gene, involved in d-glucarate utilization. The deduced palindromic DNA motifs of UdhR and GudR are characteristic of DNA-binding sites of the LacI family regulators (25). The LysR family regulator, GulR, was identified in only two Ralstonia species, in which it is predicted to control the hexuronate utilization operon via two palindromic sites located in the common upstream region of the divergently transcribed kdgD and gulR genes.
(v) Sugar uptake transporters.
Orthologues of the known hexuronate transporters UxuPQM and ExuT were found in 35 studied genomes of proteobacteria (Table S1), in which they are commonly regulated by GguR, UdhR, and GulR TFs (Fig. 2). An additional candidate hexuronate transporter, only ∼30% identical to ExuT (named ExuT-II), was identified as a member of the reconstructed GguR regulons in four Pseudomonas species, including P. fluorescens strain FW300-N1B4. These strains lack the known hexuronate transporters but possess the complete hexuronate catabolic pathway (Fig. 1 and 2). The hexuronate utilization gene loci in many Burkholderia, Pseudomonas, and Ralstonia spp. contain an additional GguR regulon gene encoding a hypothetical outer membrane porin (Omp), which is presumably involved in the outer membrane uptake of hexuronates and/or polygalacturonates. Orthologues of the known aldarate transporter, GarP, belong to the reconstructed GguR regulons in most Burkholderia, Cupriavidus, and Pseudomonas spp., whereas in both Ralstonia species, it belongs to the GulR-regulated hexuronate/aldarate utilization gene cluster (Fig. 2). The GguR regulons in Halomonas and two Pseudomonas spp. contain a hypothetical tripartite transporter from the TTT family (named TctABC), which was proposed to be involved in aldarate uptake. Orthologues of the solute-binding TctC proteins belong to the reconstructed GguR regulons in all studied Comamonadaceae and Cupriavidus species, and most of these species possess two to four TctC paralogues that are coregulated by the GguR (and sometimes GudR) regulators. The identified tctC transporter genes often cluster with the aldarate dehydratase genes gudD and garD. Another candidate transporter from the MFS family (named GguT) was identified in the GguR regulons of the Burkholderia and Pseudomonas species that lack TctC transporters. Novel gguT transporter genes are clustered on the chromosomes with either gudD dehydratase or uxuF lactonase genes (Fig. 2), suggesting that they are potentially involved in uptake of aldarates or hexuronate metabolic intermediates.
(vi) Additional enzymatic components of regulons.
The GguR-regulated operons in several studied genomes include additional candidate regulon members. These include another previously unknown enzyme from the aldose 1-epimerase family (PF01263, named AldE). The enzymes presumably catalyze the mutarotation of the α- and β-anomers of d-GlcA and d-GalA after transport of the monosaccharide or after hydrolysis by a uronidase (e.g., pectinase or glucuronidase) in the cytosol, upstream of the uronate dehydrogenase (the uronate dehydrogenase is specific for the β-anomer). The novel hexuronic acid mutarotase gene, aldE, is found in the majority of Pseudomonas species, both Ralstonia species, and Polaromonas. Two GguR-regulated glycohydrolytic enzymes, namely, polygalacturonase (Pgl) in the Burkholderiaceae and alpha-glucosidase (Agl) in the two Ralstonia species, are likely involved in cytoplasmic degradation of polygalacturonate and glucuronides.
Experimental validation of novel transcriptional regulators. (i) In vitro DNA binding assays.
We further assessed the reconstructed hexuronate catabolic regulons by a combination of in vitro and in vivo techniques. First, we used the fluorescence polarization assay (FPA) to assess the specific binding of two GguR regulators (Bpro_3110 and Rpic_0945) to their predicted DNA sites in R. pickettii and Polaromonas sp. JS666. Both recombinant GguR proteins specifically bind to the synthetic 27-bp DNA fragments containing GguR operators at the udh (Bpro_3109) and gudD (Rpic_0946) genes (Fig. 3A and B). The apparent dissociation constant (Kd) values for Bpro_3110 and Rpic_0945 proteins interacting with their corresponding DNA sites were 0.05 mM and 0.1 mM, respectively. By testing the influence of potential sugar effectors on protein-DNA interaction, we identified that KDG (5-keto-4-deoxy-d-glucarate/galactarate), a common hexuronate/aldarate catabolic pathway intermediate, has a disruptive effect on GguR-DNA binding. However, the addition of d-glucuronate, d-galacturonate, d-glucarate, or meso-galactarate had no effect on the complex formation (data not shown). Using FPA, we determined that KDG at minimal concentration of 0.15 mM can substantially disrupt the binding of Bpro_3110 to its target DNA site (Fig. 3C). Electrophoretic mobility shift assay (EMSA) confirmed specific binding of Rpic_0945 (1 μM) to the synthetic 50-bp DNA fragment containing GguR-binding site at the gudD (Rpic_0946) gene (Fig. 3D). We also tested the interaction between GudR (Bpro_3418) regulator and its predicted DNA operator at the kdgD (Bpro_3419) gene in Polaromonas sp. JS666. EMSA demonstrated specific binding of the GudR protein (1 μM) to the synthetic 63-bp DNA fragment containing two tandem GudR-binding sites (Fig. 3D). Addition of hexuronates, aldarates, or KDG had no effect on the GudR-DNA interaction (data not shown); thus, a specific effector of GudR remains unknown.
FIG 3.

In vitro DNA binding assays of GguR and GudR regulators from Ralstonia and Polaromonas spp. (A) Interaction between the Rpic_0945-encoded protein (0.15 to 1.0 mM) and an upstream DNA fragment of the Rpic_0946 gene that contains a predicted GguR-binding site; also performed with reshuffled fragments of the Rpic_0946 and Bpro_3109 gene upstream regions (as negative controls; no GguR-binding sites). (B) Interaction between Bpro_3110 protein (0.05 to 1.5 mM) and an upstream DNA fragment of the Bpro_3109 gene that contains a predicted GguR-binding site. (C) Interaction between Bpro_3110 protein (0.15 mM) and an upstream DNA fragment for the Bpro_3109 gene in the presence of increasing concentrations of KDG (0.15 to 2.5 mM). (D) Electrophoretic mobility shift assay (EMSA) with GguR and GudR regulators and their DNA sites. Lanes 1 to 4, interaction between increasing concentrations of Rpic_0945/GguR protein (0, 0.01, 0.1, and 1 mM) and an upstream DNA fragment of Rpic_0946 (0.5 nM); lane 5, testing the effect of 10 mM meso-galactarate with 1 μM Rpic_0945 protein (no effect); lanes 6 to 10, interaction between increasing concentrations of Bpro_3418/GudR (0, 0.01, 0.1, 1 mM) and an upstream DNA fragment of Bpro_3419 (0.5 nM); lane 11, as a negative control, Bpro_3418 (1 mM) does not bind to an upstream DNA fragment of Rpic_0946 (0.5 nM).
(ii) In vivo gene expression analysis.
R. pickettii 12J contains two operons predicted to be involved in catabolism of hexuronates and aldarates. The gudD-exuT-agl-omp-aldE (Rpic_0946 to Rpic_0950) operon is controlled by the KDG-responsive GntR family regulator GguR (Rpic_0945), while the kdgD-kgsD-garP-gudD2-garD-udh-uxuL-exuT2 (Rpic_4452 to Rpic_4445) operon is regulated by the LysR family regulator GulR (Rpic_4453) (Fig. 2). To investigate sugar acid-specific induction of the predicted GguR- and GulR-regulated gene clusters, we performed reverse transcription-quantitative PCR (RT-qPCR) with specific primers designed for each gene from these loci. Total RNA was isolated from R. pickettii grown in medium supplied with d-GlcA, d-GalA, d-GlcAA, or meso-GalAA and compared to the same from cells grown on d-glucose (Fig. 4). The GulR regulon genes were upregulated 16- to 64-fold in the cells grown on either d-GalA, meso-GalAA, or d-GlcAA, whereas the effect of d-GlcA on expression of genes from the GulR-regulated operon was negligible. The GguR regulon genes demonstrated the highest induction (57- to 6,000-fold) on d-GlcAA and a moderate or small upregulation on d-GlcA (3- to 132-fold) and meso-GalAA (2- to 15-fold), whereas d-GalA had no effect on the GguR-regulated operon. Thus, the two catabolic operons have different patterns of induction by hexuronates and aldarates, suggesting that GulR responds to a different effector than GguR, which likely senses the KDG intermediate.
FIG 4.

In vivo gene expression analysis in Ralstonia pickettii grown on hexuronates and aldarates. (A) Log2 fold changes of genes from GulR-regulated cluster in R. pickettii grown on d-galacturonate (d-GalA), d-glucuronate (d-GlcA), meso-galactarate (meso-GalAA) and d-glucarate (d-GlcAA) relative to those grown on d-glucose. Individual transcript levels were measured by RT-qPCR with a set of specific primers. A housekeeping gene was used as a normalization control. Genes in the gene cluster are highly upregulated when grown on d-galacturonate. (B) Log2 fold changes of genes from GguR-regulated cluster in R. pickettii grown on the same sugar acids/diacids relative to those grown on d-glucose.
Experimental validation of novel hexuronic acid catabolic lactonases. (i) In vitro biochemical characterization of UxuF and UxuL lactonases.
To test the proposed lactone hydrolysis reaction identified through regulon and pathway reconstruction, four putative UxuF orthologues and two UxuL proteins from Burkholderia, Ralstonia, and Pseudomonas species were selected for further analysis (Table 1). Proteins from each family were cloned, heterologously expressed, and purified to homogeneity. Because the putative substrates (the δ-lactones, galactaro-1,5-lactone, and glucaro-1,5-lactone) are not commercially available, they were prepared chemically as previously described for galactaro-1,5-lactone. Purified proteins from both families were incubated for 2 min in reaction mixtures containing either d-galactaro-1,5-lactone (representative nuclear magnetic resonance [NMR] data shown in Fig. S4) or d-glucaro-1,5-lactone (data not shown). Enzymes that did not exhibit complete conversion of either lactone by proton NMR were monitored using a polarimeter. For the UxuL protein from R. pickettii, full kinetic characterization, including kcat, Km, and kcat/Km, was performed, while kcat/Km was determined for the other five proteins tested in this work for comparison to the R. pickettii UxuL (Table 1). Despite comparable Km values, of around 1 mM for both substrates, the R. pickettii UxuL enzyme hydrolyzed galactarolactone twice as fast as glucarolactone. The catalytic efficiencies of other enzymes tested in this work were in the range of 7.0 × 105 M−1 s−1 to 4.5 × 106 M−1 s−1. Note that P. syringae has enzymes from both lactonase families described in this report (UxuL and UxuF). The UxuL lactonase has a higher catalytic efficiency with glucarolactone (kcat/Km, 2.2 × 106 M−1 s−1) as the substrate, while the UxuF lactonase has a higher catalytic efficiency with galactarolactone (kcat/Km, 3.2 × 106 M−1 s−1) as the substrate. Those results may suggest a preference for one substrate over another along with a single, dedicated function for each enzyme. Interestingly, the UxuF lactonase from P. syringae contains a signal peptide suggesting its localization to the periplasm, and the corresponding gene PSPTO2765 is not a member of the reconstructed GguR regulon, while all other tested lactonases are conserved members of the GguR regulons in the Burkholderiaceae and Pseudomonadaceae (Fig. S1). Although no putative lactone hydrolases from the Comamonadaceae family were tested in vitro, on the basis of protein sequence similarity and conserved regulon and pathway features, confident functional annotation can be assigned to all identified UxuL and UxuF orthologues from that taxonomic group also. None of the enzymes tested in this work were active on the gamma lactone of either sugar or on gluconolactone (data not shown).
TABLE 1.
Kinetic parameters of the hexuronic acid catabolic lactonases UxuF and UxuL
| Gene locus tag | Organism |
d-Galactaro-1,5-lactone |
d-Glucaro-1,5-lactone |
||||
|---|---|---|---|---|---|---|---|
| kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | ||
| UxuL (PF08450) | |||||||
| Rpic_4446 | Ralstonia pickettii 12J | 720 ± 60 | 1.1 ± 0.3 | 6.5 × 105 | 310 ± 30 | 0.88 ± 0.3 | 3.5 × 105 |
| PSPTO_1052 |
Pseudomonas syringae pv. tomato DC3000 |
7.0 × 105 | 2.2 × 106 | ||||
| UxuF (PF10282) | |||||||
| Bcep1808_2255 | Burkholderia vietnamiensis G4 | 3.7 × 106 | 4.1 × 105 | ||||
| BMULJ_02167 |
Burkholderia multivorans ATCC 17616 |
4.5 × 106 | 4.2 × 105 | ||||
| Bcep18194_A5499 | Burkholderia lata ATCC 17760 | 5.9 × 105 | 3.4 × 105 | ||||
| PSPTO_2765 |
Pseudomonas syringae pv. tomato DC3000 |
3.2 × 106 | 7.0 × 104 | ||||
TABLE 2.
Functional roles of genes involved in hexuronate/aldarate utilization in proteobacteria
| Proteina | Role |
|---|---|
| Regulators | |
| GguR* | Predicted transcriptional regulator for hexuronate/aldarate utilization, GntR family |
| GudR* | Predicted transcriptional regulator for glucarate utilization, LacI family |
| GulR* | Predicted transcriptional regulator for hexuronate/aldarate utilization, LysR family |
| UdhR* | Predicted transcriptional regulator for hexuronate utilization, LacI family |
| Enzymes | |
| Udh | Uronate dehydrogenase (EC 1.1.1.203) |
| UxuL* | Predicted uronate lactonase, PF08450 family |
| UxuF* | Predicted uronate lactonase, PF10282 family |
| Gci | Galactarolactone cycloisomerase (EC 5.5.1.27) |
| Gli | Galactarolactone isomerase (EC 5.4.1.4) |
| GarD | meso-Galactarate dehydratase (EC 4.2.1.42) |
| GudD | d-Glucarate dehydratase (EC 4.2.1.40) |
| KdgD | 5-Dehydro-4-deoxyglucarate dehydratase (EC 4.2.1.41) |
| KgsD | Alpha-ketoglutaric semialdehyde dehydrogenase (EC 1.2.1.26) |
| KgsD2* | Alpha-ketoglutaric semialdehyde dehydrogenase (EC 1.2.1.26), alternative |
| GarL | 2-Dehydro-3-deoxyglucarate aldolase (EC 4.1.2.20) |
| GarR | 2-Hydroxy-3-oxopropionate reductase (EC 1.1.1.60) |
| Transporters | |
| UxuPQM | Hexuronate transporter, TRAP family |
| TctC* | Substrate binding protein of predicted aldarate transporter, TTT family |
| GarP (GudP) | d-Glucarate/meso-galactarate (aldarate) permease, MFS superfamily |
| GguT* | Predicted aldarate transporter, MFS superfamily |
| ExuT | Hexuronate transporter, MFS superfamily |
| ExuT-II* | Predicted hexuronate transporter, MFS superfamily |
| Additional | |
| Pgl | Polygalacturonase (EC 3.2.1.15), GH28 family |
| Agl | Alpha-glucosidase (EC 3.2.1.20), GH31 family |
| Omp* | Outer membrane porin precursor, possibly involved in polygalacturonate uptake |
| AldE* | d-Glucuronate/d-galacturonate mutarotase |
Proteins with novel functional roles predicted in this work are marked with an asterisk; proteins whose functions were experimentally validated in this study are underlined.
(ii) Metal testing.
The novel lactone hydrolases, UxuL and UxuF, identified in this work belong to the beta-propeller Pfam clan (CL0186) and Pfam families PF08450 (SMP-30/gluconolactonase/LRE-like) and PF10282 (lactonase and 7-bladed beta-propeller), respectively. Members of those families require a divalent metal cation for activity, primarily Ca2+. The activity for one representative clan member, Rpic_4446 (UxuL) from R. pickettii 12J, was tested in the presence of Ca2+, Co2+, Mg2+, Mn2+, Ni2+, Zn2+, and EDTA (Fig. S5). In the presence of EDTA, the reaction progress curve matched that of the progress curve for the uncatalyzed reaction, consistent with the enzyme requiring a metal for activity. The enzyme showed the highest activity when Zn2+ was the divalent metal. The order from least to most active is Ca2+ > Mg2+ > Ni2+ > Co2+ > Mn2+ > Zn2+. Despite other members of these enzyme families having been described as using Ca2+ as the preferred metal cation, the lactone hydrolases had the highest activity with Zn2+ as the metal, while Ca2+ yielded the least activity.
Experimental validation of a novel TTT family transporter via differential scanning fluorimetry.
Bpro_3101 (TctC; 510340) was by screened by DSF against a 189-compound library containing a large number of sugar ligands, including all variants of 3- to 6-carbon sugar acids (aldonic, aldaric, and uronic acids). The only ligand which yielded significant stabilization to Bpro_3101 was d-glucarate (differential melting temperature [ΔTm] = 6.1°C), while an alternate putative ligand, meso-galactarate, did not stabilize Bpro_3101 (ΔTm = 0.25°C) (Fig. 5). The DSF results suggest an alternative entry point into the uronic acid utilization pathway for Polaromonas sp. JS666 and related organisms. The entry point would be through the uptake of d-glucarate by the TctC solute-binding protein Bpro_3101 and its related TctAB membrane component, though the membrane component is not colocated in Polaromonas sp. JS666.
FIG 5.
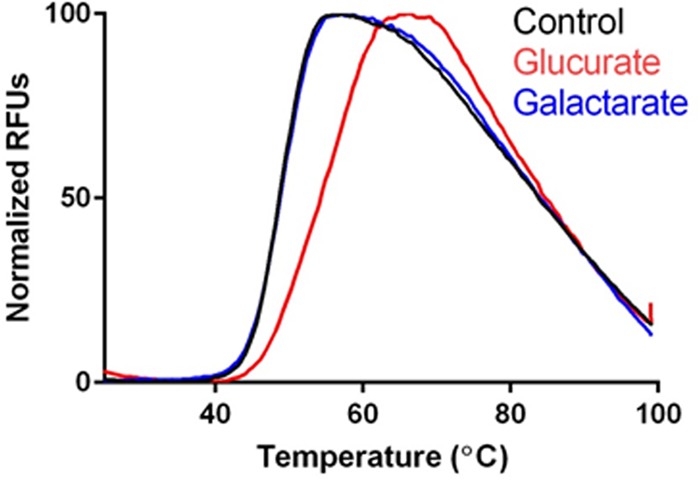
In vitro characterization of aldarate binding to the TctC protein Bpro_3101 from Polaromonas using differential scanning fluorimetry (DSF). Heat stabilization of Bpro_3101 by d-glucarate (ΔTm = 6.1°C) and not meso-galactarate (ΔTm = 0.25°C) versus a control reaction is shown. RFUs, relative fluorescence units.
DISCUSSION
Carbohydrate catabolic pathways involving committed enzymes, transporters, and transcriptional regulators are highly diverse and differ significantly between different taxonomic groups of bacteria (6, 26, 27). Hexuronic and aldaric acids are naturally abundant six-carbon sugar acids that are often utilized as carbon and energy sources by diverse species of heterotrophic bacteria. Glucuronic and galacturonic acids are common components of polysaccharides (e.g., gums and pectin), mucins, and peptidoglycans constituting the cell wall. d-Glucaric acid and meso-galactaric acid (also known as mucic acid) are natural products found in a variety of plants, including fruits and vegetables. The former is also produced and excreted in urine by mammals. In previous studies, we identified and characterized novel components of the hexuronate catabolic networks, including the UxuPQM family of hexuronate-specific TRAP transporters (21). In the current study, we have examined the genomic and functional context of the UxuPQM transporters in four families of gamma- and betaproteobacteria. Comparative genomics analysis and metabolic reconstruction identified a novel variant of the hexuronate catabolic pathway involving two new subfamilies of lactonases (UxuF and UxuL), as well as a new subfamily of aldaric acid transporters (TctC). The predicted functions of the novel enzymes and transporters were experimentally confirmed biochemically.
In the previously characterized variant of the oxidative pathway in A. tumefaciens, the uronate dehydrogenase, Udh, oxidizes d-GalA, resulting in the formation of galactaro-1,5-lactone, which is further isomerized to KDG in two enzymatic steps (Gli and Gci) with galactaro-1,4-lactone as an intermediate (10, 11). The two novel lactonases, UxuL and UxuF, identified in this study provide an alternative biochemical route directly merging the products of Udh with the aldaric acid catabolic pathway (Fig. 1). Thus, bacteria possessing this novel pathway variant are capable of metabolizing both hexuronic and aldaric acids pending the presence of their respective transporters. The novel hexuronic acid catabolic pathway is commonly present among the studied species of beta- and gammaproteobacteria. In contrast to the Gli/Gci enzymes, which were found only in the Halomonadaceae family, orthologues of UxuL/UxuF were identified in the majority of Burkholderia, Pseudomonas, Ralstonia, and Comamonadaceae species. The genome context analysis of the uxuL and uxuF genes revealed their strong association with the udh and uxuPQM genes, encoding the first enzymatic and uptake steps of hexuronate utilization. In addition, the uxuL genes are often colocalized with the garD gene, encoding galactarate dehydratase, which is required for the following enzymatic step after formation of meso-galactarate by a lactonase. The experimentally tested UxuL and UxuF lactonases from different species were active on both glucaro- and galactaro-1,5-lactones, although their kinetic parameters differed in some cases. That finding suggests individual members of these enzymatic families may have one or another preferable substrate. These results are in agreement with the observed redundant distribution of uxuL and uxuF genes across the analyzed genomes. For example, Variovorax spp. and Acidovorax sp. strain KKS102 contain both uxuL and uxuF genes, whereas Acidovorax avenae and Burkholderia ambifaria have two paralogues of uxuL and uxuF, respectively. In the public databases, uxuL- and uxuF-like genes are annotated as hypothetical proteins or misannotated as 6-phosphogluconolactonase. The combined bioinformatics and experimental results obtained in this study support reannotation of the UxuL and UxuF enzymes and their orthologues to uronate lactonases from the PF08450 and PF10282 families, respectively (see Uniprot and GenBank protein identifiers in Table S3).
Using bioinformatics analyses of hexuronate utilization genes, we identified a new GntR family regulator (GguR) in four lineages of beta- and gammaproteobacteria and three other lineage-specific regulators (GudR, GulR, and UdhR) that were predicted to coregulate the overlapping hexuronate and aldarate catabolic pathways and uptake transporters (Fig. 1). Prior to this study, two paralogous GntR family regulators, UxuR and ExuR, were known to control the isomerase pathways of hexuronate utilization in enterobacteria and two other lineages of gammaproteobacteria (Vibrionaceae and Pasteurellaceae), whereas the mechanism of regulation of the oxidative pathway had not been studied. The GguR and other regulons were reconstructed by identification and comparative analysis of candidate binding sites upstream of the hexuronate/aldarate utilization genes. The reconstructed GguR regulons include various hexuronate transporters and enzymes from both variants of the oxidative pathway of hexuronate utilization, as well as the downstream aldarate catabolic pathway enzymes. The expanded GguR regulon is in agreement with reconstructed hexuronic acid catabolic pathways that have aldaric acids as intermediates (Fig. 1). Interestingly, the GguR regulators were also found in several species missing hexuronate-specific transporters and the upstream part of the hexuronate catabolic pathway (e.g., Cupriavidus, Comamonas, Alicycliphilus, and two Pseudomonas spp.). The aldarate transporters and catabolic enzymes belong to the reconstructed GguR regulons in those species. These observations suggest that the content of GguR regulons and hexuronic and aldaric acid catabolic capabilities of certain proteobacterial species were reduced due to potential gene loss events. Experimental characterization of the GguR regulon confirmed the predicted DNA binding motif and identified its molecular effector, KDG, which is a common intermediate of the hexuronate and aldarate catabolic pathways. These results suggest that GguR functions as a negative regulator of its target operons and is derepressed by elevated levels of KDG during utilization of hexuronic or aldaric acids. Other identified regulators in the reconstructed hexuronate catabolic network are possibly responsible for fine-tuned expression of certain enzymes and transporters in small subsets of closely related bacteria. For instance, the LysR family regulator GulR replaces GguR in the control of the major hexuronate utilization operon in two Ralstonia spp., whereas the LacI family regulator UdhR controls the exuT and udh genes in Burkholderia spp.
Based on genomic context analysis of the reconstructed regulons, we have identified several novel transporters involved in utilization of hexuronic and aldaric acids. The solute-binding component, TctC, of a novel aldarate-specific transporter from the TTT family was identified in all studied species of Comamonadaceae, as well as in Halomonas, Cupriavidus, and two Pseudomonas spp. The TctC solute-binding proteins belong to the reconstructed GguR or GudR regulons and are often represented by two or more paralogues. Interestingly, the TctC and GarP (GudD) transporters demonstrate complementary patterns of occurrence in the analyzed genomes, suggesting their involvement in aldarate uptake. Experimental testing of a representative TctC transporter from Polaromonas confirmed its specific affinity to d-glucarate but not to meso-galactarate. Involvement of two other TctC paralogues in meso-galactarate uptake in Polaromonas remains to be tested. The membrane components, TctA and TctB, do not show genomic association with the majority of identified aldarate-related TctC components, with exception of two Pseudomonas species containing the gudD-tctCBA gene locus in the reconstructed GguR regulon (Fig. 2). Since the analyzed proteobacterial genomes encode a large number of TctC family transporters, we propose that the universal TctAB complex is shared by many substrate-specific TctC components. Several other transport systems from the MFS family were identified within the reconstructed GguR regulons in Burkholderia and Pseudomonas spp., in which they are possibly involved in aldarate and/or hexuronate uptake. The exact specificities of these transporters remain to be determined in the future studies.
The exact source of hexuronic and aldaric acids the bacteria discussed in this study metabolize in nature remains unclear, yet there are several possible sources. The presence of putative polygalacturonase and alpha-glucosidase genes in the reconstructed GguR regulons of Burkholderia spp. suggests that these species can acquire galacturonate from pectin. Indeed, several Pseudomonas spp., Polaromonas, and Chromohalobacter salexigens are capable of pectin degradation (28). Yet bacteria in nature do not exist in isolation; thus, another source of uronic acids is the monomers released through the action of hydrolases secreted by other soil organisms, such as fungi. Algae are another major source of polysaccharides (e.g., polyglucuronic acid from green algae and seaweed). Hyaluronic acid, another source of glucuronic acid, is abundant in connective and epithelial tissues of vertebrates (29, 30). The aldaric acids are likely less naturally abundant yet are certainly formed as intermediates in catabolizing the uronic acids.
In summary, we combined bioinformatics and experimental techniques to reconstruct and validate hexuronate and aldarate utilization pathways and associated transcriptional regulons in proteobacteria. The established functional roles of novel enzymes, regulators, and transporters have allowed us to improve the quality of genomic annotations and to accurately infer the detailed metabolic and regulatory networks in four lineages of beta- and gammaproteobacteria. Comparative genomics and metabolic context analyses conducted in this work and previously revealed a substantial diversity of hexuronate utilization pathway variants, including the catabolic enzymes, uptake transporters, and regulators, thus expanding our knowledge of sugar catabolism in bacteria.
MATERIALS AND METHODS
Bioinformatics tools and databases.
The analyzed proteobacterial genomes (see Table S1 in the supplemental material) were downloaded from GenBank. Locus tag gene identifiers are used throughout. Orthologues between proteins from different taxonomic groups were defined as bidirectional best hits, with a 30% identity threshold using the Smith-Waterman algorithm implemented in the GenomeExplorer program (31). In dubious cases, orthologues were confirmed by construction of phylogenetic trees and comparative analysis of gene neighborhoods using the MicrobesOnline tree browser tool (32). Functional gene assignments and metabolic subsystem analysis were performed using the SEED database and Web tools that combine protein similarity search, positional gene clustering, and phylogenetic profiling of genes (33). The Swiss-Prot/UniProt Database (34) and Transporter Classification Database (TCDB) (35) of previously characterized enzymes and transporters were used for annotation of the previously characterized protein families. The PFAM protein domain database was used to verify protein functional and structural annotations (36). The analyzed functional roles of known and predicted hexuronate/aldarate catabolic enzymes, transporters, and regulators are summarized in Table 2. Gene locus tags for all identified orthologues are available in Table S1. Identification of signal peptide sequences and protein localization predictions were preformed using the SignalP Web server (37). Multiple-sequence alignments were constructed by MUSCLE (38). Phylogenetic trees were built using a maximum likelihood algorithm implemented in the PhyML Web tool (39) and visualized in iTOL (40).
Regulon identification and analysis.
For genomic reconstruction of novel transcription factor (TF) regulons, we used the comparative genomics approach based on identification of candidate TF-binding sites in closely related bacterial genomes (41, 42). First, we revealed orthologues of each regulator and analyzed the genomic context of their genes to reveal colocalizations with other catabolic pathway genes. Phylogenetic analysis of the GguR regulator family revealed several major groups of orthologues. For each group of GguR orthologues, as well as for the taxonomy-specific groups of GudR, UdhR, and GulR orthologues, we identified their putative DNA-binding motifs using a de novo motif discovery tool implemented in the RegPredict Web server (http://regpredict.lbl.gov/) (43). First, we collected training sets of orthologous upstream gene regions for each prospective regulator-controlled operon determined via the genome context analysis of the respective regulator-encoding genes. The collected sets of upstream regions were analyzed using a DNA motif recognition program (the “Discover Profile” procedure implemented in RegPredict) to identify conserved palindromic DNA motifs. After construction of a positional-weight matrix for each identified DNA motif, the studied genomes were scanned with recognition matrices to determine additional candidate binding sites and reconstruct the respective regulons using the comparative genomics approach implemented in the RegPredict and GenomeExplorer tools (31). The scores of sites were calculated as a sum of nucleotide weights for each position. Sequence logos were built using the WebLogo package (44). The content of reconstructed regulons and identified TF-binding sites are summarized in Table S1.
Cloning, expression, and purification of lactone hydrolases and the TctC protein.
The lactonase hydrolases and Bpro_3101 from Polaromonas sp. JS666 were cloned, expressed, and purified as previously described (21). E. coli strains XL1-Blue and BL21(DE3) were used for gene cloning and protein overexpression, respectively. Briefly, genes of interest were PCR amplified from genomic DNA using KOD Hot Start DNA polymerase, with the resultant amplified fragment ligated into the N-terminal, tobacco etch virus (TEV)-cleavable, 6×His tag vector pNIC28-Bsa4 (45), by ligation independent cloning (46). Vectors containing the genes of interest were transformed into BL21(DE3) containing the pRIL plasmid (Stratagene), and proteins were expressed using ZYP-5052 autoinduction medium in an LEX48 airlift fermentor with overnight growth at 22°C for 16 to 22 h. Proteins were purified by HisTrap nickel-nitrilotriacetic acid (Ni-NTA) chromatography with the eluted target injected onto an inline HiLoad S200 16/60 pg (GE Healthcare) gel filtration column equilibrated with 20 mM HEPES (pH 7.5), 150 mM NaCl, 5% glycerol, and 5 mM dithiothreitol (DTT). Eluted proteins were analyzed by SDS-PAGE, snap-frozen in liquid nitrogen, and stored at −80°C.
Cloning, expression, and purification of regulators.
Bpro_3110/GguR and Bpro_3418/GudR from Polaromonas sp. JS666 and Rpic_0945/GguR from R. pickettii 12J were amplified from genomic DNA using PCR and specific primers synthesized by Integrated DNA Technologies (Table S2). Recombinant GguR and GudR proteins were overexpressed as N-terminal fusion proteins with a 6×His tag in BL21(DE3) cells. Cells were grown in LB medium to an optical density at 600 nm (OD600) equal to 0.8 at 37°C, induced with 0.2 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and harvested after 12 h of shaking at 20°C. Protein purification was performed using the rapid Ni-NTA–agarose minicolumn protocol as previously described (47). Briefly, harvested cells were resuspended in 20 mM HEPES buffer (pH 7.0) containing 100 mM NaCl, 0.03% Brij 35 detergent, and 2 mM β-mercaptoethanol supplemented with 2 mM phenylmethylsulfonyl fluoride and a protease inhibitor cocktail (Sigma-Aldrich). Lysozyme was added to 1 mg/ml, and the cells were lysed by freeze-thawing followed by sonication. After centrifugation at 18,000 rpm, Tris buffer (pH 8.0) was added to the supernatant to a final concentration of 50 mM, and the mixture was loaded onto an Ni-NTA–agarose column (0.2 ml). After washing with the starting buffer containing 1 M NaCl and 0.3% Brij 35 detergent, bound proteins were eluted with 0.3 ml of the starting buffer containing 250 mM imidazole. Protein size, expression level, distribution between soluble and insoluble forms, and purity were monitored by SDS-PAGE.
DNA binding assays.
The interaction of the purified recombinant GguR and GudR regulator proteins (containing the 6×His tag) with their cognate DNA-binding sites in Polaromonas sp. JS666 and Ralstonia pickettii 12J was assessed using two techniques: electrophoretic mobility shift assay (EMSA) and fluorescence polarization assay (FPA). The oligonucleotides containing the predicted binding sites were synthesized by Integrated DNA Technologies (Table S2). The double-stranded DNA fragments were obtained by annealing synthesized complementary oligonucleotides at a 1:10 ratio of 5′ labeled with 6-carboxyfluorescein (for FPA) or biotin (for EMSA) to unlabeled complementary oligonucleotides.
Using the FPA, we tested two GguR proteins (Rpic_0945 and Bpro_3110) and their cognate DNA-binding sites upstream of the Rpic_0946 and Bpro_3109 genes. The 6-carboxyfluorescein-labeled 30-bp DNA fragments (10 nM) were incubated with increasing concentrations of the purified proteins (50 to 1,500 nM) in a total volume of 100 μl of the binding buffer containing 100 mM Tris (pH 8), 50 mM NaCl, 10 mM MgCl2, 2 mM DTT, 5% glycerol, and 0.5 mM EDTA at 25°C for 1 h. Poly(dI-dC) was added to the reaction mixture as a nonspecific competitor DNA at 1 μg to suppress nonspecific binding. The fluorescence-labeled DNA was detected with the FLA-5100 fluorescent image analyzer (Fujifilm). To identify effectors of GguR, additional FPA experiments were performed to test the effects of d-GlcA, d-GalA, d-GlcAA, meso-GalAA, and KDG at concentrations of 2 mM each. For effector titration, the protein and DNA fragment were incubated with increasing concentrations of KDG in the incubation mixture (0.15 to 2.5 mM).
Using the EMSA, we tested the GguR (Rpic_0945) and GudR (Bpro_3418) regulators and their cognate DNA sites. The biotin-labeled DNA fragments (0.5 nM) were incubated with increasing concentrations of the recombinant purified proteins in a total volume of 20 μl. The binding buffer contained 50 mM Tris (pH 8.0), 0.15 M NaCl, 5 mM MgCl2, 1 mM DTT, 0.05% NP-40, 2.5% glycerol, and 1 μg of herring sperm DNA. After 25 min of incubation at 37°C, the reaction mixtures were separated by electrophoresis on a 1.5% agarose gel (30 min, 90 V, and room temperature). The DNA was transferred by electrophoresis onto a Hybond-N+ membrane (Amersham) and fixed by UV cross-linking. The biotin-labeled DNA was detected with a LightShift chemiluminescent EMSA kit (Thermo Fisher Scientific Inc., Rockford, IL).
Differential scanning fluorimetry.
DSF was performed using an Applied Biosystems 7900HT fast real-time PCR system with excitation at 490 nm and emission at 530 nm. Reaction mixtures (20-µl final volume) contained 10 µM protein, 1 mM ligand, and 5× Sypro Orange (5,000× stock; Invitrogen) in 100 mM HEPES (pH 7.5) plus 150 mM NaCl. Samples were heated from 22°C to 99°C at a rate of 3°C min−1 with each sample in duplicate. Bpro_3101 (TctC) was screened against a 189-compound library of primarily carbohydrates (21). Melting temperatures (midpoint of unfolding) were calculated from fitting of the melting curve to a Boltzmann equation. The average of eight control wells (no ligands) was used to calculate the differential Tm (ΔTm).
Lactone hydrolase activity screening by 1H NMR spectroscopy and polarimetry.
The δ-lactones (1,5-lactones) of d-GlcA and d-GalA were prepared from the sodium salts of the pyranuronates using the procedure described by Isbell and Frush (48), with the following modifications: at 1/2 the scale in D2O, 2× bromine, a reaction time of 22 min 30 s, and separation in a separatory funnel at 4°C. d-Glucaro-1,4-lactone was purchased from Toronto Research Chemicals Inc. d-Galactaro-1,4-lactone was formed by incubating the δ-lactone overnight at room temperature. To determine if the lactone hydrolases screened in this work catalyzed the conversion of d-glucaro-1,5-lactone and/or d-galactaro-1,5-lactone to d-GlcAA and/or meso-GalAA, spectra of the reaction mixtures 2 min after addition of enzyme were recorded by 1H NMR. A 650-μl reaction contained a 2.5 mM concentration of either δ-lactone, 1 μl of enzyme stock (300 to 3,000 nM), 50 mM sodium phosphate buffer, and 2 mM MgCl2 at pD 6.4 in D2O. All NMR spectra were collected on an Agilent 600-MHz spectrometer.
To test the effects of divalent cations on the lactone hydrolase activity, Ca2+, Co2+, Mg2+, Mn2+, Ni2+, or Zn2+ was added to the incubation mixture at a final concentration of 2 mM and activity was monitored in a continuous polarimetric assay. The change in optical rotation was monitored using a polarimeter (Jasco P-1010) and a mercury line filter (405 nm). A 1,300-μl reaction mixture containing 100 nM enzyme, 50 mM MES buffer, 2.5 mM substrate, and 2 mM metal at pD 6.4 in D2O was monitored for 2 min at room temperature. The continuous polarimetric assay also was used to determine the kinetic constants of the lactone hydrolases as described above with addition of 2 mM MnCl2 and various substrate concentrations (0.24 mM to 5.85 mM). Although Zn2+ turned out to be the best activator of Rpic_4446, MnCl2 was used in kinetic experiments due to precipitation problems encountered with ZnCl2 in the buffer during the prolonged amount of time required to complete kinetic experiments; MnCl2 was more stable than ZnCl2. The rate of the uncatalyzed reaction was determined using the same reaction conditions and monitoring for an additional 1 min 30 s. Data were fit to the Michaelis-Menten equation in Sigma Plot using the Enzyme Kinetics Module.
Gene expression analysis.
Ralstonia pickettii 12J was streaked onto nutrient broth agar plates and incubated at 30°C overnight aerobically. A single colony was picked and used to inoculate 3 ml of nutrient broth and incubated in a tube roller at 30°C overnight. Cells were rinsed at room temperature 3 times with 1× phosphate-buffered saline (PBS), used to inoculate a 3-ml culture of M9 minimal medium supplemented with 20 mM d-glucose to an OD600 equal to 0.05, and incubated in a tube roller at 30°C until reaching an OD600 equal to 0.8. Cells were rinsed at room temperature 3 times with 1× PBS and used to inoculate 3 ml of M9 minimal medium supplemented with either 20 mM d-glucose, d-GlcA, d-GalA, d-GlcAA, or meso-GalAA to an OD600 equal to 0.05. Cultures were incubated in a tube roller at 30°C until they reached an OD600 equal to 0.4. RNAprotect bacterial reagent (6 ml) from Qiagen was added to each culture. Mixtures then were vortexed and incubated at room temperature for 5 min. Next, the mixtures were centrifuged at 5,000 × g for 10 min, the supernatants were discarded, and RNA was isolated from the cells using an RNeasy minikit (Qiagen) according to the manufacturer’s instructions. RNA preps were treated with RNase-free DNase (Qiagen) to remove genomic DNA. RNA preps were deemed clear of any contaminating genomic DNA when PCRs with each primer pair failed to yield a band on an agarose gel having used an aliquot of the RNA prep as the template. The RNA was reverse transcribed using a ProtoScript first-strand cDNA synthesis kit (New England BioLabs) according to the manufacturer’s instructions. The gene-specific primers for each gene tested are shown in Table S2. The qPCR mixtures were was set up using FastStart Universal SYBR green master (ROX) (Roche) according to the manufacturer’s instructions, and the reactions were carried out on a LightCycler 480 II (Roche). Fold changes in gene expression were calculated using the formula 2−(ΔCp sample − ΔCp control), where Cp is the crossing point.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by grant U54GM093342 (to S.C.A. and J.A.G.) from the U.S. National Institutes of Health. D.A.R. was supported by the Russian Science Foundation, grant no. 14-14-00289.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00431-18.
For a commentary on this article, see https://doi.org/10.1128/JB.00628-18.
REFERENCES
- 1.Garron ML, Cygler M. 2010. Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20:1547–1573. doi: 10.1093/glycob/cwq122. [DOI] [PubMed] [Google Scholar]
- 2.Benz J, Protzko RJ, Andrich JMS, Bauer S, Dueber JE, Somerville CR. 2014. Identification and characterization of a galacturonic acid transporter from Neurospora crassa and its application for Saccharomyces cerevisiae fermentation processes. Biotechnol Biofuels 7:20. doi: 10.1186/1754-6834-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suvorova IA, Tutukina MN, Ravcheev DA, Rodionov DA, Ozoline ON, Gelfand MS. 2011. Comparative genomic analysis of the hexuronate metabolism genes and their regulation in gammaproteobacteria. J Bacteriol 193:3956–3963. doi: 10.1128/JB.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hugouvieux-Cotte-Pattat N, Robert-Baudouy J. 1987. Hexuronate catabolism in Erwinia chrysanthemi. J Bacteriol 169:1223–1231. doi: 10.1128/jb.169.3.1223-1231.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mekjian KR, Bryan EM, Beall BW, Moran CP Jr.. 1999. Regulation of hexuronate utilization in Bacillus subtilis. J Bacteriol 181:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodionova IA, Scott DA, Grishin NV, Osterman AL, Rodionov DA. 2012. Tagaturonate-fructuronate epimerase UxaE, a novel enzyme in the hexuronate catabolic network in Thermotoga maritima. Environ Microbiol 14:2920–2934. doi: 10.1111/j.1462-2920.2012.02856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon SH, Moon TS, Iranpour P, Lanza AM, Prather KJ. 2009. Cloning and characterization of uronate dehydrogenases from two pseudomonads and Agrobacterium tumefaciens strain C58. J Bacteriol 191:1565–1573. doi: 10.1128/JB.00586-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parkkinen T, Boer H, Jänis J, Andberg M, Penttilä M, Koivula A, Rouvinen J. 2011. Crystal structure of uronate dehydrogenase from Agrobacterium tumefaciens. J Biol Chem 286:27294–27300. doi: 10.1074/jbc.M111.254854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pick A, Schmid J, Sieber V. 2015. Characterization of uronate dehydrogenase catalyzing the initial step in an oxidative pathway. Microb Biotechnol 8:633–643. doi: 10.1111/1751-7915.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andberg M, Maaheimo H, Boer H, Penttilä M, Koivula A, Richard P. 2012. Characterization of a novel Agrobacterium tumefaciens galactarolactone cycloisomerase enzyme for direct conversion of D-galactarolactone to 3-deoxy-2-keto-L-threo-hexarate. J Biol Chem 287:17662–17671. doi: 10.1074/jbc.M111.335240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouvier JT, Groninger-Poe FP, Vetting M, Almo SC, Gerlt JA. 2014. Galactaro δ-lactone isomerase: lactone isomerization by a member of the amidohydrolase superfamily. Biochemistry 53:614–616. doi: 10.1021/bi5000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taberman H, Andberg M, Parkkinen T, Jänis J, Penttilä M, Hakulinen N, Koivula A, Rouvinen J. 2014. Structure and function of a decarboxylating Agrobacterium tumefaciens keto-deoxy-D-galactarate dehydratase. Biochemistry 53:8052–8060. doi: 10.1021/bi501290k. [DOI] [PubMed] [Google Scholar]
- 13.Hubbard BK, Koch M, Palmer DR, Babbitt PC, Gerlt JA. 1998. Evolution of enzymatic activities in the enolase superfamily: characterization of the (D)-glucarate/galactarate catabolic pathway in Escherichia coli. Biochemistry 37:14369–14375. doi: 10.1021/bi981124f. [DOI] [PubMed] [Google Scholar]
- 14.Aghaie A, Lechaplais C, Sirven P, Tricot S, Besnard-Gonnet M, Muselet D, de Berardinis V, Kreimeyer A, Gyapay G, Salanoubat M, Perret A. 2008. New insights into the alternative D-glucarate degradation pathway. J Biol Chem 283:15638–15646. doi: 10.1074/jbc.M800487200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodionov DA, Rodionova IA, Li X, Ravcheev DA, Tarasova Y, Portnoy VA, Zengler K, Osterman AL. 2013. Transcriptional regulation of the carbohydrate utilization network in Thermotoga maritima. Front Microbiol 4:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodionov DA, Yang C, Li X, Rodionova IA, Wang Y, Obraztsova AY, Zagnitko OP, Overbeek R, Romine MF, Reed S, Fredrickson JK, Nealson KH, Osterman AL. 2010. Genomic encyclopedia of sugar utilization pathways in the Shewanella genus. BMC Genomics 11:494. doi: 10.1186/1471-2164-11-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tutukina MN, Potapova AV, Cole JA, Ozoline ON. 2016. Control of hexuronate metabolism in Escherichia coli by the two interdependent regulators, ExuR and UxuR: derepression by heterodimer formation. Microbiology 162:1220–1231. doi: 10.1099/mic.0.000297. [DOI] [PubMed] [Google Scholar]
- 18.Nemoz G, Robert-Baudouy J, Stoeber F. 1976. Physiological and genetic regulation of the aldohexuronate transport system in Escherichia coli. J Bacteriol 2:706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valmeekam V, Loh YL, San Francisco MJ. 2001. Control of exuT activity for galacturonate transport by the negative regulator ExuR in Erwinia chrysanthemi EC16. Mol Plant Microbe Interact 14:816–820. doi: 10.1094/MPMI.2001.14.6.816. [DOI] [PubMed] [Google Scholar]
- 20.González ET, Allen C. 2003. Characterization of a Ralstonia solanacearum operon required for polygalacturonate degradation and uptake of galacturonic acid. Mol Plant Microbe Interact 16:536–544. doi: 10.1094/MPMI.2003.16.6.536. [DOI] [PubMed] [Google Scholar]
- 21.Vetting MW, Al-Obaidi N, Zhao S, San Francisco B, Kim J, Wichelecki DJ, Bouvier JT, Solbiati JO, Vu H, Zhang X, Rodionov DA, Love JD, Hillerich BS, Seidel RD, Quinn RJ, Osterman AL, Cronan JE, Jacobson MP, Gerlt JA, Almo SC. 2015. Experimental strategies for functional annotation and metabolism discovery: targeted screening of solute binding proteins and unbiased panning of metabolomes. Biochemistry 54:909–931. doi: 10.1021/bi501388y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao J, Binns AN. 2015. Involvement of Agrobacterium tumefaciens galacturonate tripartite ATP-independent periplasmic (TRAP) transporter GaaPQM in virulence gene expression. Appl Environ Microbiol 82:1136–1146. doi: 10.1128/AEM.02891-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monterrubio R, Baldoma L, Obradors N, Aguilar J, Badia J. 2000. A common regulator for the operons encoding the enzymes involved in D-galactarate, D-glucarate, and D-glycerate utilization in Escherichia coli. J Bacteriol 182:2672–2674. doi: 10.1128/JB.182.9.2672-2674.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boer H, Maaheimo H, Koivula A, Penttilä M, Richard P. 2010. Identification in Agrobacterium tumefaciens of the D-galacturonic acid dehydrogenase gene. Appl Microbiol Biotechnol 86:901–909. doi: 10.1007/s00253-009-2333-9. [DOI] [PubMed] [Google Scholar]
- 25.Ravcheev DA, Khoroshkin MS, Laikova ON, Tsoy OV, Sernova NV, Petrova SA, Rakhmaninova AB, Novichkov PS, Gelfand MS, Rodionov DA. 2014. Comparative genomics and evolution of regulons of the LacI-family transcription factors. Front Microbiol 5:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodionova IA, Li X, Thiel V, Stolyar S, Stanton K, Fredrickson JK, Bryant DA, Osterman AL, Best AA, Roidonov DA. 2013. Comparative genomics and functional analysis of rhamnose catabolic pathways and regulons in bacteria. Front Microbiol 4:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodionova IA, Leyn SA, Burkart MD, Boucher N, Noll KM, Osterman AL, Rodionov DA. 2013. Novel inositol catabolic pathway in Thermotoga maritima. Environ Microbiol 15:2254–2266. doi: 10.1111/1462-2920.12096. [DOI] [PubMed] [Google Scholar]
- 28.Wagschal K, Jordan DB, Lee CC, Younger A, Braker JD, Chan VJ. 2015. Biochemical characterization of uronate dehydrogenase from three pseudomonads, Chromohalobacter salixigens, and Polaromonas napthalenivorans. Enzyme Microb Technol 69:62–68. doi: 10.1016/j.enzmictec.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 29.Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH. (ed) 2017. Essentials of glycobiology, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 30.Fraser JR, Laurent TC, Laurent UB. 1997. Hyaluronan: its nature, distribution, functions, and turnover. J Intern Med 242:27–33. doi: 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- 31.Mironov AA, Vinokurova NP, Gel’fand MS. 2000. Software for analyzing bacterial genomes. Mol Biol (Mosk) 34:253–262. (In Russian.) [PubMed] [Google Scholar]
- 32.Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, Friedland GD, Huang KH, Keller K, Novichkov PS, Dubchak IL, Alm EJ, Arkin AP. 2010. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res 38:D396–D400. doi: 10.1093/nar/gkp919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, Crécy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen J, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Rückert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.UniProt Consortium. 2015. UniProt: a hub for protein information. Nucleic Acids Res 43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saier MH Jr, Reddy VS, Tsu BV, Ahmed MS, Li C, Moreno-Hagelsieb G. 2016. The Transporter Classification Database (TCDB): recent advances. Nucleic Acids Res 44:D372–D379. doi: 10.1093/nar/gkv1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen H. 2017. Predicting secretory proteins with SignalP. Methods Mol Biol 1611:59–73. doi: 10.1007/978-1-4939-7015-5_6. [DOI] [PubMed] [Google Scholar]
- 38.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodionov DA. 2007. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem Rev 107:3467–3497. doi: 10.1021/cr068309+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leyn SA, Suvorova IA, Kazakov AE, Ravcheev DA, Stepanova VV, Novichkov PS, Rodionov DA. 2016. Comparative genomics and evolution of transcriptional regulons in Proteobacteria. Microb Genom 2:e000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novichkov PS, Laikova ON, Novichkova ES, Gelfand MS, Arkin AP, Dubchak I, Rodionov DA. 2010. RegPrecise: a database of curated genomic inferences of transcriptional regulatory interactions in prokaryotes. Nucleic Acids Res 38:D111–D118. doi: 10.1093/nar/gkp894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savitsky P, Bray J, Cooper CD, Marsden BD, Mahajan P, Burgess-Brown NA, Gileadi O. 2010. High-throughput production of human proteins for crystallization: the SGC experience. J Struct Biol 172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aslanidis C, de Jong PJ. 1990. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodionov DA, Novichkov PS, Stavrovskaya ED, Rodionova IA, Li X, Kazanov MD, Ravcheev DA, Gerasimova AV, Kazakov AE, Kovaleva GY, Permina EA, Laikova ON, Overbeek R, Romine MF, Fredrickson JK, Arkin AP, Dubchak I, Osterman AL, Gelfand MS. 2011. Comparative genomic reconstruction of transcriptional networks controlling central metabolism in the Shewanella genus. BMC Genomics 12:S3. doi: 10.1186/1471-2164-12-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Isbell HS, Frush HL. 1943. Ring structures and mutarotations of the modifications of D-galacturonic acid. J Res Natl Bur Stand 31:33–44. doi: 10.6028/jres.031.021. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.