

Abstract
Background
Metabolic alterations as hyperglycemia and inflammation induce myocardial molecular events enhancing oxidative stress and mitochondrial dysfunction. Those alterations are responsible for a progressive loss of cardiomyocytes, cardiac stem cells, and consequent cardiovascular complications. Currently, there are no effective pharmacological measures to protect the heart from these metabolic modifications, and the development of new therapeutic approaches, focused on improvement of the oxidative stress condition, is pivotal. The protective effects of levocarnitine (LC) in patients with ischemic heart disease are related to the attenuation of oxidative stress, but LC mechanisms have yet to be fully understood.
Objective
The aim of this work was to investigate LC's role in oxidative stress condition, on ROS production and mitochondrial detoxifying function in H9c2 rat cardiomyocytes during hyperglycemia.
Methods
H9c2 cells in the hyperglycemic state (25 mmol/L glucose) were exposed to 0.5 or 5 mM LC for 48 and 72 h: LC effects on signaling pathways involved in oxidative stress condition were studied by Western blot and immunofluorescence analysis. To evaluate ROS production, H9c2 cells were exposed to H2O2 after LC pretreatment.
Results
Our in vitro study indicates how LC supplementation might protect cardiomyocytes from oxidative stress-related damage, preventing ROS formation and activating antioxidant signaling pathways in hyperglycemic conditions. In particular, LC promotes STAT3 activation and significantly increases the expression of antioxidant protein SOD2. Hyperglycemic cardiac cells are characterized by impairment in mitochondrial dysfunction and the CaMKII signal: LC promotes CaMKII expression and activation and enhancement of AMPK protein synthesis. Our results suggest that LC might ameliorate metabolic aspects of hyperglycemic cardiac cells. Finally, LC doses herein used did not modify H9c2 growth rate and viability.
Conclusions
Our novel study demonstrates that LC improves the microenvironment damaged by oxidative stress (induced by hyperglycemia), thus proposing this nutraceutical compound as an adjuvant in diabetic cardiac regenerative medicine.
1. Introduction
Cardiovascular complications are recognized as the primary cause of mortality in subjects with diabetes mellitus (DM) [1, 2], characterized by hyperglycemia which is determined by a defect of insulin secretion, insulin action, or both [3]. Moreover, DM is associated with inflammation condition.
Chronic hyperglycemia, characterizing overt diabetes, or fluctuant hyperglycemia, present in the prediabetic condition, are responsible for the activation of numerous signaling pathways that exacerbate the systemic inflammation and lead to the development of diabetic complications [4, 5]. Recent evidence establishes how hyperglycemia is involved in the regulation of sirtuin (SIRT) transcription factors. The deregulation of SIRT expression is strictly correlated with the progression of inflammation and atherosclerotic disease. In particular, Balestrieri et al. demonstrated that SIRT6 protein expression is downregulated in atherosclerotic plaques of diabetics, and this defect is linked to the chronic oxidative stress condition [6]. Those evidences indicate that chronic inflammation, oxidative stress condition and predisposition to ischemic heart disease are higher in patients with DM than in nondiabetics [7, 8].
Traditional therapeutic approaches as well as innovative promising strategies [7] (i.e., stem/progenitor cell therapy, existing cardiomyocyte proliferation, and reprogramming noncardiac cells) are limited in patients with DM [8–11]. Periprocedural intensive glycemic control, during early percutaneous coronary intervention in diabetic patients, was shown to improve myocardial protection by increasing SIRT1 expression, endothelial progenitor cell number, and their capability to differentiate in mature cardiomyocytes [12]. Hyperglycemia in diabetic subjects is the major factor responsible for the failure of regenerative myocardial therapeutic strategies. Recent data indicate that the overproduction of reactive oxygen species (ROS) and the oxidative stress condition are the main causes involved in diabetic cardiac injury and in the lack of success in cardiac regenerative therapies [13–17]. Hyperglycemia enhances ROS production impairing cardiac microenvironment and regeneration capacity [16, 17]. In particular, several publications showed that the hyperglycemic and oxidative microenvironment induces mitochondrial abnormalities and cellular damage, eventually leading to senescence and apoptosis of cardiac progenitors [18, 19].
Thus, to optimize regenerative strategies for diabetic patients, the development of new therapeutic approaches focused on the reduction of oxidative stress condition is fundamental.
Noteworthy, in recent years, L-carnitine (LC) has been proposed as a nutraceutical integrator in the treatment of numerous cardiac syndromes, including coronary disease, atherosclerosis, and toxic myocardial injury [20–22]. It is well established that LC, facilitating transport of long-chain fatty acids into the mitochondrial matrix, plays an important role in supporting cardiac energy homeostasis [23–25]. Most importantly, some studies successfully showed LC's ability to reduce oxidative stress, hypoxic cellular damage, and apoptosis of cardiac cells in normal glycemic condition [26, 27]. In particular, Mao et al. have recently demonstrated that LC pretreatment ameliorated cellular damage induced by H2O2 in H9c2 rat cardiomyocytes, enhancing mitochondrial function [28].
LC effects on cardiac metabolism and function have been demonstrated under a variety of clinical conditions [29, 30], while LC antioxidative effects in hyperglycemic cardiomyocites were not investigated, yet.
The aim of this study is to investigate in vitro LC ability to counteract oxidative stress condition, modifying ROS production and promoting mitochondrial detoxifying function in H9c2 rat cardiomyocytes under hyperglycemic condition (25 mmol/L glucose). The goal is to identify in LC a novel adjuvant agent in cell therapy able to ameliorate the microenvironment of the hyperglycemic heart, thereby supporting cardiac progenitor cell proliferation and differentiation.
2. Materials and Methods
2.1. Chemicals and Reagents
All utilized reagents were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Primary antibodies against GAPDH (FL-335), p-CaMKII α (22B1), CaMKII (M-176), AMPK α1/2 (H-300), SOD2 (FL-222), p21 (C-19), peroxidase-conjugated secondary antibodies for Western blot analysis, and FITC-conjugated antibodies for immunofluorescence study were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). STAT3 (124H6) and p-STAT3 (Ser727) were purchased from Cell Signaling Technology (Danvers, MA, USA).
The CellROX® Oxidative Stress Reagent kit (C10443) was purchased from Thermo Fisher Scientific, Life Technologies Italia (Monza, Italy).
2.2. Cell Line and Culture Conditions
The American Type Culture Collection (ATCC, Manassas, VA, USA) offered the cardiomyoblast H9c2 cell line of rat embryo. The H9c2 cell line was regularly cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, Grand Island, NY, USA) containing 25 mmol/L glucose, 100 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO, USA), 100 U/mL penicillin, and 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA) in 75 cm2 tissue culture flasks and then incubated at 37°C by adopting 5% CO2. H9c2 cells exposed to high glucose (25 mmol/L) represent a validated in vitro model in order to mimic cardiac hyperglycemic/diabetic condition [31, 32]. Cells were fed every day; following literature indications, H9c2 cells were split when reaching 70–80% confluence in order to prevent the loss of the differentiation potential [28, 32]. Preliminarily, we evaluated the effective concentration of LC based on our previous work performed on skeletal myoblasts and literature evidence (data not shown) [28, 33]. We identified the correct LC concentrations as the lower concentrations that activated STAT3 signaling. Then, H9c2 cells in the active proliferation phase were treated with 0.5 or 5 mM LC as indicated in Figure 1.
Figure 1.
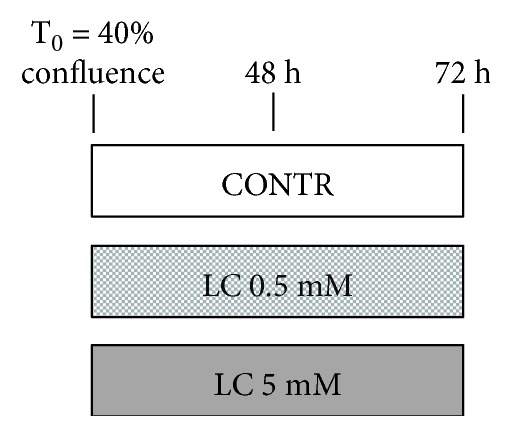
Scheme of treatment. Upon reaching 40% confluence, H9c2 cells were treated for 48 and 72 h with 0.5 or 5 mM LC.
2.3. Growth Curve and Cell Viability Test
H9c2 cells were plated on 60 mm × 15 mm culture dishes at 20% confluence and grown in DMEM. After an overnight phase, the cells were treated or not with 0.5 or 5 mM LC. At 24, 48, and 72 h after treatments, cells were trypsinized and stained with trypan blue and were counted using hemocytometer. The average values for each single day were used to plot a growth curve. Cell viability was calculated by dividing the unstained viable cell count by the total cell count. In addition, morphological changes were observed daily by phase contrast microscopy.
2.4. Western Blot Analysis
Western blot analysis was performed as described previously [34]. Cell lysates were prepared using RIPA buffer implemented with protease inhibitors. 30 μg of proteins was separated by SDS-polyacrylamide gel electrophoreses (SDS-PAGE) and electrophoretically transferred to nitrocellulose membranes (Protran®, Whatman® Schleicher & Schuell). The blots were then blocked and incubated with specific primary antibodies, followed by incubation with anti-species-specific secondary antibodies. To confirm equal protein loading per sample, we used GAPDH protein as housekeeping protein. Finally, detection of specific proteins was performed by enhanced chemoluminescence reagent (Western Lightning ECL Pro, Perkin Elmer). Quantitative measurement of immunoreactive band intensities was performed by densitometric analysis using the Scion Image software (Scion Corporation, Frederick, MD, USA). Data were then converted into fold changes (FC) of the control.
2.5. Immunofluorescence Studies
H9c2 cells were grown on coverslips with or without LC 0.5 or 5 mM LC. After 48 and 72 h of treatment, cells were washed 3 times with PBS, then fixed in prepared 4% paraformaldehyde for 20 minutes. The cells on the coverslips were washed with PBS and incubated for 30 minutes at room temperature with 1% bovine serum albumin in PBS with 0.2% Triton X-100. At that point, H9c2 cells were incubated with primary antibodies for 120 minutes. To detect the primary antibody, binding site cells were washed three times in PBS and followed by incubation with specific antibodies FITC-conjugated for 90 minutes. Nuclei were revealed with DAPI staining. Coverslips with cells were mounted and observed using Nikon Eclipse 50I microscopy. The images were captured using Nis-Elements D 4.00 software. Immunofluorescence signals were estimated using the ImageJ program (http://imagej.nih.gov/ij/). Data were displayed and analyzed using Adobe Photoshop CS4®.
Automated quantification on the immunofluorescence signal was performed by using the ImageJ program (http://imagej.nih.gov/ij/) [34]. For each analyzed protein, the quantified signal was normalized for the total nuclei number.
2.6. Intracellular ROS Determination in H9c2 Cells after H2O2 Injury
Intracellular ROS levels were valued using the CellROX® Oxidative Stress Reagent kit. Briefly, H9c2 cells were pretreated with 0.5 or 5 mM LC for 48 h, and then they were exposed to 500 μM H2O2 for 30 min. At the end of the H202 injury, the fluorogenic probe of the kit was added (Figure 2(a)). CellROX® Oxidative Stress Reagents are fluorogenic probes designed to reliably measure ROS in live cells. The cell-permeable reagents are nonfluorescent or very faintly fluorescent while in a reduced state and during oxidation exhibit a strong fluorogenic signal. The CellROX® Orange Reagent signal is localized in the cytoplasm.
Figure 2.

Attenuation of ROS production induced by H2O2 in LC-pretreated H9c2 cells. (a) Scheme of treatment: H9c2 cardiomyocytes were exposed to a stressful insult with 500 μM of H2O2 after pretreatment for 48 h with 0.5 or 5 mM LC. (b) Using the CellROX reagent assay, LC action was evaluated under oxidative stress conditions. The pretreatment with 0.5 or 5 mM LC causes a reduction in ROS production after stimulus with 500 μM of H2O2. (c) Quantification of ROS production. The quantified signal was normalized for the total nuclei number. From the DAPI images, it was possible to observe that the cells pretreated with both doses of LC show higher survival compared to the control cells. (d) Quantification of nuclei number pre- and after H2O2 injury in LC-pretreated H9c2 cells. Data are expressed as fold changes (FC) of mean ± SD. Significance: ∗∗p ≤ 0.01 vs. CONTR.
The images were captured using Nis-Elements D 4.00 software. Data were displayed and analyzed using Adobe Photoshop CS4®.
Automated quantification on the immunofluorescence signal was performed by using the ImageJ program (http://imagej.nih.gov/ij/) [35]. For each analyzed protein, the quantified signal was normalized for the total nuclei number.
2.7. Statistical Analysis
All experiments were performed three times. The data are expressed as the means ± standard deviation, and statistical comparisons were performed with specific statistical packages (Prism v 7.00 GraphPad Software, San Diego, CA, USA). Differences were analyzed by one- or two-way analysis of variance (ANOVA) followed by Tukey's multiple comparison post hoc test. p < 0.05 was considered statistically significant.
3. Results
3.1. Attenuation of ROS Production Induced by H2O2 in LC Pretreated H9c2 Cells
The diabetic heart usually has ROS levels that exceed normal quantities, and ROS overproduction likely contributes to cardiomyopathy [15, 36, 37]. After stimulus with 500 μM H2O2, 48 h pretreatment with 0.5 or 5 mM LC markedly reduced ROS levels with respect to the CONTR condition (Figures 2(b) and 2(c)). Furthermore, DAPI images showed that pretreated H9c2 cells with LC, in particular with 5 mM LC, showed a higher number of nuclei than the control condition (Figures 2(b) and 2(d)): These results suggested that LC pretreatment improved cellular survival after H2O2 injury.
3.2. LC Improves Antioxidant Response in H9c2 Cells under Hyperglycemic Condition
Signal transducer and activator of transcription 3 (STAT3) activation thought phosphorylation on Ser727 is an important protective mechanism to prevent ROS generation in the setting of oxidative stress [38–40]. Under normoxic conditions, the treatment with 0.5 or 5 mM LC for 48 and 72 h significantly enhanced myocardial STAT3 phosphorylation on Ser727 (Figure 3(a)).
Figure 3.

LC improves antioxidant response in H9c2 cells under hyperglycemic condition. (a) Representative Western blot and relevant quantification: LC stimuli for 48 and 72 h significantly enhanced STAT3 activation (ratio pSTAT3/STAT3). (b) Western blot analysis and relevant quantification: 5 mM LC improved SOD2 protein content in H9c2 cells after 48 and 72 h of treatment with respect to control cells. Data are expressed as fold changes (FC) of mean ± SD. Significance: ∗p ≤ 0.05 vs. CONTR; ∗∗p ≤ 0.01 vs. CONTR; §§p ≤ 0.01 vs. LC 0.5 mM.
Moreover, after 48 and 72 h of treatment, exposure of cells to 0.5 mM LC caused rapid increase of SOD2, a well-known antioxidant enzyme [41]. This effect was significantly augmented with the 5 mM LC dose (Figure 3(b)).
3.3. LC Downregulation of CaMKII Pathway
Ca2+/calmodulin-dependent protein kinase II (CaMKII), a multifunctional serine/threonine protein kinase, is implicated in the pathogenesis of cardiac diseases [42, 43] promoting ROS overproduction [44, 45]. As shown by immunofluorescence analysis (Figure 4(a)), compared with untreated H9c2 cells, LC treatments decreased CaMKII and pCaMKII-positive cell numbers after 72 h of stimuli: immunofluorescence quantification shows a greater effect with the 5 mM LC dose. Furthermore, after 72 h, either 0.5 or 5 mM doses of LC significantly decreased the phosphorylation of CaMKII α isoform, as shown by Western blot analysis (Figure 4(b)).
Figure 4.

LC downregulation of CaMKII pathway. (a) LC treatments decreased CaMKII-positive cell number and its α phosphorylated form after 72 h of stimuli: this effect was more marked for the 5 mM LC dose. The quantified signal was normalized for the total nuclei number. (b) Representative Western blot and relevant quantification: LC stimuli for 72 h promote CaMKII α isoform activation. Data are expressed as fold changes (FC) of mean ± SD. Significance: ∗p ≤ 0.05 vs. CONTR.
3.4. LC Action on AMPK Protein Expression
5′-AMP-activated kinase (AMPK) has become a strategic cellular target for the cure of cardiovascular disease correlated with DM [46–48]: it is likely that AMPK activity in the diabetic heart may ameliorate cardiac function. After LC stimuli, immunofluorescence for AMPK showed an increase in AMPK-positive cells in a dose- and time-dependent manner (Figure 5).
Figure 5.

LC action on AMPK protein expression. Immunofluorescence assay and relevant quantification showed an increase in the number of AMPK-positive cells in LC condition at 48 and 72 h. The quantified signal was normalized for the total nuclei number. Data are expressed as fold changes (FC) of mean ± SD. Significance: ∗∗∗p ≤ 0.001 vs. CONTR; §§§p ≤ 0.001 vs. LC 5 mM.
3.5. LC Action on Cardiomyocyte Viability
An uncontrolled consequence of sustained hyperglycemia is the induction of cardiomyocyte death that causes a loss of contractile units, which declines organ function and provokes hypertrophy of vital cardiomyocytes [49–51]. We investigated viability and morphologic features of H9c2 cells after exposure to 0.5 or 5 mM LC (Figure 6(a)). The growth curve showed that LC treatments did not induce a change of cellular proliferation with respect to untreated control cells (Figure 6(b)). Moreover, the viability graph showed the absence of cell mortality in all treatment conditions (Figure 6(b)). To support these data, phase contrast images, collected at day 3 of the growth curve, confirmed the absence of morphological changes in cells treated with 0.5 or 5 mM LC respect to control (Figure 6(c)).
Figure 6.

LC action on cardiomyocyte viability. (a) Experimental scheme for growth curve and viability determination. (b) 0.5 or 5 mM LC did not alter the H9c2 proliferative potential. Viability graph shows the absence of cell mortality in all treatment conditions. (c) Phase contrast images show how treatment with 0.5 or 5 mM LC do not modify the morphology of H9c2 cells. Furthermore, there is no p21 translocation from cytoplasm to nuclei following treatment with both doses of LC. Data are expressed as fold changes (FC) of mean ± SD.
In opposition to its antiproliferative functions, p21 can also play proproliferative and survival roles when it is localized in the cytosol [51]. Figure 6(c) shows that the protein content of p21 in cardiomyocytes treated with 0.5 or 5 mM LC was superimposable to control cells, and this result highlights that there is not p21 translocation from cytoplasm to nuclei.
4. Discussion
In the present study, we demonstrated that LC supplementation significantly decreases ROS production in cardiomyocytes during hyperglycemia (Figure 2). ROS generation induced by high glucose causes apoptosis of cardiac cells and important decrease in growth factor secretion [15, 16, 52–55]. In the heart microenvironment, the oxidative stress induced by hyperglycemia can lead to stem cell senescence which is characterized by the production and secretion of soluble factor SASP (senescence-associated secretory phenotype). Those factors, responsible for the onset of chronic inflammation and oxidative stress, are considered the pathophysiological link between aging and diabetes in cardiovascular diseases [56].
The association hyperglycemia/ROS/cellular senescence represents a major cause of inefficiency of regenerative medicine [9, 10, 56, 57]. Further, clinical trials established that an altered glycemic control is equal to therapeutic failure [13, 14]. Our data point out that LC could counteract oxidative stress in the hyperglycemic condition. Those observations are consistent with several previous works that reported that LC stimuli decrease ROS generation in the skeletal muscle, bone and cardiac cells grown under normoglycemic condition [26, 27, 58, 59]. Furthermore, as shown in Figure 6, both doses of LC did not modify the H9c2 growth rate and did not induce cellular damages.
Studying the mechanism by which LC may modulate ROS formation, we observed an increase in the serine 727 (Ser727) phosphorylation of STAT3, after 48 and 72 hours of treatment (Figure 3(a)). Recent evidence stresses the critical role of STAT3 in modulating mitochondrial respiratory chain function and ROS production [39, 40]. Serine 727 (Ser727) phosphorylation has a primary part in STAT3 influence on mitochondria: its decrement is associated with the development of cardiac hypertrophy and dilated cardiomyopathy [60]. Moreover, several data showed that STAT3 conserves complex I activity in ischemic condition enhancing cell viability and plays an important role in heart protection from chronic stress induced by hyperglycemia [61]. STAT3 activation promotes the expression of SOD2, the principal enzyme effective in reducing mitochondrial oxidative species [41, 62]. LC treatment significantly increased SOD2 expression in in vitro cardiomyocytes, confirming LC capability to counteract oxidative stress in the hyperglycemic condition (Figure 3(b)).
Interestingly, a number of studies have indicated that CaMKII enhances mitochondrial dysfunction, ROS formation, and apoptosis [63], eventually causing cardiomyocyte death both following hyperglycemia [64, 65] and oxidative stress conditions [43, 45, 66].
Hyperglycemic cardiac cells are characterized by a vicious circle between ROS/mitochondrial dysfunction/CaMKII that triggers cellular damages and apoptosis phenomena [42–44]. Taking into consideration that literature evidence, we investigated LC action on CaMKII protein content. As reported in Figure 4, LC treatments reduce CaMKII activation. Remarkably, recent experiments hypothesized that CaMKII activation is related to Ca2+ movement from the endoplasmic reticulum to mitochondria.
Recently, LC was shown to promote Ca2+ availability which is needed for proliferation and differentiation of human osteoblast-like cells, via a depolarization of L-type calcium channels [59]. Despite the Ca2+ supply via the L-type channels being essential to ensure appropriate cardiac cell contraction, an excessive Ca2+ influx increases mitochondrial ROS production in cardiomyocytes under oxidative stress. Various authors suggested that cardiac damage, induced by CaMKII activation, is caused by an increase in L-type Ca2+ current [67].
LC capability of modulating CaMKII in cardiac cells could make LC a molecule appropriate for impeding the cross-talk between L-type Ca2+ channels and ROS production and restoring normal mitochondrial function in cardiac cells. Further investigations are necessary to unravel LC action on cardiac calcium channels.
AMPK represents a strategic target in cardiovascular alterations associated with DM. It is well known that impairment of AMPK activation characterizes hyperglycemic cardiomyocytes. In particular, during hypoxia/reoxygenation (H/R) injury in diabetic patients, AMPK deficiency is associated with an increase in ROS production and apoptosis [46, 47]. As shown in Figure 5, LC stimuli enhanced AMPK protein synthesis, suggesting that LC not only could ameliorate the oxidative microenvironment but also could improve metabolic functions of cardiac cells. With this in mind, future studies could investigate LC's possible action on the protein kinase C (PKC) signaling pathway, implicated in diabetic damage. As known, in hyperglycemic condition, the decrease in AMPK activity is associated with an increase of diacylglycerol (DAG) production that activates PKC and NADPH-oxidase causing an abnormal production of ROS [68, 69]. Then, LC ability to stimulate AMPK activation and to regulate the production of acyl-CoA could counteract hyperinsulinemia-correlated oxidative stress.
Whereas the higher risk of developing atrial fibrillation (AF) in diabetic subjects compared to healthy ones is associated with mitochondrial ROS overproduction, ATP depletion, and abnormal calcium homeostasis [70, 71], the key modulator role shown by LC suggests the potential use of this micronutrient as adjuvant therapy in different cardiac pathologies associated with DM.
In conclusion, LC described effects could be useful in association with other cardioactive drugs. Ranolazine is an antianginal drug with hypoglycemic action used in AF treatment, which was shown to enhance skeletal muscle differentiation [35]. In cardiac regenerative medicine, antioxidative LC action, in association with ranolazine, might lead to the improvement of the hyperglycemic-oxidative microenvironment, prevention of apoptosis, and preservation of cell viability. Equally interesting could be the synergistic effect of LC with allopurinol [72] or the α-lipoic acid antioxidant [73] in the prevention and treatment of AF. Remarkable therapeutic perspectives could arise from the association of LC with new hypoglycemic drugs, such as incretins, whose effects on the hyperglycemia control, inflammation, and atherosclerotic plaque progression have recently been demonstrated [74, 75].
Taken together, our results argue that the use of LC as a coadjuvant in therapeutic treatments is very promising, although extensive studies focused on the determination of the effective LC dose and route of administration in humans should be undertaken. Anyway, LC doses used in our study (0.5–5 mM) are compatible with ranges used in humans receiving LC therapy [76, 77].
5. Conclusion
The results of this work (Table 1) provide, for the first time, fundamental in vitro cellular evidence that treatment with LC could be a potential strategy to improve the cardiac oxidative microenvironment caused by hyperglycemia.
Table 1.
Summary of LC action on hyperglycemic cardiomyocytes (H9c2).
| Hyperglycemic cardiomyocites (H9c2) | |||||
|---|---|---|---|---|---|
| L-Carnitine treatment | ROS | STAT3 | CaMKII | AMPK | Cellular growth and vitality |
|
|
|
|

|
|
According to the findings of the present study, demonstrating the LC ability to decrease production of ROS, activate STAT3 and AMPK, and downregulate CaMKII, LC could represent a potential adjuvant therapeutic strategy in diabetic cardiac regenerative medicine.
This adjuvant therapy might be recommended for diabetic patients with high risk of cardiac injury as it exerts a cardioprotective effect by restoring the microenvironmental equilibrium by strengthening cardiomyocytes' defense mechanisms through the stimulation of endogenous antioxidants (SOD2).
Further, in vitro and in vivo studies on this topic are essential to improve the knowledge on the effects of LC in cardiac diseases associated with diabetes and on its potential synergic action with other antioxidative and hypoglycemic agents.
Acknowledgments
This study was partially supported by the Ricerca Corrente funding from the Italian Ministry of Health to IRCCS Policlinico San Donato.
Data Availability
The authors confirm that the data supporting the findings of this study are openly available within the article.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
FV, PS, and AM are coauthors and contributed equally to this work.
References
- 1.Garcia M. J., McNamara P. M., Gordon T., Kannell W. B. Morbidity and mortality in diabetics in the Framingham population: sixteen year follow-up study. Diabetes. 1974;23(2):105–111. doi: 10.2337/diab.23.2.105. [DOI] [PubMed] [Google Scholar]
- 2.Jia G., Whaley-Connell A., Sowers J. R. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia. 2018;61(1):21–28. doi: 10.1007/s00125-017-4390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33(Supplement 1):S62–S69. doi: 10.2337/dc10-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pickering R. J., Rosado C. J., Sharma A., Buksh S., Tate M., de Haan J. B. Recent novel approaches to limit oxidative stress and inflammation in diabetic complications. Clinical & Translational Immunology. 2018;7(4, article e1016) doi: 10.1002/cti2.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 6.Balestrieri M. L., Rizzo M. R., Barbieri M., et al. Sirtuin 6 expression and inflammatory activity in diabetic atherosclerotic plaques: effects of incretin treatment. Diabetes. 2015;64(4):1395–1406. doi: 10.2337/db14-1149. [DOI] [PubMed] [Google Scholar]
- 7.Tajik A. A., Dobre D., Aguilar D., et al. A history of diabetes predicts outcomes following myocardial infarction: an analysis of the 28 771 patients in the high-risk MI database. European Journal of Heart Failure. 2017;19(5):635–642. doi: 10.1002/ejhf.797. [DOI] [PubMed] [Google Scholar]
- 8.Jorsal A., Wiggers H., McMurray J. J. V. Heart failure: epidemiology, pathophysiology, and management of heart failure in diabetes mellitus. Endocrinology and Metabolism Clinics of North America. 2018;47(1):117–135. doi: 10.1016/j.ecl.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Lin Z., Pu W. T. Strategies for cardiac regeneration and repair. Science Translational Medicine. 2014;6(239, article 239rv1) doi: 10.1126/scitranslmed.3006681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satthenapalli V. R., Lamberts R. R., Katare R. G. Concise review: challenges in regenerating the diabetic heart: a comprehensive review. Stem Cells. 2017;35(9):2009–2026. doi: 10.1002/stem.2661. [DOI] [PubMed] [Google Scholar]
- 11.Sanganalmath S. K., Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circulation Research. 2013;113(6):810–834. doi: 10.1161/CIRCRESAHA.113.300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marfella R., Rizzo M. R., Siniscalchi M., et al. Peri-procedural tight glycemic control during early percutaneous coronary intervention up-regulates endothelial progenitor cell level and differentiation during acute ST-elevation myocardial infarction: effects on myocardial salvage. International Journal of Cardiology. 2013;168(4):3954–3962. doi: 10.1016/j.ijcard.2013.06.053. [DOI] [PubMed] [Google Scholar]
- 13.Cai L., Keller B. B. Cardiac regeneration and diabetes. Regenerative Medicine Research. 2014;2(1):p. 1. doi: 10.1186/2050-490X-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ofstad A. P., Atar D., Gullestad L., Langslet G., Johansen O. E. The heart failure burden of type 2 diabetes mellitus—a review of pathophysiology and interventions. Heart Failure Reviews. 2018;23(3):303–323. doi: 10.1007/s10741-018-9685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volpe C. M. O., Villar-Delfino P. H., dos Anjos P. M. F., Nogueira-Machado J. A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death & Disease. 2018;9(2):p. 119. doi: 10.1038/s41419-017-0135-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faria A., Persaud S. J. Cardiac oxidative stress in diabetes: mechanisms and therapeutic potential. Pharmacology & Therapeutics. 2017;172:50–62. doi: 10.1016/j.pharmthera.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Shah M. S., Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circulation Research. 2016;118(11):1808–1829. doi: 10.1161/CIRCRESAHA.116.306923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diogo C. V., Suski J. M., Lebiedzinska M., et al. Cardiac mitochondrial dysfunction during hyperglycemia--the role of oxidative stress and p66Shc signaling. The International Journal of Biochemistry & Cell Biology. 2013;45(1):114–122. doi: 10.1016/j.biocel.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Salpea K. D., Maubaret C. G., Kathagen A., Ken-Dror G., Gilroy D. W., Humphries S. E. The effect of pro-inflammatory conditioning and/or high glucose on telomere shortening of aging fibroblasts. PLoS One. 2013;8(9, article e73756) doi: 10.1371/journal.pone.0073756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z. Y., Liu Y. Y., Liu G. H., Lu H. B., Mao C. Y. L-Carnitine and heart disease. Life Sciences. 2018;194:88–97. doi: 10.1016/j.lfs.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 21.Dinicolantonio J. J., Niazi A. K., McCarty M., Lavie C. J., Liberopoulos E., O'Keefe J. H. L-Carnitine for the treatment of acute myocardial infarction. Reviews in Cardiovascular Medicine. 2014;15(1):52–62. doi: 10.3909/ricm0710. [DOI] [PubMed] [Google Scholar]
- 22.DiNicolantonio J. J., Lavie C. J., Fares H., Menezes A. R., O'Keefe J. H. L-Carnitine in the secondary prevention of cardiovascular disease: systematic review and meta-analysis. Mayo Clinic Proceedings. 2013;88(6):544–551. doi: 10.1016/j.mayocp.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Fiorentino T. V., Prioletta A., Zuo P., Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Current Pharmaceutical Design. 2013;19(32):5695–5703. doi: 10.2174/1381612811319320005. [DOI] [PubMed] [Google Scholar]
- 24.da Silva Guimarães S., de Souza Cruz W., da Silva L., et al. Effect of L-carnitine supplementation on reverse remodeling in patients with ischemic heart disease undergoing coronary artery bypass grafting: a randomized, placebo-controlled trial. Annals of Nutrition & Metabolism. 2017;70(2):106–110. doi: 10.1159/000465531. [DOI] [PubMed] [Google Scholar]
- 25.Wong A. P., Niedzwiecki A., Rath M. Myocardial energetics and the role of micronutrients in heart failure: a critical review. American Journal of Cardiovascular Disease. 2016;6(3):81–92. [PMC free article] [PubMed] [Google Scholar]
- 26.Xue M., Chen X., Guo Z., et al. L-Carnitine attenuates cardiac dysfunction by ischemic insults through Akt signaling pathway. Toxicological Sciences. 2017;160(2):341–350. doi: 10.1093/toxsci/kfx193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanca A. J., Ruiz-Armenta M. V., Zambrano S., et al. Inflammatory and fibrotic processes are involved in the cardiotoxic effect of sunitinib: protective role of L-carnitine. Toxicology Letters. 2016;241:9–18. doi: 10.1016/j.toxlet.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Mao C. Y., Lu H. B., Kong N., et al. Levocarnitine protects H9c2 rat cardiomyocytes from H2O2-induced mitochondrial dysfunction and apoptosis. International Journal of Medical Sciences. 2014;11(11):1107–1115. doi: 10.7150/ijms.9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrari R., Merli E., Cicchitelli G., Mele D., Fucili A., Ceconi C. Therapeutic effects of L-carnitine and propionyl-L-carnitine on cardiovascular diseases: a review. Annals of the New York Academy of Sciences. 2004;1033(1):79–91. doi: 10.1196/annals.1320.007. [DOI] [PubMed] [Google Scholar]
- 30.Lango R., Smolenski R. T., Narkiewicz M., Suchorzewska J., Lysiak-Szydlowska W. Influence of L-carnitine and its derivatives on myocardial metabolism and function in ischemic heart disease and during cardiopulmonary bypass. Cardiovascular Research. 2001;51(1):21–29. doi: 10.1016/S0008-6363(01)00313-3. [DOI] [PubMed] [Google Scholar]
- 31.Zhang C., Zhang L., Chen S., et al. The prevention of diabetic cardiomyopathy by non-mitogenic acidic fibroblast growth factor is probably mediated by the suppression of oxidative stress and damage. PLoS One. 2013;8(12, article e82287) doi: 10.1371/journal.pone.0082287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun S., Yang S., Dai M., et al. The effect of Astragalus polysaccharides on attenuation of diabetic cardiomyopathy through inhibiting the extrinsic and intrinsic apoptotic pathways in high glucose-stimulated H9C2 cells. BMC Complementary and Alternative Medicine. 2017;17(1):p. 310. doi: 10.1186/s12906-017-1828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Montesano A., Senesi P., Luzi L., Benedini S., Terruzzi I. Potential therapeutic role of L-carnitine in skeletal muscle oxidative stress and atrophy conditions. Oxidative Medicine and Cellular Longevity. 2015;2015:13. doi: 10.1155/2015/646171.646171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villa I., Senesi P., Montesano A., et al. Betaine promotes cell differentiation of human osteoblasts in primary culture. Journal of Translational Medicine. 2017;15(1):p. 132. doi: 10.1186/s12967-017-1233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terruzzi I., Montesano A., Senesi P., Vacante F., Benedini S., Luzi L. Erratum to: Ranolazine promotes muscle differentiation and reduces oxidative stress in C2C12 skeletal muscle cells. Endocrine. 2017;58(1):p. 46. doi: 10.1007/s12020-017-1243-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen E., Li Y., Li Y., et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. 2009;58(10):2386–2395. doi: 10.2337/db08-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Modesti A., Bertolozzi I., Gamberi T., et al. Hyperglycemia activates JAK2 signaling pathway in human failing myocytes via angiotensin II-mediated oxidative stress. Diabetes. 2005;54(2):394–401. doi: 10.2337/diabetes.54.2.394. [DOI] [PubMed] [Google Scholar]
- 38.Meier J. A., Larner A. C. Toward a new STATe: the role of STATs in mitochondrial function. Seminars in Immunology. 2014;26(1):20–28. doi: 10.1016/j.smim.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szczepanek K., Chen Q., Derecka M., et al. Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. The Journal of Biological Chemistry. 2011;286(34):29610–29620. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meier J. A., Hyun M., Cantwell M., et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Science Signaling. 2017;10(472, article eaag2588) doi: 10.1126/scisignal.aag2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen X., Zheng S., Metreveli N. S., Epstein P. N. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55(3):798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 42.Couchonnal L. F., Anderson M. E. The role of calmodulin kinase II in myocardial physiology and disease. Physiology. 2008;23(3):151–159. doi: 10.1152/physiol.00043.2007. [DOI] [PubMed] [Google Scholar]
- 43.Luczak E. D., Anderson M. E. CaMKII oxidative activation and the pathogenesis of cardiac disease. Journal of Molecular and Cellular Cardiology. 2014;73:112–116. doi: 10.1016/j.yjmcc.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Odagiri K., Katoh H., Kawashima H., et al. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. Journal of Molecular and Cellular Cardiology. 2009;46(6):989–997. doi: 10.1016/j.yjmcc.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 45.Singh M. V., Kapoun A., Higgins L., et al. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. The Journal of Clinical Investigation. 2009;119(4):986–996. doi: 10.1172/JCI35814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruderman N. B., Carling D., Prentki M., Cacicedo J. M. AMPK, insulin resistance, and the metabolic syndrome. The Journal of Clinical Investigation. 2013;123(7):2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beauloye C., Bertrand L., Horman S., Hue L. AMPK activation, a preventive therapeutic target in the transition from cardiac injury to heart failure. Cardiovascular Research. 2011;90(2):224–233. doi: 10.1093/cvr/cvr034. [DOI] [PubMed] [Google Scholar]
- 48.Chang W., Zhang M., Li J., et al. Berberine improves insulin resistance in cardiomyocytes via activation of 5′-adenosine monophosphate-activated protein kinase. Metabolism. 2013;62(8):1159–1167. doi: 10.1016/j.metabol.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 49.Leonardini A., Avogaro A. Abnormalities of the cardiac stem and progenitor cell compartment in experimental and human diabetes. Archives of Physiology and Biochemistry. 2013;119(4):179–187. doi: 10.3109/13813455.2013.798334. [DOI] [PubMed] [Google Scholar]
- 50.Cai L., Kang Y. J. Cell death and diabetic cardiomyopathy. Cardiovascular Toxicology. 2003;3(3):219–228. doi: 10.1385/CT:3:3:219. [DOI] [PubMed] [Google Scholar]
- 51.Child E. S., Mann D. J. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5(12):1313–1319. doi: 10.4161/cc.5.12.2863. [DOI] [PubMed] [Google Scholar]
- 52.Wu H., Cai L., de Haan J. B., Giacconi R. Targeting oxidative stress in diabetic complications: new insights. Journal of Diabetes Research. 2018;2018:2. doi: 10.1155/2018/1909675.1909675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masi S., D’Aiuto F., Cooper J., et al. Telomere length, antioxidant status and incidence of ischaemic heart disease in type 2 diabetes. International Journal of Cardiology. 2016;216:159–164. doi: 10.1016/j.ijcard.2016.04.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lal N., Chiu A. P. L., Wang F., et al. Loss of VEGFB and its signaling in the diabetic heart is associated with increased cell death signaling. American Journal of Physiology-Heart and Circulatory Physiology. 2017;312(6):H1163–H1175. doi: 10.1152/ajpheart.00659.2016. [DOI] [PubMed] [Google Scholar]
- 55.Aliwarga T., Evangelista E., Sotoodehnia N., Lemaitre R., Totah R. Regulation of CYP2J2 and EET levels in cardiac disease and diabetes. International Journal of Molecular Sciences. 2018;19(7, article 1916) doi: 10.3390/ijms19071916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shakeri H., Lemmens K., Gevaert A. B., de Meyer G. R. Y., Segers V. F. M. Cellular senescence links aging and diabetes in cardiovascular disease. American Journal of Physiology-Heart and Circulatory Physiology. 2018;315(3):H448–H462. doi: 10.1152/ajpheart.00287.2018. [DOI] [PubMed] [Google Scholar]
- 57.Molgat A. S. D., Tilokee E. L., Rafatian G., et al. Hyperglycemia inhibits cardiac stem cell–mediated cardiac repair and angiogenic capacity. Circulation. 2014;130(11_Supplement_1):S70–S76. doi: 10.1161/CIRCULATIONAHA.113.007908. [DOI] [PubMed] [Google Scholar]
- 58.Le Borgne F., Ravaut G., Bernard A., Demarquoy J. L-Carnitine protects C2C12 cells against mitochondrial superoxide overproduction and cell death. World Journal of Biological Chemistry. 2017;8(1):86–94. doi: 10.4331/wjbc.v8.i1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferraretto A., Bottani M., Villa I., et al. L-Carnitine activates calcium signaling in human osteoblasts. Journal of Functional Foods. 2018;47:270–278. doi: 10.1016/j.jff.2018.05.068. [DOI] [Google Scholar]
- 60.Camporeale A., Marino F., Papageorgiou A., et al. STAT3 activity is necessary and sufficient for the development of immune-mediated myocarditis in mice and promotes progression to dilated cardiomyopathy. EMBO Molecular Medicine. 2013;5(4):572–590. doi: 10.1002/emmm.201201876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H., Yao W., Liu Z., et al. Hyperglycemia abrogates ischemic postconditioning cardioprotection by impairing AdipoR1/Caveolin-3/STAT3 signaling in diabetic rats. Diabetes. 2016;65(4):942–955. doi: 10.2337/db15-0782. [DOI] [PubMed] [Google Scholar]
- 62.Luo S., Gu X., Ma F., et al. ZYZ451 protects cardiomyocytes from hypoxia-induced apoptosis via enhancing MnSOD and STAT3 interaction. Free Radical Biology & Medicine. 2016;92:1–14. doi: 10.1016/j.freeradbiomed.2015.12.026. [DOI] [PubMed] [Google Scholar]
- 63.Feng N., Anderson M. E. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. Journal of Molecular and Cellular Cardiology. 2017;103:102–109. doi: 10.1016/j.yjmcc.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu W., Tsang S., Browe D. M., et al. Interaction of β1-adrenoceptor with RAGE mediates cardiomyopathy via CaMKII signaling. JCI Insight. 2016;1(1, article e84969) doi: 10.1172/jci.insight.84969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo M., Guan X., Luczak E. D., et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. The Journal of Clinical Investigation. 2013;123(3):1262–1274. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Timmins J. M., Ozcan L., Seimon T. A., et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. The Journal of Clinical Investigation. 2009;119(10):2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tonegawa K., Otsuka W., Kumagai S., et al. Caveolae-specific activation loop between CaMKII and L-type Ca2+ channel aggravates cardiac hypertrophy in α1-adrenergic stimulation. American Journal of Physiology. Heart and Circulatory Physiology. 2017;312(3):H501–H514. doi: 10.1152/ajpheart.00601.2016. [DOI] [PubMed] [Google Scholar]
- 68.Kraegen E. W., Saha A. K., Preston E., et al. Increased malonyl-CoA and diacylglycerol content and reduced AMPK activity accompany insulin resistance induced by glucose infusion in muscle and liver of rats. American Journal of Physiology. Endocrinology and Metabolism. 2006;290(3):E471–E479. doi: 10.1152/ajpendo.00316.2005. [DOI] [PubMed] [Google Scholar]
- 69.Itani S. I., Ruderman N. B., Schmieder F., Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002;51(7):2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 70.Xie W., Santulli G., Reiken S. R., et al. Mitochondrial oxidative stress promotes atrial fibrillation. Scientific Reports. 2015;5(1, article 11427) doi: 10.1038/srep11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gambardella J., Sorriento D., Ciccarelli M., et al. Functional role of mitochondria in arrhythmogenesis. Advances in Experimental Medicine and Biology. 2017;982:191–202. doi: 10.1007/978-3-319-55330-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Y., Zhao J., Qiu J., et al. Xanthine oxidase inhibitor allopurinol prevents oxidative stress‐mediated atrial remodeling in alloxan‐induced diabetes mellitus rabbits. Journal of the American Heart Association. 2018;7(10) doi: 10.1161/JAHA.118.008807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sardu C., Santulli G., Santamaria M., et al. Effects of alpha lipoic acid on multiple cytokines and biomarkers and recurrence of atrial fibrillation within 1 year of catheter ablation. The American Journal of Cardiology. 2017;119(9):1382–1386. doi: 10.1016/j.amjcard.2017.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marfella R., Sardu C., Balestrieri M. L., et al. Effects of incretin treatment on cardiovascular outcomes in diabetic STEMI-patients with culprit obstructive and multivessel non obstructive-coronary-stenosis. Diabetology and Metabolic Syndrome. 2018;10(1):p. 1. doi: 10.1186/s13098-017-0304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marfella R., Sardu C., Calabrò P., et al. Non-ST-elevation myocardial infarction outcomes in patients with type 2 diabetes with non-obstructive coronary artery stenosis: effects of incretin treatment. Diabetes, Obesity and Metabolism. 2018;20(3):723–729. doi: 10.1111/dom.13122. [DOI] [PubMed] [Google Scholar]
- 76.Fielding R., Riede L., Lugo J., Bellamine A. l-Carnitine supplementation in recovery after exercise. Nutrients. 2018;10(3):p. 349. doi: 10.3390/nu10030349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sawicka A., Hartmane D., Lipinska P., Wojtowicz E., Lysiak-Szydlowska W., Olek R. l-Carnitine supplementation in older women. A pilot study on aging skeletal muscle mass and function. Nutrients. 2018;10(2):p. 255. doi: 10.3390/nu10020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are openly available within the article.