

Abstract
Background
Historically, three founder mutations in the BRCA1/2 (OMIM 113705; OMIM 600185) genes have been the focus of cancer risks within the Ashkenazi Jewish (AJ) population. However, there are several additional mutations associated with increased susceptibility to cancer in individuals of AJ ancestry.
Methods
We report three patients who exemplify the need to keep these additional founder mutations in mind when pursuing hereditary cancer genetic testing of individuals in this population. All gene sequences in this paper were aligned to reference sequences based on human genome build GRCh37/UCSC hg19.
Results
review of the literature discusses that the combined risk is 12.36%–20.83% forhaving 1 of the 10 hereditary cancer AJ founder mutations in the BRCA1, BRCA2, CHEK2 (OMIM 604373), APC (OMIM 611731), MSH2 (OMIM 609309), MSH6 (OMIM 600678), and GREM1 (OMIM 603054) genes for individuals of AJ ancestry.
Conclusion
We recommend testing for all 10 of these AJ founder cancer susceptibility mutations for individuals within this population as standard screening in order to ensure appropriate cancer risk management and cascade testing.
Keywords: APC, Ashkenazi, BRCA1, BRCA2, breast cancer, cascade testing, CHEK2, colon cancer, founder mutations, GREM1, Jewish, MSH2, MSH6
1. INTRODUCTION
Individuals of Ashkenazi Jewish (AJ) ancestry are at increased risks for carrying certain genetic mutations due to the founder effect. Founder mutations can occur in isolated populations with limited influx of new genetic variants, leading to an increased prevalence of rare mutations (Ferla et al., 2007). The AJ population has multiple known founder mutations associated with hereditary conditions in the pediatric population which are typically evaluated through prenatal, pediatric, and adult genetics clinics. However, this paper focuses on genes associated with hereditary cancer risks (BRCA1, BRCA2, CHEK2, APC, MSH2, MSH6, and GREM1) that do not have pathognomonic features (i.e., the BLM gene which is associated with growth deficiency and a butterfly rash). Recognizing that an individual coming to a hereditary cancer clinic is of AJ descent can lead genetic counselors and other healthcare providers to offer genetic testing for all of the relevant AJ hereditary cancer founder mutations.
2. BREAST CANCER AND AJ ANCESTRY
Historically, individuals with AJ ancestry were offered BRCA1/2 genetic analysis for three founder mutations, and if one of these was found, this was followed by site specific testing of other family members. These AJ founder mutations include two pathogenic mutations of the BRCA1 gene (c.185delAG and c.5382insC) and one pathogenic mutation within the BRCA2 gene (c.6174delT), these three mutations are found in approximately 1 in 40 individuals of AJ ancestry (Bahar et al., 2001; Ferla et al., 2007; Frank et al., 2002; Rosenthal, Moyes, Arnell, Evans, & Wenstrup, 2015).
One in 40 AJ individuals will have one of the three BRCA1/2 founder mutations. These three mutations have an approximate 2%–3% frequency in AJ individuals : the BRCA1 185delAG mutation has a reported frequency of 0.96%–1.14%, the BRCA1 5382insC mutation has a 0.13%–0.28% frequency and BRCA2 6174delT has a 0.6%–1.52% frequency (Bahar et al., 2001; Ferla et al., 2007). Given the high frequency of these three founder mutations in the AJ population, testing for all three mutations is always recommended, even when a family has another known pathogenic hereditary cancer mutation.
With the advent of next‐generation panels which can perform analysis of multiple genes at once, sequencing and deletion duplication analysis of all variants of the BRCA1/2 genes are now often offered to those with AJ ancestry. This testing can also include full analysis of other genes associated with hereditary breast cancer as well as genes with increased susceptibility to hereditary colon and other cancers.
There are also two AJ founder mutations in the CHEK2 gene. Routine analysis of the CHEK2 gene was initially limited to the c.1100delC mutation in this gene. Current screening guidelines for those who are gene positive refer primarily to this specific mutation which has a 0.06% frequency in the Caucasian population (Leedom et al., 2016). The frequency of this mutation has been seen equally in AJ and non‐AJ individuals (Offit et al., 2003; Shaag et al., 2005); and thus does not appear to have a founder effect in the AJ population.
However another CHEK2 mutation, the c.1283C>T (p.Ser428Phe) mutation does have an increased frequency in the AJ population of between 2.4% and 5% (Laitman, Kaufman, Lahad, Papa, & Friedman, 2007; Leedom et al., 2016; Shaag et al., 2005; Walsh et al., 2017). Shaag et al. (2005) concluded that the S428F mutation conferred a 2‐fold increased breast cancer risk among AJ women. A second CHEK2 mutation with an increased frequency within the AJ population is the c.470T>C (p.Ile157Thr) mutation which is found in 0.7% of the general Caucasian population, but it is reported in 0.46%–1.2% of the AJ population (Laitman et al., 2007; Leedom et al., 2016). Leedom et al. (2016) also concluded that the Ile157Thr mutation is a moderate risk mutation which confers a 1.5 fold increased risk for breast cancer compared to other CHEK2 mutations. These two CHEK2 founder mutations have an approximate 3%–4% frequency in the AJ population equating to approximately 1 in 30 AJ individuals having one of these two mutations. The National Comprehensive Cancer Network's (NCCN) guidelines regarding cancer risk and management for the CHEK2 gene are based on frameshift mutations (Daly et al., 2018; Provenzale et al., 2017). The risks for missense mutations are unclear, but the breast cancer risk with the Ile157Thr mutation appears to be lower. Since the NCCN guidelines have not been established for all variants of CHEK2 mutations, it may be prudent to consider screening recommendations for the known mutation, with consideration of personal and family history. Knowing which individuals inherited an AJ CHEK2 founder mutation could help determine which family members could benefit from increased screening or surveillance. This knowledge will also become impactful in the future when further information regarding non frameshift pathogenic CHEK2 mutation management guidelines become available.
Our first patient was a 63 year old male with no personal history of cancer who presented to our clinic with a family history of a pathogenic CHEK2 mutation (Figure 1). The patient's 61 year old sister was diagnosed with breast cancer at 51 years of age, she had previous multi‐gene panel testing performed, which showed the c.1283C>T pathogenic AJ founder mutation in the CHEK2 gene. The relevant family history included a mother diagnosed with breast cancer at 45 years of age who had passed away and both maternal and paternal AJ lineages. We pursued BRCA1, BRCA2 and CHEK2 gene testing through Invitae Laboratory which found the c.1283C>T pathogenic CHEK2 mutation as well as a c.5946delT pathogenic BRCA2 mutation (aka 6174delT). If testing had pursued only the known familial pathogenic mutation this additional BRCA2 founder mutation would have been missed.
Figure 1.
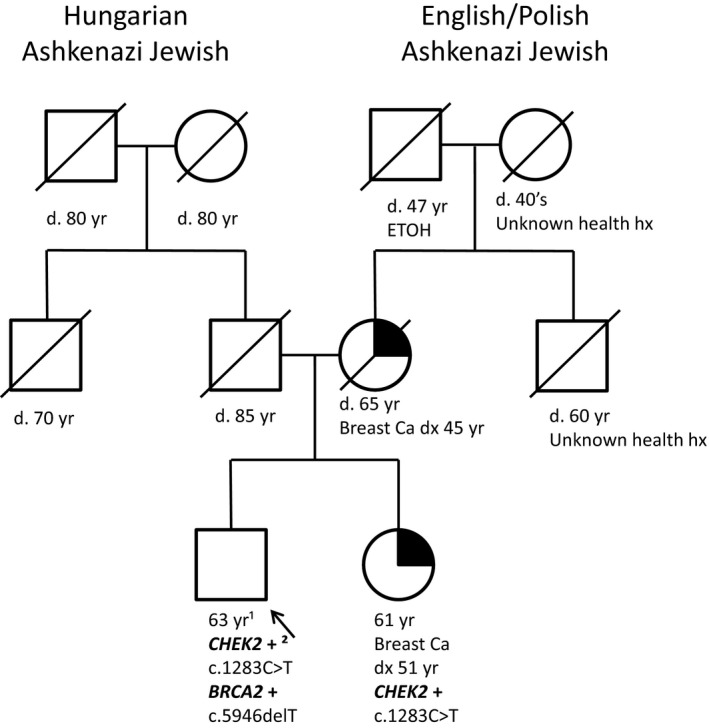
Pedigree of Patient 1. ¹Yr, ¹years; +², ²positive for pathogenic mutation
Our second patient was a 56 year old female who was diagnosed with pancreas cancer at 55 years of age who never smoked (Figure 2). The reported family history included a maternal great aunt with gastric cancer diagnosed at 75 years of age, a father diagnosed with lymphatic cancer at 61 years of age and prostate cancer at 70 years of age, and a paternal great uncle diagnosed with gastric cancer. Both maternal and paternal lineages included AJ ancestry. Several years prior to her diagnosis the patient was offered AJ founder mutation testing for the BRCA1 and BRCA2 genes but declined testing. Following her diagnosis of pancreatic cancer, she now elected to pursue genetic testing analysis including 79 genes on a multi cancer next generation panel through Invitae Laboratory. This test found the pathogenic AJ founder mutation in the CHEK2 gene (c.1283C>T) as well as a variant of uncertain clinical significance in the BLM gene (c.696C>A). This patient illustrates the need for AJ population screening to include more than just the BRCA1 and BRCA2 AJ founder mutations. Had this patient only pursued the traditional BRCA1/2 AJ founder mutations she may not have pursued additional genetic testing after being diagnosed with pancreas cancer and her CHEK2 mutation could have been missed. This patient also demonstrates the importance of pursuing AJ population based screening as her family history is not indicative of the CHEK2 mutation which was found.
Figure 2.
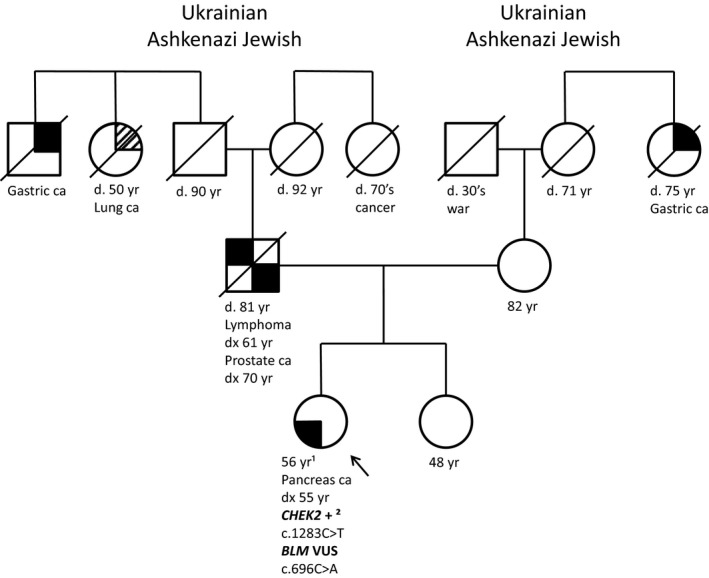
Pedigree of Patient 2. ¹Yr, ¹years; +², ²positive for pathogenic mutation
It is important to not only offer the AJ population screening for the three known BRCA1/2 AJ founder mutations, but also other pathogenic mutations associated with hereditary cancer within this population. Walsh et al. (2017) reported that AJ women diagnosed with invasive breast cancer, without a BRCA1/2 founder mutation, have a remaining 1% chance of having a non‐founder BRCA1/2 pathogenic mutation; if she is diagnosed before 40 years of age this chance increases to 3% (Walsh et al., 2017). Frank et al. (2002) found that 16 of the 74 (21.64%) deleterious BRCA1/2 mutations found in AJ individuals were non‐founder mutations, supporting the need for full BRCA1/2 gene sequencing in AJ individuals.
Clinical questions arise from these risk estimates. If an individual of AJ ancestry presents to a hereditary cancer clinic for population based genetic testing how many genes, or which founder mutations should be offered? Several papers provide data for consideration. Fifty percent of BRCA1/2 positive women diagnosed with breast cancer had no known immediate family history of cancer, indicating family history alone may not be enough to determine appropriate testing (Shkedi‐Rafid, Gabai‐Kapara, Grinshpun‐Cohen, & Levy‐Lahad, 2012). Individuals from AJ families may present unique challenges in determining appropriate testing, in part due to smaller family sizes and lack of medical information concerning past generations due to factors such as the Holocaust (Shkedi‐Rafid et al., 2012). Specifically, no family historians may be living or available and family members may have died at young ages, prior to developing cancer. We therefore recommend for adding AJ founder mutations for genes associated with breast and colorectal cancer (APC, MSH2, MSH6, and GREM1) regardless of family history as standard screening within the AJ population.
3. COLORECTAL CANCER AND AJ ANCESTRY
Currently, the NCCN guidelines do not address testing AJ individuals for founder mutations in the colorectal cancer genes,even though, according to Raskin et al. (2011) approximately 14%–16% of all colorectal cancers within the AJ population can be attributed to founder mutations in the APC, BLM, MSH2 and MSH6 genes. The GREM1 AJ founder mutation is a 40 kb duplication upstream of the gene and is present in 0.7% of AJ individuals meeting Lynch syndrome criteria (Laitman, Jaeger, Katz, Tomlinson, & Friedman, 2015). Laitman's (2015) single patient with this mutation did not fit the criteria for GREM1 as they had no increased polyp production. The MSH2 AJ founder mutation c.1906G>C (p.Ala636Pro) has a frequency of 0.4%–0.7% (Foulkes et al., 2002; Guillem et al., 2007; Lavie, Gruber, Lejbkowicz, Dishon, & Rennert, 2008). According to Foulkes et al. (2002) this mutation accounts for 2%–3% of all colorectal cancers diagnosed before 60 years of age within the AJ population, while Guillem et al. (2007) indicates that this mutation accounts for two thirds of the Lynch syndrome mutations within the AJ population. Foulkes et al. (2002) reported that 271 AJ women diagnosed with breast cancer and having a family history of either colorectal or ovarian cancer were tested for this founder mutation and no mutations were identified. The MSH6 gene has two separate AJ founder mutations; the c.3984_3987dupGTCA has a 0.3% frequency in this population while the c.3959_3962delCAAG has a 0.11% frequency (Raskin et al., 2011). Raskin et al. (2011) found none of the 22 unrelated families with these two founder mutations met Amsterdam I criteria, and only two of these families met the Amsterdam II criteria.
The AJ APC founder mutation c.3920T>A (p.Ile1307Lys) has a frequency of 6.1%–12% (Bahar et al., 2001; Foulkes et al., 2002; Raskin et al., 2011). The APC AJ founder mutation is unique as it does not present with the typical presentation of familial adenomatous polyposis (FAP) syndrome or even of attenuated FAP syndrome, but rather appears to only increase the risk for colorectal cancers in the AJ population by creating a hyper mutable region of the gene (Bahar et al., 2001; Laken et al., 1997). The risk for individuals in the AJ population to have colorectal cancer with this APC AJ founder mutation is estimated to double the general population risk, but does not appear to increase the risk for non AJ individuals (Boursi et al., 2013; Liang et al., 2013). According to a study by Laken et al. (1997) the APC AJ founder mutation was found in 28% of AJ families with a family history of colorectal cancer.
The NCCN guidelines have screening and management recommendations for all of the AJ founder mutation in the colorectal cancer genes (APC, GREM1, MSH2 and MSH6). Therefore, knowing an individual inherited one of these mutations would direct future medical management and surveillance. This would include beginning colonoscopy screenings earlier and repeating them more often than the average individual (Daly et al., 2018; Provenzale et al., 2017).
Our third patient had an incidental APC AJ founder mutation identified on genetic testing after he was diagnosed with pancreas cancer at 38 years of age (Figure 3). His family history included a brother with thymus cancer diagnosed at 39 years of age, a maternal aunt with breast cancer diagnosed at 42 years of age, a maternal great aunt with breast cancer who passed away in her 60s, a paternal aunt with ovarian cancer who passed away at 64 years of age, another paternal aunt with breast cancer diagnosed at 69 years of age; paternal grandfather had prostate cancer diagnosed at 68 years of age, a paternal great aunt had ovarian cancer and passed away in her 70's. The maternal ancestry was Russian and Hungarian with AJ ancestry. The paternal ancestry was United Kingdom with no known AJ ancestry. Results of the patient's 79 multi gene cancer panel through Invitae Laboratory found the AJ APC founder mutation (c.3920T>A) as well as a variant of uncertain clinical significance in the SDHB gene (c.158G>A). His mother (with AJ ancestry) subsequently had genetic testing from GeneDx Laboratory for the 20 gene breast ovarian cancer panel with the APC gene added on and was found to have the AJ APC founder mutation (c.3920T>A). Therefore, testing for the APC AJ founder mutation in AJ families is warranted, even without a family history of colorectal cancer.
Figure 3.
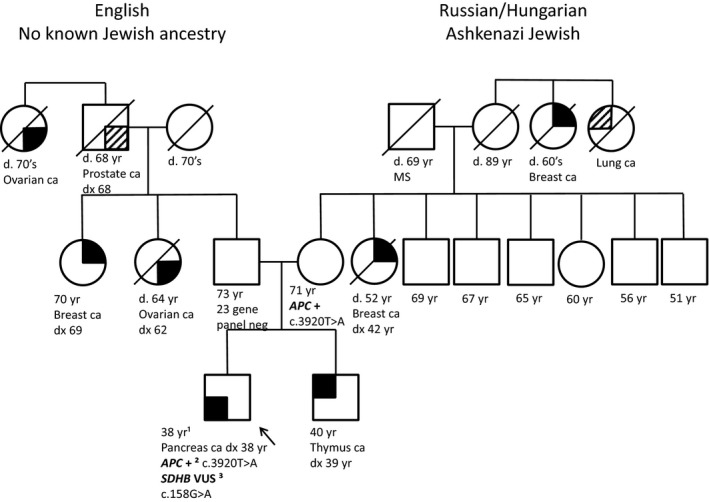
Pedigree of Patient 3. ¹Yr, ¹years; +², ²positive for pathogenic mutation; VUS³, ³variant of uncertain clinical significance
4. DISCUSSION
AJ founder mutations for genes associated with hereditary cancer are prevalent in the AJ population with a frequency of approximately 12.36%–20.83% (Table 1). The incidence of BRCA1, BRCA2 and CHEK2 founder mutations are approximately 4.75%–7.02% and the GREM1, MSH2, MSH6, and APC founder mutations are approximately 7.61%–13.81%.
Table 1.
Frequencies of AJ founder mutations in genes associated with cancer
| Breast Cancer Genes | Colorectal Cancer Genes | ||
|---|---|---|---|
| Mutation | Frequency | Mutation | Frequency |
| BRCA1 185delAG | 0.96–1.14% | GREM1 40 kb dup | 0.70% |
| BRCA1 5382insC | 0.13–0.28% | MSH2 1906G>C | 0.4–0.7% |
| BRCA2 6174delT | 0.6–1.52% | MSH6 3984_3987dupGTCA | 0.30% |
| CHEK2 1283C>T | 2.6–2.88% | MSH6 3959_3962delCAAG | 0.11% |
| CHEK2 470T>C | 0.46–1.2% | APC 3920T>A | 6.1–12% |
| Total | 4.75–7.02% | Total | 7.61–13.81% |
| Overall total mutation frequency 12.36%–20.83%a | |||
For genes associated with increased risk for breast, ovary, uterus, colon, and other cancers.
These high gene frequencies, and our patient reports, illustrate the importance of incorporating evaluation and testing for common risk alleles within the AJ population as a part of hereditary cancer gene analysis as standard screening. These mutation are not only fairly frequent in this population, they can directly impact medical management and care for these families. Population based screening may provide early detection, screening, management and treatment for these associated hereditary cancer syndromes. Additional cascade testing of other family members can lead to effective risk‐reducing management and preventative decisions by providers. Seeing a genetic counselor or other provider with expertise in hereditary cancer syndromes is important for people with AJ ancestry. Those with expertise in hereditary cancer syndromes will be aware of these founder mutations and will discuss the option of pursuing a larger panel based genetic test which could include all of the AJ founder mutations. Genetic counselors and genetic specialists will also emphasize how screening and surveillance recommendations continue to evolve for these AJ founder mutations.
CONFLICTS OF INTEREST
All authors have read and approved the submission to the journal. The authors report no conflicts of interest.
Cox DM, Nelson KL, Clytone M, Collins DL. Hereditary cancer screening: Case reports and review of literature on ten Ashkenazi Jewish founder mutations. Mol Genet Genomic Med. 2018;6:1236–1242. 10.1002/mgg3.460
REFERENCES
- Bahar, A. Y. , Taylor, P. J. , Andrews, L. , Proos, A. , Burnett, L. , Tucker, K. , … Buckley, M. F. (2001). The frequency of founder mutations in the BRCA1, BRCA2 and APC genes in Australian Ashkenazi Jews: Implications for the generality of the U.S. population data. American Cancer Society, 92, 440–445. [DOI] [PubMed] [Google Scholar]
- Boursi, B. , Sella, T. , Liberman, E. , Shapira, S. , David, M. , Kazanov, D. , … Kraus, S. (2013). The APC p. I1307K polymorphism is a significant risk factor for CGC in average risk Ashkenazi Jews. European Journal of Cancer, 49, 3680–3685. 10.1016/j.ejca/2013.03.040 [DOI] [PubMed] [Google Scholar]
- Daly, M. B. , Pilarski, R. , Berry, M. , Buys, S. S. , Farmer, M. , Friedman, S. , … Darlow, S. National Comprehensive Cancer Network Guidelines (2018). Genetic / Familial High‐Risk Assessment: Breast and Ovarian (Version 1.2018). Retrieved from http://www.nccn.org/ Accessed May 14, 2018
- Ferla, R. , Calo, V. , Cascio, S. , Rinaldi, G. , Badalamenti, G. , Carreca, I. , … Russo, A. (2007). Founder mutations in BRCA1 and BRCA2 genes. Annals of Oncology, 18(6), vi93–vi96. 10.1093/annonc/mdm234 [DOI] [PubMed] [Google Scholar]
- Foulkes, W. D. , Thiffault, I. , Gruber, S. B. , Horwitz, M. , Hamel, N. , Lee, C. , … Ellis, N. A. (2002). The founder mutation MSH2*1906G>C is an important cause of hereditary nonpolyposis colorectal cancer in the Ashkenazi jewish population. American Journal of Human Genetics, 71, 1395–1412. 10.1086/345075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, T. S. , Deffenbaugh, A. M. , Reid, J. E. , Hulick, M. , Ward, B. E. , Linqenfelter, B. , … Critchfield, G. C. (2002). Clinical Characteristics of individuals with germline mutations in BRCA1 and BRCA2: Analysis of 10,000 individuals. Journal of Clinical Oncology, 20(6), 1480–1490. 10.1200/JCO.2002.20.6.1480 [DOI] [PubMed] [Google Scholar]
- Guillem, J. G. , Glogowski, E. , Moore, H. G. , Nafa, K. , Markowitz, A. J. , Shia, J. , … Ellis, N. A. (2007). Single‐amplicon MSH2 A636P mutation testing in Askenazi Jewish patients with colorectal cancer: Role in presurgical management. Annals of Surger, 245(4), 560–565. 10.1097/01.sla.0000252589.26244.d4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitman, Y. , Jaeger, E. , Katz, L. , Tomlinson, I. , & Friedman, E. (2015). GREM1 germline mutation screening in Ashkenazi Jewish patients with familial colorectal cancer. Genetics Research (Camb), 97, e11 10.1017/S0016672315000105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitman, Y. , Kaufman, B. , Lahad, E. L. , Papa, M. Z. , & Friedman, E. (2007). Germline CHEK2 mutations in Jewish Ashkenazi women at high risk for breast cancer. The Israel Medical Association Journal, 9, 791–796. [PubMed] [Google Scholar]
- Laken, S. J. , Petersen, G. M. , Gruber, S. B. , Oddoux, C. , Ostrer, H. , Giardiello, F. M. , … Vogelstein, B. (1997). Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC . Nature Genetics, 17, 79–83. 10.1038/ng0997-79 [DOI] [PubMed] [Google Scholar]
- Lavie, O. , Gruber, S. B. , Lejbkowicz, F. , Dishon, S. , & Rennert, G. (2008). Gynecologic malignancies in Ashkenazi families with the MSH2 A636P founder mutation. American Journal of Obstetrics & Gynecology, 199, 148e1‐148.e3. 10.1016/j.ajob.2008.02.018 [DOI] [PubMed] [Google Scholar]
- Leedom, T. P. , LaDuca, H. , McFarland, R. , Li, S. , Dolinsky, J. S. , & Chao, E. C. (2016). Breast cancer risk is similar for CHEK2 founder and non‐founder mutation carriers. Cancer Genetics, 209, 403–407. 10.1016/j.cancergen.2016.08.005 [DOI] [PubMed] [Google Scholar]
- Liang, J. , Lin, C. , Hu, F. , Wang, F. , Zhu, L. , Yao, X. , … Zhao, Y. (2013). APC Polymorphisms and the risk of colorectal neoplasia: A HuGE review and meta‐analysis. American Journal of Epidemiology, 177(11), 1169–1179. 10.1093/aje/kws382 [DOI] [PubMed] [Google Scholar]
- Offit, K. , Pierce, H. , Kirchhoff, T. , Kolachana, P. , Rapaport, B. , Gregersen, P. , … Ellis, N. (2003). Frequency of CHEK2*1100delC in New York breast cancer cases and controls. BioMed Central Medical Genetics, 4(1), 10.1186/1471-2350-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzale, D. , Gupta, S. , & Ahnen, D.J. , et al. National Comprehensive Cancer Network Guidelines (2017). Genetic / Familial High‐Risk Assessment: Colorectal (Version 3.2017). http://www.nccn.org/ Accessed May 14, 2018 [DOI] [PubMed]
- Raskin, L. , Schwenter, F. , Freytsis, M. , Tischkowitz, M. , Wong, N. , Chong, G. , … Foulkes, W. D. (2011). Characterization of two Ashkenazi Jewish founder mutations in MSH6 gene causing Lynch syndrome. Clinical Genetics, 79, 512–522. 10.1111/j.1399-004.2010.01594.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal, E. , Moyes, K. , Arnell, C. , Evans, B. , & Wenstrup, R. J. (2015). Incidence of BRCA1 and BRCA2 non‐founder mutations in patients of Ashkenazi Jewish ancestry. Breast Cancer Research and Treatment, 149, 223–227. 10.1007/s10549-014-3218-x [DOI] [PubMed] [Google Scholar]
- Shaag, A. , Walsh, T. , Renbaum, P. , Kirchhoff, T. , Nafa, K. , Shiovitz, S. , … King, M. C. (2005). Functional and genomic approaches reveal an ancient CHEK2 allele associated with breast cancer in the Ashkenazi Jewish population. Human Molecular Genetics, 14(4), 555–563. 10.1093/hmg/ddi052 [DOI] [PubMed] [Google Scholar]
- Shkedi‐Rafid, S. , Gabai‐Kapara, E. , Grinshpun‐Cohen, J. , & Levy‐Lahad, E. (2012). BRCA genetic testing of individuals from families with low prevalence of cancer: Experiences of carriers and implications for population screening. Genetics in Medicine, 14(7), 688–694. 10.1038/gim.2012.31 [DOI] [PubMed] [Google Scholar]
- Walsh, T. , Mandell, J. B. , Norquist, B. N. , Casadei, S. , Gulsuner, S. , Lee, M. K. , & King, M. C. (2017). Genetic predisposition to breast cancer due to mutations other than BRCA1 and BRCA2 founder alleles among Ashkenazi Jewish women. JAMA, 3(12), 1647–1653. 10.1001/jamaoncol.2017.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]