

Abstract
Background
Early‐onset Parkinson's disease (PD) is the most common inherited form of parkinsonism, with the PRKN gene being the most frequently identified mutated. Exon rearrangements, identified in about 43.2% of the reported PD patients and with higher frequency in specific ethnicities, are the most prevalent PRKN mutations reported to date in PD patients.
Methods
In this study, three consanguineous families with early‐onset PD were subjected to whole‐genome sequencing (WGS) analyses that were followed by Sanger sequencing and droplet digital PCR to validate and confirm the disease segregation of the identified genomic variations and to determine their parental origin.
Results
Five different PRKN structural variations (SVs) were identified. Because the genomic sequences surrounding the break points of the identified SVs might hold important information about their genesis, these were also characterized for the presence of homology and repeated sequences.
Conclusion
We concluded that all identified PRKN SVs might originate through retrotransposition events.
Keywords: Parkinson’s disease, PRKN, retrotransposition, structural variations, whole‐genome sequencing
1. INTRODUCTION
Mutations in the PRKN gene (OMIM #600,116) are the most common cause of autosomal recessive Parkinson's disease (PD). The PRKN gene is located on chromosome 6q26 and its larger transcript (transcript variant 1; NM_004562.2) contains 12 coding exons and encodes a protein of 465 amino acids (NP_004553.2). All types of mutations, including missense, nonsense, splice site, frameshift, and structural variations (SVs), have been reported in PD patients carrying PRKN mutations. SVs (exon rearrangements) are the most common type of mutations, being identified in about 43.2% of the reported patients (Kasten, et al., 2018). In a large multicenter study, where we identified PRKN mutations in 71.42% of the examined patients, SVs were the most prevalent mutations identified in the Iranian PD population (Taghavi et al., 2018). The most common PRKN SV identified to date in the PD population is the c.(171+1_172‐1)_(412+1_413‐1)del mutation, which consists of a deletion encompassing the entire exon 3 of the PRKN gene (https://www.mdsgene.org) (Kasten, et al., 2018). The Multiplex Ligation‐Dependent Probe Amplification (MPLA; MRC Holland), which allows the detection of DNA copy number changes (CNVs) of up to 40 sequences in a single reaction, is the most frequently used technique to identify SVs in the PRKN gene, despite the fact that it does not determine the genomic localization of the deletion/insertion break points. On the other hand, whole‐genome sequencing (WGS), considered the most comprehensive genetic screening as it captures both coding and noncoding genetic variation, enables us to identify gene fusions, CNVs, and other complex SVs (Royer‐Bertrand & Rivolta, 2014). The continuous progress of read coverage uniformity and reduced allele bias in WGS (Meynert, Ansari, FitzPatrick, & Taylor, 2014) has led to improved detection of copy number changes and de novo variations (Gilissen, et al., 2014; Ritter, et al., 2015).
2. CLINICAL REPORT
We here described the clinical characteristics of three different families carrying PRKN SVs. All patients’ clinical details are summarized in Table 1. Briefly, all patients began the disease in childhood, with the youngest patient developing the disease at the age of 10 years. Most of the patients showed slow disease progression with the exception of patient FC‐P2, who onset the disease at the age of 17 and showed severe disease progression (Table 1). Only one family, consisted of three affected siblings, showed additional symptoms (Table 1).
Table 1.
Clinical details of patients carrying PRKN SVs
| Patient (gender) | Age at onset | Age | PRKNMutations | Start location | First symptom | Tremor | Progression | Parkinsonism | Rigidity | Postural instability | Dystonia | Bradykinesia | Hypokinesia | Autonomic dysfunction | Pyramidal signs | Stride and sleep apnea | REM sleep behavior disorder | Falling | Response to levodopa | Other symptoms |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FA_P1 (Male) | 13 | 62 | Delx5/Delx5 | Limbs | Resting tremor | Rest, intention, limbs | Slow | Sy | Y | La | Y | Y | Y | Y | N | N | N | Y | Y | Eye lid apraxia or blepharospasm, incontinence, sensory polyneuropathy, impaired smell |
| FA_P2 (Male) | 13 | 54 | Limbs | Resting tremor | Intention, limbs, chin | Slow | Sy | N | La | N | Y | Y | N | N | N | N | Y | Y | Incontinence, sensory polyneuropathy | |
| FA_P3 (Male) | 13 | 50 | Whole body | Whole body tremor | Rest, intention, limbs | Slow | Sy | Y | La | N | Y | Y | N | N | N | N | N | Y | Sensory polyneuropathy | |
| FB_P1 (Female) | 10 | 24 | Delx2/Delx3 | Limbs | Rigidity | Rest | Slow | Asy | Y | Y | Y | Y | N | N | N | N | N | Y | Y | None |
| FC_P1 (Female) | 17 | 27 | Delx4–6/Delx4 | Whole body | Resting tremor | Rest | Fast | Sy | Y | Y | Y | Y | N | N | N | N | N | Y | N | None |
Family A was previously reported in Taghavi et al., 2018 (Family 3).
As: asymmetric; Ea: early; FA_P1: family A, patient 1; La: late; N: No; Sy: symmetric; Y: yes.
3. METHODS
Three different families with early‐onset PD were clinically examined and subjected to WGS analyses. The local ethics committee at each participating medical center approved this study, and informed consent, according to the Declaration of Helsinki, was obtained from all participants. DNA samples from all participants were isolated from whole blood, using standard procedures. WGS was performed as previously described (Sanchez, et al., 2016). Specifically, deletions were called by using GenomeSTRiP (v2.0) (Handsaker, Korn, Nemesh, & McCarroll, 2011) and were jointly called by using 17 HapMap individuals (CEPH Platinum Genomes pedigree). All deletions annotated as PASS in the GenomeSTRiP results were further filtered by using custom scripts to remove redundant calls and break points overlapping repeat regions, or with extensive mapping ambiguity. Identified deletions affecting coding areas were further analyzed through SplazerS, which identifies and split‐aligns reads that cross‐structural variant break points (Emde, et al., 2012). First, all reads mapping to the candidate region were extracted, and then by using SplazerS, they were mapped back to the region to identify and confirm the break point locations. Subsequently, Sanger sequencing, as described elsewhere (Krebs, et al., 2013) and by using primers flanking the cut‐off points previously determined by the WGS analyses, was used to validate the identified deletions and to determine the genomic localization of the deletions’ break points. Primer sequences were designed by using a public primer design website (https://ihg.gsf.de/ihg/ExonPrimer.html; primer sequences available upon request) using the NM_004562.2 gene sequence as a reference. The validated CNVs were later quantified through the ddPCR QX100 system (Bio‐rad, USA) by using TaqMan probes targeting PRKN exons 2–6 as well as a reference gene (TERT) (Hindson, et al., 2011). Taqman probes were acquired from Applied Biosystems (Life Technologies, USA), and a DNA sample from a healthy individual as well as a non‐template control were, respectively, used as reference control DNA and negative control. All CNV scores were calculated using the Quantasoft software according to the manufacturer's instructions (Bio‐Rad, USA). These analyses were done in all available family members in order to examine the disease segregation of the deletions and to determine their parental origin (Figure 1).
Figure 1.
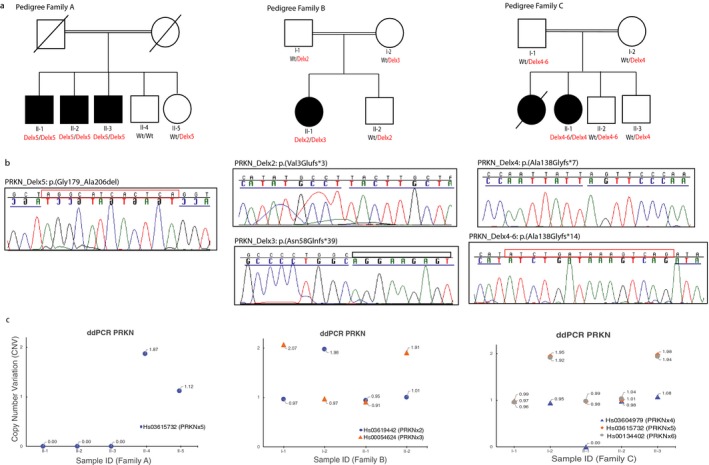
Identification of five different PRKN SVs in three families with PD. (a) Pedigree structure of families carrying PRKN SVs. Dark squares (males) and circles (females) indicate PD. Wt indicates wild‐type allele. (b) Sanger chromatograms of the PRKN gene (NM_004562.2) showing the deletions’ break points (highlighted with a blue line). MIs observed in Pedigree A and C are highlighted with a red rectangle. (c) CNVs plots of PRKN exons 2, 3, 4, 5, 6 obtained through ddPCR QX100 system, Bio‐rad). Two exon copies (homozygous wt allele) are represented with a CNV score close to 2, one exon copy (heterozygous mutant allele) with a CNV score close to 1, and no copies with a CNV score of 0
The formation of SV is a complex phenomenon that is not well understood. Long homologies around break point suggest SV formation by nonallelic homologous recombination (NAHR); short homologies, with high mobile element content within SV regions, indicate that they originated through transposable element insertions (TEI); while little or no homology suggests SV formation by a nonhomologous end‐joining (NHEJ) or by a template‐switching mechanism during replication. We therefore examined the proximal and distal sequences (~2 kb) to the break points to determine their homology and the presence of repeated sequences, as they are known to affect the genomic integrity through recombination involving insertion, deletion, and rearrangements (Abyzov, et al., 2015; Kaer & Speek, 2013; Lupski & Stankiewicz, 2005). First, pairwise sequence comparisons were carried out to determine the homology between the sequences flanking the deletions’ break points. Both proximal and distal sequences to the deletions’ break points were aligned through Clustal Omega software (https://www.ebi.ac.uk/Tools/msa/clustalo/). Data from the NIH Roadmap Epigenomics project (https://www.roadmapepigenomics.org/) were used to examine the deletions’ break points for the presence of chromatin marks and repeated elements, such as retrotransposons, including the long terminal repeat (LTR) and the non‐LTR retrotransposons (i.e.; long interspersed elements (LINEs or L1) and short interspersed element (SINEs)) (Kaer & Speek, 2013). Data for the chromatin states using a multivariate Hidden Markov Model (HMM; ChromHMM analysis) were also investigated (Ernst, et al., 2011).
4. RESULTS AND DISCUSSION
We identified five different PRKN deletions. Family A was previously reported and shown to carry a homozygous PRKN exon 5 deletion (NG_008289.2: g.574615_702745del) that was identified through WGS (Taghavi et al., 2018). Here, we described the identified mutation (c.534+42943_618+24033delinsAGGCATCACTCA) and its effect on the protein (p.(Gly179_Ala206del)) following the guidelines of the Human Genome Variation Society (https://www.hgvs.org/) and the Mutalyzer program (https://www.LOVD.nl/mutalyzer/) (Figure 1, Table 2) (https://databases.lovd.nl/shared/variants/0,000,368,892).
Table 2.
PRKN deletions identified through whole‐genome sequencing in three different PD families
| Family | Chr | Break points | Mutations (NG_008289.2 (PRKN_ NM_004562.2) | Implicated exons | Size (bp) | ||
|---|---|---|---|---|---|---|---|
| Position (5′) | Position (3′) | Nucleotide change | Protein change | ||||
| Ped. A | 6 | 162,451,090 | 162,579,220 | c.534+42943_618+24033delinsAGGCATCACTCA | p.(Gly179_Ala206del) | Exon 5 | 128,130 |
| Ped. B | 6 | 162,837,521 | 162,925,282 | c.8‐60777_171+26821del | p.(Val3Glufs*3) | Exon 2 | 87,761 |
| 6 | 162,655,261 | 162,724,846 | c.172‐41049_412+28296del | p.(Asn58Glnfs*39) | Exon 3 | 69,585 | |
| Ped. C | 6 | 162,605,414 | 162,641,899 | c.413‐19615_534+16749del | p.(Ala138Glyfs*7) | Exon 4 | 36,485 |
| 6 | 162,340,003 | 162,667,521 | c.412+16037_734+54332delinsATCTGATAAAGTCAG | p.(Ala138Glyfs*14) | Exons 4, 5, 6 | 327,518 | |
Additionally, we identified two isolated PD cases carrying compound heterozygous PRKN deletions and characterized all five PRKN deletions. In the new cases, the performed WGS analyses led to the identification of 683 and 592 coding (including missense, nonsense, and frameshift) and splice site nucleotide variations for patient B_II‐1 and patient C_II‐1, respectively. Because both patients were born to consanguineous marriages, a recessive pattern of inheritance was suspected (Figure 1). However, no rare (with a frequency <0.5% for a recessive model) or novel homozygous or compound heterozygous coding variations were identified in the patients’ genomes, meaning that all variations identified were present in heterozygosis and therefore were not compatible with a recessive pattern of inheritance. All known coding variations (known and unknown) identified in the known PD genes were as well examined, but no pathogenic mutation was identified. We then also examined all SVs identified through WGS in the patients’ genomes. Patient B_II‐1 was shown to carry 50 SVs while 56 different SVs were identified in the patient C_II‐1. Interestingly, we found that both patients carried two different heterozygous SNVs at the PRKN locus. The PD patient from Family B was shown to carry heterozygous PRKN deletions involving exon 2 (NG_008289.2:g.228553_316314del; https://databases.lovd.nl/shared/variants/0,000,368,960) and exon 3 (NG_008289.2:g.428989_498574del; https://databases.lovd.nl/shared/variants/0,000,368,961), respectively, while the patient from Family C was shown to carry a heterozygous PRKN exon 4 deletion (NG_008289.2:g.511936_548421del; https://databases.lovd.nl/shared/variants/0000368999) and another heterozygous deletion involving PRKN exons 4–6 (NG_008289.2:g.486315_813833del; https://databases.lovd.nl/shared/variants/0000368998) (Table 2; Figure 1b,c). In addition, we identified through Sanger sequencing small micro‐insertions (MIs) of 12 and 15 bp, respectively, in the patients carrying the two largest deletions (~128 and ~327 kb; Figure 1b, Table 2). All deletions segregated with disease status as only the patients were carriers of two mutant alleles, as expected for an autosomal recessive inheritance (Figure 1a–c; Table 2). The nomenclature of each identified deletion was checked in the Mutalyzer program, with which we examined their effect on the protein. All deletions but one were predicted to cause premature stop codons (Table 2), thus resulting in truncated, nonfunctional proteins.
We found long terminal repeat (LTR) and non‐LTR retrotransposons at both proximal and distal break point regions along with short DNA homologies around the deletions’ break points (the majority of them close to the deletions’ break points or <1 kb away [Tables 2 and 3]), indicating that all these deletions might originate through TEI (Lupski & Stankiewicz, 2005). All proximal and distal regions showed repressive marks, such as tri‐methylation at H3K9 and H3K27 (H3K9me3, H3K27me3). Although the majority of genomic regions surrounding the deletions’ break points also showed methylation and/or tri‐methylation at H3K4 (H3K4me3, H3K4me1), which influences transcriptional activation, depletion of active marks was observed in some of the break points, as it has been observed in other TEI break points (Abyzov, et al., 2015) (Table 3).
Table 3.
Retrotransposons identified within 1–2 Kb upstream and downstream regions to the deletions’ break points
| Fam | Mutation (NG_008289.2) | Upstream Repeats | Downstream Repeats | Mechanisms | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Position | Size (bp) | Type | Chromatin marks | Position | Size (bp) | Type | Chromatin marks | |||
| Ped. A | g.574615_702745del | 162,450,885–162,451,171 | 286 | SINE (AluJr4) | H3K9me3 H3K27me3 | 162,579,449–162,579,761 | 312 | SINE (AluSz) | H3K4me1 H3K4me3 H3K9me3 H3K27me3 | TEI |
| Ped. B | g.228553_316314del | 162,836,570–162,836,852 | 282 | SINE (AluSz) | H3K4me1 H3K4me3 H3K9me3 H3K27me3 | 162,925,457–162,926,411 | 954 | LINE (L1ME3C) | H3K4me1 H3K4me3 H3K9me3 H3K27me3 | TEI |
| g.428989_498574del | 162,654,701–162,655,043 | 342 | LTR (MSTD) | H3K4me1 H3K4me3 H3K9me3 H3K27me3 | 162,724,757–162,725,149 | 392 | LINE (L1MC4A) | H3K9me3 H3K27me3 | TEI | |
| Ped. C | g.511936_548421del | 162,605,380–162,605,745 | 365 | LTR (THE1C) | H3K4me1 H3K4me3 H3K9me3 H3K27me3 | 162,643,141–162,643,834 | 693 | LTR (MLT1E1A) | H3K4me1 H3K4me3 H3K9me3 H3K27me3 | TEI |
| g.486315_813833del | 162,339,759–162,340,554 | 795 | LINE (L1ME1) | H3K4me1 H3K4me3 H3K9me3 H3K27me3 | 162,669,915–162,670,199 | 284 | SINE (AluSc) | H3K4me1 H3K9me3 K27me3 | TEI | |
We concluded that WGS is the preferred technique to well characterize the copy number changes observed in the PRKN gene as well as other parkinsonism genes, as it enables us to characterize the bases around their break points that are thought to hold important information about their genesis (Kidd, et al., 2010). Because exon rearrangements are the most common mutations identified in the PRKN gene and in recessive PD, since the PRKN gene is the most frequently mutated gene, the characterization of PRKN SVs is essential for understanding their formation mechanisms as well as for examining and interpreting their functional effects in model organisms. Taken together, precise mapping of deletion break points and localization of the repeated elements is important because they might reveal common disease signatures that will, in turn, lead to novel genomic editing strategies for gene therapy (Esposito, et al., 2017; Kaer & Speek, 2013; Lupski & Stankiewicz, 2005).
DISCLOSURE STATEMENT
The authors declare no conflict of interest.
ACKNOWLEDGMENT
Authors thank all patients and relatives for their generous contribution to this study.
Bravo P, Darvish H, Tafakhori A, et al. Molecular characterization of PRKN structural variations identified through whole‐genome sequencing. Mol Genet Genomic Med. 2018;6:1243–1248. 10.1002/mgg3.482
Funding information
This work was in part supported by the Shahid Beheshti University of Medical Sciences, the American Parkinson Disease Association (APDA; CPR), and the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS079388; CPR).
REFERENCES
- Abyzov, A. , Li, S. , Kim, D. R. , Mohiyuddin, M. , Stutz, A. M. , Parrish, N. F. , … Hurles, M. (2015). Analysis of deletion breakpoints from 1,092 humans reveals details of mutation mechanisms. Nature Communications, 6, 7256 10.1038/ncomms8256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emde, A. K. , Schulz, M. H. , Weese, D. , Sun, R. , Vingron, M. , Kalscheuer, V. M. , … Reinert, K. (2012). Detecting genomic indel variants with exact breakpoints in single‐ and paired‐end sequencing data using SplazerS. Bioinformatics, 28(5), 619–627. 10.1093/bioinformatics/bts019 [DOI] [PubMed] [Google Scholar]
- Ernst, J. , Kheradpour, P. , Mikkelsen, T. S. , Shoresh, N. , Ward, L. D. , Epstein, C. B. , … Coyne, M. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature, 473(7345), 43–49. 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito, G. , Tremolaterra, M. R. , Marsocci, E. , Tandurella, I. C. , Fioretti, T. , Savarese, M. , & Carsana, A. (2017). Precise mapping of 17 deletion breakpoints within the central hotspot deletion region (introns 50 and 51) of the DMD gene. Journal of Human Genetics, 62(12), 1057–1063. 10.1038/jhg.2017.84 [DOI] [PubMed] [Google Scholar]
- Gilissen, C. , Hehir‐Kwa, J. Y. , Thung, D. T. , van de Vorst, M. , van Bon, B. W. , Willemsen, M. H. , … Schenck, A. (2014). Genome sequencing identifies major causes of severe intellectual disability. Nature, 511(7509), 344–347. 10.1038/nature13394 [DOI] [PubMed] [Google Scholar]
- Handsaker, R. E. , Korn, J. M. , Nemesh, J. , & McCarroll, S. A. (2011). Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nature Genetics, 43(3), 269–276. 10.1038/ng.768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson, B. J. , Ness, K. D. , Masquelier, D. A. , Belgrader, P. , Heredia, N. J. , Makarewicz, A. J. , … Legler, T. C. (2011). High‐throughput droplet digital PCR system for absolute quantitation of DNA copy number. Analytical Chemistry, 83(22), 8604–8610. 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaer, K. , & Speek, M. (2013). Retroelements in human disease. Gene, 518(2), 231–241. 10.1016/j.gene.2013.01.008 [DOI] [PubMed] [Google Scholar]
- Kasten, M. , Hartmann, C. , Hampf, J. , Schaake, S. , Westenberger, A. , Vollstedt, E. J. , … Dulovic, M. (2018). Genotype‐phenotype relations for the Parkinson's Disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Movement Disorders, 33, 730–741. 10.1002/mds.27352 [DOI] [PubMed] [Google Scholar]
- Kidd, J. M. , Graves, T. , Newman, T. L. , Fulton, R. , Hayden, H. S. , Malig, M. , … Eichler, E. E. (2010). A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell, 143(5), 837–847. 10.1016/j.cell.2010.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs, C. E. , Karkheiran, S. , Powell, J. C. , Cao, M. , Makarov, V. , Darvish, H. , … Buxbaum, J. D. (2013). The Sac1 domain of SYNJ1 identified mutated in a family with early‐onset progressive Parkinsonism with generalized seizures. Human Mutation, 34(9), 1200–1207. 10.1002/humu.22372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski, J. R. , & Stankiewicz, P. (2005). Genomic disorders: Molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genetics, 1(6), e49 10.1371/journal.pgen.0010049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynert, A. M. , Ansari, M. , FitzPatrick, D. R. , & Taylor, M. S. (2014). Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics, 15, 247 10.1186/1471-2105-15-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter, D. I. , Haines, K. , Cheung, H. , Davis, C. F. , Lau, C. C. , Berg, J. S. , … Wheeler, D. A. (2015). Identifying gene disruptions in novel balanced de novo constitutional translocations in childhood cancer patients by whole‐genome sequencing. Genetics in Medicine, 17, 831–835. 10.1038/gim.2014.189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer‐Bertrand, B. , & Rivolta, C. (2014). Whole genome sequencing as a means to assess pathogenic mutations in medical genetics and cancer. Cellular and Molecular Life Sciences, 72(8), 1463–1471. 10.1007/s00018-014-1807-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez, E. , Darvish, H. , Mesias, R. , Taghavi, S. , Firouzabadi, S. G. , Walker, R. H. , … Paisan‐Ruiz, C. (2016). Identification of a Large DNAJB2 Deletion in a Family with Spinal Muscular Atrophy and Parkinsonism. Human Mutation, 37(11), 1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghavi, S. , Chaouni, R. , Tafakhori, A. , Azcona, L. J. , Firouzabadi, S. G. , Omrani, M. D. , … Ahmadi, M. (2018). A Clinical and molecular genetic study of 50 families with autosomal recessive Parkinsonism revealed known and novel gene mutations. Molecular Neurobiology, 55(4), 3477–3489. 10.1007/s12035-017-0535-1 [DOI] [PMC free article] [PubMed] [Google Scholar]