

Abstract
Particulate matter (PM) increases levels of pro-inflammatory cytokines, but its effects on the skin remain largely unknown. We investigated the signal transduction pathway and epigenetic regulatory mechanisms underlying cellular inflammation induced by PM with a diameter of ≤ 2.5 (PM2.5) in vitro and in vivo. PM2.5-treated skin keratinocytes produced various inflammatory cytokines, including IL-6. The binding of PM2.5 to TLR5 initiated intracellular signaling through MyD88, and led to the translocation of NFκB to the nucleus, where it bound the NFκB site within IL-6 promoter. Furthermore, PM2.5 induced a direct interaction between TLR5 and NOX4, and in turn induced the production of ROS and activated NFκB-IL-6 downstream, which was prevented by siRNA-mediated knockdown of NOX4 or antioxidant treatment. Furthermore, expression of TLR5, MyD88, NOX4, phospho-NFκB, and IL-6 was increased in skin tissue of PM2.5-treated flaky tail mice. PM2.5-induced increased transcription of IL-6 was regulated via DNA methylation and histone methylation by epigenetic modification; the binding of DNA demethylase and histone methyltransferase to the IL-6 promoter regions resulted in increased IL-6 mRNA expression. Our findings provide deep insight into the pathogenesis of PM2.5 exposure and can be used as a therapeutic strategy to treat inflammatory skin diseases caused by PM2.5 exposure.
Keywords: Particulate matter, Interleukin-6, Reactive oxygen species, Toll like receptor, Epigenetic modification
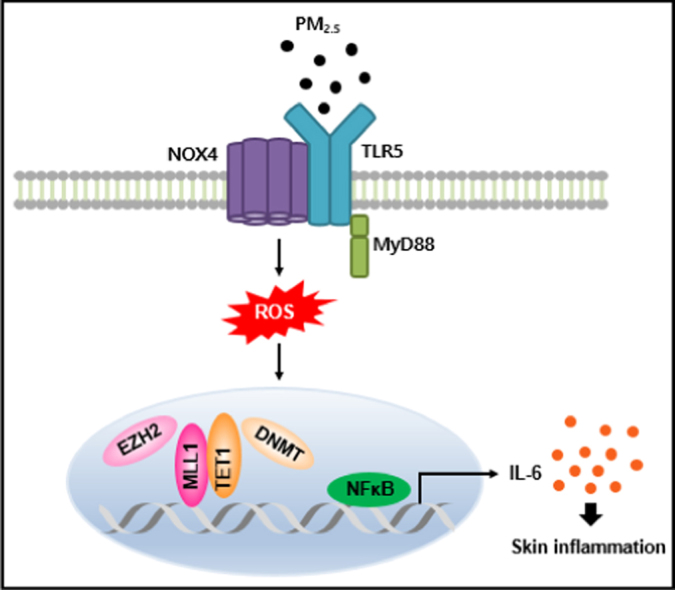
Graphical abstract
Ligation of TLR5 with PM2.5 stimulates NOX4 to generate ROS, which in turn activates NFκB and promotes the secretion of pro-inflammatory cytokines, including IL-6, in skin keratinocytes. In addition, epigenetic changes prompt the binding of a DNA demethylase TET1 and a histone methyltransferase MLL1 to the IL-6 locus in place of a DNA methyltransferase DNMT and a histone methyltransferase EZH2. This binding induces the expression of IL-6 and pro-inflammatory mediators, which induce monocyte accumulation, leading to skin inflammation.
1. Introduction
Ambient air pollution has become a major worldwide problem; especially, fine particulate matter (PM2.5), which has an aerodynamic diameter of less than 2.5 µm, is a threat to health in Korea [1]. Many reports have demonstrated that PM containing polyaromatic hydrocarbons can trigger an inflammatory response in the respiratory system and may lead to systemic inflammatory reactions [2], [3], [4]. The human skin serves as the first line of defense against air pollutants. However, studies on the inflammatory effects of PM2.5 on the skin and the underlying mechanisms are limited.
Local innate immunity plays a key role in initiating and coordinating homeostasis and resistance to foreign particles or pathogen infection by regulating effector cytokines and other mediators of inflammation [5]. Toll-like receptors (TLRs) have been identified as pivotal immune receptors; following pathogen detection, they mediate the activation of innate and adaptive immune responses through modulation of gene expression [6]. It is reported that human keratinocytes constitutively express TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, and TLR9 [7]. Keratinocytes of skin cells play an essential role in immune responses by producing chemokines following TLR engagement [8]. These chemokines, in turn, stimulate the migration of leucocytes to the site of injury or infection. TLRs specifically recognize highly conserved pathogen-associated motifs: triacyl lipopeptide is recognized by TLR1-TLR2 heterodimer, diacyl lipopeptide by TLR2-TLR6 heterodimer, double-stranded RNA by TLR3, lipopolysaccharide by TLR4, and flagellin by TLR5 [9]. Recent studies have demonstrated a molecular link between TLR activation and the activity of NADPH oxidase (NOX) isozymes (NOX1–5 and DUOX1, 2) in the innate immune (inflammation) response, suggesting that NOX activation is indispensable in TLR-mediated inflammation responses [10]. For example, DUOX2-mediated H2O2 generation activated by the flagellin–TLR5 axis induces interleukin (IL)-8 production and MUC5AC expression in the nasal epithelium [10]. In addition, flagellin-induced interaction between the COOH region of NOX4 and the Toll/interleukin-1 receptor domain of TLR5 leads to H2O2 generation, which in turn promotes the secretion of pro-inflammatory cytokines, including IL-8, as well as the expression of ICAM-1 in human aortic endothelial cells [11]. The recognition by TLRs leads to the activation of downstream signaling molecules, such as nuclear factor kappa B (NFκB). NFκB promotes the expression of target genes that mediate inflammatory cytokines, such as IL-1β and IL-6 [12].
DNA methylation, the most studied of the epigenetic mechanisms, is a natural process that suppresses gene expression via the addition of methyl groups to the DNA. PM‐related hypomethylation of the repetitive Alu transposable element and the pro-inflammatory gene, TLR4, has been observed, and it is correlated with increased blood pressure [13]. This repetitive element- and gene‐specific hypomethylation can be induced by PM-induced biological processes, such as oxidative stress. Oxidative DNA damage can interfere with the ability of methyltransferases to interact with DNA, resulting in hypomethylation of cytosine residues at CpG sites [14]. Recently, we reported that HaCaT keratinocytes can internalize PM2.5, as observed with a transmission electron microscope. In this study, we showed that the internalized PM2.5 triggered oxidative stress and that mouse skin exposed to PM2.5 underwent large structural changes in the epidermis, strongly suggesting that PM2.5 can penetrate the superficial skin layers [15].
In the present study, biomarkers associated with inflammation and oxidative stress were investigated in vitro and in vivo to elucidate PM2.5-induced skin inflammation and the underlying mechanisms in terms of TLR signaling and epigenetic alteration.
2. Materials and methods
2.1. PM2.5 preparation
PM2.5, which is a standard diesel PM (SRM 1650b) issued by the National Institute of Standards and Technology (NIST, USA), was purchased from Sigma-Aldrich (St. Louis, MO, USA). The 1650b diesel PM, with a mean diameter of 0.18 µm, was predominantly composed of polycyclic aromatic hydrocarbons (PAHs) and nitro-PAHs. The certified mass fraction values of PAHs and nitro-PAHs in SRM 1650b are provided in Supplementary Table 1 and 2. PM2.5 was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 100%, and when applied to cells, DMSO % was not above 0.01%.
2.2. Keratinocyte culture
The human keratinocyte cell lines HaCaT and HEK-001 were obtained from Cell Lines Service (Heidelberg, Germany) and the American Type Culture Collection (Manassas, VA, USA), respectively. NHEK were obtained from Thermo Fisher Scientific (Waltham, MA, USA). HaCaT cells were cultured in RPMI-1640 medium containing 10% heat-inactivated fetal calf serum. HEK-001 cells were cultured in keratinocyte-SFM medium with 5 ng/ml human recombinant epidermal growth factor (Thermo Fisher Scientific). Normal human epidermal keratinocytes (NHEK) were cultured in EpiLife serum-free medium containing EpiLife undefined growth supplement (Thermo Fisher Scientific) and were used at the third or fourth passage. All cells were grown in a 5% CO2 incubator at 37 °C.
2.3. Mouse experiment
All experimental procedures using hairless mice were conducted in accordance with the guidelines for the care and use of laboratory animals of Jeju National University (Jeju, Republic of Korea) (permit number: 2017-0026). HR-1 hairless male mice were randomly divided into four animals per group. PM2.5 was administered together with propylene glycol and NAC. PM2.5 was dispersed in propylene glycol at a concentration of 100 μg/ml, spread on a nonwoven polyethylene pad over an area of 1 cm2, and then applied to the dorsal region of mice for 7 consecutive days. The skin tissue was immediately dissected for histological and molecular analyses [16]. All experimental procedures using homozygous flaky tail mice (a/a ma ft/ma ft/J), which carry double-homozygous ft and ma mutations, were approved by the Animal Research Ethics Board of Yonsei University (Seoul, Republic of Korea) (permit number: 2016-0007). C57BL/6 J (wild type) mice and flaky tail mice were randomly divided into three animals per group; control group for C57BL/6 J, PM2.5-treated C57BL/6 J group, control group for flaky tail mice, and PM2.5-treated flaky tail mice group. The hairs on the upper back were removed from anesthetized mice using an electric shaver and depilatory protocol. After hair removal, all procedures were the same as those for hairless mice.
2.4. Histological analysis
Skin sections were fixed in 4% paraformaldehyde and routinely processed for paraffin embedding. Sections (5 µm) of paraffin-embedded skin were deparaffinized and stained with hematoxylin and eosin. The thickness of the epidermis (from the stratum basale to the stratum corneum) was measured in approximately 10 randomly chosen fields taken from 3 representative sections per group by microscopic evaluation at an optical magnification of 100 × with a digital camera. Immunohistochemistry was carried out using the ABC Elite kit (Vector Laboratories, Burlingame, CA, USA). The skin sections were incubated with primary antibody for 1 h. The sections were counterstained with hematoxylin before mounting.
2.5. Human protein cytokine array
Cells were cultured for 3 h and 24 h in the presence of PM2.5 and the supernatants were collected. Cytokines in the supernatants were measured using a RayBio human cytokine antibody array C5 (RayBiotech, Norcross, GA, USA), according to the manufacturer's instructions. Chemiluminescence signals were detected on an ImageQuant LAS-4000 system using a charge-coupled-device camera.
2.6. Enzyme-linked immunosorbent assay (ELISA)
After the 24 h incubation of cells with PM2.5, culture supernatants were collected and stored at −20 °C. IL-1β and IL-6 released in the supernatants were measured by using a Human Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
2.7. Western blot (WB) analysis
Crude protein in cell lysates was electrophoresed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane that was incubated with primary antibodies. Anti-IL-1β, anti-IL-6, and anti-TET1 antibodies were purchased from Thermo Fisher Scientific; anti-TLR5, anti-phospho-NFκB (p65) and anti-DNMT1 antibodies were from Abcam (Cambridge, UK); anti-MyD88, anti-EZH2, anti-H3K27Me3, and anti-H3K4Me3 antibodies were from Cell Signaling Technology (Danvers, MA, USA); anti-NOX4 and anti-DNMT3B antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); and anti-MLL1 antibody was from Active Motif (Carlsbad, CA, USA). Then, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Pierce, Rockland, IL, USA), and protein bands were detected using an enhanced chemiluminescence western blotting detection kit (Amersham, Little Chalfont, Buckinghamshire, UK).
2.8. Reverse transcription-polymerase chain reaction (RT-PCR)
The RT-PCR conditions for forward primer 5′-GAACTCCTTCTCCACAAGCGCCTT-3′ and reverse primer 5′-CAAAAGACCAGTGATGATTTTCACCAGG-3′ for IL-6 and forward primer 5′-AAGGTCGGAGTCAACGGATTT-3′ and reverse primer 5′-GCAGTGAGGGTCTCTCTCCT-3′ for GAPDH were as follows: 5 min at 94 °C; 35 cycles of 1 min at 94 °C, 1 min at 58 °C, and 1 min at 72 °C; and 7 min at 72 °C.
2.9. Flow cytometric analysis
Cells were harvested, washed in FACS buffer (PBS, 0.5% BSA, 0.01% sodium azide), pelleted, suspended in permeabilization/fixation buffer (Thermo Fisher Scientific), and maintained for 20 min at room temperature. The cells were then washed in permeabilization buffer, pelleted, and incubated with anti-TLR5 antibody or anti-MyD88 antibody at 4 °C for 1 h. FITC-conjugated secondary antibody was added at 4 °C for 1 h. Finally, the samples were washed, suspended in FACS buffer, and then analyzed using a FACS LSRII flow cytometer (BD Biosciences) for detection of TLR5 and MyD88. Captured data from gated live populations were analyzed using CellQuest software.
2.10. Quantitative real-time PCR (qRT-PCR)
The qRT-PCR primers were as follows: forward primer 5′-CTATACACCAAGTTGTCAGC-3' and reverse primer 5′-GTCTCCAACTCAGTAAGGTG-3′ for TLR1, forward primer 5'-GCCAAAGTCTTGATTGATTG-3' and reverse primer 5′-TTGAAGTTCTCCAGCTCCTG-3' for TLR2, forward primer 5'-GATCTGTCTCATAATGGCTTG-3′ and reverse primer 5'-GACAGATTCCGAATGCTTGTG-3′ for TLR3, forward primer 5′-TGGATACGTTTCCTTATAAG-3′ and reverse primer 5′-GAAATGGAGGCACCCCTTC-3′ for TLR4, forward primer 5′-ATTGCCAATATCCAGGATGC-3′ and reverse primer 5'-CACCACCATGATGAGAGCAC-3′ for TLR5, forward primer 5′-GCTTACCTCCGAGGATCACA-3′ and reverse primer 5′-CGGGAGGGTGGGTATCTAA-3′ for NOX4, and forward primer 5′-CACCTTCTACAATGAGCTGCGTGT-3′ and reverse primer 5′-CACAGCCTGGATAGCAACGTACA-3′ for actin. The qRT-PCR conditions were as follows: 10 min at 95 °C; 40 cycles of 15 s at 95 °C and 1 min at 60 °C; followed by 7 min at 72 °C. PCR amplification was carried out in a programmable thermal cycler.
2.11. Proximity ligation assay (PLA)
The mouse/rabbit red starter Duolink kit (Sigma-Aldrich) was used for this experiment. Cells were permeabilized according to the manufacturer's instructions and were incubated with primary antibodies, mouse anti-TLR5 (1:100) and rabbit anti-MyD88 (1:100) or rabbit anti-NOX4 (1:100) diluted in fluorescence dilution buffer (5% fetal calf serum, 5% normal donkey serum, 2% bovine serum albumin in PBSCM, pH 7.6) for 2 h at room temperature. Then, they were incubated with the PLA probes for 1 h at 37 °C in a humid chamber. The ligation reaction was carried out at 37 °C for 1 h in a humid chamber. The cells were then incubated with the amplification mix for 2 h at 37 °C in a darkened humidified chamber, mounted using mounting medium, and imaged using a Zeiss confocal microscope and the LSM 510 program.
2.12. Immunoprecipitation and western blot analysis (IP-WB)
Cells were lysed in 0.5% NP-40, 10 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 5 mM MgCl2. Lysates were incubated overnight at 4 °C with antibodies targeting TLR5, MyD88 or NOX4, and IgG. Immune complexes were collected with protein A/G PLUS Beads (Santa Cruz Biotechnology) overnight at 4 °C and were washed with immunoprecipitation buffer. Equal amounts of the precipitates were separated by SDS-PAGE, followed by western blot analysis with antibodies specific for TLR5, MyD88, or NOX4.
2.13. Transfection of small interfering RNA (siRNA)
Cells were transfected with 20 nM of control small interfering RNA (siRNA) or siRNAs against TLR1, TLR3, TLR4, TLR5, MyD88, NOX4, TET1, or MLL1 using Lipofectamine RNAiMax reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.
2.14. Drug affinity responsive target stability (DARTS) assay
The DARTS assay was conducted according to a previously described protocol [17]. HaCaT cells were grown to 80% confluence in 10 mm dishes, treated with PM2.5 or DMSO for 1 h, and lysed in M-PER buffer (Pierce, Rockford, IL, USA) containing protease and phosphatase inhibitors. After centrifugation (12,000 ×g, 10 min, 4 °C), 10 × TNC buffer [500 mM Tris·HCl (pH 8.0), 500 mM NaCl, 100 mM CaCl2] was added to the lysates, and the protein concentration was determined with a dye-binding protein assay kit (Bio-Rad Laboratories) following the manufacturer's instructions. The lysates (50 μg/μl) were digested with pronase (protein-to-pronase ratio of 1:2500 or 1:20,000) for 30 min. Digestion was stopped by adding 5 ×Laemmli sample buffer. Then, the samples were subjected to western blot analysis.
2.15. Detection of intracellular ROS
Cells were treated with 25 µM dichlorodihydrofluorescein diacetate. The fluorescence of 2′,7′-dichlorofluorescein was detected using a flow cytometer.
2.16. Immunocytochemistry
Cells plated on chamber slides were fixed with 4% paraformaldehyde for 30 min and permeabilized with PBS containing 0.1% Triton X-100 for 2.5 min. The cells were treated with blocking medium (PBS containing 3% bovine serum albumin) for 1 h and incubated for 2 h with the primary antibody diluted in blocking medium. Then, the cells were incubated with FITC- or Alexa 594-conjugated secondary antibody (Santa Cruz Biotechnology) for 1 h. Stained cells were washed with PBS and mounted on microscope slides in mounting medium containing DAPI and imaged on a Zeiss confocal microscope using the LSM 510 software.
2.17. Protein carbonylation assay
Total protein of the skin tissues was isolated using protein lysis buffer and quantified. The OxiSelect™ protein carbonyl ELISA kit (Cell Biolabs, San Diego, CA, USA) was used in accordance with the manufacturer's instructions.
2.18. Quantitative methylation-specific PCR (qMSP) and bisulfite sequencing
For methylation analysis, DNA was extracted using a standard phenol-chloroform method. Two micrograms of DNA was modified by bisulfite with the EZ DNA methylation kit™ (Zymo Research, Orange, CA, USA), which guarantees a > 99% conversion rate (non-methylated cytosine to uracil; protection of methylated cytosine residues). Promoter methylation was analyzed using MSP primer pairs located close to the putative transcription start site in the 5′ CpG island, 2 μl of bisulfite-treated DNA as the template, and JumpStart REDTaq DNA polymerase (Sigma-Aldrich), as previously described [18]. For bisulfite sequence analysis, PCR amplicons were separated by 2% agarose gel electrophoresis, purified with a gel extraction kit (Qiagen GmbH, Hilden, Germany), and cloned using the TOPO TA vector system (Invitrogen, Carlsbad, CA, USA). Individual clones were isolated and purified using the NucleoSpin plasmid isolation kit (Macherey-Nagel, Düren, Germany). Randomly selected positive clones (10–15 from each sample) were sequenced using the M13F primer, and the methylation status of each CpG dinucleotide was analyzed. For quantification of IL-6 methylation, bisulfite-treated samples were subjected to qMSP. The methylation levels were normalized based on Alu element amplification. qPCR was performed using a CFX96TM real-time system (Bio-Rad, Hercules, CA, USA).
2.19. Chromatin immunoprecipitation (ChIP) and quantitative PCR (qPCR)
ChIP assays were performed with a simple ChIP™ kit (Cell Signaling Technology) according to the manufacturer's instructions, with slight modifications. DNMT1, DNMT3B, TET1, EZH2, H3K27Me3, MLL1, or H3K4Me3 antibodies and normal rabbit IgG were used. DNA recovered from the immune-precipitated complexes was subjected to qPCR.
2.20. Statistical analysis
Mean ± standard error of the mean is reported. Data were subjected to analysis of variance, followed by Tukey's test to analyze differences between conditions. In each case, p < 0.05 was considered statistically significant.
3. Results
3.1. PM2.5 stimulates pro-inflammatory production in human keratinocytes
To examine pro-inflammatory cytokine production in human keratinocytes, supernatants from HaCaT cells cultured at 3 h and at 24 h after PM2.5 stimulation were assessed using cytokine arrays. The assay revealed that six cytokines, IL-6, MDC, MIP-1β, EGF, IGF-1, and MIF were strongly upregulated at 3 h in PM2.5-treated HaCaT cells as compared to control cells, whereas five cytokines, IL-1β, IL-6, IL-8, IL-16, and TGF-β2 were strongly upregulated at 24 h in PM2.5-treated HaCaT cells as compared to control cells (Fig. 1A). The levels of the pro-inflammatory cytokines IL-1β and IL-6 were confirmed to be increased upon exposure to PM2.5 in HaCaT, HEK-001 keratinocyte cell lines, and (NHEK) (Fig. 1B), as well as in murine epidermis (Fig. 1C and D). We selected IL-6 as a major mediator of inflammation induced by PM2.5 for further study. IL-6 mRNA expression was induced between 3 h and 72 h after PM2.5 exposure of HaCaT cells (Fig. 1E). In full agreement with the RT-PCR results, western blotting (WB) revealed a clear induction of IL-6 protein in HaCaT, HEK-001, and NHEK cells (Fig. 1F).
Fig. 1.

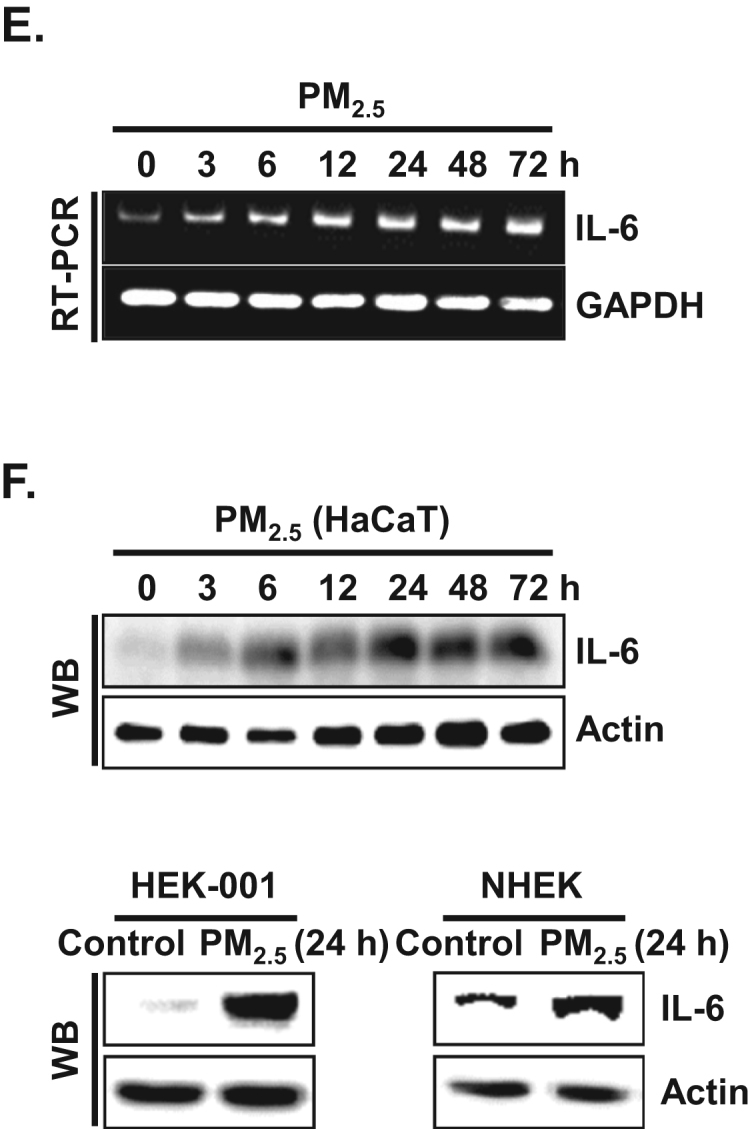
PM2.5 induces cytokine production in keratinocytes. A. Cells were incubated in the presence of PM2.5 for 3 h and 24 h. Cytokine secretion into the supernatants was analyzed using cytokine antibody arrays. Each spot indicates the expression level of a cytokine detected by each primary antibody. B. IL-1β and IL-6 concentrations in HaCaT, HEK-001, and NHEK were assessed using a human IL-1β and an IL-6 Quantikine ELISA kit, respectively. C. Skin sections were stained with IL-1β antibody and counterstained with hematoxylin. D. IL-6 expression was detected by western blotting using IL-6 antibody. The expression of E. IL-6 mRNA by RT-PCR and F. protein by western blot assay was assessed at indicated time points. *Significantly different from control group (p < 0.05).,.
3.2. PM2.5 activates TLR5 early and induces the release of IL-6 in human keratinocytes and mouse skin
We analyzed whether PM2.5 could induce the gene expression of various TLRs in keratinocytes. HaCaT cells were incubated with PM2.5 for 0.5 h or 24 h, and TLR gene expression was analyzed by quantitative RT-PCR (qRT-PCR). PM2.5 treatment induced a significant increase in the expression of TLR1 and TLR5 at 0.5 h and of TLR3 at 24 h (Fig. 2A). TLR5 was highly expressed at 0.5 h and maintained up to 24 h compared to TLR3. In addition, TLR5 was highly expressed compared to TLR1 at 0.5 h and 24 h (Fig. 2B). The mRNA expression pattern of TLR5 in HaCaT cells was consistent with protein expression in murine epidermis (Fig. 2C) and with protein expression in HaCaT cells (Fig. 2D). In addition, the expression pattern of TLR5 at 0.5 h after PM2.5 treatment in HaCaT cells was confirmed using flow cytometry (Fig. 2E).
Fig. 2.


PM2.5 activates TLR5-mediated IL-6 production. The mRNA expression of A. various TLRs and B. TLR1 and TLR5 at indicated time points was assessed by qRT-PCR. C. Skin sections were stained with TLR5 antibody and counterstained with hematoxylin. Protein expression of TRL5 and MyD88 was detected by D. western blotting and E. flow cytometry with the corresponding antibodies. Interaction between TLR5 and MyD88 was examined by F. PLA and G. IP. Cells were transfected with H. siTLR5 RNA or I. siMyD88 RNA, respectively, and incubated for 24 h. IL-6 was detected by western blotting with IL-6 antibody. *Significantly different from control group (p < 0.05).,.
TLRs recruit a specific combination of Toll/IL-1 receptor-containing adaptors, including myeloid differentiation primary response gene 88 (MyD88). As shown in Fig. 2D and E, MyD88 protein expression was consistent with TLR5 protein expression. In addition, proximity ligation assay (PLA) and immunoprecipitation (IP)-WB analysis showed that TLR5 strongly interacted with MyD88 in PM2.5-treated cells as compared to PM2.5-untreated cells, suggesting that downstream signaling might be induced (Fig. 2F and G). To test whether TLR5 mediated IL-6 generation in PM2.5-treated human keratinocytes, HaCaT cells were transfected with siRNA specific to TLR5 or MyD88, and IL-6 expression was analyzed by western blot assay. IL-6 expression was abolished in HaCaT cells transfected with TLR5 or MyD88 siRNA (Fig. 2H and I).
3.3. PM2.5 induces the TLR5–NFκB–IL-6 pathway
To investigate whether PM2.5 binds to TLR5 and/or MyD88, we performed a drug affinity responsive target stability (DARTS) assay using HaCaT cells. The DARTS assay is based on the principle that the binding of a ligand and its receptor leads to a thermodynamically more stable state in which resistance to protease degradation is markedly increased [19]. To test PM2.5 interaction with TLR5, lysates from HaCaT cells were incubated with PM2.5 or flagellin (FL, positive control). Pronase-digested cell lysates were subjected to western blotting using TLR5-specific antibody. As shown in Fig. 3A, protease susceptibility of TLR5, which has a molecular weight around 103 kDa, was clearly reduced in cell lysates pretreated with PM2.5 and flagellin as compared to the control 1:20,000–1:2500 pronase treatments. These observations indicated that PM2.5 promotes IL-6 production by binding to TLR5. In addition, when we assessed PM2.5 interaction with MyD88, there was no significant interaction of MyD88 (around 33 kDa) with PM2.5 and flagellin (Fig. 3A). These observations indicated that PM2.5 promotes IL-6 production by binding to TLR5 but not to MyD88. To assess whether TLR5 can activate the canonical NFκB pathway to induce IL-6, we detected the phosphorylation of NFκB p65 subunit at indicated time points. As shown in Fig. 3B, phospho-NFκB p65 was expressed between 0.5 h and 24 h after treatment with PM2.5. In addition, the phospho-NFκB p65 subunit translocated into the nucleus at 3 h in the presence of PM2.5 and bound to promoter region of IL-6 (Fig. 3C and D). Transfection of HaCaT cells with siRNA specific to NFκB p65 suppressed the increase in IL-6 by PM2.5 treatment (Fig. 3E). In addition, transfection with siRNA specific to TLR5 or MyD88 reduced the increase in phospho-NFκB p65 expression by PM2.5 treatment, resulting in decreased IL-6 expression (Fig. 3F and G).
Fig. 3.

PM2.5 induces the TLR5–NFκB–IL-6 pathway. A. Binding of PM2.5 to TLR5 or MyD88 was determined by DARTS assay. Cell lysates were prepared, incubated with PM2.5 and flagellin for 1 h, and digested with pronase to protein ratios (1:2500 to 1:20,000) for 10 min. TLR5 and MyD88 were detected by western blotting with the corresponding antibodies. B. The phospho-NFκB p65 subunit was detected by western blotting with the corresponding antibody. C. The nuclear localization of phospho-NFκB p65 was detected by confocal microscopy after FITC labeling with the corresponding antibodies. D. Phospho-NFκB p65 binding to the IL-6 promoter was analyzed using ChIP followed by PCR. Cells were transfected with E. siNFκB p65 RNA, F. siTLR5 RNA, and G. siMyD88 RNA, incubated for 24 h, and IL-6 and NFκB were detected by western blotting with the corresponding antibodies.
3.4. PM2.5 activates TLR5 and NOX4 interaction and induces ROS production in human keratinocytes
Recently, Kim et al. [11] reported that flagellin-induced interaction between TLR5 and NOX4 led to ROS generation, which in turn promoted the secretion of pro-inflammatory cytokines, including IL-6 [11]. In the current study, the expression of NOX4 mRNA and protein was increased between 0.5 h and 24 h after PM2.5 treatment (Fig. 4A and B). In addition, TLR5 strongly interacted with NOX4 at 0.5 h in PM2.5-treated cells as compared to non-treated cells, as indicated by PLA assay and IP-WB analysis (Fig. 4C and D). Furthermore, in HaCaT cells transfected with siRNA specific to NOX4, the increase in IL-6 by PM2.5 treatment was suppressed (Fig. 4E). The interaction of TLR5 and NOX4 in the presence of PM2.5 led to an increase in ROS at 0.5 h, while this response was attenuated by N-acetylcysteine (NAC), a well-known antioxidant (Fig. 4F). In HaCaT cells transfected with siRNA specific to TLR5 or NOX4, the increase in ROS by PM2.5 treatment was suppressed (Fig. 4G and H). However, siRNA-mediated silencing of TLR5 did not influence NOX4 expression (Fig. 4I). In addition, to test whether other TLRs induced in PM2.5-treated human keratinocytes (Fig. 2A) affect the ROS–NFκB–IL-6 pathway, cells were transfected with siRNA specific to TLR1, TLR3, and TLR4 (Fig. 4J). We found that ROS levels and phospho-NFκB and IL-6 expression were abolished in HaCaT cells transfected with siTLR1, siTLR3, and siTLR4 RNAs (Fig. 4K, L, and M).
Fig. 4.




PM2.5 activates TLR5 and NOX4 interaction and induces ROS production in keratinocytes. The expression of A. NOX4 mRNA and B. protein at indicated time points was assessed by qRT-PCR and western blotting, respectively. Interaction between TLR5 and NOX4 was examined by C. PLA and D. IP-WB. E. Cells were transfected with siNOX4 RNA and incubated for 24 h. IL-6 was detected by western blotting with an IL-6 antibody. F. The intracellular ROS level was determined by flow cytometry after staining of the cells with DCF-DA. Cells were transfected with siRNA specific to G. TLR5 and H. NOX4, and ROS production was assessed by flow cytometry after DCF-DA staining. I. Cells were transfected with TLR5 siRNA and incubated for 24 h. NOX4 was detected by western blotting with a NOX4 antibody. J. Cells were transfected with siTLR1 RNA, siTLR3 RNA, and siTLR4 RNA. K. The intracellular ROS production was assessed by flow cytometry after DCF-DA staining. L. Phospho-NFκB p65 and M. IL-6 were detected by western blotting with phospho-NFκB (p65) and IL-6 antibodies, respectively. *Significantly different from control group (p < 0.05); #significantly different from PM2.5-treated group (p < 0.05).,.
3.5. The NFκB–IL-6 pathway is mediated by PM2.5-TLR5-mediated oxidative stress
For a better understanding of the importance of ROS in TLR5-mediated NFκB activation, and synthesis of IL-6, HaCaT cells were transfected with siRNA specific to NOX4. The transfected cells showed decreased protein expression of the phospho-NFκB p65 subunit (Fig. 5A). In addition, NAC pretreatment suppressed the increase in phospho-NFκB p65 at 3 h in PM2.5-treated HaCaT cells compared to the control (Fig. 5B), leading to reduced production of IL-6 (Fig. 5C). In addition, in the skin of PM2.5-treated mice, protein expression of phospho-NFκB p65 and IL-6 was increased, whereas NAC pretreatment of PM2.5-treated mice reduced these responses (Fig. 5D and E). Thus, PM2.5 induced skin cellular inflammation through ROS-dependent, TLR5-initiated activation of the NFkB signaling pathway that results in IL-6 expression.
Fig. 5.

PM2.5 induces the NFκB-IL-6 pathway mediated by oxidative stress. A. Cells were transfected with NOX4 siRNA and were incubated for 24 h, and phospho-NFκB p65 was detected by western blotting with a phospho-NFκB p65 antibody. B. Phospho-NFκB p65 was detected by western blotting with the corresponding antibody in HaCaT cells. C. The concentration of IL-6 in HaCaT cells was assessed using a human IL-6 Quantikine ELISA kit. D. Phospho-NFκB p65 and IL-6 in HaCaT cells were detected by western blotting with the corresponding antibodies in mouse skin cells. E. Mouse skin sections were stained with phospho-NFκB p65 subunit and IL-6 and were counterstained with hematoxylin. *Significantly different from control group (p < 0.05); #significantly different from PM2.5-treated group (p < 0.05).
3.6. Expression levels of inflammatory mediators and oxidative stress are increased in PM2.5-treated flaky tail mice
To evaluate whether inflammatory pathway (TLR5-NOX4-NFκB-IL6) in PM2.5-treated cells occurs in flaky tail mice, we detected the expression level of TLR5, MyD88, NOX4, phospho-NFκB, and IL-6 upon PM2.5 in skin tissues of wild type (C57BL/6 J) mice and flaky tail mice. The expression level of TLR5, MyD88, NOX4, phospho-NFκB, and IL-6 in PM2.5-treated skin tissues of wild type mice was increased compared to that in untreated-skin tissues of wild type mice. In addition, the levels of these inflammatory mediators in the PM2.5-treated skin tissues of flaky tail mice were increased compared to those in untreated-skin tissues of flaky tail mice, which showed a high level of inflammatory mediators compared to that in C57BL/6 J with PM2.5 or without PM2.5 (Fig. 6A).
Fig. 6.

PM2.5 increased expressions of inflammatory mediators and oxidative stress in flaky tail mice. A. The protein expression level of skin tissues was detected by western blotting using each antibody. B. Protein oxidation was assayed by measuring carbonyl formation. *Significantly different from control group of C57BL/6 J mice (p < 0.05); **significantly different from PM2.5-treated group of C57BL/6 J mice (p < 0.05); #significantly different from control group of flaky tail mice (p < 0.05).
To obtain further evidence regarding oxidative stress induced by PM2.5 in wild type mice and flaky tail mice, protein peroxidation was evaluated by detecting protein carbonylation, a marker of protein peroxidation, which revealed a significant elevation in protein carbonylation in skin tissues of PM2.5-treated flaky tail mice compared to the three other mice groups (Fig. 6B).
3.7. IL-6 transcription is regulated epigenetically via promoter DNA methylation in PM2.5-induced inflammation
Recently, Yang et al. [20] reported that the human IL-6 promoter contains abundant CpG sites around the transcription start site, including region A (-1469 to −1238 bp, 8 CpG sites), region B (-638 to −346 bp, 7 CpG sites), and region C (+150 to +347 bp, 10 CpG sites) [20]. Using ChIP-qPCR analysis, we assessed whether DNA methyltransferases DNMT1 and DNMT3B, and DNA demethylase TET1 can directly bind to the IL-6 promoter region (Fig. 7A). The binding of DNMT1 and DNMT3B to the IL-6 promoter region occurred in the absence of PM2.5. However, binding at the A and B regions decreased upon PM2.5 treatment (Fig. 7B and C). The loss of binding of DNMT1 and DNMT3B by PM2.5 prompted us to investigate whether this is accompanied by the appearance of TET1. We found that concomitant with the decreases in DNMT1 and DNMT3B, TET1 strongly bound the A and B regions of the IL-6 promoter region in PM2.5-treated cells (Fig. 7D). Next, we examined the methylation status of region A by quantitative methylation-specific PCR (qMSP) and bisulfite sequencing analysis. As shown in Fig. 7E, the DNA methylation level of the IL-6 promoter region was significantly decreased in PM2.5-treated cells compared to the controls. This was confirmed by bisulfite sequencing, which revealed lower methylation in PM2.5-treated cells than in the controls (59% vs. 38%) (Fig. 7F). Thus, the change in IL-6 transcription in PM2.5-treated cells was due to promoter DNA methylation. Collectively, these findings showed that during PM2.5-mediated IL-6 expression, TET can replace DNMT, leading to IL-6 expression. Since the increase in TET expression and hypomethylation of the IL-6 promoter preceded the induction of IL-6 during PM2.5-induced inflammation, we elucidated whether the expression of IL-6 depended on TET. Transfection with siTET1 RNA in PM2.5-treated cells reduced the expression level of IL-6 compared with that in cells treated with PM2.5 alone (Fig. 7G). Thus, TET1 markedly contributed to the transcriptional expression of the IL-6 locus, leading to skin inflammation.
Fig. 7.

IL-6 is regulated epigenetically via DNA methylation in PM2.5-induced inflammation. A. ChIP-qPCR was performed using primer sets specific for the regions indicated as A (-1469 bp to −1238 bp), B (-638 bp to −346 bp), and C (+150 bp to +347 bp) along the IL-6 promoter region. ChIP was conducted using antibodies against B. DNMT1, C. DNMT3B, and D. TET1, and the precipitates were analyzed by qPCR. E. Gene methylation levels were normalized to that of the Alu element. F. Bisulfite sequencing analysis of the IL-6 promoter. Black circles represent methylated cytosine resides; white circles represent non-methylated cytosine residues. G. Cells were transfected with siTET1 RNA and incubated for 24 h. TET1 and IL-6 were detected by western blotting with the corresponding antibodies. *Significantly different from control group (p < 0.05).
3.8. IL-6 is regulated epigenetically via histone methylation in PM2.5-induced inflammation
Depending on the site and degree of histone methylation, it can lead to either gene expression or repression. To investigate whether epigenetic histone methylation is involved in PM2.5-induced IL-6 expression, we assessed whether EZH2, which is a transcriptional repressor that has H3K27 methyltransferase activity, occupies the IL-6 locus in HaCaT cells by ChIP-qPCR. We observed the binding of EZH2 and its substrate, H3K27Me3, to the IL-6 promoter region in the absence of PM2.5. However, binding of EZH2 to the A and B regions of the IL-6 promoter was decreased by PM2.5 treatment (Fig. 8A), resulting in a decrease in H3K27Me3 (Fig. 8B). The loss of EZH2 and the H3K27Me3-repressive histone mark by PM2.5 prompted us to investigate whether this is accompanied by the appearance of MLL1, a transcriptional activator that has H3K4 methyltransferase activity, and the H3K4Me3-positive histone mark. We found that, concomitant with the decreases in EZH2 and H3K27Me3, MLL1 and the active H3K4Me3 mark strongly bound to the A and B regions of the IL-6 promoter in PM2.5-treated cells (Fig. 8C and D). Collectively, these results showed that during PM2.5-mediated IL-6 expression, the transcriptional activator MLL1 can replace the transcriptional silencer EZH2. As the increase in MLL1 expression preceded the induction of IL-6 during PM2.5-induced inflammation, we examined whether IL-6 expression depended on MLL1. Transfection with siMLL1 RNA in PM2.5-treated cells reduced the expression level of IL-6 compared to that in cells treated with PM2.5 alone (Fig. 8E). Thus, MLL1 contributes significantly to the transcriptional expression of the IL-6 locus.
Fig. 8.

IL-6 is regulated epigenetically via histone methylation in PM2.5-induced inflammation. ChIP was conducted using antibodies directed against A. EZH2, B. H3K27Me3, C. MLL1, and D. H3K4Me3, and the precipitates were analyzed by qPCR. Cells were transfected with siMLL1 RNA and incubated for 24 h. E. MLL1 and IL-6 were detected by western blotting with the corresponding antibodies. *Significantly different from control group (p < 0.05). F. Proposed model for the involvement of the TLR5-NOX4-IL-6 pathway in PM2.5-induced inflammation. Ligation of TLR5 with PM2.5 stimulates NOX4 to generate ROS, which in turn promotes the secretion of pro-inflammatory cytokines, including IL-6, in skin keratinocytes. In addition, epigenetic changes prompt the binding of TET1 and MLL1 to the IL-6 locus in place of DNMT and EZH2. This binding induces the expression of IL-6 and of pro-inflammatory mediators, which induce monocyte accumulation, leading to skin inflammation.
4. Discussion
Pro-inflammatory cytokines, including ILs, initiate inflammatory reactions in skin keratinocytes [21]. Several reports have shown that various stimulators induce an increase in the expression of IL-1β, IL-6, IL-8, MCP-1, MIP-1β, and TNF-α in HaCaT cells [22], [23], [24]. In the present study, the expression of various cytokines, including IL-6, was induced by PM2.5 treatment. IL-6 is a pleiotropic cytokine involved in the physiology of virtually every organ system, and its production is promptly increased in acute inflammatory responses associated with allergy, asthma and atopy [25], [26]. Therefore, the inhibition of IL-6 production is helpful for preventing inflammation.
TLRs are a family of receptors that predominantly recognize pathogen-associated molecular patterns (PAMP), and binding of PAMP to TLR on host cells leads to an inflammatory response, including the secretion of cytokines and chemokines [27]. The present study showed that TLR5- and MyD88-dependent signaling is essential for IL-6 production in response to PM2.5, as this response was prevented by siRNA targeting TLR5 or its associated adaptor MyD88. In addition, the NFκB signaling pathway downstream of TLR5–MyD88 was activated for induction of IL-6 by PM2.5; NFκB signaling was prevented by siRNA targeting TLR5 or MyD88 and resulted in a decrease in IL-6 expression. From these findings, we conclude that the binding of PM2.5 to TLR5 initiates intracellular signaling through MyD88, and this TLR5 and MyD88 signaling results in the activation of downstream kinases, leading to translocation of free NFκB to the nucleus, where it binds to the promoter region of IL-6.
Excessive TLR expression reportedly marks an active inflammatory reaction and triggers the downstream NFκB signaling pathway [28]. NFκB p65 is a critical regulatory transcription factor involved in major inflammatory pathways downstream of TLRs that regulates the expression of many inflammatory genes, including IL-6, and the production of cytokines [12]. NFκB exists in the cytoplasm in an inactive form bound to the IκB inhibitor. Upon stimulation, the NFκB p65 subunit separates from IκBα and translocates into the nucleus, triggering the transcription of multiple inflammatory genes [29]. The results of the present study showed that PM2.5 stimulated phosphorylation of NFκB p65 and its subsequent translocation into nucleus and binding to promoter site of IL-6.
Recently, Kim et al. (2016) reported that NOX4 plays a vital role in TLR5-dependent inflammatory responses, including IL-8 production, and is involved in atherosclerosis [11]. In addition, they reported that flagellin stimulated H2O2 generation through direct TLR5–NOX4 interaction. In our study, interestingly, TLR5 and NOX4 interacted directly and moreover, cells in which NOX4 expression was knocked down failed to induce IL-6. This interaction of TLR5 and NOX4 induced ROS, which was prevented by siRNA targeting TLR5 or NOX4. However, TLR5 did not affect NOX4 expression, as siRNA targeting TLR5 did not change NOX4 expression. Furthermore, the TLR5-NOX4-ROS signal pathway activated NFκB-IL-6 downstream, which was prevented by siRNA targeting NOX4 or by the antioxidant NAC in vitro as well as in vivo.
Several recent studies showed that exposure to PM is associated with the development as well as the exacerbation of atopic dermatitis [30], [31], [32]. Flaky tail mice are essentially deficient in filaggrin, which is the main skin barrier protein, and is related to the incidence of atopic dermatitis and the response to inflammation [33]. In the present study, we showed the activation of the inflammatory pathway (TLR5-NOX4-NFκB-IL6) by PM2.5 in wild type mice and flaky tail mice, suggesting that PM2.5-dependent inflammation can cause development as well as exacerbation of atopic dermatitis. Interestingly, Liu et al. [34] demonstrated that PM2.5-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6-AKT-STAT3-NFκB-dependent pathway, in which IL-6 is upstream of NFκB [34].
To elucidate the molecular mechanisms underlying IL-6 overproduction in PM2.5-treated cells, we assessed the epigenetic modification of DNA and histones. Mammalian gene expression requires a loosening of the chromatin structure, exposure of the promoter region, and the binding of transcription factors and RNA polymerase, which is regulated by epigenetic modifications. DNA methylation determines the accessibility of the promoter region and histone modifications, including methylation, determine the loosening of chromatin. Thus, investigating how epigenetic modifications regulate gene expression can provide deeper insight into gene expression regulation. In the present study, PM2.5 treatment induced DNA hypomethylation of and H3K4Me3 binding to the IL-6 promoter. Bellavia et al. [13] demonstrated that ambient PM induced DNA hypomethylation of long interspersed nuclear element-1 and TLR4 in human blood; however, no significant differences in IL-6 methylation were observed [13]. This may be due to different experimental conditions, including PM composition and the cell system used. In mammals, DNA methylation primarily occurs at CpG dinucleotides, which are often observed in clusters called CpG islands that are generally located in the gene promoter. The methylation of CpG islands leads to stable and heritable transcriptional silencing through the binding of methyl-DNA-specific proteins to methylated CpG islands. Further, methyl-DNA-specific proteins attract histone-modifying enzymes, which establish a silenced chromatin state. We noticed that the binding of DNMT as well as of EZH2 in regions A (-1469 to −1238 bp) and B (-638 to −346 bp) of the IL-6 promoter was reduced, whereas the binding of TET as well as of MLL1 in these two regions was increased under PM2.5 treatment. In addition, using siRNA, we showed that IL-6 expression is dependent on TET1 and MLL1.
To our knowledge, this is the first report that verified a link between PM2.5 and the TLR5 signaling pathway; the TLR5-NOX4-ROS-NFκB-IL-6 pathway plays a significant role in mediating PM2.5-induced inflammation. Targeting the TLR5–NOX4 cascade in skin keratinocytes might be a therapeutic strategy for mitigating the harmful effects of PM2.5 (Fig. 8F).
Acknowledgments
This work was supported by a grant from the Basic Research Laboratory Program (NRF-2017R1A4A1014512) by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.101080.
Appendix A. Supplementary material
Supplementary material
References
- 1.Jung S.J., Lim S., Kwon S., Jeon J., Kim J., Lee J., Kim S. Characterization of particulate matter from diesel passenger cars tested on chassis dynamometers. J. Environ. Sci. 2017;54:21–32. doi: 10.1016/j.jes.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 2.Ma Q.Y., Huang D.Y., Zhang H.J., Wang S., Chen X.F. Exposure to particulate matter 2.5 (PM2.5) induced macrophage-dependent inflammation, characterized by increased Th1/Th17 cytokine secretion and cytotoxicity. Int. Immunopharmacol. 2017;50:139–145. doi: 10.1016/j.intimp.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 3.Miyake T., Wang D., Matsuoka H., Morita K., Yasuda H., Yatera K., Kanazawa T., Yoshida Y. Endocytosis of particulate matter induces cytokine production by neutrophil via Toll-like receptor 4. Int. Immunopharmacol. 2018;57:190–199. doi: 10.1016/j.intimp.2018.02.020. [DOI] [PubMed] [Google Scholar]
- 4.Yoon S., Han S., Jeon K.J., Kwon S. Effects of collected road dusts on cell viability, inflammatory response, and oxidative stress in cultured human corneal epithelial cells. Toxicol. Lett. 2018;284:152–160. doi: 10.1016/j.toxlet.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Shang Y., Smith S., Hu X. Role of Notch signaling in regulation innate immunity and inflammation in health and disease. Protein Cell. 2016;7:159–174. doi: 10.1007/s13238-016-0250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukherjee S., Karmakar S., Babu S.P. TLR2 and TLR4 mediated host immune response in major infectious disease: a review. Braz. J. Infect. Dis. 2016;20:193–204. doi: 10.1016/j.bjid.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuki T., Yoshida H., Akazawa Y., Komiya A., Sugiyama Y., Inoue S. Activation of TLR2 enhances tight junction barrier in epidermal keratinocytes. J. Immunol. 2011;187:3230–3237. doi: 10.4049/jimmunol.1100058. [DOI] [PubMed] [Google Scholar]
- 8.Chen L., DiPietro L.A. Toll-like receptor function in acute wounds. Adv. Wound Care. 2017;6:344–355. doi: 10.1089/wound.2017.0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goulopoulou S., McCarthy C.G., Webb R.C. Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol. Rev. 2016;68:142–167. doi: 10.1124/pr.114.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joo J.H., Ryu J.H., Kim C.H., Kim H.J., Suh M.S., Kim J.O., Chung S.Y., Lee S.N., Kim H.M., Bae Y.S., Yoon J.H. Dual oxidase 2 is essential for the toll-like receptor 5-mediated inflammatory response in airway mucosa. Antioxid. Redox Signal. 2012;16:57–70. doi: 10.1089/ars.2011.3898. [DOI] [PubMed] [Google Scholar]
- 11.Kim J., Seo M., Kim S.K., Bae Y.S. Flagellin-induced NADPH oxidase 4 activation is involved in atherosclerosis. Sci. Rep. 2016;6:25437. doi: 10.1038/srep25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu T., Zhang L., Joo D., Sun S.C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017 doi: 10.1038/sigtrans.2017.23. (pii: 17023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellavia A., Urch B., Speck M., Brook R.D., Scott J.A., Albetti B., Behbod B., North M., Valeri L., Bertazzi P.A., Silverman F., Gold D., Baccarelli A.A. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J. Am. Heart Assoc. 2013;2:e000212. doi: 10.1161/JAHA.113.000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valinluck V., Tsai H.H., Rogstad D.K., Burdzy A., Bird A., Sowers L.C. Oxidative damage to methyl‐CpG sequences inhibits the binding of the methyl‐CpG binding domain (MBD) of methyl‐CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piao M.J., Ahn M.J., Kang K.A., Ryu Y.S., Hyun Y.J., Shilnikova K., Zhen A.X., Jeong J.W., Choi Y.H., Kang H.K., Koh Y.S., Hyun J.W. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol. 2018;92:2077–2091. doi: 10.1007/s00204-018-2197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee C.W., Lin Z.C., Hu S.C., Chiang Y.C., Hsu L.F., Lin Y.C., Lee I.T., Tsai M.H., Fang J.Y. Urban particulate matter down-regulates filaggrin via COX2 expression/PGE2 production leading to skin barrier dysfunction. Sci. Rep. 2016;6:27995. doi: 10.1038/srep27995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lomenick B., Jung G., Wohlschlegel J.A., Huang J. Target identification using drug affinity responsive target stability (DARTS) Curr. Protoc. Chem. Biol. 2011;3:163–180. doi: 10.1002/9780470559277.ch110180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herman J.G., Graff J.R., Myöhänen S., Nelkin B.D., Baylin S.B., Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleta-Soriano E., Martinez J.P., Hinkelmann B., Gerth K., Washausen P., Diez J., Frank R., Sasse F., Meyerhans A. The myxobacterial metabolite ratjadone A inhibits HIV infection by blocking the Rev/CRM1-mediated nuclear export pathway. Microb. Cell Fact. 2014;13:17. doi: 10.1186/1475-2859-13-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang F., Zhou S., Wang C., Huang Y., Li H., Wang Y., Zhu Z., Tang J., Yan M. Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci. Rep. 2017;7:43592. doi: 10.1038/srep43592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernard F.X., Morel F., Camus M., Pedretti N., Barrault C., Garnier J., Lecron J.C. Keratinocytes under fire of proinflammatory cytokines: bone fide innate immune cells involved in the physiopathology of chronic atopic dermatitis and psoriasis. J. Allergy. 2012;2012:718725. doi: 10.1155/2012/718725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kee J.Y., Jeon Y.D., Kim D.S., Han Y.H., Park J., Youn D.H., Kim S.J., Ahn K.S., Um J.Y., Hong S.H. Korean red ginseng improves atopic dermatitis-like skin lesions by suppressing expression of proinflammatory cytokines and chemokines in vivo and in vitro. J. Ginseng Res. 2017;41:134–143. doi: 10.1016/j.jgr.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park K., Lee J.H., Cho H.C., Cho S.Y., Cho J.W. Down-regulation of IL-6, IL-8, TNF-α and IL-1β by glucosamine in HaCaT cells, but not in the presence of TNF-α. Oncol. Lett. 2010;1:289–292. doi: 10.3892/ol_00000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seo W.Y., Youn G.S., Choi S.Y., Park J. Butein, a tetrahydroxychalcone, suppresses pro-inflammatory responses in HaCaT keratinocytes. BMB Rep. 2015;48:495–500. doi: 10.5483/BMBRep.2015.48.9.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slaats J., Ten Oever J., van de Veerdonk F.L., Netea M.G. IL-1β/IL-6/CRP and IL-18/ferritin: distinct inflammatory programs in infections. PLoS Pathog. 2016;12:e1005973. doi: 10.1371/journal.ppat.1005973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strzelak A., Ratajczak A., Adamiec A., Feleszko W. Tobacco smoke induces and alters immune responses in the lung triggering inflammation, allergy, asthma and other lung diseases: a mechanistic review. Int. J. Environ. Res. Public. Health. 2018;15 doi: 10.3390/ijerph15051033. (pii: E1033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cronin J.G., Turner M.L., Goetze L., Bryant C.E., Sheldon I.M. Toll-like receptor 4 and MYD88-dependent signaling mechanisms of the innate immune system are essential for the response to lipopolysaccharide by epithelial and stromal cells of the bovine endometrium. Biol. Reprod. 2012;86:51. doi: 10.1095/biolreprod.111.092718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pahwa R., Jialal I. Hyperglycemia induces Toll-like receptor activity through increased oxidative stress. Metab. Syndr. Relat. Disord. 2016;14:239–241. doi: 10.1089/met.2016.29006.pah. [DOI] [PubMed] [Google Scholar]
- 29.Konrath F., Witt J., Sauter T., Kulms D. Identification of new IκBα complexes by and iterative experimental and mathematical modeling approach. PLoS Comput. Biol. 2014;10:e1003528. doi: 10.1371/journal.pcbi.1003528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cecchi L., D'Amato G., Annesi-Maesano I. External exposome and allergic respiratory and skin diseases. J. Allergy Clin. Immunol. 2018;141:846–857. doi: 10.1016/j.jaci.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 31.Oh I., Lee J., Ahn K., Kim J., Kim Y.M., Sun Sim C., Kim Y. Association between particulate matter concentration and symptoms of atopic dermatitis in children living in an industrial urban area of South Korea. Environ. Res. 2018;160:462–468. doi: 10.1016/j.envres.2017.10.030. [DOI] [PubMed] [Google Scholar]
- 32.Ngoc L.T.N., Park D., Lee Y., Lee Y.C. Systematic review and meta-analysis of human skin diseases due to particulate matter. Int. J. Environ. Res. Public Health. 2017;14 doi: 10.3390/ijerph14121458. (pii: E1458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moniaga C.S., Egawa G., Kawasaki H., Hara-Chikuma M., Honda T., Tanizake H., Nakajima S., Otsuka A., Matsuoka H., Kubo A., Sakabe J., Tokura Y., Miyachi Y., Amagai M., Kabashima K. Flaky tail mouse denotes human atopic dermatitis in the steady state and by topical application with Dermatophagoides pteronyssinus extract. Am. J. Pathol. 2010;176:2385–2393. doi: 10.2353/ajpath.2010.090957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu C.W., Lee T.J., Chen Y.C., Liang C.J., Wang S.H., Lue J.H., Tsai J.S., Lee S.W., Chen S.H., Yang Y.F., Chuang T.Y., Chen Y.L. PM2.5-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6/AKT/STAT3/NF-κB-dependent pathway. Part. Fibre Toxicol. 2018;15:4. doi: 10.1186/s12989-018-0240-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material