

Abstract
To estimate the functions of mitochondria of diverse eukaryotic nonmodel organisms in which the mitochondrial proteomes are not available, it is necessary to predict the protein sequence features of the mitochondrial proteins computationally. Various prediction methods that are trained using the proteins of model organisms belonging particularly to animals, plants, and fungi exist. However, such methods may not be suitable for predicting the proteins derived from nonmodel organisms because the sequence features of the mitochondrial proteins of diversified nonmodel organisms can differ from those of model organisms that are present only in restricted parts of the tree of eukaryotes. Here, we proposed NommPred, which predicts the mitochondrial proteins of nonmodel organisms that are widely distributed over eukaryotes. We used a gradient boosting machine to develop 2 predictors—one for predicting the proteins of mitochondria and the other for predicting the proteins of mitochondrion-related organelles that are highly reduced mitochondria. The performance of both predictors was found to be better than that of the best method available.
Keywords: gradient boosting machine, machine learning, mitochondrial proteins, mitochondrion-related organelle proteins, nonmodel organisms
Introduction
Almost all the eukaryotes possess mitochondria in their cells. Although the origin of mitochondria is still debatable, it is widely accepted that a bacterium that was closely related to extant alpha-proteobacteria1 had been engulfed by an ancestral eukaryotic host and it gave rise to mitochondria. The endosymbiotic event had a notable influence on eukaryotic cellular evolution. Because mitochondria perform functions such as energy metabolism, amino acid metabolism, lipid metabolism, Fe-S cluster biogenesis, apoptosis, and oxidation stress reaction that are essential for eukaryotic cells, elucidation of the diversity and evolution of mitochondrial functions is a crucial research subject in eukaryotic evolutionary biology.
Mitochondria are separated from other cellular components by a double membrane, resulting in the concentration of mitochondrial proteins inside the membrane. In general, the functions of an organelle are determined by the protein repertory of the organelle. Therefore, the estimation of the function of mitochondria needs to determine the repertory of mitochondrial proteins, most of which are nuclear encoded, expressed in cytosol, and finally transported into mitochondria.2
To determine a repertory of mitochondrial proteins, the proteomic analysis of mitochondria is essential. For model organisms in mammals, yeasts, and plants, experimental methods for the proteomic analysis of mitochondria have already been established during their long research histories3–7; however, for nonmodel organisms, there are no general strategies for the proteomic analysis of mitochondria. Even in nonmodel organisms, information on the amino acid sequences of proteins is indirectly obtained from the nucleotide sequences of the genome or transcriptome analysis, and these are useful tools for studying the cellular and molecular biological research subjects of nonmodel organisms of which proteins are difficult to treat directly during experiments. Recently, high-throughput sequencing, the so-called next-generation sequencing (NGS), has allowed us to easily obtain the entire genome or transcriptome data even from nonmodel organisms at a low cost and in a short time. Therefore, transcriptome analysis is performed for the entire cell extracts of nonmodel organisms including mitochondria and the other cellular components, and the mitochondrial proteins are predicted using an amino acid sequence–based computational method instead of purifying mitochondria and determining the repertory of mitochondrial proteins directly. Such a bioinformatics approach needs to discriminate mitochondrial proteins from all the proteins that are deduced from the entire cell transcriptome data.
A machine learning approach has been often used to classify mitochondrial/nonmitochondrial proteins. Various software programs based on machine learning are available; these programs predict whether an input protein sequence is a mitochondrial protein. For example, TPpred38 and MitoFates9 are prediction software programs based on support vector machines, whereas TargetP10 is a software program based on neural network techniques.
Most of the current prediction software programs, including TPpred3, MitoFates, and TargetP, are trained only with the data derived from model organisms, which belong, in particular, to the taxonomic groups—metazoa (animals), embryophyta (land plants), and fungi—and these programs are designed for application to the proteins of model organisms and their relatives. Model organisms have been studied experimentally at an enormous cost because of their basic biological, medical, or industrial importance. This has resulted in the accumulation of vast biochemical experimental data of protein localization to cellular compartments including mitochondria.
However, in the case of nonmodel organisms, except for those that are closely related to the known model organisms in metazoa, embryophyta, and fungi, very few experimental data are available because of the shortage of basic experimental procedures, although they exhibit most parts of the eukaryotic diversities.11 Hereafter, we refer to such nonmodel organisms that do not belong to metazoa, embryophyta, and fungi as nonmodel organisms. Therefore, for the study of the mitochondrial proteins derived from nonmodel organisms, the sequence data of genome or transcriptome that are produced using the NGS approach are mainly used to predict the proteins that would be mitochondrially localized. In general, the prediction tools designed for model organisms are usually applied for these analyses; however, these tools do not necessarily guarantee accuracy of prediction because the N-terminal sequence features important for the prediction of the mitochondrial proteins could be far divergent in nonmodel organisms compared with those of the model organisms. In particular, in the case of highly reduced mitochondria, the so-called mitochondrion-related organelles (MRO), in anaerobic or microaerophilic organisms, the prediction of the mitochondrial protein using the prediction tools that are currently available is highly inaccurate.12 Therefore, in general, for predicting mitochondrial/MRO proteins in nonmodel organisms, the consensus of the results from multiple predictors is considered to avoid false predictions. However, this cannot be validated.
To resolve this problem, here, we propose a software program, NommPred (nonmodel organismal mitochondrial/MRO protein predictor), which predicts the mitochondrial/MRO proteins derived from nonmodel organisms. To develop this software, we prepared a data set including the mitochondrial or MRO proteins derived widely from nonmodel organisms and adopted a gradient boosting machine (GBM)13–15 as a classifier. GBM, which is one of the ensemble classifiers, was used instead of the support vector machine,16 which was adopted in the previous predictors MitoFates9 and TPpred3.8 NommPred could resolve the problem due to the inconsistency between the origins of the training and input data when predicting the mitochondrial/MRO proteins of nonmodel organisms. The performance of NommPred is superior to MitoFates, which was demonstrated to be the best among the alternative methods,9 in predicting the mitochondrial/MRO proteins derived from nonmodel organisms. Therefore, NommPred is the best predictor for the mitochondrial/MRO proteins of nonmodel organisms.
Materials and Methods
Scheme of NommPred
A flow chart and a message sequence chart of the newly developed software, NommPred, are illustrated in Figures 1 and 2, respectively. The software takes as input both the protein sequence in FASTA format (definition is available from www.ncbi.nlm.nih.gov/books/NBK53702/) and organismal information from which the protein sequence is derived. The feature of each protein was extracted based on MitoFates9 to create a 920-dimensional feature vector (Figure 1 and Supplementary Table 1). The vector is subjected to the GBM predictor (Mit Predictor for mitochondrial proteins or MRO Predictor for MRO proteins as described below), and the predictor outputs the prediction results.
Figure 1.

Flow chart of NommPred. The closed circle represents the starting point of the program, and the closed circle surrounded by a larger open circle represents the end point. The user input data (input data) include the protein sequence in FASTA format and information of the protein sequence origin (taxonomic group). The input data are classified into (the first black bar in User action step) protein sequence, which is used for feature extraction, and organismal information, which is used for the selection of an appropriate GBM Predictor: Mit Predictor, MRO Predictor, or others. In the feature extraction step, the 920 calculated features (Supplementary Table 1) are integrated, and a 920-dimensional feature vector is obtained as the output. In the figure, only 6 feature categories are depicted with the number of individual features. This vector is subjected to a selected GBM Predictor as the input data, and then the prediction result is shown (output results).
Figure 2.

Message sequence chart of NommPred. The software for NommPred is console user interface (CUI) and it runs on the terminal. The software accepts a protein sequence file in multi-FASTA format and a text file with information of the origins of the sequences and outputs the prediction results at last.
Data sets
The data set used for the training and test is shown in Table 1. The mitochondrial or MRO proteins are treated as positive samples and the others as negative samples. The sequence data were obtained from UniProt Consortium17 (www.uniprot.org/), GiardiaDB18,19 (giardiadb.org/giardiadb/), TrichDB18 (trichdb.org/trichdb/), and ApiLoc20 (apiloc.biochem.unimelb.edu.au/apiloc/apiloc). Although these databases sometimes annotate mitochondrial or MRO proteins based on computational prediction, we used only those proteins whose localization was confirmed experimentally (eg, Western blotting, immunoblotting, or fluorescence microscope analysis) to mitochondria or MROs by investigating the literature. Then, we applied protein sequence redundancy reduction using the BLASTClust program from the NCBI BLAST packages.21 We adopted the criteria of being redundant at >95% sequence identity. Finally, we prepared 392 positive mitochondrial or MRO protein sequences and 3739 negative sequences. We classified the entire data set into mitochondrial and MRO data sets, Mit and MRO. Then, we created a predictor for each data set; one is the predictor for the mitochondrial protein trained with the mitochondrial proteins of 7 nonmodel organismal taxonomic groups (Mit Predictor), whereas the other is the predictor for the MRO protein trained with the MRO proteins of 3 nonmodel organismal taxonomic groups that possess MRO (groups marked with asterisks in Table 1) (MRO Predictor) because these 2 data sets were expected to be apparently different in the N-terminal sequence features of the mitochondrial/MRO protein sequences. The N-terminal sequence features of the MRO proteins are generally considered to be extremely divergent from those of the mitochondrial proteins.12
Table 1.
Entire data set used for training and test.
| Taxonomic groupa | Positive samplesb | Negative samplesc |
|---|---|---|
| Chlorophyta | 60 | 81 |
| Dictyostelium | 52 | 622 |
| Piroplasma | 7 | 387 |
| Plasmodium | 42 | 435 |
| Stramenopiles | 44 | 1029 |
| Toxoplasma | 30 | 125 |
| Trypanosomatida | 48 | 587 |
| *Entamoeba | 7 | 94 |
| *Giardia | 20 | 271 |
| *Trichomonas | 82 | 108 |
| Total | 392 | 3739 |
If the taxonomic group corresponds exactly to the genus, the name of the genus is represented in italic form. “Stramenopiles” is not a formal taxonomic rank but is generally used for the name of the group.
The “positive samples” column lists the number of sequences of the mitochondrial or MRO proteins.
The “negative samples” column lists the number of sequences of the nonmitochondrial or non-MRO proteins. The groups that possess MRO are represented with asterisks.
Training and prediction method
We adopted GBM, one of the ensemble learning algorithms, and created predictors using XGBoost22 package in R Core Team23 for the Mit and MRO data sets (Mit Predictor and MRO predictor). We searched for optimal values of logical variables employed in the XGBoost algorithm. Parameters for tree boosting, learning rate (eta), maximum depth of a tree (max_depth), minimum sum of instance weight (min_child_weight), maximum delta step (max_delta_step), and gamma were tuned with grid search, and finally we determined to set the default values for these variables. In addition, we optimized the parameter of the number of trees to the model by cross-validation. For other parameters, we used the default value. For the extraction of features in MitoFates, we used the method described in the work by Fukasawa et al.9
Performance measures
To evaluate the performances of both the NommPred predictors—Mit Predictor and MRO Predictor—a receiver operating characteristic (ROC) curve and an ROC area under the curve24 (AUC) were used. In the R system, the ROC curve was drawn by plotting the true-positive rate (y-axis) against the false-positive rate (x-axis) for different cutoff values, and the ROC AUC was drawn based on the ROC curve.
To evaluate the robustness of the ROC AUC measures, we randomly divided the Mit or MRO data set into 3 subsets (3-fold cross-validation), and we used 2 of them for the training data, and the other for the test data. This process was repeated 100 times.
To compare NommPred with a previous predictor, MitoFates, we used the same test data as that of NommPred for MitoFates to evaluate its performance. In this performance comparison, we performed the paired t test and Wilcoxon signed rank test to evaluate the difference between the means of these 100 paired ROC AUC scores.
Search for best prediction models
In the prediction of the mitochondrial proteins, if we know the taxonomic group from which the sequence data are derived, we may be able to use a preferable predictor instead of the general Mit Predictor.
To search for a combination of taxonomic groups for the best prediction of the mitochondrial proteins of a given taxonomic group, we prepared the predictors trained with the data from all possible combinations of taxonomic groups at least including the given taxonomic group. There are 7 taxonomic groups possessing mitochondria, and we created 26 predictors (Figure 3, step 2) for each taxonomic group. For the preparation of each predictor, we performed 3-fold cross-validation 100 times with random repeats. These predictors were evaluated by means of the ROC AUC scores in R. We processed the following steps (see also Figure 3).
Figure 3.

Procedure for searching the best predictor of mitochondrial protein for each taxonomic group. Colored square boxes represent sequence data sets of the taxonomic groups. The area covered with slanted lines indicates that it contains mitochondrial protein sequences. For details, see text.
To compare these taxonomic group–specific predictors with a previous predictor, we also computed 100 ROC AUC scores of MitoFates for the prediction of mitochondrial proteins on each taxonomic group. Each test data set was randomly generated by the same procedure (Figure 3, steps 4.2-4.5) from a given taxonomic group. In this performance comparison, we performed the t test and Wilcoxon-Mann-Whitney test to evaluate the difference of mean scores between the taxonomic group–specific predictor which achieved the best score and MitoFates. We also performed these tests for the difference of mean scores between best scoring predictor and second best scoring predictor:
Select one taxonomic group from the 7 groups (eg, Piroplasma in Figure 3) as the given group.
Create all possible 26 combinations of the other taxonomic groups (eg, 6 taxonomic groups except Piroplasma).
For each of the 26 combinations, add the given group (eg, Piroplasma) as each combination contains the given group (eg, [Chlorophyta] + Piroplasma, [Dictyostelium] + Piroplasma, . . ., [Chlorophyta + Plasmodium] + Piroplasma, . . ., [Chlorophyta + Plasmodium + Toxoplasma + Trypanosomatida] + Piroplasma, . . . in Figure 3).
- Repeat 100 times steps 4.1 to 4.5.
- 4.1. For each combination, combine the data sets of the taxonomic groups other than the given group (eg, Piroplasma).
- 4.2. For the data set of the given group (eg, Piroplasma) and the combined data set, divide each data set randomly into 3 subsets.
- 4.3. Randomly select 1 of the 3 subsets of the data set of the given group (eg, Piroplasma) and use it as a test data set. Randomly select 2 subsets from the 3 subsets of the combined data set, merge these subsets with the other 2 subsets of the data set of the given group (eg, Piroplasma), and use it as a training data set.
- 4.4. Train the predictor using the above training data set.
- 4.5. Calculate the ROC AUC score using the test data set.
Calculate the mean of 100 ROC AUC scores for each of the 26 predictors and plot the 26 mean values.
Repeat steps 1 to 5 until the 7th taxonomic group (eg, Trypanosomatida in Figure 3) is selected in step 1.
Evaluation of the influence of a taxonomic group on the others
To evaluate the degree of influence of a taxonomic group on the others, we performed the following analyses in R (see also Supplementary Figure 1).
Select one taxonomic group from the 7 groups (eg, Chlorophyta in Supplementary Figure 1) as the factor group and select another group as the target group (eg, Piroplasma in Supplementary Figure 1) on which the degree of influence of the factor group is evaluated.
To prepare the control data sets, create all 25 combinations from the 5 taxonomic groups except for the above target and factor groups and add the data set of a target group to a combined data set for 1 of the combinations of the 5 taxonomic groups to make a control data set (then, 25 control data sets each of which excludes the factor group are generated).
Add the data set of the factor group to each control data set to generate an experimental data set (then, 25 experimental data sets each of which includes the factor group are generated).
- Repeat 100 times steps 4.1 to 4.4 on the control and experimental data sets.
- 4.1. For each of the 25 combinations, combine the data of the taxonomic groups other than the target group (eg, Piroplasma).
- 4.2. For the data set of the target group and the combined data set, divide each data set randomly into 3 subsets.
- 4.3. Randomly select 1 of the 3 subsets of the data set of the target group and use it as a test data set. Randomly select 2 subsets from 3 subsets of the combined data set, merge these subsets with the other 2 subsets of the data set of the target group, and use it as a training data set.
- 4.4. Develop a predictor trained with the above training data set in 3-fold cross-validation and then using the above test data set, calculate the ROC AUC score for the predictor.
Calculate the mean of 100 ROC AUC scores for each of the 25 + 25 predictors from the control and experimental data sets.
Perform the Wilcoxon signed rank test to evaluate the difference between the mean scores of the above 25 paired predictors from the control and experimental data sets.
Tabulate the results (P value and difference of scores [“Score Influence”]; Supplementary Table 2).
Repeat steps 1 to 7 until all combinations of the factor and target groups have been selected in step 1.
Principal component analysis of the features
To investigate the multidimensional trends of the features in Mit data set, we performed a principal component analysis (PCA) in the R system. We used a 283 (mitochondrial protein sequences) × 920 (extracted features) matrix as the input data.
Cluster analysis of the features
To cluster the 7 taxonomic groups based on the features in the Mit data set, we used the k-means++ method25 (k = 2) for a 283 × 141 (principal component [PC] scores) matrix in the R system because the components 1 through 141 in the PCA could account for 80% of the total variance of the data matrix as shown in the “Results” section.
Results
Performance comparison analysis
Prediction of mitochondrial proteins
We performed the performance comparison analysis between NommPred and a previous method, MitoFates. A data set including the mitochondrial and nonmitochondrial proteins of 7 nonmodel organismal taxonomic groups was used for the preparation of the training and test data sets (as described in the “Materials and Methods” section), resulting in the creation of Mit Predictor. Performance measure scores are listed in Table 2 (also see Supplementary Figure 2).
Table 2.
Comparison of the mean ROC AUC scores between NommPred and MitoFates. In NommPred, mitochondrial proteins were predicted by Mit Predictor, whereas MRO proteins were by MRO Predictor.
| Prediction target | ROC AUC | P value | ||
|---|---|---|---|---|
| NommPred | MitoFates | Paired t test | Wilcoxon signed rank test | |
| Mitochondrial protein | 0.9463 | 0.9080 | 1.618e−42 | 0.00e+00 |
| MRO protein | 0.9041 | 0.8021 | 6.855e−40 | 0.00e+00 |
The 100 randomly generated data sets (n = 100) of mitochondrial or MRO proteins were used for cross-validation (see “Materials and Methods” section).
For the mean ROC AUC scores (sample size n = 100), MitoFates achieved 0.9080, whereas the performance of Mit Predictor of NommPred was superior with a value of 0.9463 (Table 2). Moreover, the difference between the 2 mean ROC AUC scores was significant (paired t test: P = 1.618 × 10−42, Wilcoxon signed rank test: P = ~0).
Generally, the ROC AUC score ranging between 0.5 and 0.7 is regarded as less accurate, between 0.7 and 0.9 as moderately accurate, and more than 0.9 as highly accurate.26 Based on these criteria, MitoFates still showed sufficient accuracy in the prediction of the mitochondrial proteins derived from nonmodel organisms. However, for the prediction of those proteins, Mit Predictor with a higher ROC AUC score was preferred.
Prediction of MRO proteins
As described in the “Materials and Methods” section, we classified the entire data set into 2—Mit and MRO (Table 1). The MRO data set including the MRO and non-MRO proteins of 3 nonmodel organismal groups was used for the preparation of the training and test data sets (described in the “Materials and Methods” section), resulting in the creation of MRO Predictor. We performed a similar comparison analysis between the performance of MRO Predictor and that of MitoFates for the prediction of the MRO proteins. The performance measure scores are listed in Table 2.
MitoFates achieved a mean ROC AUC score (sample size n = 100) of 0.8021, whereas the performance of the MRO predictor of NommPred was far better with a mean value of 0.9041 (paired t test: P = 6.855 × 10−40, Wilcoxon signed rank test: P = ~0; Table 2). Based on these results, MRO Predictor of NommPred is suitable for the MRO proteins.
Searching for best prediction model
To search for the best combination of taxonomic groups for the best prediction of the mitochondrial proteins of a given taxonomic group, we prepared all possible 26 predictors for each taxonomic group, and then we calculated the ROC AUC scores of each predictor (Figure 3) and plotted it using the boxplot (Figure 4). The best scoring predictor for each of the 7 taxonomic groups is indicated in Supplementary Table 3 (also see Supplementary Figure 3).
Figure 4.

Boxplot showing the performance of all possible predictors of the mitochondrial proteins for each taxonomic group. The ROC AUC scores (y-axis) of the 7 taxonomic groups possessing mitochondria (x-axis) are plotted. Each closed circle represents the ROC AUC score (y-axis) of each predictor for predicting the mitochondrial proteins of each taxonomic group. Lines within the boxplot indicate the median, the lower/higher quartile (Q1/Q3), and lower/higher whiskers. The star symbols correspond to the scores of the best scoring predictor for each taxonomic group (see Supplementary Table 3). The triangle symbols represent the scores of the predictor trained with the data of all the 7 taxonomic groups. The cross symbols represent the scores of the predictor trained only with the data of the corresponding taxonomic group. The diamond symbols represent the scores of MitoFates.
The predictor trained using all the data from the 7 taxonomic groups corresponds to Mit Predictor. In the prediction of the proteins derived from each taxonomic group, the performance of Mit Predictor was average or superior to the other predictors. The scores of these predictions are represented by triangles in Figure 4. Based on the results, if the taxonomic origin of proteins were unknown, the predictor trained using all the data from the 7 taxonomic groups, that is, Mit Predictor, would be practically the best choice.
The predictors that were trained using only the data from the corresponding taxonomic group clearly showed poor performance in each prediction (represented by the cross symbols in Figure 4), whereas the predictors represented by the star symbols in Figure 4 are the best scoring predictors for predicting the mitochondrial proteins of each taxonomic group.
On each of the 7 taxonomic group–specific predictions, the difference of the mean ROC AUC score between the best scoring predictor and the second best scoring predictor was not statistically significant (Supplementary Table 4), whereas the difference between the best scoring predictor and MitoFates (represented by the diamond symbol in Figure 4) was significant (Table 3). Therefore, if the taxonomic origin of the protein to be predicted was known in advance, NommPred could provide the best scoring predictor as most appropriate predictor.
Table 3.
Comparison of the mean ROC AUC scores between the best scoring predictor of mitochondrial proteins for each taxonomic group in NommPred and MitoFates.
| Prediction target: mitochondrial proteins of | ROC AUC | P value | ||
|---|---|---|---|---|
| NommPred (best scoring predictor) | MitoFates | Welch t test | WMW test | |
| Piroplasma | 0.9729 | 0.9335 | 2.597e−11 | 1.725e−11 |
| Chlorophyta | 0.9631 | 0.9444 | 3.067e−06 | 8.982e−06 |
| Dictyostelium | 0.9506 | 0.9381 | 0.003263 | 0.006537 |
| Plasmodium | 0.9436 | 0.9022 | 2.347e−12 | 1.479e−11 |
| Stramenopiles | 0.9809 | 0.9311 | 0.00e+00 | 0.00e+00 |
| Toxoplasma | 0.8899 | 0.8255 | 1.599e−10 | 2.682e−09 |
| Trypanosomatida | 0.9490 | 0.9036 | 2.822e−15 | 1.468e−13 |
The 100 randomly generated data sets (n = 100) of mitochondrial proteins were used for cross-validation (see “Materials and Methods” section).
Evaluation of the influence of a taxonomic group on the others
We performed an experiment to evaluate the influence of a taxonomic group on the others, as described in the “Materials and Methods” section. Significant relationships were identified for 28 pairs of the taxonomic groups (Supplementary Table 2, P < .05: Wilcoxon signed rank test) out of the 49 pairs examined. Hereafter in this text, when the data from a taxonomic group A (Factor group in Supplementary Table 2) significantly increased or decreased with respect to the performance of prediction of another taxonomic group B (Target group in Supplementary Table 2), we represent the relationship as A → +B or A → −B, respectively. Concerning the group Piroplasma, significant relationships (P < .01) were identified as Trypanosomatida → +Piroplasma, stramenopiles → +Piroplasma, Plasmodium → +Piroplasma, and Chlorophyta → +Piroplasma. The difference between the scores corresponding to these relationships was clearly higher than the others in Supplementary Table 2. Piroplasma seems to be most sensitive to the influence of the other taxonomic groups. Toxoplasma is likely to have a negative influence on other groups significantly (P < .01) such as Toxoplasma → −stramenopiles, Toxoplasma → −Piroplasma, and Toxoplasma → −Dictyostelium. Chlorophyta and Toxoplasma seem to have a significant positive influence on each other (P < .01), whereas Dictyostelium and Plasmodium seem to have a highly significant positive influence on each other (P < 10−5). Based on these results, some taxonomic groups were suggested to form clusters, and thus we analyzed the clustering of the groups using the mitochondrial sequence data from the 7 taxonomic groups.
PCA of features
The PCA was performed on the vectors of the 920-dimensional features extracted from the 283 mitochondrial protein sequences that are derived from the 7 taxonomic groups. The cumulative contribution ratio was 80% after the 141st PC was included. We scattered PC 1, PC 2, and PC 3 on a 3-dimensional plot (Figure 5). Two clusters were observed in the plot: one is the Dictyostelium and Plasmodium cluster, whereas the other cluster includes the other taxonomic groups Chlorophyta, Piroplasma, stramenopiles, Toxoplasma, and Trypanosomatida.
Figure 5.

Three-dimensional plot of PCA. PC 1, PC 2, and PC 3 scores were derived from the PCA on the 920-dimensional features of the mitochondrial protein sequences derived from the 7 taxonomic groups possessing mitochondria. Each taxonomic group is distinguished by the color shown in the legend.
Cluster analysis of the features
To evaluate the clustering by PCs 1 to 3 in higher dimensions, we performed cluster analysis using the k++ method (k = 2) and observed that for any PC dimension ranging from 2 to 141, the clusters formed by clustering PCs 1 to 3 still remained (Figure 6, the case using up to 141st dimensions of PC): one includes the Dictyostelium and Plasmodium mitochondrial proteins, whereas the other includes those from Chlorophyta, stramenopiles, Toxoplasma, and Trypanosomatida. The number of mitochondrial proteins of Piroplasma was less, and these were not clearly clustered into either of the 2 groups.
Figure 6.
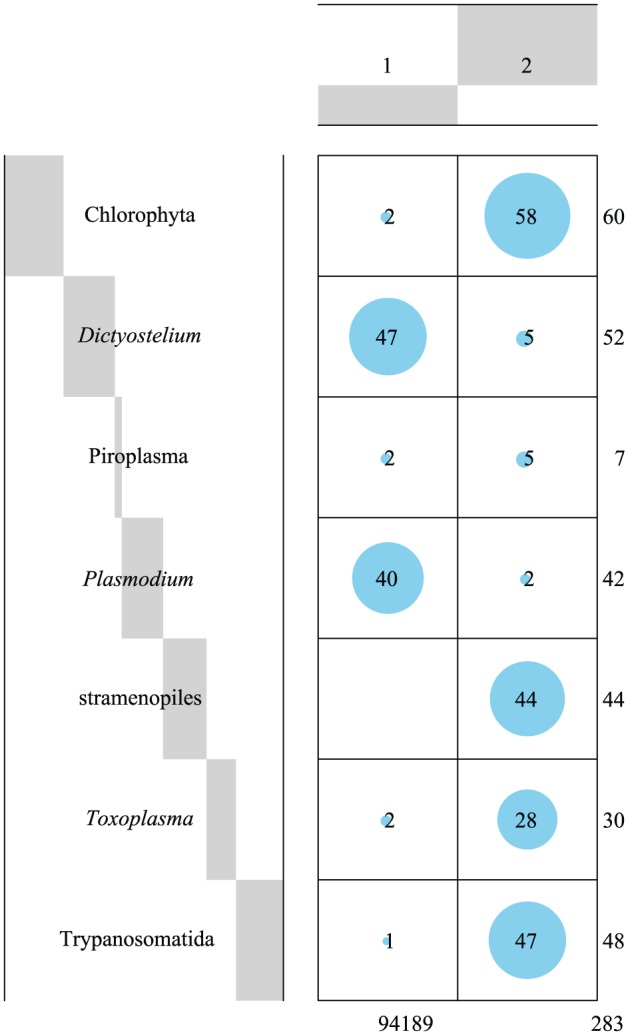
Balloon plot showing the clustering of the mitochondrial protein sequences. The balloon plot suggests the presence of 2 clusters of mitochondrial protein sequences derived from 7 taxonomic groups. Each balloon represents the relative magnitude of the value. The value inside the cell represents the number of sequences. Each value outside the cell represents the sum of each column or row. Gray bars on the column or row headers represent the proportions of the categories for the column or row against the total.
Discussion
We succeeded in developing NommPred, the predictors for the mitochondrial and MRO proteins derived from diverse nonmodel organisms, except for those belonging to metazoa, embryophyta, and fungi. Previously, the protein sequence data derived from nonmodel organisms were subjected to the predictor trained only using the data from model organisms. NommPred could resolve the problem resulted from such inconsistency between the origins of the training data (model organisms) and the input data (nonmodel organisms).
Performance comparison analysis
The results of the statistical analysis (Tables 2 and 3) clearly supported the superiority of NommPred in the performance of predicting the mitochondrial proteins of nonmodel organisms when compared with the existing best method, MitoFates. In particular, NommPred is the first software that is expected to be used for predicting the MRO proteins. NommPred would be useful for the prediction of metabolic pathways relating to the mitochondria/MROs from nonmodel organisms, the NGS data of which can be available.
Searching for best prediction model
The predictor for the mitochondrial proteins trained using the data from all the 7 taxonomic groups in Figure 4 does not exhibit the best score when compared with various alternative predictors examined in Figure 4 and Supplementary Table 3 in predicting the proteins of a given taxonomic group. However, the performance of the predictor is not poor for any taxonomic groups. Thus, Mit Predictor, which corresponds to the predictor trained using the data from all the 7 taxonomic groups, can be applied for general usage. The performance of the predictor that was trained using the data derived only from a given taxonomic group was inferior to several predictors that were trained using the data from the various combinations of the taxonomic groups (Figure 4). When a taxonomic group of the protein to be predicted is known in advance, NommPred can provide taxonomic group–specific predictors of the mitochondrial proteins with the best performance (Supplementary Table 3) in addition to Mit Predictor.
In the mitochondrial proteins of model organisms of which the mitochondrial targeting signals were experimentally investigated, the N-terminal signal sequences are generally divergent27,28; however, the previous predictors can accurately distinguish between the mitochondrial and nonmitochondrial proteins based on the N-terminal sequence features.9,28 In our present analyses for nonmodel organisms, the performance of the predictors of each taxonomic group was unexceptionally improved by the addition of protein sequences from some other taxonomic groups (Figure 4). This improvement in the performance indicates that the N-terminal sequence features of the mitochondrial proteins still share some common features even in nonmodel organisms. However, depending on the case, adding the protein sequences of some taxonomic groups negatively influenced the performance of prediction. The inclusion of Toxoplasma or Plasmodium was shown to have a negative influence on some of the other taxonomic groups in the performance of prediction (Supplementary Table 2). It might be because of the divergent nature of their protein sequences including the N-terminal portion because of the high evolutionary rate resulted probably from their parasitic lifestyle.29 However, the inclusion of the other parasitic group, Piroplasma or Trypanosomatida, did not have any negative influence on the other taxonomic groups. The N-terminal sequence features of these groups might be less divergent than those of Toxoplasma or Plasmodium.
PCA and cluster analysis of the features
Interestingly, the result of the PCA (Figure 5) and cluster analysis (Figure 6) showed the presence of 2 clusters of taxonomic groups, one including Trypanosomatida, Toxoplasma, stramenopiles, and Chlorophyta and the other including Dictyostelium and Plasmodium. We tried to investigate the kinds of N-terminal sequence features that are significant for the classification of the mitochondrial protein sequences into 2 clusters by referring to the contribution-weighted loading values of PCs 1 through 141, the cumulative contribution ratio of which amounted to 80%. However, we could not interpret biologically because we could not narrow the repertory of the feature variables with extremely high absolute loading values compared with the other variables (data not shown).
Taxonomic groups that influence each other positively are (Supplementary Table 2) likely to belong to 1 of the 2 clusters obtained from the above cluster analysis, suggesting that the taxonomic groups in the same cluster share the trends of the N-terminal sequence features that are used for training.
Piroplasma was influenced significantly by all the other taxonomic groups with a large difference in the mean ROC AUC scores (“Score Influence” in Supplementary Table 2), indicating that the training of the predictor of Piroplasma could be sensitive to the data from the other taxonomic groups. The sensitivity corresponds to the high variability of its ROC AUC scores (Figure 4). Such instability of the predictor of Piroplasma could be the result of the shortage of the mitochondrial protein sequence data used in the present analyses. The inclusion of more data from Piroplasma in the future will improve the stability of the predictor.
The inclusion of Plasmodium was shown to have a positive influence on the prediction of Dictyostelium mitochondrial proteins and vice versa (Supplementary Table 2). Dictyostelium is not a parasitic organismal group; however, it is a highly divergent free-living amoeba in the higher order group, Amoebozoa,30 and thus is phylogenetically far distant from Plasmodium that belongs to the other higher order group, Alveolata. In these 2 taxonomic groups, Dictyostelium and Plasmodium, the convergence of the substitution patterns might occur by chance, leading to a situation in which similar N-terminal sequence features in their mitochondrial or nonmitochondrial proteins are shared.
The N-terminal sequence features of the MRO proteins of the parasitic groups Trichomonas, Giardia, and Entamoeba are extremely divergent and reduced.31–33 We segregated in advance these groups with MROs from the other taxonomic groups with mitochondria because the inclusion of these groups might have a negative influence on other taxonomic groups in their performance of prediction. Owing to the separation of the MRO proteins from the mitochondrial ones, we could develop an independent predictor (MRO Predictor) and achieve high performance in predicting the MRO proteins. Because the import machinery of proteins of these MROs could be significantly different from that of mitochondria,34-36 the development of a predictor specific to the MRO proteins is reasonable.
Conclusions and Future Plan
We developed a software program, NommPred, which includes Mit Predictor and MRO Predictor for the predictions of mitochondrial and MRO proteins derived from diverse nonmodel organisms. Both predictors were shown to significantly exceed in performance, compared with the previously best method, MitoFates, in the prediction of mitochondrial/MRO proteins from nonmodel organisms. Because there is no other predictor suitable for the prediction of MRO proteins, MRO predictor in NommPred is useful tool to search for putative MRO proteins.
In this study, we retrieved almost all protein sequence data whose cellular localization was experimentally verified to mitochondria/MROs from various sequence databases. However, the origins of the sequence data of mitochondrial/MRO proteins in the entire data set (Table 1) are biased for those of the parasitic organisms. Therefore, taxon sampling of our data set is still very sparse. The accumulation of more data of the mitochondrial/MRO proteins of nonmodel organisms, especially from the free-living ones whose localization was confirmed experimentally, is essential to further improve the predictors presented in this work. We should continuously make efforts toward updating the data set to provide more accurate predictors. Although NommPred may still have some problems that need to be improved in the future, we hope it will be helpful for the prediction of the mitochondrial/MRO proteins of nonmodel organisms.
Software Distribution
NommPred, the software developed in this study, is distributed in GitLab (https://gitlab.com/kkei/NommPred.git). The values of the default parameters are shown here. This software requires 2 inputs. One is the sequence information: Mit Predictor and MRO Predictor of NommPred accept a multi FASTA format file as the input data. The other is taxonomic information of the input sequences: NommPred would select the best predictor automatically based on this information (the user can manually select a predictor trained specifically with the proteins of a given taxonomic group or a general predictor, Mit Predictor, trained using the proteins of all the 7 taxonomic groups). Finally, NommPred outputs the estimated probabilities that the input sequences are mitochondrial/MRO proteins. The schemes of this software are illustrated in Figure 1 (flow chart) and Figure 2 (message sequence chart). More details or usage instructions are available in the documents at the GitLab page.
Supplemental Material
Supplemental material, Supplementary_Figure_1_xyz11796ba3b28fd for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics
Supplemental Material
{kind=link}
Supplemental material, Supplementary_Figure_2_xyz117963bb647c2 for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics
Supplemental Material
{kind=link}
Supplemental material, Supplementary_Figure_3_xyz11796e5b0f92d for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics
Supplemental Material
{kind=link}
Supplemental material, Supplementary_Figure_4_xyz117968f5495fe for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics
Acknowledgments
The authors would like to thank Editage (www.editage.jp) for English language editing.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the “Tree of Life” research project of University of Tsukuba and grants from the Japan Society for Promotion of Sciences (15H04406 and 15H05231 awarded to T.H.).
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: KK and TA conceived and designed the experiments. KK analyzed the data. KK and TH wrote the first draft of the manuscript. TA and HK contributed to the writing of the manuscript. KK, TA, TH, and HK agree with manuscript results and conclusions; jointly developed the structure and arguments for the paper; and made critical revisions and approved final version. All authors reviewed and approved the final manuscript.
Disclosures and Ethics: As a requirement of publication, authors have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality, and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. The external blind peer reviewers report no conflicts of interest.
Supplemental material: Supplemental material for this article is available online.
References
- 1. Gray MW, Archibald JM. Origins of mitochondria and plastids In: Bock R, Knoop V, eds. Genomics of Chloroplasts and Mitochondria. 1st ed Dordrecht, The Netherlands: Springer; 2012:1–30. [Google Scholar]
- 2. Gonczarowska-Jorge H, Zahedi RP, Sickmann A. The proteome of Baker’s yeast mitochondria. Mitochondrion. 2017;33:15–21. doi: 10.1016/j.mito.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 3. Kumar A, Agarwal S, Heyman JA, et al. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sickmann A, Reinders J, Wagner Y, et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci U S A. 2003;100:13207–13212. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reinders J, Zahedi RP, Pfanner N, Meisinger C, Sickmann A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res. 2006;5:1543–1554. doi: 10.1021/pr050477f. [DOI] [PubMed] [Google Scholar]
- 6. Cherry JM, Hong EL, Amundsen C, et al. Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen X, Li J, Hou J, Xie Z, Yang F. Mammalian mitochondrial proteomics: insights into mitochondrial functions and mitochondria-related diseases. Expert Rev Proteomics. 2010;7:333–345. doi: 10.1586/epr.10.22. [DOI] [PubMed] [Google Scholar]
- 8. Savojardo C, Martelli PL, Fariselli P, Casadio R. TPpred3 detects and discriminates mitochondrial and chloroplastic targeting peptides in eukaryotic proteins. Bioinformatics. 2015;31:3269–3275. doi: 10.1093/bioinformatics/btv367. [DOI] [PubMed] [Google Scholar]
- 9. Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K. MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics. 2015;14:1113–1126. doi: 10.1074/mcp.M114.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 11. Adl SM, Simpson AGB, Lane CE, et al. The revised classification of eukaryotes. J Eukaryot Microbiol. 2012;59:429–493. doi: 10.1111/j.1550-7408.2012.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Makiuchi T, Nozaki T. Highly divergent mitochondrion-related organelles in anaerobic parasitic protozoa. Biochimie. 2014;100:3–17. doi: 10.1016/j.biochi.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 13. Friedman J, Hastie T, Tibshirani R. Additive logistic regression: a statistical view of boosting. Ann Statist. 2000;28:337–407. doi: 10.1214/aos/1016218223. [DOI] [Google Scholar]
- 14. Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Statist. 2001;29:1189-1232. doi: 10.1214/aos/1013203451. [DOI] [Google Scholar]
- 15. Friedman JH. Stochastic gradient boosting. Comput Stat Data Anal. 2002;38:367–378. doi: 10.1016/S0167-9473(01)00065-2. [DOI] [Google Scholar]
- 16. Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20:273–297. doi: 10.1007/BF00994018. [DOI] [Google Scholar]
- 17. UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:158–169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aurrecoechea C, Brestelli J, Brunk BP, et al. GiardiaDB and TrichDB: integrated genomic resources for the eukaryotic protist pathogens Giardia lamblia and Trichomonas vaginalis. Nucleic Acids Res. 2009;37:D526–D530. doi: 10.1093/nar/gkn631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hagen KD, Hirakawa MP, House SA, et al. Novel structural components of the ventral disc and lateral crest in Giardia intestinalis. PLoS Negl Trop Dis. 2011;5:e1442. doi: 10.1371/journal.pntd.0001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Woodcroft BJ, Scanlon KL, Doyle M, Speed T, Ralph SA. ApiLoc – a database of published protein sub-cellular localisation in Apicomplexa (version 3). http://apiloc.biochem.unimelb.edu.au/apiloc/apiloc. Updated 2011. Accessed June 11, 2016.
- 21. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403-410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 22. Chen T, Guestrin C. XGBoost: a scalable tree boosting system [published online ahead of print March 9, 2016]. arXiv. doi: 10.1145/2939672.2939785. [DOI] [Google Scholar]
- 23. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: The R Foundation; 2018. https://www.R-project.org/. [Google Scholar]
- 24. Bradley AP. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997;30:1145–1159. doi: 10.1016/S0031-3203(96)00142-2. [DOI] [Google Scholar]
- 25. Arthur D, Vassilvitskii S. K-means++: The advantages of careful seeding. In: Gabow H, ed. Proceedings of the Eighteenth Annual ACM-SIAM Symposium on Discrete Algorithms. New York, NY: ACM; 2007:1027–1035. [Google Scholar]
- 26. Fischer JE, Bachmann LM, Jaeschke R. A readers’ guide to the interpretation of diagnostic test properties: clinical example of sepsis. Intensive Care Med. 2003;29:1043–1051. doi: 10.1007/s00134-003-1761-8. [DOI] [PubMed] [Google Scholar]
- 27. Williams EJ, Pal C, Hurst LD. The molecular evolution of signal peptides. Gene. 2000;253:313–322. doi: 10.1016/S0378-1119(00)00233-X. [DOI] [PubMed] [Google Scholar]
- 28. Fukasawa Y, Leung RK, Tsui SK, Horton P. Plus ça change—evolutionary sequence divergence predicts protein subcellular localization signals. BMC Genomics. 2014;15:46. doi: 10.1186/1471-2164-15-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bromham L, Cowman PF, Lanfear R. Parasitic plants have increased rates of molecular evolution across all three genomes. BMC Evol Biol. 2013;13:126. doi: 10.1186/1471-2148-13-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bapteste E, Brinkmann H, Lee JA, et al. The analysis of 100 genes supports the grouping of three highly divergent amoebae: Dictyostelium, Entamoeba, and Mastigamoeba. Proc Natl Acad Sci U S A. 2002;99:1414–1419. doi: 10.1073/pnas.032662799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zimorski V, Major P, Hoffmann K, Brás XP, Martin WF, Gould SB. The N-terminal sequences of four major hydrogenosomal proteins are not essential for import into hydrogenosomes of Trichomonas vaginalis. J Eukaryot Microbiol. 2012;60:89–97. doi: 10.1111/jeu.12012. [DOI] [PubMed] [Google Scholar]
- 32. Garg S, Stölting J, Zimorski V, et al. Conservation of transit peptide-independent protein import into the mitochondrial and hydrogenosomal matrix. Genome Biol Evol. 2015;7:2716–2726. doi: 10.1093/gbe/evv175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mi-ichi F, Abu Yousuf M, Nakada-Tsukui K, Nozaki T. Mitosomes in Entamoeba histolytica contain a sulfate activation pathway. Proc Natl Acad Sci U S A. 2009;106:21731–21736. doi: 10.1073/pnas.0907106106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jedelský PL, Doležal P, Rada P, et al. The minimal proteome in the reduced mitochondrion of the parasitic protist Giardia intestinalis. PLoS ONE. 2011;6:e17285. doi: 10.1371/journal.pone.0017285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schneider RE, Brown MT, Shiflett AM, et al. The Trichomonas vaginalis hydrogenosome proteome is highly reduced relative to mitochondria, yet complex compared with mitosomes. Int J Parasitol. 2011;41:1421–1434. doi: 10.1016/j.ijpara.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rada P, Doležal P, Jedelský PL, et al. The core components of organelle biogenesis and membrane transport in the hydrogenosomes of Trichomonas vaginalis. PLoS ONE. 2011;6:e24428. doi: 10.1371/journal.pone.0024428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplementary_Figure_1_xyz11796ba3b28fd for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics
Supplemental material, Supplementary_Figure_2_xyz117963bb647c2 for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics
Supplemental material, Supplementary_Figure_3_xyz11796e5b0f92d for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics
Supplemental material, Supplementary_Figure_4_xyz117968f5495fe for NommPred: Prediction of Mitochondrial and Mitochondrion-Related Organelle Proteins of Nonmodel Organisms by Keitaro Kume, Toshiyuki Amagasa, Tetsuo Hashimoto and Hiroyuki Kitagawa in Evolutionary Bioinformatics