

Abstract
Background
The renal tubules, which have distant metabolic features and functions in different segments, reabsorb >99% of approximately 180 l of water and 25,000 mmol of Na + daily. Defective metabolism in renal tubules is involved in the pathobiology of kidney diseases. However, the mechanisms underlying the metabolic regulation in renal tubules remain to be defined.
Methods
We quantitatively compared the proteomes of the isolated proximal tubules (PT) and distal tubules (DT) from C57BL/6 mouse using tandem mass tag (TMT) labeling-based quantitative mass spectrometry. Bioinformatics analysis of the differentially expressed proteins revealed the significant differences between PT and DT in metabolism pathway. We also performed in vitro and in vivo assays to investigate the molecular mechanism underlying the distant metabolic features in PT and DT.
Findings
We demonstrate that the renal proximal tubule (PT) has high expression of lipid metabolism enzymes, which is transcriptionally upregulated by abundantly expressed PPARα/γ. In contrast, the renal distal tubule (DT) has elevated glycolytic enzyme expression, which is mediated by highly expressed c-Myc. Importantly, PPARγ transcriptionally enhances the protease iRhom2 expression in PT, which suppresses EGF expression and secretion and subsequent EGFR-dependent glycolytic gene expression and glycolysis. PPARγ inhibition reduces iRhom2 expression and increases EGF and GLUT1 expression in PT in mice, resulting in renal tubule hypertrophy, tubulointerstitial fibrosis and damaged kidney functions, which are rescued by 2-deoxy-d-glucose treatment.
Interpretation
These findings delineate instrumental mechanisms underlying the active lipid metabolism and suppressed glycolysis in PT and active glycolysis in DT and reveal critical roles for PPARs and c-Myc in maintaining renal metabolic homeostasis. FUND: This work was supported by the National Natural Science Foundation of China (grants 81572076 and 81873932; to Q.Z.), the Applied Development Program of the Science and Technology Committee of Chongqing (cstc2014yykfB10003; Q.Z.), the Program of Populace Creativities Workshops of the Science and Technology Committee of Chongqing (Q.Z.), the special demonstration programs for innovation and application of techniques (cstc2018jscx-mszdX0022) from the Science and Technology Committee of Chongqing (Q.Z.).
Keywords: PPARγ, PPARα, C-Myc, Nrf2, Lipid metabolism, Glycolysis, Renal proximal tubules, Renal distal tubules, Kidney
Research in context.
Evidence before this study
The renal tubules, which have distant metabolic features and functions in different segments. Defective metabolism in renal tubules is involved in the pathobiology of kidney diseases. However, the mechanisms underlying the metabolic regulation in renal tubules remain to be defined.
Added value of this study
This study found that highly expressed PPARs and c-Myc increases lipid and glycolytic enzyme expression in the renal proximal and distal tubules, respectively. PPARγ enhances iRhom2 expression to suppress EGF expression and EGFR-dependent glycolytic gene expression and glycolysis in the renal proximal and distal tubules. PPARγ inhibition results in renal tubule hypertrophy, tubulointerstitial fibrosis and damaged kidney functions, which could be rescued by 2-deoxy-d-glucose treatment.
Implications of all the available evidence
Our findings delineate instrumental molecular mechanisms underlying the active lipid metabolism and suppressed glycolysis in the PT and active glycolysis in the DT and reveal critical roles for PPARs and c-Myc in maintaining metabolic homeostasis in the kidney. Given that PPARs are implicated to have roles in many renal pathophysiological conditions, including diabetic nephropathy, glomerulosclerosis, chronic kidney disease, and kidney fibrosis, our study elucidates the potential to treat PPARγ function-defective kidney diseases with currently available EGF receptor antagonists.
Alt-text: Unlabelled Box
1. Introduction
The mammalian kidney contains thousands of individual nephron units, each of which consists of a glomerulus that generates filtrate of blood and a long epithelial tubule that modifies the filtrate by transporting substances into and out of it to form the final urine. The renal tubule, which reabsorbs >99% of approximately 180 l of water and 25,000 mmol of Na+ daily, is comprised of many segments, including the proximal (convoluted) tubule (PT) and distal (convoluted) tubule (DT), and the loop of Henle, each having distinct cell types and functions [1,2]. PT and DT are derived from nephron progenitor cells (metanephric mesenchymal cells) and develop into two segments with varied morphologies and functions [[1], [2], [3]]. PT is responsible for recovering the majority of filtrate, including nearly 80% of fluid and electrolytes, >99% of glucose and amino acids, and endocytosis-dependent massive protein and macromolecular substances. DT further fine-tunes the composition of filtrate and plays a key role in maintaining potassium, calcium, magnesium homeostasis, and water absorption [1,2]. To achieve these different functions, PT and DT have distinct types of energy metabolism. Specifically, PT metabolizes fatty acids instead of glucose to produce energy, whereas DT oxidizes glucose at a rate three times greater than that by PT [1,2]. Normally regulated energy metabolism plays an instrumental role in maintaining kidney functions, and abnormal alteration of this metabolism contributes to many renal pathophysiological states. For instance, inhibition of fatty acid oxidation in kidney tubule cells promotes kidney fibrosis development in patients with chronic kidney disease, whereas defective activation of aerobic glycolysis deregulates the balance between proliferation and apoptosis of kidney tubule cells in patients with autosomal dominant polycystic kidney disease [4,5]. Despite the distant features of cell metabolism in different segments of the renal tubules, the molecular mechanisms underlying these observations remain to be elucidated. In this study, we showed that the nuclear receptor peroxisome proliferator- activated receptor (PPAR)α/γ and c-Myc are highly expressed in PT and DT, respectively. (PPAR)α/γ upregulate expression of the enzymes for active lipid metabolism, whereas PPARγ increases expression of iRhom2 to suppress EGF expression and secretion and EGFR activation-dependent glycolytic gene expression and glycolysis in PT. In contrast, c-Myc upregulates glycolytic gene expression, increasing glycolysis in DT. PPARγ inhibition resulted in enhanced glycolysis-related hypertrophy and tubulointerstitial fibrosis of renal tubules and impaired kidney functions.
2. Materials and methods
2.1. Materials
Rabbit polyclonal antibodies recognizing PPARα, PPARγ, Glut1, NCC, iRhom2 and EGF (for neutralization) and mouse monoclonal antibody against c-Myc were obtained from Abcam (Cambridge, MA). Rabbit polyclonal antibodies recognizing PFKL, β-tubulin, CD28K, EGFR, phospho-EGFR, phospho-ERK, phospho-AKT, and AKT were obtained from Cell Signaling Technology (Beverly, MA). Rabbit polyclonal antibodies recognizing PPARγ, ACOX1, ACSL1, ACAA1, ACAA2, HADH, HK2, and LDHA were obtained from Proteintech (Wuhan, China). Rabbit polyclonal antibodies recognizing EGF and CD36 were obtained from Bioworld (St. Louis Park, MN). Rabbit polyclonal antibody recognizing Podocin and ERK1 and mouse monoclonal antibody against EGF was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibody against CD-28 K was purchased from Boster (Wuhan, China). Recombinant human and rat EGF proteins were obtained from Peprotech (Rocky Hill, NJ). PPARγ inhibitor GW9662 was purchased from Sigma (St. Louis, MO). 2-deoxy-d-glucose was obtained from Sangon biotech (Shanghai, China). Lipofectamine™2000 transfection reagents and BODIPY® 500/510 C1, C12 were from Invitrogen (Carlsbad, CA, USA). Mouse KIM-1 Quantikine ELISA kit was purchased from R&D (Minneapolis, MN).
2.2. Cell lines and cell culture conditions
NRK-52E, MDCK, and HEK 293FT cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin antibiotics. HK2 cells were maintained in DMEM/F-12 medium supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin antibiotics. The cells were cultured in 37 °C with 5% CO2 condition. Before transfection, the culture medium was replaced by medium supplemented with 0.5% fetal bovine serum.
2.3. Isolation of mouse renal proximal and distal tubules
Renal Proximal and distal tubules were separated from C57BL/6 male mouse using a method adapted from a previous publication [6]. Briefly, the kidneys were instantly put in the ice-cold modified Krebs-Henseleit buffer (KHB) (118 mM NaCl, 4.0 mM KCl, 1.0 mM KH2PO4, 27.2 mM NaHCO3, 1.25 mM CaCl2, 1.20 mM MgCl2, 5.0 mM glucose, and 10 mM N-2-hydroxyethylpiperazine- N′-2-ethanesulfonic acid [HEPES]), and the capsules were stripped. The renal cortices were excised, minced, and incubated in 10 ml KHB-enzyme solution (9 ml KHB, 1 ml collagenase I) for 35 min at 37 °C. The tubules were collected by centrifugation at 750g for 2 min and then filtered through a 100 μm mesh and a 74 μm mesh to remove the undissociated tissues and the glomeruli, respectively. Separation of the tubules was achieved by Percoll gradient centrifugation. Suspended tubules in 35% isosmotic Percoll solution were centrifuged at 4 °C for 10 min at 17,540g. The upper bands were enriched with distal tubules, and the lower bands were enriched with proximal tubules. The proximal and distal fractions were washed three times to remove Percoll and were then stored at −80 °C.
2.4. Proteomics
For the quantitative proteomic analysis, the isolated renal proximal tubules (three groups) and distal tubules (three groups) were analyzed as described previously [7]. Briefly, tryptic peptides from three biological replicates of the C57 adult mouse renal proximal tubules and distal tubules were labeled with Tandem Mass Tag™ 6-plex (TMT sixplex™) Reagents (Thermo scientific, Rockford, USA). The labeled peptides were mixed together with an equal molar ratio and separated into 10 fractions by RP-HPLC before LC-MS/MS analysis. Raw mass spectrometric data were analyzed in the MaxQuant environment (version 1.4.1.2) [8]. MS/MS spectra were searched using the Andromeda search engine against the decoy UniProt- Mus musculus database (77,129 entries,released on 05/03/2014) [9], The false discovery rate (FDR) was set to 0.01 for both peptide and protein identifications (Benjamini Hochberg). Statistical and bioinformatics analyses were mainly performed by the software Perseus version 1.4.0.17 [10]. A paired t-test was performed to determine statistical significance of differential expression of proteins between the C57 adult mouse renal proximal tubules and distal tubules with a false discovery rate value of 0.05. KEGG metabolism pathway enrichment analyses were performed using the DAVID Bioinformatics Resources 6.7 [11]. Upstream regulators of gene expression were analyzed using QIAGEN's Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity). A p-value < .05 was used as the cut-off for all statistical analyses.
2.5. DNA constructs and mutagenesis
PCR-amplified Rat PPARα, Rat RHBDF2, and Mouse PPARγ were inserted into pCDNA3.1(+). Species-specific RHBDF2 promoters were amplified by PCR and cloned into pGL3.basic vector. The mutant RHBDF2 promoter vector was constructed, as described before [12]. The primers used in DNA constructs were listed in Supplementary Table 1.
2.6. Transfection
Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) was used to transfect overexpression vectors and small interfering RNA (siRNA) into cells. The transfection efficiency was confirmed by real time PCR and western blot. Cells were transfected with 200 nM siRNA. The target sites of siRNA were listed in Supplementary Table 2.
2.7. Promoter activity assay
The promoter activity of RHBDF2 was determined by Dual-luciferase reporter assay system. Briefly, pGL3-RHBDF2 promoter, pCDNA3.1(+) or pCDNA3.1(+)-PPARγ and pRL-SV40 renilla were transfected into HEK293FT cells in 24-well plate via Lipofectamine™ 2000. pRL-SV40 renilla vector was cotransfected for normalization of transfection efficiency. The HEK293FT cells were further treated for 24 h after transfections with or without PPARγ ligand (50 μM Rosiglitazone(TZD)). The luciferase activity was detected 36 h after transfections by Dual-Luciferase Reporter Assay System (Promega, Madi-son, WI, USA).
2.8. ChIP assay
ChIP was performed by SimpleChIP Enzymatic Chromatin IP kit (Cell Signaling Technology). Chromatin prepared from NRK-52E cells in a 10-cm dish was used to determine total DNA input and for overnight incubation with an anti-PPARγ antibody (Abcam) or normal mouse IgG. The Rat RHBDF2 promoter-specific primers used in PCR were listed in Supplementary Table 1.
2.9. Real time PCR
RNA was extracted using the RNAprep pure Cell/Bacteria Kit and RNAprep pure Tissue Kit (TIANGEN, Beijing, China) following the manufacture's instructions. 1 μg RNA was reverse transcripted into cDNA by the First Strand cDNA Synthesis Kit (Thermo Scientific, USA). The mRNA level was detected by SYBR-Green qRT-PCR kit (Takara, Japan) and normalized relative to the PPIB (peptidylprolyl isomerase B) mRNA levels. The primers used in real time PCR were listed in Supplementary Table 1.
2.10. Immunoblot analysis
Extraction of proteins from cultured cells using a modified buffer was followed by immunoblot analyses with antibodies, as described previously [13]. We extracted the protein using RIPA lysis buffer and boiled with 5× SDS loading buffer 95 °C for 10 min. we adjusted the total protein with BCA protein concentration determination kit (Thermo scientific, Rockford, USA) to ensure the same consistence of total protein and internal control. 20 μg of protein of each sample was separated by 12% SDS–PAGE, and transferred to NC membrane (Millipore, USA), The transferred membrane was stained with Ponceau S to ensure the same loading of each lines on the membrane. Subsequently, the membrane was washed with 1 × PBS to remove Ponceau S and blocked with 5% (w/v) fat-free milk in TBST at room temperature for 1 h. Then the membrane was probed with primary antibodies (anti-PPARα (1:1000 dilution, Abcam),anti-Glut1 (1:1000 dilution, Abcam), anti-NCC (1:1000 dilution, Abcam), anti-iRhom2(1:500 dilution, Abcam), anti-c-Myc(1:1000 dilution, Abcam) anti-CD28K(1:1000 dilution, Cell Signaling Technology) anti-PFKL(1:1000 dilution, Cell Signaling Technology), anti-β-tubulin(1:2000 dilution, Cell Signaling Technology), anti-EGFR(1:2000 dilution, Cell Signaling Technology), anti-phospho-EGFR(1:2000 dilution, Cell Signaling Technology), anti-phospho-ERK(1:2000 dilution, Cell Signaling Technology), anti-phospho-AKT(1:2000 dilution, Cell Signaling Technology), anti-AKT(1:2000 dilution, Cell Signaling Technology), anti-PPARγ(1:1000 dilution, Proteintech), anti-ACOX1(1:1000 dilution, Proteintech), anti-ACSL1(1:1000 dilution, Proteintech), anti-ACAA1(1:1000 dilution, Proteintech), anti-ACAA2(1:1000 dilution, Proteintech), anti-HADH(1:1000 dilution, Proteintech), anti-HK2(1:1000 dilution, Proteintech), anti-LDHA(1:5000 dilution, Proteintech) anti-EGF(1:1000 dilution, Bioworld), anti-CD36(1:1000 dilution, Bioworld), anti-Podocin(1:1000 dilution, Santa Cruz Biotechnology) and anti-ERK1(1:1000 dilution, Santa Cruz Biotechnology)) at 4 °C overnight, The membrane was then washed with 1 × TBST at room temperature 3 times for 5 min and probed with corresponding second antibody at room temperature for 1 h. The final signals of WB fragments were developed by Chemiluminescent Horseradish Peroxidase (HRP) Substrate Reagent (Millipore, Billerica, MA, USA) and then were detected with ChemiDoc™ XRS+ (Bio-Rad, Hercules, CA, USA). After that, we stripped the membranes (no more than two times), re-blocked the stripped membranes using 5% fat-free milk at room temperature for 2 h and probed with other primary antibodies at room temperature for 2 h.
2.11. In vivo experiments
C57BL/6 male mice (6-week old) were divided into four groups: 1) Control group (DMSO + normal saline (NS); n = 10); 2) 2-DG group (DMSO +2-DG; n = 10); 3) GW9662 group (GW9662 + NS; n = 10); 4) GW9662 and 2-DG group (GW9662 + 2-DG; n = 10). Mice were injected intraperitoneally with GW9662 (10 mg/kg body weight) in DMSO solvent and 2-DG (500 mg/kg body weight) in normal saline (NS). Preferentially injected GW9662 or DMSO was followed by injection with 2-DG or NS one h later. After continuously injection for four weeks, the urine was collected and all mice were sacrificed and the kidneys were harvested and fixed in 4% paraformaldehyde for the subsequent experiments.
2.12. Histological and immunostaining analysis of kidney
Kidney specimens were fixed in 4% paraformaldehyde and then embedded by paraffin or O.C.T (SAKURA, Tissue-Tek). The paraffin-embedded tissues were cut into 4-μm-thick sections for hematoxylin and eosin (H&E) staining, Sirius red staining and Masson's trichrome staining, and the O.C.T-embedded tissues were cut into 10-μm-thick sections for immunofluorescence staining. For immunofluorescence staining, sections were air-dried, washed, and blocked with 5% goat serum with 0.1% Triton X-100 and 0.1% saponin for 1 h at room temperature. Incubation with anti-EGF (1:100, Santa Cruz), or anti-Glut1 (1:50, Abcam), anti- PPARγ (1:50, Proteintech), anti-PPARα (1:100, Abcam), anti-CD28K (1:100, Boster), anti-CD28K (1:200, Cell Signaling Technology),anti-c-Myc (1:100, Abcam), anti-NRF2 (1:100, Abcam) antibody was performed at 4 °C overnight. After washed 3 times with 1× PBS, the sections were incubated with an goat anti-rabbit 555 (1:500, Invitrogen), goat anti-mouse 488 (1:500, Invitrogen) or LTL (1:300, Vector Laboratories), and DAPI (1:5000, Life Technologies). Fluorescence images were collected using a fluorescence microscope (Nikon).
2.13. Measurements of glucose consumption and lactic acid production
The levels of glucose and lactic acid in culture medium were measured by the glucose Assay Kit and the Lactic Acid assay kit (Nanjing Jiancheng Bioengineering Institute, China). In brief, 48 h after transfection, the cells were counted and the media were collected for the detection of glucose and lactic acid concentration. The results were normalized by cell numbers.
2.14. Fatty acid uptake assays
Fatty acid uptake was performed in NRK-52E cells with BODIPY® 500/510 C1, C12 probe (Invitrogen Life Sciences, Carlsbad, CA) dissolved in DMSO. 48 h after transfections, the cells were incubated for 5 min with the fatty acid probes to a final concentration of 2 μM. And then the cells were rinsed with ice-cold PBS for three times to remove the external fluorescence probes. Fixed the cell with 4% paraformaldehyde for 10 min at room temperature and washed with ice-cold PBS to remove the paraformaldehyde. The images were collected and scanned using a fluorescence microscope (Nikon). The fluorescence signal was normalized to the cell number.
2.15. ElISA
The levels of secreted EGF and urine KIM-1 were detected by the corresponding Elisa kits. 48 h after transfection, the culture media of NRK-52E cells were collected for the detection of secreted EGF. The results were normalized to cell numbers. The urine of mice was collected for the detection of KIM-1 level.
2.16. Measurements of serum creatinine and urea nitrogen
Serum was collected from mice and serum creatinine and urea nitrogen was determined using an automatic biochemical analyzer (enzyme methods)in Clinical Laboratory of the Second affiliated Hospital of Chongqing Medical University.
2.17. NADP/NADPH quantitation assays
NADP/NADPH quantitation assays were performed in MDCK cell line following the instruction of NADP/NADPH quantitation kit (Sigma, St. Louis, MO). The transfected MDCK cells were wash with cold PBS, digested with trypsin and pelleted 4 × 106 cells for each assay by centrifuging at 2000 rpm for 5 min. The pelleted cells were extracted with 800 μL of NADP/NADPH Extraction Buffer followed by 10 min standing on ice. The samples were centrifuged for 10 min at 10,000 ×g to remove insoluble material. Then 50 μL supernatant was transferred into a labeled 96 well plate in duplicate. The Master Reaction Mix was set up following the manual and was added into each sample followed by incubation of 5 min at room temperature. 10 μL of NADPH developer was added and incubated at room temperature for 1–4 h according to the color. The absorbance was measure at 450 nm. 10 μL Stop Solution was added into the reactions and the color was stable within 48 h. The results were calculated according to the instructions.
2.18. Statistical analysis
All the experiments data were performed in triplicate at least. There were 10 mice in each group of the in vivo assay. We used the Prism 5 software (GraphPad, San Diego, CA, USA) to calculate the statistical data. The results were expressed as mean ± SD and unpaired student's t -test was introduced to analysis data between two groups, while ANOVA was used for multiple comparison groups. p value ≤.05 was considered significant.
3. Results
3.1. Lipid metabolism-regulating proteins and glycolytic enzymes are highly expressed in PT and DT, respectively
To understand the regulatory mechanisms underlying the different metabolic features of PT and DT, we isolated PT and DT fractions from adult mice via density gradient centrifugation (Fig. S1A). Purity of PT and DT fractions was verified via immnunoblotting, showing enriched-expression of megalin in PT fractions and enriched-expression of CD28K [14] in DT fractions but no expression of the glomerulus protein podocin in either fraction (Fig. S1B). Quantitative proteomic analysis of both PT and DT fractions using tandem mass tag labeling coupled with liquid chromatography (LC)-tandem mass spectrometry identified a total of 4445 proteins with a 1% false-positive protein identification rate at both the protein and peptide level. We quantified 3326 of these proteins and showed that 2247 of them had markedly different expression levels in PT and DT fractions (Fig. 1A), and these results had high overall reproducibility (Fig. S1C). Using protein abundance changes (log2 [PT/DT]) >0.26 and less than −0.26 (1.2-fold change) as criteria, we analyzed 2208 of 2247 proteins meeting these criteria and found that 835 proteins were overexpressed in PT fractions (log2 [PT/DT] > 0.26) and that 1373 proteins were overexpressed in DT fractions (log2 [PT/DT] < −0.26) (Fig. 1B). In addition, many of these proteins were PT- or DT-enriched proteins identified previously (Fig. S1D) [[14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33]] strongly supporting the accuracy and efficiency of our approaches.
Fig. 1.

Lipid metabolism-regulating proteins and glycolytic enzymes are highly expressed in PT and DT, respectively.
(A) Distributions and quantifications of the proteins in renal PT and DT. Dots in blue, grey and purple indicate that 2247 proteins are differentially expressed between PT and DT fractions (t-test P < .05) whereas dots in green indicate 1079 proteins with no statistical significance in their expression between PT and DT fractions. Dots in blue represent the proteins with protein abundance change PT/DT > 1.2. Dots in purple indicate proteins with abundance change PT/DT < 0.83. The x axis shows the log2 of protein abundance change between PT and DT, whereas the y axis shows the -log10 of the t-test P value.
(B) Bar graph represents the number of proteins differentially expressed in PT (left bar) and DT (right bar).
(C, D) KEGG metabolism pathway enrichment analysis was performed. The 15 most significant enriched metabolism pathways in PT (C) and DT (D) fractions were sorted according to the p value and shown. The y axis shows the -log10 of the t-test P value.
(E) A diagram shows the fatty acid β-oxidation pathway and the enriched expression of proteins in this pathway in PT (blue) fractions.
(F) A diagram shows the glycolysis pathway and the enriched expression of proteins in this pathway in DT (red) and PT (blue) fractions.
We next subjected the differentially expressed proteins to Kyoto Encyclopedia of Genes and Genomes metabolism pathway enrichment analysis and revealed that proteins involved in fatty acid metabolism, amino acid metabolism, the PPAR signaling pathway, and the citrate cycle were enriched in PT fractions (Fig. 1C). In contrast, the proteins that regulate mRNA splicing, protein synthesis, pentose phosphate pathways, cell cycle progression, and glycolysis were highly expressed in DT fractions (Fig. 1D). The connection of the protein expression with fatty acid metabolism in PT was reflected by high expression of CD36, ACOX1, ACSL1, CPT2, ACAD1, ACADV1, HADH, ACAA1a/1b and ACAA2, which are involved in fatty acid degradation (Fig. 1E). In addition, G6PC was highly expressed in PT (Fig. 1F). In contrast to PT, DT had overexpression of enzymes in the glycolytic pathway, such as GLUT1, HK2, PFKL, PFKP, GAPDH, PGAM, ENO1, and LDHA (Fig. 1F). Of note, antioxidative proteins such as G6PD and PGD in the pentose phosphate pathway and ME1, which maintain the NADPH level, were enriched in DT (Figs. 1F, data not shown). In line with previously reported metabolic features of PT and DT [34,35], these findings supported that highly expressed enzymes involved in fatty acid and lipid metabolism provide major energy sources in PT and that highly expressed glycolytic enzymes supply major energy sources in DT.
3.2. PPAR and c-Myc regulate lipid metabolism and glycolysis in PT and DT, respectively
To determine the regulatory mechanisms underlying the different protein expression patterns in PT and DT, we performed Ingenuity Pathway Analysis-based upstream regulator analysis (QIAGEN, Valencia, CA, USA) to identify the upstream regulators of gene expression in PT and DT. We found that PPARα and PPARγ were highly scored activated upstream transcription factors in PT (Fig. 2A) but not in DT (Fig. 2B). In agreement with this finding and previous reports [[36], [37], [38]], immunofluorescent staining of kidney cortex tissue samples demonstrated that the expression patterns of PPARα and PPARγ matched with that of PT marker LTL but not with that of DT marker CD28K (Fig. 2C), supporting that PPARα and PPARγ have a higher expression in PT than DT. PPARα and PPARγ are lipid sensors and regulate the expression of genes involved in lipid metabolism [39]. As expected, depletion of PPARα or PPARγ in NRK-52E (rat proximal tubular epithelial cells) reduced the mRNA (Fig. 2D) and protein (Fig. 2E) expressions of CD36, ACOX1, HADH, ACAA1, and ACAA2 [40,41]. In addition, reconstituted expression of PPARα and PPARγ restored the protein expression of these genes (Fig. 2F). We observed similar effects in HK2 human proximal tubular epithelial cells (Fig. S3A) [42]. In line with PPARγ-dependent regulation of the expression of CD36, which is involved in fatty acid uptake [43,44], depletion of PPARγ (Fig. 2G) or PPARα (Fig. S3B) reduced the fatty acid uptake in NRK-52E cells; and the observed reduction was rescued by reconstituted expression of PPARγ and PPARα. These results combined with previous reports [30,36,37], indicated that PPARα and PPARγ are upregulated in PT cells and play an instrumental role in lipid metabolism in PT.
Fig. 2.

PPAR and c-Myc regulate lipid metabolism and glycolysis in PT and DT, respectively.
(A) Upstream regulators of gene expression in PT identified by IPA upstream regulator analysis.
(B) Upstream regulators of gene expression in DT identified by IPA upstream regulator analysis.
(C) Immunofluorescence analyses of PPARα and PPARγ in the adult C57 mouse kidney sections were performed with the indicated antibodies. Scale bar = 50 μm.
(D) Real time PCR analyses of mRNA levels of CD36, ACOX1, HADH, ACAA1, and ACAA2 in NRK-52E cells with or without PPARα (left) and PPARγ (right) knockdown were performed. The data represent the mean ± SD from n = 3 independent experiments. *** P ≤ .001.
(E) NRK-52E cells with or without PPARα and PPARγ depletion were analyzed using an immunoblotting assay with the indicated antibodies including PPARα, PPARγ, CD36, ACOX1, ACAA1, ACAA2 and HADH. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(F) Endogenous PPARα- or PPARγ-depleted NRK-52E cells were reconstituted with expression of PPARα or PPARγ, respectively. Immunoblotting analyses were performed with the indicated antibodies including PPARα, PPARγ, CD36, ACOX1, ACAA1, ACAA2 and HADH. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(G) Fatty acid uptake of NRK-52E cells with or without PPARγ depletion and reconstituted expression of PPARγ was determined. Represented pictures were shown. The bar graph below represents the fluorescence scanning of the fatty acid uptake images. Data represent the means ± SD of three independent experiments. * P ≤ .05.
(H) Immunofluorescence analyses of MYC and NRF2 in the adult C57 mouse kidney sections were performed with the indicated antibodies. Scale bar = 50 μm.
(I) MDCK cells with or without MYC depletion were analyzed using immunoblot analyses with the indicated antibodies including MYC, HK2, PFKL, GLUT1 and LDHA. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(J) MDCK cells with or without NRF2 depletion were analyzed using real time PCR of the mRNA levels of the indicated genes including ME1, G6PD, PGD, NRF2. The data represent the mean ± SD from n = 3 independent experiments. *** P ≤ .001.
(K, L) The media of MDCK cells with or without MYC depletion were collected for analysis of glucose consumption (K) and lactate production (L). The data represent the mean ± SD from n = 3 independent experiments. **P ≤ .01, ***P ≤ .001. (M) NADPH:NADP ratios in MDCK cells with or without NRF2 depletion were measured. The data represent the mean ± SD from n = 3 independent experiments. ** P ≤ .01.
In contrast to the gene expression patterns in PTs, our Ingenuity Pathway Analysis-based upstream analysis revealed that c-Myc and the Nrf2 (also known as NFE2L2) were the highly scored transcriptional factors in DT (Fig. 2B). Immunofluorescent staining of kidney tissue samples demonstrated that the expression patterns of c-Myc and Nrf2 matched with that of CD28K but not with that of LTL (Fig. 2H), supporting that c-Myc and Nrf2 have a higher expression in DT than PT. C-Myc is a critical regulator of glycolytic gene expression, whereas Nrf2 is important for regulation of antioxidant protein expression [45,46]. As predicted, depletion of c-Myc in MDCK cells (distal tubular epithelial cells) reduced GLUT1, HK2, PFKL, and LDHA expressions (Fig. 2I), whereas Nrf2 depletion reduced the mRNA levels of G6PD, PGD, and ME1 (Fig. 2J), which are directly regulated by Nrf2 [47]. Consistent with the effect of c-Myc depletion on glycolytic gene expression, c-Myc depletion also inhibited glucose uptake (Fig. 2K) and lactate production of MDCK cells (Fig. 2L). In addition, the NADPH:NADP ratio decreased in the presence of Nrf2 depletion in MDCK cells (Fig. 2M). These results strongly suggested that expression of c-Myc and Nrf2 is upregulated in DT and that they play important roles in glycolysis and NADPH production in DT cells.
Taken together, our findings indicated that PPARα/γ and c-Myc regulate lipid metabolism and glycolysis in PT and DT, respectively.
3.3. PPARγ suppresses glycolysis via inhibition of EGFR activation in PT cells
PT is known to metabolize glucose poorly [48,49]. Intriguingly, we found that PPARγ depletion in NRK-52E cells or HK2 cells increased the expression of glycolytic proteins, including GLUT1, HK2, PFKL, and LDHA (Fig. 3A, Fig. S4A); and the increased protein expressions were abrogated by reconstituted expression of PPARγ (Fig. 3A). In contrast, PPARα depletion reduced the expression of these proteins (Fig. S4B). However, PPARα overexpression in NRK-52E cells did not dramatically increase the expression of these proteins (Fig. S4C). These results suggested that PPARγ and PPARα have opposite roles in regulation of glycolysis and that PPARα expression is necessary but not sufficient for increasing glycolytic protein expression. These results also suggested that PT-highly expressed PPARγ, which has inhibitory function in glycolysis, is critical for a low glycolysis level in PT. Consistent with the effect of PPARγ above, PPARγ depletion increased glucose consumption (Fig. 3B) and lactate production (Fig. 3C) in NRK-52E cells and were rescued by reconstituted expression of PPARγ. These results indicated that PPARγ suppresses glycolysis in PT cells.
Fig. 3.

PPARγ suppresses glycolysis via inhibition of EGFR in PT cells.
(A) NRK-52E cells with or without PPARγ depletion were transfected with a control vector or a vector expressing PPARγ. Immunoblotting analyses were performed with the indicated antibodies including PPARγ, HK2, PFKL, GLUT, LDHA. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(B, C) The media of serum-starved NRK-52E cells with or without PPARγ depletion and with or without reconstituted expression of PPARγ were collected for analysis of glucose consumption (B) and lactate production(C). The data represent the mean ± SD from n = 3 independent experiments. *** P ≤ .001.
(D) NRK-52E cells with or without PPARγ depletion were lysed. Immunoblotting analyses were performed with the indicated antibodies including PPARγ, P-EGFR, Total-EGFR, P-ERK1/2, total-ERK1, P-AKT, Total-AKT. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(E) NRK-52E cells with or without PPARγ depletion were treated with or without EGFR inhibitor AG1478 (2 μM) for 48 h. Immunoblotting analyses were performed with the indicated antibodies including PPARγ, P-EGFR, HK2, PFKL, GLUT1, LDHA. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(F) NRK-52E cells with or without PPARγ overexpression were treated with or without EGF (100 ng/ml) for 24 h. Immunoblotting analyses were performed with the indicated antibodies including PPARγ, HK2, PFKL, GLUT1, GAPDH, LDHA. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(G, H) The media of serum-starved NRK-52E cells with or without PPARγ depletion in the presence or absence of AG1478 (2 μM) for 48 h were collected for analysis of glucose consumption (G) and lactate production (H). The data represent the mean ± SD from n = 3 independent experiments. * P ≤ .05;** P ≤ .01; *** P ≤ .001.
Growth factor receptor activation promotes glycolysis, and our previous studies demonstrated that activation of EGFR increases glycolytic gene expression [45,50,51]. Of note, we found that PPARγ depletion in NRK-52E cells resulted in increased phosphorylation of EGFR as well as the downstream signaling molecules extracellular signal-regulated kinase (ERK) and AKT (Fig. 3D). Treated with the EGFR inhibitor AG1478 could block PPARγ depletion-upregulated expression of HK2, PFKL, GLUT1, and LDHA (Fig. 3E). In contrast, EGF treatment-induced expression of these proteins was not affected by overexpression of PPARγ in NRK-52E (Fig. 3F) or HK2 (Fig. S4D) cells. These results strongly suggested that EGF is a downstream molecule of PPARγ and that EGFR activation is suppressed by PPARγ expression. In line with the effect of treatment with AG1478 on glycolytic gene expression, AG1478 also inhibited PPARγ depletion-enhanced glucose consumption (Fig. 3G) and lactate production (Fig. 3H) in NRK-52E cells, further supporting that PPARγ suppresses glycolysis via inhibition of EGFR activation.
3.4. PPARγ suppresses glycolysis in PT via iRhom2-mediated downregulation of EGF expression and secretion
To identify the mechanism underlying PPARγ-suppressed EGFR activation, we performed Realtime PCR and it revealed that PPARγ depletion did not affect the mRNA expression of EGF and EGFR (Fig. 4A). However, PPARγ depletion increased both intracellular and secreted EGF levels (Fig. 4B) but not cellular EGFR expression (Fig. 3D). Notably, incubation of NRK-52E (Fig. 4C) and HK2 (Fig. S5A) cells with a neutralizing antibody against EGF inhibited PPARγ depletion-induced EGFR phosphorylation. These results strongly suggested that PPARγ inhibits EGFR activation by reducing EGF stability and secretion.
Fig. 4.

PPARγ suppresses the glycolysis in PT through iRhom2-mediated downregulation of EGF expression and secretion.
(A) Real time PCR analyses of mRNA levels of the indicated gene in NRK-52E cells with or without PPARγ depletion were performed. The data represent the mean ± SD from n = 3 independent experiments. *** P ≤ .001. ns represents not significant difference between the indicated sample and the counterpart in the absence of PPARγ depletion.
(B) The intracellular and secreted EGF levels of NRK-52E cells with or without PPARγ depletion were analyzed by immunoblotting assay with the indicated antibodies including PPARγ and EGF (left) and ELISA (right), respectively. * P ≤ .05.
(C) NRK-52E cells with or without PPARγ depletion were treated with or without EGF neutralizing antibody for 48 h. Immunoblotting analyses were performed with the indicated antibodies including PPARγ, P-EGFR, Total-EGFR. All the experiments data were performed in triplicate at least and the representative imagines were shown.
(D) The mRNA levels of PPARγ and RHBDF2 of NRK-52E cells with or without PPARγ depletion were determined by real time PCR analysis. The data represent the mean ± SD from n = 3 independent experiments. *** P ≤ .001.
(E, F) NRK-52E cells with or without PPARγ depletion were transfected with or without a vector expressing iRhom2. Immunoblotting analyses with the indicated antibodies including PPARγ, iRhom2, EGF, P-EGFR, Total-EGFR, P-ERK1/2, Total-ERK1, P-AKT, Total-AKT, HK2, PFKL, GLUT1, LDHA (E) and ELISA analyses of secreted EGF levels (F) were performed. The data represent the mean ± SD from n = 3 independent experiments. ** P ≤ .01.
(G) Sequence alignment of the putative PPRE within the human, rat, and mouse RHBDF2 promoters. The varied nucleotides in WT RHBDF2 promoter of different species and mutated nucleotides in the RHBDF2 promoter used for luciferase assay were labeled in red.
(H) ChIP analyses of NRK-52E cells were performed with an anti-PPARγ antibody and the primers for the putative PPRE region of RHBDF2 promoter.
(I) Luciferase reporter vectors with species-specific or mutated RHBDF2 promoter were co-transfected with the vector expressing PPARγ into HEK293FT cells treated with or without PPARγ ligand Rosiglitazone (TZD) (50 μM). Luciferase reporter analyses were performed. The data represent the mean ± SD from n = 3 independent experiments. ** P ≤ .01; *** P ≤ .001.
(J, K) Serum-starved NRK52-E cells with or without PPARγ depletion or iRhom2 overexpression were treated with or without EGF (100 ng/ml) for 24 h. The media were collected for analysis of glucose consumption (j) and lactate production (k). The data represent the mean ± SD from n = 3 independent experiments. * P ≤ .05; ** P ≤ .01.
It was reported that iRhom2 can bind to EGF in the endoplasmic reticulum (ER) to induce proteasomal destruction of EGF in a process called ER-associated degradation [52]. As expected, PPARγ depletion reduced the mRNA (Fig. 4D) and protein (Fig. 4E) level of RHBDF2, which correlated with increased EGF expression and EGFR phosphorylation (Fig. 4E). In contrast, iRhom2 overexpression in NRK-52E and HK2 cells (Fig. S5B) suppressed the effect of PPARγ depletion on enhanced phosphorylation of EGFR and downstream ERK and AKT; increased expression of HK2, PFKL, GLUT1, and LDHA; and upregulated expression (Fig. 4E) and secretion (Fig. 4F) of EGF. These results indicated that PPARγ inhibits EGF expression and secretion via upregulation of iRhom2 expression.
We next sought to identify the mechanism underlying PPARγ-regulated iRhom2 expression by examining whether PPARγ directly regulates RHBDF2 gene transcription. It is known that PPARγ binds to the PPAR response element (PPRE; AGGTCA-N-AGGTCA) of a targeted gene promoter [53]. Sequence analysis of the human, rat, and mouse RHBDF2 promoter regions revealed the putative PPRE with a 2- or 3-nucleotide variation (Fig. 4G). Chromatin immunoprecipitation (ChIP) analysis with an anti-PPARγ antibody revealed that PPARγ bound specifically to the putative PPRE region of the RHBDF2 promoter in NRK-52E cells (Fig. 4H). Expression of PPARγ induced transcriptional activity of the RHBDF2 promoter of different species (human, mouse, rat) as measured using a luciferase reporter assay with a vector expressing RHBDF2 promoter-driven luciferase (Fig. 4I); and the effect induced by PPARγ expression was further enhanced by PPARγ ligand Rosiglitazone (TZD) treatment. In contrast, mutation of the putative PPRE of the RHBDF2 promoter (Fig. 4G) abrogated the promoter's response to PPARγ-induced transcription (Fig. 4I). These results indicated that PPARγ binds to the PPRE of the RHBDF2 promoter and induces iRhom2 expression. As expected, iRhom2 overexpression in NRK-52E cells inhibited PPARγ depletion-enhanced glucose consumption (Fig. 4J) and lactate production (Fig. 4K), and this inhibition was abrogated by exogenous EGF stimulation. Taken together, these findings revealed that PPARγ suppresses glycolysis in PT via iRhom2-mediated downregulation of EGF expression and secretion.
3.5. PPARγ inhibition results in renal tubule hypertrophy and kidney dysfunction
To determine the functional consequences of the regulation of PPARγ in renal PTs, we intraperitoneally injected mice with GW9662, a selective PPARγ inhibitor [54]. Immunofluorescence (IF) analysis demonstrated that the treatment markedly increased EGF and GLUT1 expression in PTs (Fig. 5A). In addition, H&E, Masson and Sirius red staining of renal tubules revealed that GW9662 treatment induced hypertrophy (Fig. 5B, top panel) and tubulointerstitial fibrosis of them (Fig. 5C, D, top panel), suggesting that PPARγ inhibition resulted in pathologically phenotypic changes in the renal tubules. In line with this finding, renal functional analyses demonstrated that mice given GW9662 had elevated KIM-1 mRNA expression in the kidneys (Fig. 5E), KIM-1 protein expression in urine (Fig. 5F) and serum creatinine and urea nitrogen (Fig. S6A,B); these results were in agreement with previous findings showing that KIM-1 was considerably expressed in the kidneys, specifically, in proximal tubular cells in humans and in the urine after proximal tubular injury and kidney fibrosis [2,55].
Fig. 5.

PPARγ inhibition results in hypertrophy of renal tubules and dysfunction of kidney.
(A-D) GW9662 with or without 2-DG were intraperitoneally injected into C57 adult male mice daily for four weeks. The mice were sacrificed and examined for glycolysis protein expression and renal tubule structure and functions.
(A) Immunofluorescence staining of renal cortex sections of the mice with the indicated treatment was performed with indicated antibodies.
(B) H&E-stained renal cortex sections showed representative hypertrophy of renal tubules in mice injected with DMSO, GW9662, 2-DG, or GW9662 with 2-DG. Scale bar = 50 μm.
(C) Masson-stained renal cortex sections showed representative tubulointerstitial collagen fibers in mice injected with DMSO, GW9662, 2-DG, or GW9662 with 2-DG. Scale bar = 50 μm.
(D) Sirius red-stained renal cortex sections showed representative tubulointerstitial collagen fibers in mice injected with DMSO, GW9662, 2-DG, or GW9662 with 2-DG. Scale bar = 50 μm.
(E) Real time PCR analyses were performed to measure the KIM-1, CD36, RHBDF2 mRNA levels in kidney of the mice with the indicated treatment. ** P ≤ .01; *** P ≤ .001.
(F) The KIM-1 levels in urine of the mice with the indicated treatment were measured to assess renal PT dysfunction. *** P ≤ .001.
To determine whether PPARγ inhibition-enhanced glycolysis contributes to pathologically phenotypic and functional changes in renal tubules, we intraperitoneally injected mice with 2-deoxy-d-glucose (2-DG), an effective inhibitor of glycolysis [56], and found that it prevented GW9662-induced hypertrophy and tubulointerstitial fibrosis of the tubules (Fig. 5B,C,D, bottom panel) and increased KIM-1 mRNA expression in the kidneys (Fig. 5E) and KIM-1 expression in urine (Fig. 5F). In contrast, PPARγ-dependent CD36 and RHBDF2 mRNA expression in the kidneys was reduced by GW9662 treatment but was not affected by 2-DG treatment (Fig. 5E), implying that 2-DG does not directly regulate PPARγ activity. These results suggested that PPARγ suppresses glycolysis in PT cells to maintain normal kidney function and that disrupted PPARγ function leads to pathologic changes and dysfunction in the kidneys.
4. Discussion
The kidney, which accounts for approximately 20% of cardiac output in humans, consumes a large amount of energy to transport large amounts of water, iron, and proteins and form urine. Different segments of renal tubules have distinct metabolic features and functions. We performed quantitative proteomic analysis of PT and DT and demonstrated that enzymes involved in fatty acid and lipid metabolism are highly expressed in PTs and responsible for active lipid metabolism in them, whereas glycolytic enzyme expression is increased in DT to produce high rates of glycolysis. Importantly, we demonstrated that metabolism of fatty acids and lipids in PT is regulated by abundantly expressed PPARα/γ via transcriptional regulation of lipid-metabolic enzymes. In contrast, in DTs, c-Myc upregulates the expression of glycolytic genes to enhance glycolysis, and Nrf2 upregulates that of G6PD, PGD, and ME1 to maintain antioxidation functions in cells. In addition, we revealed that PPARγ transcriptionally increased iRhom2 expression in PTs, which suppressed EGF expression and secretion, thereby inhibiting EGFR activation-dependent glycolytic gene expression and maintaining a low level of glycolysis (Fig. 6, model). Inhibition of PPARγ expression or function reduced iRhom2 expression and increased EGF and GLUT1 expression in PTs in mice, resulting in hypertrophy of renal tubules and kidney dysfunctions, which were rescued by glycolysis inhibition via treatment with 2-DG. Thus, our findings delineated an instrumental molecular mechanism underlying the active fatty acid and lipid metabolism and suppressed glycolysis in PTs and high rate of glycolysis in DTs and revealed critical roles for PPARα/γ and c-Myc in maintaining metabolic homeostasis in the kidneys.
Fig. 6.
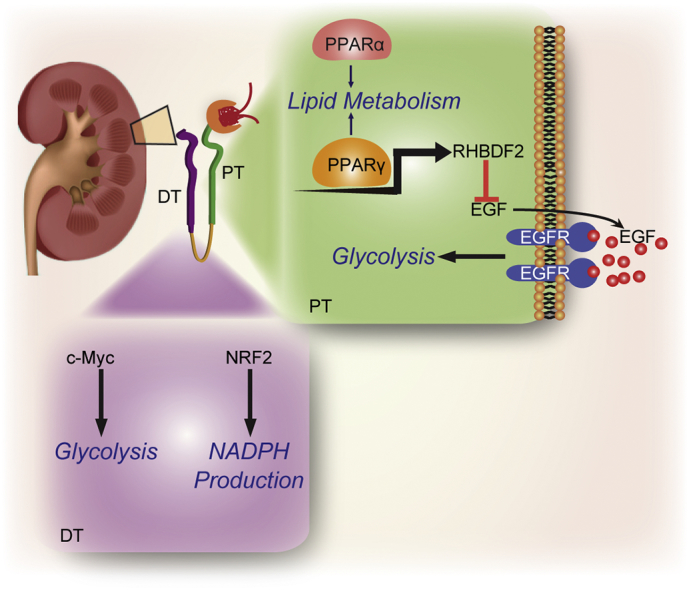
A model of the regulation of metabolism in PT and DT by PPAR, c-Myc, and NRF2.
In previous studies, PPARα and PPARγ were implicated to have roles in many aspects of metabolic disorders, including obesity, insulin resistance, dyslipidemia, inflammation, and hypertension. In addition, they were implicated to be involved in many renal pathophysiological conditions, including diabetic nephropathy, glomerulosclerosis, chronic kidney disease, and kidney fibrosis [3,15]. In line with these previous findings demonstrating that PPARα and PPARγ are critical metabolism regulators, we found that in addition to playing critical roles in lipid metabolism in PT, they have distinct roles in regulation of glycolysis in PT. Although the mechanism underlying PPARα-mediated glycolysis warrants further investigation, we demonstrated that PPARγ suppresses glycolysis via iRhom2-mediated EGF degradation in PT. The present study elucidated the potential to treat PPARγ function-inhibition kidney diseases with currently available EGFR antagonists.
It has been previously documented that the expression pattern of EGF and EGFR is not consistent in renal tubule segments. EGF is mainly expressed in the thick ascending limb of Henle and DT [57], whereas EGFR is observed in PT [58]. Of interest, early study [59] demonstrated that the amount of EGF protein in urine (which is mainly contributed by DT) positively correlated with renal function in chronic kidney disease (CKD), while our current study showed that the aberrant high EGF expression level in PT, which is mediated by PPARγ inhibition, led to the renal tubule dysfunction. The previous studies and our data suggest that the EGF and EGFR may differentially regulate PT and DT function. Besides, EGF-EGFR axis was reported to play an important but bidirectional role in renal tubule diseases [60]. On one hand, Activation of EGFR by EGF, can promote renal regeneration and functional recovery in the early stage of acute kidney injury [[60], [61], [62]]. On the other hand, EGFR activation is also involved in the initiation and progression of renal diseases, including renal fibrosis in various animal models of CKD, diabetic nephropathy, hypertensive nephropathy, or polycystic kidney disease [60,63], which suggests that EGF-EGFR axis probably have distinct function in the different stage of renal diseases. Given that the PT is more easily injured than DT in renal tubule diseases [64,65], the specific function of EGF-EGFR axis in PT during the progress of renal diseases should not be overlooked in further studies.
Another interesting finding in our dataset is highly enriched functional pathways, like Ribosome, Spliceosome, and Proteasome in DT, which reveals the highly anabolicly active status of DT. Previous evidence demonstrated that DT plays crucial roles in potassium, sodium, and divalent cation homeostasis (1,2). However, little is known about the link between the identified molecules enriched in these pathways and the function and regulation mechanism of DT. Nevertheless, our current findings reveal the potential role of metabolism in DT and give some clues for our future studies.
Animal study statement
The study was approved by the ethics committee of Chongqing Medical University.
Conflicts of interest
The authors declare no competing financial interests.
Author contributions
Q.Z. conceived the study. Q.Z., Z. Lu, Z. Lyu and Z.M. designed the study and wrote the manuscript with comments from all authors. Z. Lyu, Z.M., Q.L., performed experiments. Q.H., and Y.L. provided reagents. Y.W., H.Z. provided conceptual advice.
Acknowledgments
Acknowledgments
We thank John R. Speakman (Institute of Genetics and Developmental Biology, Chinese Academy of Sciences) for providing the Ingenuity Pathway Analysis platform; Xiahe Huang, Haitao Ge, Yuanya Zhang, Yajun Xie, and Jinlong Wang (Institute of Genetics and Developmental Biology, Chinese Academy of Sciences) for technical support in mass spectrometry analyses; and Don Norwood for critical reading of this manuscript. Z. Lu is a Ruby E. Rutherford Distinguished Professor.
Funding sources
This work was supported by the National Natural Science Foundation of China (grants 81572076 and 81873932; to Q.Z.), the Applied Development Program of the Science and Technology Committee of Chongqing (cstc2014yykfB10003; Q.Z.), the Program of Populace Creativities Workshops of the Science and Technology Committee of Chongqing (Q.Z.), the special demonstration programs for innovation and application of techniques (cstc2018jscx-mszdX0022) from the Science and Technology committee of Chongqing (Q.Z.).
Data availability
We have deposited the MS raw files to the ProteomeXchange Consortium through the PRIDE partner repository (the dataset identifier PXD007828; http://www.proteomexchange.org) [66,67].
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.10.072.
Appendix A. Supplementary data
Supplementary material 1
Supplementary material 2
Supplementary material 3
Supplementary material 4
Supplementary material 5
References
- 1.Luyckx V.A., Brenner B.M. Birth weight, malnutrition and kidney-associated outcomes--a global concern. Nat Rev Nephrol. 2015;11(3):135–149. doi: 10.1038/nrneph.2014.251. [DOI] [PubMed] [Google Scholar]
- 2.Taal M.W. Elsevier LTD; Oxford: 2011. Brenner and Rector's the Kidney. [Google Scholar]
- 3.Mh L., Ap M. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012;4(5):1–18. doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang H.M. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21(1):37. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe I. Defective Glucose Metabolism in Polycystic Kidney Disease Identifies a Novel Therapeutic Paradigm. Nat Med. 2013;19(4):488. doi: 10.1038/nm.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gesek F.A., Wolff D.W., Strandhoy J.W. Improved separation method for rat proximal and distal renal tubules. Am J Phys. 1987;253(2):358–365. doi: 10.1152/ajprenal.1987.253.2.F358. [DOI] [PubMed] [Google Scholar]
- 7.Ge H. Translating Divergent Environmental Stresses into a Common Proteome Response through the Histidine Kinase 33 (Hik33) in a Model Cyanobacterium. Mol Cell Proteomics. 2017;16(7):1258–1274. doi: 10.1074/mcp.M116.068080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.J C., M M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 9.The UniProt Consortium UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45(D1):D158. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox J., Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. Bmc Bioinformatics. 2012;13(Suppl. 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.W, H.d., S. BT, and L. RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 12.Bryksin A.V., Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques. 2010;48(6):463–464. doi: 10.2144/000113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Z. Activation of Protein Kinase C Triggers its Ubiquitination and Degradation. Mol Cell Biol. 1998;18(2):839. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Georgas K.M. Expression of metanephric nephron-patterning genes in differentiating mesonephric tubules. Dev Dyn. 2011;240(6):1600. doi: 10.1002/dvdy.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J.W., Chou C.L., Knepper M.A. Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-specific Transcriptomes. J Am Soc Nephrol Jasn. 2015;26(11):2669–2677. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jászai J. Prominin-2 is a novel marker of distal tubules and collecting ducts of the human and murine kidney. Histochem Cell Biol. 2010;133(5):527–539. doi: 10.1007/s00418-010-0690-1. [DOI] [PubMed] [Google Scholar]
- 17.Feric M. Large-scale phosphoproteomic analysis of membrane proteins in renal proximal and distal tubule. Am J Phys Cell Phys. 2011;300(4):C755. doi: 10.1152/ajpcell.00360.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brändli A.W. Organization of the pronephric kidney revealed by large-scale gene expression mapping. Genome Biol. 2008;9(5):R84. doi: 10.1186/gb-2008-9-5-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright J. Transcriptional adaptation to Clcn5 knockout in proximal tubules of mouse kidney. Physiol Genomics. 2008;33(3):341–354. doi: 10.1152/physiolgenomics.00024.2008. [DOI] [PubMed] [Google Scholar]
- 20.S T. A primary culture of mouse proximal tubular cells, established on collagen-coated membranes. Am J Physiol Ren Physiol. 2007;293(2):F476. doi: 10.1152/ajprenal.00363.2006. [DOI] [PubMed] [Google Scholar]
- 21.Amsellem S. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21(11):1859. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helbert M.J.F. Immunodissection of the human proximal nephron: Flow sorting of S1S2S3, S1S2 and S3 proximal tubular cells. Kidney Int. 1997;52(2):414–428. doi: 10.1038/ki.1997.348. [DOI] [PubMed] [Google Scholar]
- 23.Baer P.C. Isolation of proximal and distal tubule cells from human kidney by immunomagnetic separation. Technical note. Kidney Int. 1997;52(5):1321–1331. doi: 10.1038/ki.1997.457. [DOI] [PubMed] [Google Scholar]
- 24.Melià M.J. Identification of androgen-regulated genes in mouse kidney by representational difference analysis and random arbitrarily primed polymerase chain reaction. Endocrinology. 1998;139(2):688. doi: 10.1210/endo.139.2.5763. [DOI] [PubMed] [Google Scholar]
- 25.Birn H. Renal tubular reabsorption of folate mediated by folate binding protein 1. Journal of the American Society of Nephrology Jasn. 2005;16(3):608. doi: 10.1681/ASN.2004080711. [DOI] [PubMed] [Google Scholar]
- 26.Sweet D.H. Organic anion transporter 3 (Slc22a8) is a dicarboxylate exchanger indirectly coupled to the Na+ gradient. Am J Physiol Ren Physiol. 2003;284(4):F763. doi: 10.1152/ajprenal.00405.2002. [DOI] [PubMed] [Google Scholar]
- 27.Kusaba T. Differentiated kidney epithelial cells repair injured proximal tubule. Proc Natl Acad Sci U S A. 2014;111(4):1527. doi: 10.1073/pnas.1310653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pradervand S. A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Pflugers Arch - Eur J Physiol. 2010;460(6):925–952. doi: 10.1007/s00424-010-0863-8. [DOI] [PubMed] [Google Scholar]
- 29.Lai L.W., Yong K.C., Lien Y.H. Site-specific expression of IQGAP1, a key mediator of cytoskeleton, in mouse renal tubules. Journal of Histochemistry & Cytochemistry Official Journal of the Histochemistry Society. 2008;56(7):659–666. doi: 10.1369/jhc.2008.950113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheval L. Atlas of gene expression in the mouse kidney: new features of glomerular parietal cells. Physiol Genomics. 2011;43(3):161. doi: 10.1152/physiolgenomics.00093.2010. [DOI] [PubMed] [Google Scholar]
- 31.Shen S.S. Kidney-specific cadherin, a specific marker for the distal portion of the nephron and related renal neoplasms. Modern Pathology An Official Journal of the United States & Canadian Academy of Pathology Inc. 2005;18(7):933. doi: 10.1038/modpathol.3800373. [DOI] [PubMed] [Google Scholar]
- 32.Nowak G., Schnellmann R.G. Integrative effects of EGF on metabolism and proliferation in renal proximal tubular cells. Am J Phys. 1995;269(1):1317–1325. doi: 10.1152/ajpcell.1995.269.5.C1317. [DOI] [PubMed] [Google Scholar]
- 33.Harris R.C., Daniel T.O. Epidermal growth factor binding, stimulation of phosphorylation, and inhibition of gluconeogenesis in rat proximal tubule. J Cell Physiol. 1989;139(2):383–391. doi: 10.1002/jcp.1041390222. [DOI] [PubMed] [Google Scholar]
- 34.Ross B.D., Espinal J., Silva P. Glucose metabolism in renal tubular function. Kidney Int. 1986;29(1):54–67. doi: 10.1038/ki.1986.8. [DOI] [PubMed] [Google Scholar]
- 35.Rs B., Lj M. Metabolic substrate utilization by rabbit proximal tubule. An NADH fluorescence study. Am J Phys. 1988;254(2):407–416. doi: 10.1152/ajprenal.1988.254.3.F407. [DOI] [PubMed] [Google Scholar]
- 36.Braissant O. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, −beta, and -gamma in the adult rat. Endocrinology. 1996;137(1):354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 37.Endo Y. Thiazolidinediones enhance sodium-coupled bicarbonate absorption from renal proximal tubules via PPARγ-dependent nongenomic signaling. Cell Metab. 2011;13(5):550. doi: 10.1016/j.cmet.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 38.Min Z. Renal tubular epithelium-targeted peroxisome proliferator-activated receptor-γ maintains the epithelial phenotype and antagonizes renal fibrogenesis. Oncotarget. 2016;7(40):64690. doi: 10.18632/oncotarget.11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rm E., Gd B., Yx W. PPARs and the complex journey to obesity. Nat Med. 2004;10(4):355. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 40.Rice E.K. Induction of MIF synthesis and secretion by tubular epithelial cells: a novel action of angiotensin II. Kidney Int. 2003;63(4):1265–1275. doi: 10.1046/j.1523-1755.2003.00875.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang B. miR-200a Prevents renal fibrogenesis through repression of TGF-[beta]2 expression. Diabetes. 2011;60(1):280. doi: 10.2337/db10-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan M.J. HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1993;45(1):48–57. doi: 10.1038/ki.1994.6. [DOI] [PubMed] [Google Scholar]
- 43.Okamoto F. CD36 abnormality and impaired myocardial long-chain fatty acid uptake in patients with hypertrophic cardiomyopathy. Jpn Circ J. 1998;62(7):499–504. doi: 10.1253/jcj.62.499. [DOI] [PubMed] [Google Scholar]
- 44.Ll P., Siersbæk M., Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23(6):631. doi: 10.1016/j.semcdb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Yang W. PKM2 phosphorylates Histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150(4):685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loboda A. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cellular & Molecular Life Sciences Cmls. 2016;73(17):3221–3247. doi: 10.1007/s00018-016-2223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu K.C., Cui J.Y., Klaassen C.D. Beneficial Role of Nrf2 in Regulating NADPH Generation and Consumption. Toxicol Sci. 2011;123(2):590. doi: 10.1093/toxsci/kfr183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mather A., Pollock C. Glucose handling by the kidney. Kidney Int Suppl. 2011;79(120):S1. doi: 10.1038/ki.2010.509. [DOI] [PubMed] [Google Scholar]
- 49.F, K., A. K, and R. HE Renal sodium transport and oxygen consumption. Am J Phys. 1961;201(201):511. doi: 10.1152/ajplegacy.1961.201.3.511. [DOI] [PubMed] [Google Scholar]
- 50.Yang W. Nuclear PKM2 regulates [bgr]-catenin transactivation upon EGFR activation. Nature. 2011;480(7375):118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.W Y. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012;14(12):1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hosur V. Rhbdf2 mutations increase its protein stability and drive EGFR hyperactivation through enhanced secretion of amphiregulin. Proc Natl Acad Sci U S A. 2014;111(21):E2200. doi: 10.1073/pnas.1323908111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.M A. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li N. Regulation of neural crest cell fate by the retinoic acid and Pparg signalling pathways. Development. 2010;137(3):389–394. doi: 10.1242/dev.044164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Humphreys B.D. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest. 2013;123(9):4023–4035. doi: 10.1172/JCI45361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aghaee F., Pirayesh I.J., Baradaran B. Enhanced radiosensitivity and chemosensitivity of breast cancer cells by 2-deoxy-d-glucose in combination therapy. J Breast Cancer. 2012;15(2):141. doi: 10.4048/jbc.2012.15.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salido E.C. Expression of epidermal growth factor in the rat kidney. An immunocytochemical and in situ hybridization study. Histochemistry. 1991;96(1):65–72. doi: 10.1007/BF00266763. [DOI] [PubMed] [Google Scholar]
- 58.Norman J. EGF-induced mitogenesis in proximal tubular cells: potentiation by angiotensin II. Am J Phys. 1987;253(2 Pt 2):F299–F309. doi: 10.1152/ajprenal.1987.253.2.F299. [DOI] [PubMed] [Google Scholar]
- 59.Ju W. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med. 2015;7(316):316ra193. doi: 10.1126/scitranslmed.aac7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang J., Liu N., Zhuang S. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 2013;83(5):804–810. doi: 10.1038/ki.2012.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Humes H.D. Epidermal growth factor enhances renal tubule cell regeneration and repair and accelerates the recovery of renal function in postischemic acute renal failure. J Clin Invest. 1989;84(6):1757–1761. doi: 10.1172/JCI114359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Safirstein R., Zelent A.Z., Price P.M. Reduced renal prepro-epidermal growth factor mRNA and decreased EGF excretion in ARF. Kidney Int. 1989;36(5):810–815. doi: 10.1038/ki.1989.266. [DOI] [PubMed] [Google Scholar]
- 63.Staruschenko A. Epidermal growth factors in the kidney and relationship to hypertension. Am J Physiol Ren Physiol. 2013;305(1):F12–F20. doi: 10.1152/ajprenal.00112.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chevalier R.L. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Ren Physiol. 2016;311(1):F145–F161. doi: 10.1152/ajprenal.00164.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakhoul N., Batuman V. Role of proximal tubules in the pathogenesis of kidney disease. Contrib Nephrol. 2011;169:37–50. doi: 10.1159/000313944. [DOI] [PubMed] [Google Scholar]
- 66.Vizcaino J.A. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32(3):223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deutsch E.W. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45(Database issue):D1100–D1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1
Supplementary material 2
Supplementary material 3
Supplementary material 4
Supplementary material 5
Data Availability Statement
We have deposited the MS raw files to the ProteomeXchange Consortium through the PRIDE partner repository (the dataset identifier PXD007828; http://www.proteomexchange.org) [66,67].