

Abstract
Background
Gaucher Disease is caused by mutations of the GBA gene which encodes the lysosomal enzyme acid beta-glucosidase (GCase). GBA mutations commonly affect GCase function by perturbing its protein homeostasis rather than its catalytic activity. Heat shock proteins are well known cytoprotective molecules with functions in protein homeostasis and lysosomal function and their manipulation has been suggested as a potential therapeutic strategy for GD. The investigational drug arimoclomol, which is in phase II/III clinical trials, is a well-characterized HSP amplifier and has been extensively clinically tested. Importantly, arimoclomol efficiently crosses the blood-brain-barrier presenting an opportunity to target the neurological manifestations of GD, which remains without a disease-modifying therapy.
Methods
We used a range of biological and biochemical in vitro assays to assess the effect of arimoclomol on GCase activity in ex vivo systems of primary fibroblasts and neuronal-like cells from GD patients.
Findings
We found that arimoclomol induced relevant HSPs such as ER-resident HSP70 (BiP) and enhanced the folding, maturation, activity, and correct cellular localization of mutated GCase across several genotypes including the common L444P and N370S mutations in primary cells from GD patients. These effects where recapitulated in a human neuronal model of GD obtained by differentiation of multipotent adult stem cells.
Interpretation
These data demonstrate the potential of HSP-targeting therapies in GCase-deficiencies and strongly support the clinical development of arimoclomol as a potential therapeutic option for the neuronopathic forms of GD.
Funding
The research was funded by Orphazyme A/S, Copenhagen, Denmark.
Keywords: Gaucher disease, GBA, Heat shock response, Arimoclomol, Heat shock proteins, HSP, HSP70, Proteostasis, GCase, Lysosomes, Lysosomal storage diseases, Sphingolipidoses
Research in context.
Evidence before this study
Neuronopathic Gaucher disease is an ultra-rare, devastating monogenetic disorder without any available therapy. Gaucher Disease is caused by mutations in the GBA gene which encodes the lysosomal enzyme acid beta-glucosidase (GCase). The mutations are primarily missense mutations giving rise to misfolded variants of GCase. Most of the mutations, including all the most common, appear amenable to chaperoning strategies as previous studies have indicated that induction of molecular chaperones of the Heat shock protein 70 (HSP70) family can improve residual activity of misfolded GCase.
Added value of this study
Arimoclomol is an orally available, brain-penetrant small molecule HSP70 amplifier in late-stage clinical development in several diseases. The data reported herein provide proof-of-concept for the development of arimoclomol as a potential therapy for neuronopathic Gaucher disease and have been instrumental for the advancement of arimoclomol into the currently running phase II clinical trial in Gaucher patients.
The data herein not only offer novel mechanistic insight to how the HSP70 system can be mobilized as a potential therapeutic option for neuronopathic Gaucher disease, but by extension also holds promise for Parkinson's disease, as mutations in GBA constitute the highest genetic risk factor for the development of Parkinson's disease.
Implications of all the available evidence
In summary, the available evidence suggest that amplification of HSP70 family members might provide a therapeutic benefit to diseases associated with GCase deficiency and that arimoclomol could provide a first-in-class therapy for neuronopathic Gaucher disease.
Alt-text: Unlabelled Box
1. Introduction
Gaucher disease (GD) is one of the most prevalent human metabolic storage disorders belonging to the group of lysosomal storage diseases (LSDs) [1]. It is primarily caused by autosomal recessive mutations in the GBA gene leading to deficiency of the lysosomal enzyme acid beta-glucosidase (GCase, EC 3.2.1.45). >460 GBA mutations, the majority being missense mutations, have been identified (http://www.hgmd.cf.ac.uk) [2]. The mutations commonly lead to increased protein misfolding, premature degradation and abnormal chaperone recognition, which in turn lead to reduced GCase function. GCase dysfunction leads to the accumulation of its substrate glucosylceramide (GlcCer) and other sphingolipids, including glucosylsphingosine (GlcSph) causing cellular dysfunction and subsequent clinical manifestations primarily in the central nervous system (CNS), visceral and bone systems [3].
GD is clinically divided in visceral type I (GD1), acute neuronopathic type 2 (GD2) and sub-acute neuronopathic type 3 (GD3) forms, although the age of onset and the phenotypic expression of the disease is variable [4]. Visceral involvement in GD includes liver and spleen enlargement and dysfunction, as well as the displacement of normal bone marrow by storage cells causing anemia, thrombocytopenia, and bone disease. Although GD1 is considered a non-neuronopathic form, there is increasing evidence that neurological symptoms (i.e. Parkinson's syndrome, tremors, peripheral neuropathy) become a prominent part of the pathology as the disease progresses [[5], [6], [7], [8], [9]]. GD2 is very rare (1% of cases) and is associated with a severe and rapid neurodegeneration, leading to an early death, usually before the second year of life [10]. In GD3, visceral and biochemical signs are similar to GD1 and the first symptoms are usually due to peripheral involvement. The involvement of the CNS generally appears later and includes oculomotor apraxia, ataxia, epilepsy, and mental deterioration [11]. Currently, two types of treatments are available for GD1: enzyme replacement therapy (ERT) and substrate reduction therapy (SRT) but there are no approved disease-modifying treatments for the neurological forms of the disease.
The molecular mechanisms involved in the neurodegenerative process in GD are not fully elucidated but the disease pathology ultimately stem from the loss of function of GCase. Mutations in the GBA gene are numerous with the majority being missense mutations that affect the correct folding and maturation of the protein, but do not completely abrogate the catalytic activity [2]. In GD, misfolded GCase is retained in the endoplasmic reticulum (ER), from where it is retro-translocated back to the cytosol to be eliminated by the ubiquitin proteasome pathway [12]. This process, known as ER-associated degradation (ERAD), impede mutant GCase from reaching the lysosome, leading to compromised lysosomal GCase activity and function. In addition, the presence of misfolded GCase triggers the unfolded protein response (UPR) and interferes with the degradation of other proteins [13]. The loss of GCase function leads to accumulation of metabolites such as GlcCer and GlcSph, expansion of a compromised, destabilized lysosomal compartment, inflammation and loss of neurons potentially involving the Rip3k pathway [3,14,15].
Members of the HSP70 family of molecular chaperones include HSP70 encoded by HSPA1A and BiP/GRP78/HSP70-5 encoded by HSPA5 [16], which have been shown to be important for lysosomal and GCase function [[17], [18], [19], [20], [21]]. Studies of GCase, and its most prevalent missense mutations for non-neuronopathic (N370S) and neuronopathic (L444P) GD, have demonstrated that it binds to HSP70 and HSP90 which in concert with cochaperones such as TCP1 guide the enzyme through to either correct folding and lysosomal activity, or to the ubiquitin proteasome pathway for degradation [[21], [22], [23], [24]].
Several lines of evidence indicate that it is possible to at least partially rescue GCase folding and function by strategies aimed at increasing the levels of cellular molecular chaperones such as HSP70 or through the use of small molecules acting as chemical chaperones [[22], [23], [24], [25], [26], [27], [28], [29]].
Arimoclomol, a small molecule amplifier of HSP70 and other HSP chaperones, has emerged as a potential therapeutic agent for several LSDs including Niemann-Pick Disease Type C (NPC) [18] and is currently being evaluated in a phase II/III trial for NPC (ClinicalTrials.gov identifier NCT02612129). Importantly, previous clinical studies of other neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and sporadic inclusion body myositis (sIBM), have demonstrated a safety profile for arimoclomol compliant with chronic use, as well as penetrance to the CNS and signs of efficacy [[30], [31], [32]]. Based on the clinical safety-profile, the CNS-penetrable ability, and the HSP-inducing mechanism of action, arimoclomol may present a first-in-class treatment paradigm for GD patients – particularly patients with currently untreated neurological symptoms. We therefore investigated the effect of arimoclomol on the stability, localization and enzymatic activity of GCase across a broad range of genotypes in primary cultured GD fibroblasts and in a human neuronal model of GD obtained through differentiation of multipotent adult stem cells (MASCs).
2. Materials and methods
2.1. Cell culture and drug treatment
All human fibroblast cell lines used for this study were obtained from Coriell Biorepositories and cultured under standard cell culture conditions (37 °C and 5% CO2) in DMEM supplemented with non-essential amino acids (NEAA), 1% Pen-Strep and 12% FCS.
Cells were treated with vehicle (PBS) or arimoclomol-citrate (BRX-345) dissolved in PBS for the indicated time and medium containing fresh compound was added every 2–3 days. For experiments with imiglucerase cells were treated for 5 days with medium replenishment every 2–3 days. Fresh medium containing compounds was also added on the day before visualization by ABPs.
Arimoclomol-citrate is a proprietary compound owned by Orphazyme A/S.
2.2. Western blotting and EndoH assay
Cells were collected and lysed in 25 mM KPi buffer, pH 6.5 + 0.1% Triton X-100 including a cocktail of protease inhibitors (Roche). Protein concentration was determined using with the bicinchoninic acid protein assay (BCA) kit (Pierce). 10–20 μg total protein/sample was used for WB of protein levels or used for EndoH-treatment according to manufacturer's instructions (New England Biolabs). Western blots were quantified using the freeware program Fiji (an image processing package to ImageJ, https://fiji.sc/).
Antibodies used were: anti-GBA (# WH0002629M1, Sigma-Aldrich, RRID: AB_1841770), anti-Vinculin (# V9131, Sigma-Aldrich, RRID: AB_477629), anti-GRP78 (BiP) (# sc-13968, Santa Cruz Biotechnology, RRID: AB_2119991), anti-RPA (# sc-28709, Santa Cruz Biotechnology, RRID: AB_2238546), and anti-Beta actin (# A2066, Sigma-Aldrich, RRID: AB_476693).
2.3. GCase activity
The fluorometric GCase enzyme activity assay was performed as described previously [29]. Briefly, cells were seeded in 96 well plates and treated in biological triplicate with various concentrations of arimoclomol for the indicated time. Medium was replenished every 2–3 days.
2.4. ABP labelling and quantification
Fibroblasts were collected and lysed, and protein concentration determined. For labelling, equal amounts of protein were incubated with cy5-labeled ABP ME569 in 150 mM McIlvaine buffer, pH 5.2 for 1 h at 37 °C. Samples were denatured with 4× Laemmli buffer, boiled for 5 min at 98 °C and resolved by SDS-PAGE using the TGX gel system (Bio-Rad). The gels were placed in an imager (G:BOX ChemiXR5, Syngene) and fluorescence was detected with red LED lightning modules/705 M filter. The amount of labeled GCase was quantified using GeneTools v.4.03.01.0 (Syngene).
For visualization, fixed cells were incubated with ABP MDW933 (green) and images were acquired using the AXIO (Zeiss) equipped with AxioCam MRm (LED ex 470 nm, em 500-550 nm). Images were analyzed using freeware program Fiji (an image processing package to ImageJ, https://fiji.sc/).
2.5. MASC generation and differentiation
Human skin-derived multipotent adult stem cells (MASCs) were obtained from skin biopsies from healthy donors and patients affected by GD, who were under observation at the Regional Centre for Rare Diseases. Written consent was obtained from the subjects or from caregivers or guardians on behalf of the minors involved in the study.
MASCs enriched cultures were obtained, from already established skin fibroblast cultures at early passages (P1, P2, P3), as previously described [33]. Briefly, 1 × 106 cells obtained from confluent primary skin fibroblast cultures were seeded onto 100 mm plates coated with fibronectin and expanded at least for three passages in a selective media composed of 60% DMEM/40% MCDB-201 (Sigma-Aldrich) supplemented with 1 mg/ml Linoleic Acid-BSA (Sigma-Aldrich);10–9 M dexamethasone (Sigma-Aldrich); 10–4 M Ascorbic acid-2 phosphate (Sigma-Aldrich); 1× Insulin-transferrin‑sodium selenite (Sigma-Aldrich); 2% fetal bovine serum (FBS), (STEMCELL Technologies), 10 ng/ml human PDGF-BB (Peprotech EC); 10 ng /ml human EGF (Peprotech EC). Medium was replaced every 4 days and cells were spitted when they reached 70/80% confluence.
MASCs obtained after 3 passages in selective medium were detached and and stained with the following primary conjugated antibodies: CD13, CD49a, CD49b, CD49d, CD90, CD73, CD44, CD45, human leukocyte antigen-D related (HLA-DR), CD34, and CD271 (BD Biosciences, Franklin Lakes, NJ, USA); CD105 and kinase insert domain receptor (KDR; Serotec, Oxford, United Kingdom); and CD133 (Miltenyi Biotec, Bergisch Gladbach, Germany). The percentage of cells expressing all the antigens was determined by fluorescence-activated cell sorting (FACS) analysis (CyAn; Beckman Coulter, Brea, CA, USA). Properly conjugated isotype-matched antibodies were used as negative controls.
To induce neural differentiation, MASCs were seeded at a density of 8000 cells/cm2 into 96 multiwell plates (BD Biosciences) or on coverslips in medium containing DMEM-HG with 10% FBS (called N1 medium). After 24 h the DMEM-HG was replaced with fresh medium supplemented with 1% of B27 (Invitrogen), 10 ng/ml EGF (Peprotech) and 20 ng/ml bFGF (Peprotech) (called N2 medium) for 5 days. Thereafter, cells were incubated for 24/48 h in DMEM supplemented with 5 μg/ml insulin, 200 M of indomethacin and 0.5 mM IBMX (all from Sigma-Aldrich) without FBS (called N3 medium). The actual differentiation was determined by analyzing the expression of the neuron specific markers, NeuN and Tubulin b3.
For treatment, vehicle or 400 μΜ arimoclomol was included in the N2 medium, replenished after 3 days and added to the N3 medium for a total treatment time of 9 days.
2.6. Statistical analysis
GraphPad Prism v.7.03 was used for statistical analysis. Unless otherwise stated, repeated measures (RM) ANOVAs were calculated using a mixed model for repeated measures with treatments/timepoints as fixed effects and experiment as repeated random effect. Multiplicity was adjusted using Sidak-Holm or Dunnett's method. Welch-Satterthwaite t-tests were used to correct for unequal variance.
3. Results
3.1. Arimoclomol increases the quantity and ER to golgi maturation of mutated GCase in primary GD patient fibroblasts
We obtained a panel of primary skin-derived fibroblast cell lines from individuals diagnosed with GD covering the major genotypes and from three healthy donors. Sequencing of the GBA gene confirmed the presence of the reported pathogenic mutations of GBA in all GD cell lines (Supplementary Table 1). We also identified the presence of a T369M variant in the widely used control fibroblast cell line GM05659 [WT/T369M], which is therefore termed as a carrier cell line. T369M does not cause GD in homozygous carriers, but may be associated with an increased risk of developing Parkinson's disease [34,35].
Comparison of mRNA, protein and GCase activity levels across the WT and GD primary patient fibroblasts demonstrated no correlation between the level of GBA mRNA and disease (Fig. 1a), whereas neuronopathic GD (nGD) cell lines showed a profound decrease in the GCase protein level compared to WT fibroblasts (Fig. 1b). The GD1 cell line GM00372 [N370S/1-bp ins 84G] also displayed a clear decrease in the amount of GCase protein, which is likely caused by the premature stop codon being produced by the 1-bp ins 84G allele [36], whereas two other non-neuronopathic GD cell lines GM01607 [N370S/V394L] and ND34263 [N370S/N370S] had only slightly reduced GCase protein levels (Fig. 1b).
Fig. 1.

Arimoclomol increases the quantity and ER to golgi maturation of mutated GCase in primary GD patient fibroblasts.
a) Level of GBA mRNA expression in primary GD, WT and carrier fibroblasts relative to the expression level in GM00498 (WT/WT #1) cells (stippled line). b) WB analysis of GCase protein levels in primary GD fibroblasts, carrier and WT cell lines. Vinculin was used as loading control. c) Basal level of GCase activity (Fluorescence units (FLU) normalized to cell density) in the primary GD fibroblasts, carrier and WT cell lines. Data is reported as mean + SEM of 3–4 experiments/cell line. d-f) WB analysis of GCase in (d) nGD fibroblasts, (e) non-neuronopathic GD or (f) carrier and WT cell lines treated with the indicated concentrations of arimoclomol for 5 days. Lysates were subjected to EndoH-digestion analysis and the EndoH-resistant fraction is marked by **. RPA or Vinculin served as loading control for these experiments. Quantification of GCase bands on WBs is shown as arbitrary units (A.U.) with the EndoH sensitive part of the GCase band in gray and the EndoH resistant part in black. WBs are representative of 3 independent experiments/cell line.
Analysis of basal GCase activity showed reduced activity of the mutated GCase in all the GD cell lines investigated, as well as a slight reduction in the T369M carrier cell line (Fig. 1c).
Arimoclomol is a well-described co-inducer of the heat shock response, which includes the amplification of HSP70 chaperones [37,38]. We first tested if arimoclomol amplifies and prolongs the expression of HSP70 encoded by HSPA1A in primary L444P/L444P GD cells. We found that doses of arimoclomol of 100–400 μM increased the expression of HSPA1A suggesting a relatively high stress-threshold in the fibroblasts under standard cell culture conditions (Supplementary Fig. 1a). In addition, a time-dependent increase of HSPA1A and HSPA5 was seen by treatment with 400 μM arimoclomol (Supplementary Fig. 1a–b).
Supplementary Fig. 1.

Arimoclomol increases and prolongs the HSR in GD fibroblasts.
a) Expression of HSPA1 mRNA in GM10915 cells treated with arimoclomol for 48 h or 120 h. Each time point was measured in 2–3 independent experiments and data are shown as mean + SEM. The effect of arimoclomol was evaluated against control at each time point by a two-way ANOVA and multiplicity was adjusted by Holm-Sidak's method (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001). b) Expression of HSPA1A or HSPA5 mRNAs in GM10915 cells treated with vehicle (0 μM arimoclomol) or 400 μM arimoclomol for the indicated time. Expression levels, relative to the 0-h timepoint, are shown as mean ± SEM of 3 independent experiments. Statistical analysis was done by 2-way RM ANOVA and multiplicity was adjusted using Sidak-Holm. c) Representative WB analysis of BiP in GM02627 [G325R/C342G], GM10915 [L444P/L444P] and GM01607 [N370S/V394L] cells treated with the indicated concentrations of arimoclomol for 5 days. Vinculin was used as loading control. Quantification of WBs is shown as relative BiP levels in arimoclomol-treated versus control-treated cells and the data are reported as mean + SEM of 2–4 independent experiments. The effect of arimoclomol was evaluated against control by a one-way ANOVA followed by Dunnett's multiple comparisons test (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001). d) Expression of HSPA1 and HSPA5 mRNAs in GM10915 cells without heat shock (0 h timepoint) or at the indicted recovery times after 1 h at 41.5 °C. Prior to heat shock, cells were treated with vehicle or 400 μM arimoclomol for 72 h. Each time point was measured in 2–4 independent experiments and data are shown as mean + SEM. The effect of arimoclomol was evaluated against control at each time point by a two-way ANOVA and multiplicity was adjusted by Holm-Sidak's method (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001).
The increase of HSPA5 was confirmed by western blot analysis of BiP. The levels of BiP were significantly increased by 100 μM and 200 μM arimoclomol in GM10915 [L444P/L444P] cells, by 200 μM and 400 μM arimoclomol in GM01607 [N370S/V394L] cells, whereas the minor increases in GM02627 [G325R/C342G] cells did not reach statistical significance. (Supplementary Fig. 1c). We also confirmed that arimoclomol amplified and prolonged the heat shock response in heat-shocked GD cells (Supplementary Fig. 1d).
We then investigated the folding and ER-to-golgi transition of mutant GCase variants with or without arimoclomol treatment. Cell lysates were digested with Endoglycosidase H (EndoH), an endoglycosidase that specifically cleaves high mannose (>4 mannose residues) but not mature N-glycan complexes, allowing differentiation between immature glycoproteins that have not reached the mid-Golgi (EndoH-sensitive) and mature glycoproteins (EndoH-resistant) [39].
Arimoclomol dose-dependently increased the amount of GCase in primary GD fibroblasts across all tested genotypes, including neuronopathic and non-neuronopathic associated mutations (Fig. 1d–e). The [G325R/C342G] GCase mutations in GM02627 cells are not well characterized, and we show here that they result in low levels of GCase protein despite a normal level of GBA mRNA expression (Fig. 1a, b, d). The EndoH assay showed a clear ER-retention of GCase in this cell line, but the compound heterozygosity does not allow for analysis of each specific mutation. However, it is likely that both G325R and C342G are trapped in the ER, as almost all GCase was digested by EndoH, and incomplete GCase processing has been reported [40]. Nevertheless, arimoclomol increased the amount of both immature and mature GCase in the GM02627 cell line (Fig. 1d). Likewise, the GM01260 [L444P/P415R] cell line has been shown to produce unstable and immature GCase [40], which was confirmed by our EndoH analysis (Fig. 1d). At this point, we cannot discriminate between the P415R and L444P mutations in GM01260 cells. In contrast to the L444P mutation [12], the P415R is an uncharacterized mutation that may affect the binding of GCase to LIMP2 and thereby reduce the trafficking of the P415R GCase from the ER to the lysosomes [41]. We found a clear augmentation of immature GCase by arimoclomol, as well as small increase in the EndoH-resistant pool of GCase in the GM01260 cell line (Fig. 1d). In addition, GM10915 [L444P/L444P] cells treated with arimoclomol had a profound increase in mature GCase, as well as a small increase in the level of immature GCase (Fig. 1d). Thus, treatment with arimoclomol increased EndoH resistant levels of GCase for all three nGD cell lines (Fig. 1d).
Compared to the L444P and other nGD-associated mutations, we found the non-neuronopathic associated mutation N370S, to be less retained in the ER (Fig. 1e), as also observed by others [42]. A precise quantification of the effect of arimoclomol on the EndoH resistant levels in non-neuronopathic GD cells is therefore difficult, but in general there were slightly more EndoH-resistant GCase in arimoclomol-treated non-neuronopathic GD cells (Fig. 1e). EndoH digestion analysis of WT GCase demonstrated that WT GCase is subject to some degree of EndoH digestion as reported previously [12,42] and that arimoclomol could also augment WT GCase levels (Fig. 1f).
3.2. Arimoclomol increases residual GCase activity in GD cells
Having demonstrated a beneficial effect of arimoclomol on the maturation of mutant GCase protein in primary GD fibroblasts we next evaluated the effect of arimoclomol on GCase activity. The GM10915 [L444P/L444P] cell line was treated with 50–800 μM arimoclomol for 1–5 days and GCase activity was measured using 4-MUG as substrate. For each time point, the relative GCase activity in arimoclomol- treated cells was calculated as fold change to control (PBS treated cells). A time- and dose-dependent increase in GCase activity was seen in cells treated with arimoclomol corresponding to the observed amplification of HSP70 family members (Fig. 2a and Supplementary Fig. 1a–d). Longer exposure (28 days) at lower concentrations of arimoclomol resulted in increases of GCase activity in L444P/RecNcil cells up to the level of GCase activity in a carrier cell line (WT/RecNcil) (Fig. 2b). A beginning cytostatic effect at 400–800 μM arimoclomol was noticed, but no signs of cytotoxicity was observed (Supplementary Fig. 2a–b).
Fig. 2.

Arimoclomol augments residual GCase activity in primary GD patient fibroblasts.
a) GCase activity in GM10915 (L444P/L444P) cells treated with 50–800 μM arimoclomol for 1–5 days. Relative GCase activity is shown as fold change compared to vehicle-treated cells for each time point. Data are mean + SEM of 3 independent experiments. The effect of each arimoclomol concentration was evaluated against vehicle within the same day using a 2-way RM ANOVA model with the interaction arimoclomol concentration*day as fixed effect. Multiplicity was adjusted using Dunnett's method. (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001). b) GCase activity in GM00877 (L444P/RecNcil) and GM00878 (WT/RecNcil) cells treated with arimoclomol for 4 weeks. Data are shown as mean + SD, n = 3–6. c) Representative images of GCase labeled with ABP-green in GM10915 (L444P/L444P) cells treated with vehicle (PBS), 400 μM arimoclomol or 0.25–1.0 U/ml imiglucerase for 5 days. Scale bars are 30 μM. d) Effect of 400 μM arimoclomol (ari) and 0.125–1 U/ml Imiglucerase (Imi) on GCase activity levels in GM10915 cells treated for 5 days. Data are shown as mean + SEM, n = 4. Effects of treatments were compared using 1-way RM ANOVA with multiplicity adjusting using Dunnett's method. e) Relative GCase activity in primary GD patient fibroblasts treated with arimoclomol for 5 days. f) Relative GCase activity in WT fibroblast cell lines treated with the indicated concentrations of arimoclomol for 5 days. For e-f) data are reportedas mean ± SEM of 3–4 independent experiments/cell line. Statistical analysis was done using a 1-way RM ANOVA model. Multiplicity was adjusted using Dunnett's method (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001).
Supplementary Fig. 2.

Effect of arimoclomol on cellular growth.
a) Images of GM10915 cells treated with PBS (0 μM arimoclomol) or indicated concentrations of arimoclomol for 5 days. b) Cell density (OD570 values for cells stained with crystal violet) for GM10915 cells treated with 50–800 μM arimoclomol for 1–5 days. Data are reported as mean + SEM, n = 3.
To qualify the increases in GCase activity observed in primary GD fibroblasts treated with arimoclomol, we examined the levels of GCase activity obtainable with the current standard of care therapy for non-neuronopathic GD, enzyme replacement therapy, in the form of recombinant GCase (imiglucerase). We treated L444P/L444P cells with imiglucerase or arimoclomol for 5 days. Imiglurase is primarily taken up by cells via the mannose receptor mediated pathway, which is highly expressed by macrophages [43]. As fibroblasts express much less mannose receptor, cellular uptake and activity of imiglucerase from the cell culture medium was confirmed using activity-based probes (ABPs) [44] (Fig. 2c). A dose-dependent increase in GCase activity was obtained for imiglucerase (Fig. 2d), with maximum obtainable activity levelling out at 0.5–1.0 U/mL. The increase in GCase activity obtained by 400 μM arimoclomol was comparable to the maximum attainable activity levels with imiglucerase in L444P/L444P GD fibroblasts (Fig. 2c–d).
We then proceeded to investigate the effect of 5-days treatment with arimoclomol on residual GCase activity across our panel of primary GD fibroblasts. In line with the observed effects on HSP70 induction and GCase transcription and maturation, arimoclomol significantly increased residual GCase activity across all genotypes including both neuronopathic and non-neuronopathic alleles (Fig. 2e). We also observed significant increases in the two WT cell lines, but not in the T369M carrier cell line (Fig. 2f).
3.3. Arimoclomol improves the correct lysosomal localization of GCase in GD cells
To asses if the rescued GCase reaches the correct intracellular localization and to validate the observed improvement in maturation and activity increases, we took advantage of the highly specific fluorescent ABPs which allow labeling of active GCase molecules in cells [44]. WT, GM01607 [N370S/V394L], and GM02627 [G325R/C342G] cells were treated with 400 μM arimoclomol for 5 days. Imaging of fixed cells showed clear punctuate high intensity GCase localization in WT cells, whereas GD cell lines displayed decreased labeling (Fig. 3a). The labeling of active GCase in GM10915 [L444P/L444P] was below detection level and this cell line was therefore not included in the analysis. The GCase labelling intensity was increased in arimoclomol-treated primary GD patient fibroblasts, with a clear lysosomal distribution pattern. The primary image analysis was corroborated by automated image quantification demonstrating a significant fluorescence intensity shift towards the WT profile in the arimoclomol treated GD fibroblasts compared to untreated controls (Fig. 3b–d).
Fig. 3.

Arimoclomol increases lysosomal localization and activity of GCase in GD fibroblasts.
a) Representative images of primary WT or GD patient fibroblasts treated with arimoclomol for 5 days and labeled with green fluorescent ABPs. Scale bars = 10 μm. b-d) Image analysis quantification of active GCase labeling. The quantification of labeling is shown as the frequency distribution of the ABP labeling intensity per area in grouped intervals, n = 3, >100 cells analyzed per replicate. e-g) Gel quantification of ABP-labelling of active GCase. Representative fluorescent gel images of cell lysates labeled with ABP-cy5 ME569. Cells were treated with arimoclomol for 5 days and each concentration was evaluated in duplicate or triplicate samples. The quantification of ABP-labeling is shown in the right panel as mean + SEM, n = 3–4. The effect of arimoclomol was analyzed by RM oneway-ANOVA. Multiplicity was adjusted using Dunnett's method.
We further quantified the effect of arimoclomol treatment on active GCase labeling in GD cells by using SDS-page resolved cell lysates incubated with ABP (Fig. 3e–g). The amount of active, labeled GCase was significantly increased by arimoclomol for both non-neuronopathic and neuronopathic genotypes.
3.4. Arimoclomol increases GCase activity in neuronal-like cells from GD patients
We next evaluated the effect of arimoclomol in a human neuronal model of GD, obtained by induced neuronal differentiation of multipotent adult stem cells (MASCs) isolated from neuronopathic GD patients [33]. MASC-derived neuronal cultures were established from a healthy donor (WT) and from neuronopathic GD patients carrying three different genotypes: F213I/L444P, L444P/L444P and IVS2 + 1G > A/N188S. In addition, MASCs were obtained from one non-neuronopathic GD patient (Supplementary Table 2). GBA sequencing identified this GD individual as a compound heterozygous for the common N370S mutation and a variant not previously reported: c.516C > A that results in a codon change from tyrosine 133 (TAC) to a stop codon (TAA). However, the analysis of the GBA mRNA isolated from the patient's cells showed that only the allele carrying the normal cytosine in position 516 was expressed (Supplementary Fig. 4a). These results suggest that the Y133* mutation leads to the expression of an unstable transcript resulting in either no or very little truncated GCase protein. We characterized the surface immunophenotype of the MASCs (Supplementary Table 3) as previously described and observed no major differences between MASCs from GD patients and healthy donors [33]. The differentiation of early passage MASCs (Passage 1–3) to neuronal-like cells was ascertained by immunostaining of the neuronal markers NeuN and tubulin beta 3 (TUBB3) (Fig. 4a, b). The presence of arimoclomol during differentiation did not impact the neuronal differentiation (Fig. 4a, b).
Supplementary Fig. 4.

Sequencing analysis of GBA in MASCs carrying the N370/Y133* genotype.
a) Analysis of genomic DNA showed the presence of the c.516C > A mutation in heterozygosis (left panel), while sequencing of the cDNA synthesized from mRNA extracted from the patient's cells showed the absence of the mutation at position 516 of the cDNA indicating that expression of the mutated allele cannot be detected.
Fig. 4.

Arimoclomol increases GCase expression, lysosomal localization and activity of GCase in human neuronal-like GD cells.
a - b) Representative images of differentiated human neuronal like cells stained for expression of a) Tubulin-beta 3 and b) NeuN. The nucleus is visualized using DAPI. scale bars: 75 μM for Tubulin-beta 3 and 25 μM for NeuN. The quantifications of % positive cells derived from four GD patients with the indicated genotypes are shown to the right. c) GCase activity in human neuronal-like cells derived from healthy donors (WT/WT) or GD individuals with the indicated GBA mutations. Data are shown as mean + SEM, n = 2–4/cell line. The difference in GCase activity between WT and GD neuronal-like cells were analyzed by oneway-ANOVA and multiplicity was adjusted by Dunnet's method. d) Representative WB analysis of GCase levels in human neuronal-like cells derived from healthy donors (WT/WT) or GD individuals treated with vehicle or arimoclomol (400 μM). Beta-actin was used as loading control. e) EndoH-digestion assay of neuronal-like cells treated vehicle or 400 μM arimoclomol. A representative WB from one out of 2 experiments is shown. f) GCase activity in GD-derived human neuronal cells treated with vehicle or 400 μM arimoclomol for 9 (L444P/L444P, F213I/L444P, N370S/Y133*) or 11 (IVS2 + 1G/N188S) days. Data are shown as fold increase in arimoclomol-treated cells compared to vehicle-treated cells and represent the mean + SEM, n = 4–6. Statistical significance was assessed by unequal variance t-test.
The level of GCase activity was analyzed in the final stage of neuronal differentiation. The GD-derived neurons displayed severely diminished activity of GCase compared to healthy donor derived cells (Fig. 4c, Supplementary Table 2). In line with the results obtained in GD patient fibroblasts, low levels of GCase activity were associated with reduced levels of GCase protein in neuronally differentiated GD cells compared to healthy controls (Supplementary Fig. 5a).
Supplementary Fig. 5.
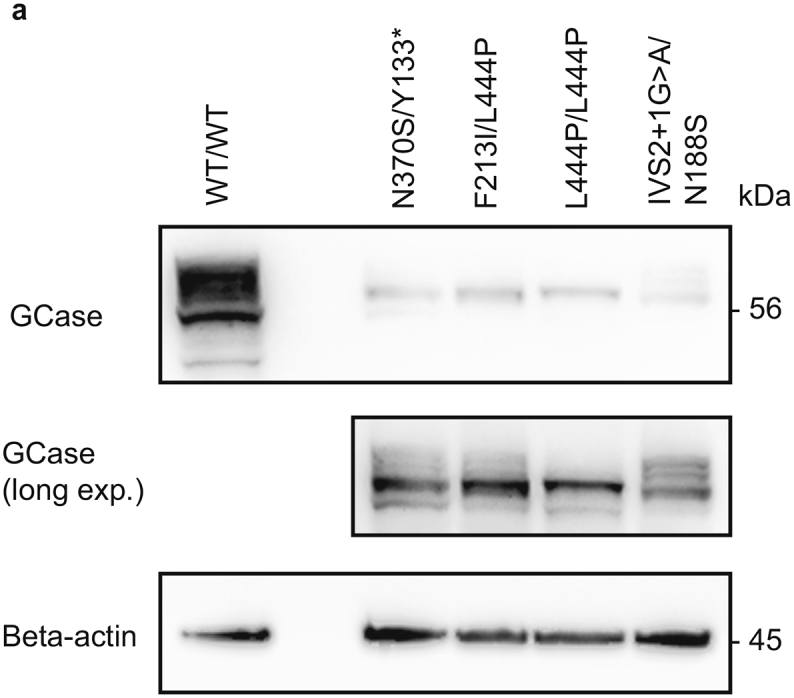
WB analysis of GCase levels in healthy and GD neuronal-like cells.
a) WB analysis of basal GCase protein levels in the final neuronal differentiation state of WT and GD-derived MASCs.
We proceeded to investigate the effect of arimoclomol on the level, maturation and activity of GCase in GD-patient derived neuronal cell cultures (Fig. 4d–f). Interestingly, EndoH treatment only had a modest effect on GCase in the WT neuronal cells which may suggest that the WT GCase in these cells is less subject to premature degradation than GCase in WT fibroblasts. Treatment with arimoclomol augmented both the level and EndoH resistant fraction of GCase in both neuronopathic and non-neuronopathic GD patient derived neuronal cells (Fig. 4d, e). In line with the WB and EndoH-assay results, treatment with arimoclomol also resulted in a corresponding significant increase in GCase activity in neuronal cells from both neuronopathic and non-neuronopathic GD patients (Fig. 4f).
4. Discussion
Our results demonstrate that arimoclomol is a heat shock protein amplifying small molecule that may be useful for the treatment of Gaucher disease including its neuronopathic forms which have no approved treatments available.
In these studies we have focused on ex vivo systems in order to address the fundamental concept of HSP-mediated refolding across the major genotypes of Gaucher disease and investigating the biological rationale for clinical development of arimoclomol for neuronopathic Gaucher disease.
We demonstrate that arimoclomol amplifies the production of disease mechanism-relevant molecular chaperones of the HSP70 family and improves mutant GCase maturation and function across major neuronopathic and non-neuronopathic genotypes in both human primary GD fibroblasts as well as in a neuronal cellular model of the disease.
As arimoclomol is a clinically enabled compound already in phase II/III clinical trials for Niemann-Pick disease type C, sporadic Inclusion Body Myositis and Amyotrophic Lateral Sclerosis (Clinicaltrials.gov identifiers NCT02612129, NCT02753530, and NCT03491462 respectively), the data reported herein provide preclinical proof-of-concept for the investigation of arimoclomol's therapeutic value in Gaucher disease.
While it is a known challenge to translate doses from in vitro studies to a human clinical setting, let alone across diseases, the arimoclomol doses used herein are congruent with the doses used for the preclinical studies in the sphingolipid storage disease Niemann-Pick type C (Kirkegaard et al., Science Transl. Med. 2016 and manuscript in prep.) which recently reported encouraging top-line results from a phase III clinical trial. Furthermore, the same type of in vitro studies has formed basis for the development of the recently approved drug migalastat, a small chemical chaperone developed for misfolded versions of the enzyme alpha-galactosidase A, in the sphingolipid storage disease Fabry disease (https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm616598.htm).
Arimoclomol amplifies the production of HSP70 family members which have been implicated in the proper folding and chaperoning of GCase as well as in maintenance of lysosomal integrity during cellular stress [[18], [19], [20],[22], [23], [24],28,29]. Studies of the induction of HSPs by small molecules in Gaucher Disease have until now made use of agents that provide proof-of-principle but due to their cytotoxic nature are not suited for development for chronic diseases [20,28,45]. These studies have however, revealed several HSP-dependent mechanisms regulating the processing of GCase. Studies in cellular models of GD have shown that HSP-induction by agents such as celastrol and MG-132 result in elevated GCase activity through increases in both cytosolic and ER-resident chaperones of the HSP70 family, through a process dependent on the UPR responsive transcription factors Ire1, ATF6 and PERK [28]. Our data strongly supports a role for HSP70 and HSPA5/BiP in the mode of action of arimoclomol and the processing and maturation of GCase, but we did not observe any transcriptional upregulaton of UPR responsive transcription factors (GM10915 cell line assessed at 48, 72 and 120 h exposure to 400 μM arimoclomol) and our attempts at knock-down of HSPA5/BiP did not result in sufficient downregulation to unequivocally resolve the potential role of individual HSP70 family members in these processes (data not shown). However, in line with the aforementioned studies and our data presented herein, additional reports have corroborated a role of the HSP70 system in GD cells, by showing that celastrol upregulates HSP70 and its associated cochaperone BCL2-associated athanogene 3 (BAG3), interrupting the HSP90-dependent degradation of GCase [23,24]. The role of HSP70 in the regulation of GCase activity has been further expanded by recent studies suggesting that HSP70 is recruited directly to GCase by progranulin (PGRN) acting as a critical cochaperone [21]. In addition, several other HSP-inducing agents such as HDAC-, proteasome- and HSP90 inhibitors as well as L-type Ca-channel blockers have demonstrated that manipulation of the HSP-systems can improve the folding and activity of mutant GCase [20,22,28,29,46]. Interestingly, parts of the mechanism of how HDAC inhibitors elicit their GCase rescuing effect might be ascribed to their role as HSP90 inhibitors, as the HDAC inhibitors vorinostat and LB-205 both bind to the middle domain of HSP90, resulting in less recognition of misfolded GCase and simultaneous upregulation of the HSP70 chaperones involved in refolding [29]. Studies of vorinostat in NPC has further expanded the knowledge on how HDAC inhibitors rescue misfolded lysosomal proteins through modulation of HSPs by a mechanism likely involving inhibition of HDAC1 [47]. This is supported by data demonstrating that HSF1, the major transcription factor for HSP70 and other HSPs, specifically interacts with HDAC1 and HDAC2 to regulate gene expression during heat shock [47,48]. It is worth noting that activation of the heat-shock response is associated with a transient downregulation of genes involved in cell cycle progression, translation and metabolism, and as such a part of the mechanism of action of HSP inducing drugs is expected to be cytostatic effects (Supplementary Fig. 2) [49,50].
Several of the small molecules that increases GCase activity in GD fibroblasts by preventing improper degradation of GCase, e.g. ambroxol, celastrol and MG132, have also been shown to transcriptionally increase GBA levels. GBA is a member of the CLEAR network (coordinated lysosomal expression and regulation) that consists of genes encoding the proteins required for lysosomal biogenesis and function [51] and ambroxol was shown to activate the CLEAR network in GD cells [52]. GBA is also a target gene of Heat Shock Factor 1 (HSF1), the master regulator of the heat shock response [49], which may be the underlying mechanism for the transcriptional upregulation of GBA seen with celastrol and MG132 [53]. We found that arimoclomol increased the expression of GBA in a time and dose dependent manner in L444P/L444P GD fibroblasts (Supplementary Fig. 3), thus suggesting that arimoclomol can increase GCase activity by stabilization of misfolded GCase, as well as by making more de novo synthesized GCase available for the stabilization, potentially through its fundamental HSF-1 activating property.
Supplementary Fig. 3.

Effect of arimoclomol on GBA mRNA levels.
a) Expression of GBA mRNA in GM10915 cells treated with arimoclomol for 48 h or 120 h. Each time point was measured in 2–3 independent experiments. Data are reported as mean + SEM. The effect of arimoclomol was evaluated against control at each time point by a two-way ANOVA and multiplicity was adjusted by Holm-Sidak's method (* < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001).
Arimoclomol holds particular promise for neuronopathic GD as it is a well-tolerated, CNS-penetrant molecule currently in a Phase II/III clinical trial for another neurodegenerative LSD, NPC [30,31] (Clinialtrials.gov identifier NCT02612129). Through its mechanism of increasing the expression of multiple HSPs, arimoclomol could potentially target GD and its CNS related events at other levels besides the HSP system's capacity to improve the folding, maturation and activity of GCase as reported herein. By amplifying cellular levels of HSP70, other HSP70 lysosome-specific cytoprotective activities such as improving the function of other sphingolipid degrading enzymes and protection against lysosomal destabilization may be achieved, as has been recently demonstrated for a number of related sphingolipid storage diseases [[18], [19], [20],54,55]. The reported capacity of HSP70 to protect against lysosomal membrane permeabilization and lysosomal cell death pathways may be particularly interesting aspects of HSP70 amplification in GD as the storage metabolite glucosylsphingosine has been shown to initiate lysosomal dysfunction and cell death [56]. We therefore also sought to analyze the levels of GCase substrate storage but this proved to be futile as we, in line with other reports, did not find any increased levels of GlcCer in the Gaucher disease fibroblasts and were technically unable to determine any amount of GlcSph(data not shown) [57]. As GlcSph is not only a storage metabolite in Gaucher Disease but also implicated in other sphingolipidoses such as Krabbe disease, it would be intriguing to explore the role of this metabolite and its response to arimoclomol further in other model systems. Not least so as this might be relevant for the reported pan-sphingolipidoses potential of HSP-based therapies [18].
Interestingly, it has recently been demonstrated that neuronal cell death in a model of GD [15] is necroptotic and involves the receptor-interacting protein Kinase-3 (RIPK3) pathway which is regulated by the HSP70 cochaperone CHIP (Carboxyl terminus of HSP70-interacting protein) [58]. Further, one of the hallmarks of necroptosis is lysosomal membrane permeabilization, a cellular event which HSP70 has been demonstrated to protect against in several disease models including sphingolipid storage diseases [3,18,19,54,[59], [60], [61], [62], [63], [64]].
Although much remains to be understood about the molecular mechanisms leading to GD and in particular its neurological manifestations, it is clear that the cytoprotective properties of the Heat shock proteins, in particular HSP70 and its cochaperones, converge with the pathogenesis of GD at several critical levels.
Neuronopathic Gaucher disease remains without any available treatment, but studies of residual activity in Gaucher disease patients indicate that the GCase activity that differentiates the manifestation of early onset neurological symptoms and hence non-neuronopathic and neuronopathic forms of the disease seems to be relatively small (Approx. 20% difference in residual activity) [65]. Although Gaucher disease is known to be heterogenic and the genotype/phenotype relationship is still not fully resolved, it seems reasonable to assume that the threshold for increases in residual GCase activity in the CNS that could translate to a clinically meaningful outcome for CNS symptoms would be of the same magnitude. This hypotheses is supported by the ongoing clinical trial of the substrate reduction therapy Venglustat/GZ-SAR 402671 for Gaucher disease type 3, in which the low residual enzyme activity levels of GCase is conceived to be adequate to resolve the storage accumulation, provided the substrate reduction is efficient enough (Clinicaltrials.gov ID: NCT02843035).
We observe significant increases in GCase activity with exposure to arimoclomol of only 5 days, but our studies in Gaucher disease fibroblasts also indicate that the longer the cells are exposed to arimoclomol, the more residual activity can be salvaged. This is an important consideration when attempting to translate these findings to a potential clinical setting. While equivalent increases where seen in the neuronal-like MASC cell system during shorter timelines (i.e. up to 9 days) it was not possible to maintain the neuronal like MASCs in their differentiated state for a longer time, precluding experiments of longer duration.
In summary, based on these observations and the data herein, we suggest that arimoclomol constitute a potential disease-modifying first-in-class compound for the treatment of Gaucher disease, in particular neuronopathic GD which is currently without efficacious treatment options.
The following are the supplementary data related to this article.
Characteristics of primary human fibroblasts
Characteristics of human MASCs
Immunophenotype of human MASCs at passage 3
List of qPCR primers and probes
Supplementary material
Acknowledgments
Acknowledgements
We thank Prof. Hermen S. Overkleeft for valuable scientific discussions.
We are also grateful for advice from Dr. Christine í Dali, Prof. Frances Platt and Mrs. Tanya Collin-Histed.
We also thank Mingshu Meng Eriksen, Anja Koustrup and Barbara Toffoletto for their technical assistance with regard to fibroblast experiments (MME & AK) and assistance in the immunophenotype characterization of MASCs (BT).
Funding sources
All work related to this manuscript was funded by Orphazyme A/S, Denmark.
Declarations of interests
TK, CKF, NHTP, CB, LMS, RM and AM are employees of Orphazyme A/S. TK is a Founder of Orphazyme A/S and hold shares in Orphazyme A/S. Orphazyme A/S funded this work. The authors have no additional financial interests.
Author contributions
TK conceived and supervised the study.
CKF, AD, CB, NHTP and TK designed the experiments with the contribution of PZ, EM, LMS, RM, AM, BB and JMFGA.
CKF, LMS, CB, RM and AM performed the experiments with human fibroblasts.
PZ, EM and PP performed the experiments with MASCs. EM and AD performed sequencing analysis.
CKF, NHTP, AD and TK wrote the manuscript. All authors approved the final manuscript.
References
- 1.Grabowski G.A. Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet. 2008 Oct;372(9645):1263–1271. doi: 10.1016/S0140-6736(08)61522-6. http://linkinghub.elsevier.com/retrieve/pii/S0140673608615226 Available from: [DOI] [PubMed] [Google Scholar]
- 2.Hruska K.S., Lamarca M.E., Scott C.R., Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567–583. doi: 10.1002/humu.20676. http://www.ncbi.nlm.nih.gov/pubmed/18338393 [cited 2015 Jun 2] Available from. [DOI] [PubMed] [Google Scholar]
- 3.Platt F.M. Sphingolipid lysosomal storage disorders. Nature. 2014 Jun;510(7503):68–75. doi: 10.1038/nature13476. 4 [cited 2014 Jun 4]. Available from: [DOI] [PubMed] [Google Scholar]
- 4.Grabowski G.A., Zimran A., Ida H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol. 2015 Jul 22;90(S12–8) doi: 10.1002/ajh.24063. http://www.ncbi.nlm.nih.gov/pubmed/26096741 [cited 2015 Oct 21]. Available from. [DOI] [PubMed] [Google Scholar]
- 5.Bembi B., Zambito Marsala S., Sidransky E., Ciana G., Carrozzi M., Zorzon M. Gaucher’s disease with Parkinson’s disease: clinical and pathological aspects. Neurol Int. 2003 Jul 8;61(1):99–101. doi: 10.1212/01.wnl.0000072482.70963.d7. http://www.ncbi.nlm.nih.gov/pubmed/12847165 [cited 2017 Aug 14] Available from. [DOI] [PubMed] [Google Scholar]
- 6.Rosenbloom B., Balwani M., Bronstein J.M., Kolodny E., Sathe S., Gwosdow A.R. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells, Mol Dis. 2011;46(1):95–102. doi: 10.1016/j.bcmd.2010.10.006. http://linkinghub.elsevier.com/retrieve/pii/S1079979610002470 Jan [cited 2017 Aug 14]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben Chetrit E., Alcalay R.N., Steiner-Birmanns B., Altarescu G., Phillips M., Elstein D. Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells Mol Dis. 2013;50(3):218–221. doi: 10.1016/j.bcmd.2012.11.011. http://linkinghub.elsevier.com/retrieve/pii/S1079979612002306 Mar [cited 2017 Aug 14]. Available from: [DOI] [PubMed] [Google Scholar]
- 8.Chérin P., Rose C., de Roux-Serratrice C., Tardy D., Dobbelaere D., Grosbois B. The neurological manifestations of Gaucher disease type 1: the French Observatoire on Gaucher disease (FROG) J Inherit Metab Dis. 2010 Aug 2;33(4):331–338. doi: 10.1007/s10545-010-9095-5. http://www.ncbi.nlm.nih.gov/pubmed/20532983 [cited 2017 Aug 23]. Available from. [DOI] [PubMed] [Google Scholar]
- 9.Devigili G., De Filippo M., Ciana G., Dardis A., Lettieri C., Rinaldo S. Chronic pain in Gaucher disease: skeletal or neuropathic origin? Orphanet J Rare Dis. 2017 Dec 31;12(1):148. doi: 10.1186/s13023-017-0700-7. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiss K., Gonzalez A.N., Lopez G., Pedoeim L., Groden C., Sidransky E. The clinical management of type 2 Gaucher disease. Mol Genet Metab. 2015 Feb;114(2):110–122. doi: 10.1016/j.ymgme.2014.11.008. http://www.ncbi.nlm.nih.gov/pubmed/25435509 [cited 2017 Aug 14]. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tylki-Szymańska A., Vellodi A., El-Beshlawy A., Cole J.A., Kolodny E. Neuronopathic Gaucher disease: demographic and clinical features of 131 patients enrolled in the International Collaborative Gaucher Group Neurological Outcomes Subregistry. J Inherit Metab Dis. 2010 Jan 19;33(4):339–346. doi: 10.1007/s10545-009-9009-6. http://www.ncbi.nlm.nih.gov/pubmed/20084461 [cited 2015 Nov 19]. Available from. [DOI] [PubMed] [Google Scholar]
- 12.Bendikov-Bar I., Ron I., Filocamo M., Horowitz M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol Dis. 2011 Jan 15;46(1):4–10. doi: 10.1016/j.bcmd.2010.10.012. http://www.ncbi.nlm.nih.gov/pubmed/21106416 [cited 2015 Aug 28]. Available from. [DOI] [PubMed] [Google Scholar]
- 13.Maor G., Rencus-Lazar S., Filocamo M., Steller H., Segal D., Horowitz M. Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet J Rare Dis. 2013;8(140) doi: 10.1186/1750-1172-8-140. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3819655&tool=pmcentrez&rendertype=abstract Jan [cited 2015 Dec 15]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Platt N., Speak A.O., Colaco A., Gray J., Smith D.A., Williams I.M. Immune dysfunction in Niemann-Pick disease type C. J Neurochem. 2016 Jan;136(Suppl):74–80. doi: 10.1111/jnc.13138. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vitner E.B., Salomon R., Farfel-Becker T., Meshcheriakova A., Ali M., Klein A.D. RIPK3 as a potential therapeutic target for Gaucher's disease. Nat Med. 2014;20(2):204–208. doi: 10.1038/nm.3449. Available from: [DOI] [PubMed] [Google Scholar]
- 16.Daugaard M., Rohde M., Jäättelä M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007 Jul 31;581(19):3702–3710. doi: 10.1016/j.febslet.2007.05.039. http://www.ncbi.nlm.nih.gov/pubmed/17544402 [cited 2013 Nov 12] Available from. [DOI] [PubMed] [Google Scholar]
- 17.Wang F., Agnello G., Sotolongo N., Segatori L. Ca2+ homeostasis modulation enhances the amenability of L444P glucosylcerebrosidase to proteostasis regulation in patient-derived fibroblasts. ACS Chem Biol. 2011 Feb 18;6(2):158–168. doi: 10.1021/cb100321m. http://www.ncbi.nlm.nih.gov/pubmed/21043486 [cited 2016 Jan 15]. Available from. [DOI] [PubMed] [Google Scholar]
- 18.Kirkegaard T., Gray J., Priestman D.A., Wallom K.-L., Atkins J., Olsen O.D. Heat shock protein-based therapy as a potential candidate for treating the sphingolipidoses. Sci Transl Med. 2016;8(355) doi: 10.1126/scitranslmed.aad9823. 355ra118-355ra118. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirkegaard T., Roth A.G., Petersen N.H.T., Mahalka A.K., Olsen O.D., Moilanen I. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010 Jan 28;463(7280):549–553. doi: 10.1038/nature08710. http://www.nature.com/nature/journal/v463/n7280/abs/nature08710.html [cited 2014 May 9]. Available from: [DOI] [PubMed] [Google Scholar]
- 20.Ingemann L., Kirkegaard T. Lysosomal storage diseases and the heat shock response: convergences and therapeutic opportunities. J Lipid Res. 2014 Nov;55(11):2198–2210. doi: 10.1194/jlr.R048090. http://www.ncbi.nlm.nih.gov/pubmed/24837749 [cited 2014 Dec 29] Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jian J., Tian Q.-Y., Hettinghouse A., Zhao S., Liu H., Wei J. Progranulin recruits HSP70 to β-glucocerebrosidase and is therapeutic against gaucher disease. EBioMedicine. 2016;13:212–224. doi: 10.1016/j.ebiom.2016.10.010. http://linkinghub.elsevier.com/retrieve/pii/S2352396416304650 Oct [cited 2016 Nov 4]. Available from: ISSN: 2352-3964; PMID: 27789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu J., Yang C., Chen M., Ye D.Y., Lonser R.R., Brady R.O. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc Natl Acad Sci U S A. 2011 Dec 27;108(52):21200–21205. doi: 10.1073/pnas.1119181109. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3248545&tool=pmcentrez&rendertype=abstract [cited 2013 Oct 30]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C., Swallows C.L., Zhang C., Lu J., Xiao H., Brady R.O. Celastrol increases glucocerebrosidase activity in Gaucher disease by modulating molecular chaperones. Proc Natl Acad Sci U S A. 2014 Jan 7;111(1):249–254. doi: 10.1073/pnas.1321341111. http://www.ncbi.nlm.nih.gov/pubmed/24351928 [cited 2014 Jan 9] Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang C., Wang H., Zhu D., Hong C.S., Dmitriev P., Zhang C. Mutant glucocerebrosidase in Gaucher disease recruits Hsp27 to the Hsp90 chaperone complex for proteasomal degradation. Proc Natl Acad Sci U S A. 2015 Jan 27;112(4):1137–1142. doi: 10.1073/pnas.1424288112. http://www.pnas.org/content/112/4/1137.long [cited 2015 Jul 28]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khanna R., Benjamin E.R., Pellegrino L., Schilling A., Rigat B.A., Soska R. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of β-glucosidase. FEBS J. 2010 Apr 10;277(7):1618–1638. doi: 10.1111/j.1742-4658.2010.07588.x. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2874831&tool=pmcentrez&rendertype=abstract [cited 2015 Oct 7]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y., Ran H., Liou B., Quinn B., Zamzow M., Zhang W. Isofagomine in vivo effects in a neuronopathic Gaucher disease mouse. PLoS One. 2011 Jan;6(4):e19037. doi: 10.1371/journal.pone.0019037. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3080394&tool=pmcentrez&rendertype=abstract [cited 2015 Oct 26]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maegawa G.H.B., Tropak M.B., Buttner J.D., Rigat B.A., Fuller M., Pandit D. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem. 2009 Aug 28;284(35):23502–23516. doi: 10.1074/jbc.M109.012393. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2749124&tool=pmcentrez&rendertype=abstract [cited 2015 Aug 28]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mu T., Ong D.S.T., Wang Y., Balch W.E., Yates J.R., Segatori L. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008 Sep 5;134(5):769–781. doi: 10.1016/j.cell.2008.06.037. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2650088&tool=pmcentrez&rendertype=abstract [cited 2014 May 9]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang C., Rahimpour S., Lu J., Pacak K., Ikejiri B., Brady R.O. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc Natl Acad Sci U S A. 2013 Jan 15;110(3):966–971. doi: 10.1073/pnas.1221046110. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3549125&tool=pmcentrez&rendertype=abstract [cited 2013 Oct 30]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cudkowicz M.E., Shefner J.M., Simpson E., Grasso D., Yu H., Zhang H. Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve. 2008;38(1):837–844. doi: 10.1002/mus.21059. http://www.ncbi.nlm.nih.gov/pubmed/18551622 Available from. [DOI] [PubMed] [Google Scholar]
- 31.Lanka V, Wieland S, Barber J, Cudkowicz M. Arimoclomol: a potential therapy under development for ALS. Expert Opin Investig Drugs. 2009 Dec;18(12):1907–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19938902 [DOI] [PubMed]
- 32.Ahmed M, Machado PM, Miller A, Spicer C, Herbelin L, He J, et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med. 2016 Mar 23;8(331):331ra41. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27009270 [DOI] [PMC free article] [PubMed]
- 33.Bergamin N., Dardis A., Beltrami A., Cesselli D., Rigo S., Zampieri S. A human neuronal model of Niemann Pick C disease developed from stem cells isolated from patient's skin. Orphanet J Rare Dis. 2013 Jan;8(1):34. doi: 10.1186/1750-1172-8-34. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3648447&tool=pmcentrez&rendertype=abstract [cited 2015 Oct 5]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alcalay R.N., Levy O.A., Waters C.C., Fahn S., Ford B., Kuo S.H. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. 2015;138(9):2648–2658. doi: 10.1093/brain/awv179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mallett V., Ross J.P., Alcalay R.N., Ambalavanan A., Sidransky E., Dion P.A. GBA p.T369M substitution in Parkinson disease: Polymorphism or association? A meta-analysis. Neurol Genet. 2016 Oct 8;2(5):e104. doi: 10.1212/NXG.0000000000000104. http://www.ncbi.nlm.nih.gov/pubmed/27648471 [cited 2017 Jul 27]. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beutler E., Gelbart T., Kuhl W., Sorge J., West C. Identification of the second common Jewish Gaucher disease mutation makes possible population-based screening for the heterozygous state. Proc Natl Acad Sci U S A. 1991 Dec 1;88(23):10544–10547. doi: 10.1073/pnas.88.23.10544. http://www.ncbi.nlm.nih.gov/pubmed/1961718 [cited 2017 Jul 27]. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vígh L., Literáti P.N., Horváth I., Török Z., Balogh G. Glatz a, et al. Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med. 1997;3(10):1150–1154. doi: 10.1038/nm1097-1150. http://www.nature.com/nm/journal/v3/n10/abs/nm1097-1150.html Available from: [DOI] [PubMed] [Google Scholar]
- 38.Kalmar B., Edet-Amana E., Greensmith L. Treatment with a coinducer of the heat shock response delays muscle denervation in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13(4):378–392. doi: 10.3109/17482968.2012.660953. [DOI] [PubMed] [Google Scholar]
- 39.Maley F., Trimble R.B., Tarentino A.L., Plummer T.H. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 1989 Aug 1;180(2):195–204. doi: 10.1016/0003-2697(89)90115-2. http://www.ncbi.nlm.nih.gov/pubmed/2510544 [cited 2016 Jan 18]. Available from. [DOI] [PubMed] [Google Scholar]
- 40.Bergmann J.E., Grabowski G.A. Posttranslational processing of human lysosomal acid beta-glucosidase: a continuum of defects in Gaucher disease type 1 and type 2 fibroblasts. Am J Hum Genet. 1989 May;44(5):741–750. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1715631&tool=pmcentrez&rendertype=abstract [cited 2015 Aug 26]. Available from: [PMC free article] [PubMed] [Google Scholar]
- 41.Reczek D., Schwake M., Schröder J., Hughes H., Blanz J., Jin X. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell. 2007;131(4):770–783. doi: 10.1016/j.cell.2007.10.018. http://www.ncbi.nlm.nih.gov/pubmed/18022370 Available from. [DOI] [PubMed] [Google Scholar]
- 42.Sawkar A.R., Kelly J.W. Chemical Chaperones and Permissive Temperatures Alter the Cellular Localization of Gaucher Disease Associated Glucocerebrosidase Variants. ACS Chem Biol. 2006;1(4):235–251. doi: 10.1021/cb600187q. [DOI] [PubMed] [Google Scholar]
- 43.Van Patten S.M., Hughes H., Huff M.R., Piepenhagen P.A., Waire J., Qiu H. Effect of mannose chain length on targeting of glucocerebrosidase for enzyme replacement therapy of Gaucher disease. Glycobiology. 2007 May 1;17(5):467–478. doi: 10.1093/glycob/cwm008. http://www.ncbi.nlm.nih.gov/pubmed/17251309 [cited 2018 Oct 10]. Available from. [DOI] [PubMed] [Google Scholar]
- 44.Witte M.D., Kallemeijn W.W., Aten J., Li K.-Y., Strijland A., Donker-Koopman W.E. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat Chem Biol. 2010 Dec;6(12):907–913. doi: 10.1038/nchembio.466. http://www.ncbi.nlm.nih.gov/pubmed/21079602 [cited 2014 Dec 24]. Available from. [DOI] [PubMed] [Google Scholar]
- 45.Kirkegaard T. Emerging therapies and therapeutic concepts for lysosomal storage diseases. Expert Opin Orphan Drugs. 2013 May 24;1(5):385–404. doi: 10.1517/21678707.2013.780970?prevSearch=allfield%3A%28thomas+kirkegaard%29&searchHistoryKey. [cited 2013 Oct 21]. Available from: [DOI] [Google Scholar]
- 46.Wang F., Segatori L. Remodeling the proteostasis network to rescue glucocerebrosidase variants by inhibiting ER-associated degradation and enhancing ER folding. PLoS One. 2013 Jan;8(4):e61418. doi: 10.1371/journal.pone.0061418. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3631227&tool=pmcentrez&rendertype=abstract [cited 2013 Nov 12]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pipalia N.H., Cosner C.C., Huang A., Chatterjee A., Bourbon P., Farley N. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc Natl Acad Sci U S A. 2011 Apr 5;108(14):5620–5625. doi: 10.1073/pnas.1014890108. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3078401&tool=pmcentrez&rendertype=abstract [cited 2013 Nov 12]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fritah S., Col E., Boyault C., Govin J., Sadoul K., Chiocca S. Heat-shock factor 1 controls genome-wide acetylation in heat-shocked cells. Mol Biol Cell. 2009 Dec;20(23):4976–4984. doi: 10.1091/mbc.E09-04-0295. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2785740&tool=pmcentrez&rendertype=abstract [cited 2014 May 9]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vihervaara A., Sergelius C., Vasara J., Blom M.A.H., Elsing A.N., Roos-Mattjus P. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci U S A. 2013 Sep 3;110(36):E3388–E3397. doi: 10.1073/pnas.1305275110. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3767495&tool=pmcentrez&rendertype=abstract [cited 2016 Aug 25]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahat D.B., Salamanca H.H., Duarte F.M., Danko C.G., Lis J.T. Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol Cell. 2016 Apr 7;62(1):63–78. doi: 10.1016/j.molcel.2016.02.025. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011 Oct 1;20(19):3852–3866. doi: 10.1093/hmg/ddr306. http://www.ncbi.nlm.nih.gov/pubmed/21752829 [cited 2012 Nov 19]. Available from. [DOI] [PubMed] [Google Scholar]
- 52.McNeill A., Magalhaes J., Shen C., Chau K.-Y., Hughes D., Mehta A. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain. 2014;137(5):1481–1495. doi: 10.1093/brain/awu020. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang F., Chou A., Segatori L. Lacidipine remodels protein folding and Ca2+ homeostasis in Gaucher's disease fibroblasts: a mechanism to rescue mutant glucocerebrosidase. Chem Biol. 2011;18(6):766–776. doi: 10.1016/j.chembiol.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 54.Zhu H, Yoshimoto T, Yamashima T. Heat shock protein 70.1 (Hsp70.1) affects neuronal cell fate by regulating lysosomal acid sphingomyelinase. J Biol Chem. 2014 Jul 29;1. [cited 2014 Aug 4] Available from: http://www.ncbi.nlm.nih.gov/pubmed/25074941 [DOI] [PMC free article] [PubMed]
- 55.Nakasone N., Nakamura Y.S., Higaki K., Oumi N., Ohno K., Ninomiya H. Endoplasmic reticulum-associated degradation of niemann-pick C1: Evidence for the role of heat shock proteins and identification of lysine residues that accept ubiquitin. J Biol Chem. 2014 Jun 2 doi: 10.1074/jbc.M114.549915. [cited 2014 Jun 3]; Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Folts C.J., Scott-Hewitt N., Pröschel C., Mayer-Pröschel M., Noble M. Lysosomal re-acidification prevents lysosphingolipid-induced lysosomal impairment and cellular toxicity. PLOS Biol. 2016;14(12):e1002583. doi: 10.1371/journal.pbio.1002583. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun Y., Florer J., Mayhew C.N., Jia Z., Zhao Z., Xu K. Properties of neurons derived from induced pluripotent stem cells of Gaucher disease type 2 patient fibroblasts: potential role in neuropathology. PLoS One. 2015 Jan;10(3):e0118771. doi: 10.1371/journal.pone.0118771. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4378893&tool=pmcentrez&rendertype=abstract [cited 2015 Oct 30]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seo J., Lee E.-W., Sung H., Seong D., Dondelinger Y., Shin J. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat Cell Biol. 2016 Mar 22;18(3):291–302. doi: 10.1038/ncb3314. http://www.nature.com/articles/ncb3314 Available from: [DOI] [PubMed] [Google Scholar]
- 59.Nylandsted J., Gyrd-Hansen M., Danielewicz A., Fehrenbacher N., Lademann U., Høyer-Hansen M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200(4):425–435. doi: 10.1084/jem.20040531. http://www.ncbi.nlm.nih.gov/pubmed/15314073 Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bivik C., Rosdahl I., Ollinger K. Hsp70 protects against UVB induced apoptosis by preventing release of cathepsins and cytochrome c in human melanocytes. Carcinogenesis. 2007 Mar;28(3):537–544. doi: 10.1093/carcin/bgl152. http://www.ncbi.nlm.nih.gov/pubmed/16950797 [cited 2013 Oct 24]. Available from. [DOI] [PubMed] [Google Scholar]
- 61.Jaattela M., Wissing D., Bauer P.A., Li G.C. Major heat shock protein hsp70 protects tumor cells from tumor necrosis factor cytotoxicity. EMBO J. 1992;11(10):3507–3512. doi: 10.1002/j.1460-2075.1992.tb05433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gyrd-Hansen M., Farkas T., Fehrenbacher N., Bastholm L., Høyer-Hansen M., Elling F. Apoptosome-independent activation of the lysosomal cell death pathway by caspase-9. Mol Cell Biol. 2006 Nov;26(21):7880–7891. doi: 10.1128/MCB.00716-06. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1636747&tool=pmcentrez&rendertype=abstract [cited 2014 Jan 24]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hwang J.-H., Ryu J.K., Yoon Y.B., Lee K.H., Park Y.-S., Kim J.-W. Spontaneous activation of pancreas trypsinogen in heat shock protein 70.1 knock-out mice. Pancreas. 2005 Nov;31(4):332–336. doi: 10.1097/01.mpa.0000183377.04295.c3. http://www.ncbi.nlm.nih.gov/pubmed/16258366 [cited 2013 Oct 24]. Available from. [DOI] [PubMed] [Google Scholar]
- 64.Kirkegaard T, Jäättelä M. Lysosomal involvement in cell death and cancer. Biochim Biophys Acta. 2009 Apr;1793(4):746–54. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18948147 [DOI] [PubMed]
- 65.Whitfield P.D., Nelson P., Sharp P.C., Bindloss C.A., Dean C., Ravenscroft E.M. Correlation among genotype, phenotype, and biochemical markers in Gaucher disease: implications for the prediction of disease severity. Mol Genet Metab. 2002;75(1):46–55. doi: 10.1006/mgme.2001.3269. http://ac.els-cdn.com/S109671920193269X/1-s2.0-S109671920193269X-main.pdf?_tid=b86fb442-24f0-11e3-8ce9-00000aacb35d&acdnat=1380010385_9b4421a621be14609912f7d700944324%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/11825063 Available from: [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characteristics of primary human fibroblasts
Characteristics of human MASCs
Immunophenotype of human MASCs at passage 3
List of qPCR primers and probes
Supplementary material