

Abstract
Microtubule-associated protein tau (MAPT) hyperphosphorylation and aggregation, are two hallmarks of a family of neurodegenerative disorders collectively referred to as tauopathies. In many tauopathies, including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP) and Pick’s disease, tau aggregates are found associated with highly sulfated polysaccharides known as heparan sulfates (HSs). In AD, amyloid beta (Aβ) peptide aggregates associated with HS are also characteristic of disease. Heparin, an HS analog, promotes misfolding, hyperphosphorylation and aggregation of tau protein in vitro. HS also provides cell surface receptors for attachment and uptake of tau seeds, enabling their propagation. These findings point to HS-tau interactions as potential therapeutic targets in tauopathies. The zebrafish genome contains genes paralogous to MAPT, genes orthologous to HS biosynthetic and chain modifier enzymes, and other genes implicated in AD. The nervous system in the zebrafish bears anatomical and chemical similarities to that in humans. These homologies, together with numerous technical advantages, make zebrafish a valuable model for investigating basic mechanisms in tauopathies and identifying therapeutic targets. Here, we comprehensively review current knowledge on the role of HSs in tau pathology and HS-targeting therapeutic approaches. We also discuss novel insights from zebrafish suggesting a role for HS 3-O-sulfated motifs in tau pathology and establishing HS antagonists as potential preventive agents or therapies for tauopathies.
Keywords: heparan sulfate, glycosaminoglycans, tauopathy, tau, Alzheimer’s disease, zebrafish, drug discovery
Heparan Sulfate Proteoglycans
Structure and Biosynthesis
Glycosaminoglycans (GAGs), formerly called mucopolysaccharides, are a major class of anionic polysaccharides, consisting of unbranched and often long polysaccharide chains made of disaccharide units. Most GAGs are bound to core proteins, forming proteoglycans (PGs), which are important components of extracellular matrices. All extracellular matrices contain PGs, but these glycoproteins are also found in membrane-bound secretory granules and in cell nuclei. PGs are evolutionarily ancient and are found in all Bilateria species (organisms with left-right symmetry). So far, five structurally different GAG species have been described: heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS) and hyaluronan or hyaluronic acid (HA). While heparin, a highly sulfated HS, is mainly produced by connective tissue-type mast cells and bipotential glial progenitor cells, HS is synthesized by all cell types (Couchman and Pataki, 2012; Lindahl et al., 2017). In the zebrafish (Danio rerio), HS is synthetized in developing embryos, larvae and adults (Zhang et al., 2009).
Glycosaminoglycans synthesis starts in the endoplasmic reticulum (ER), with the formation of a tetrasaccharide linker (Xyl-Gal-Gal-GlcA) bound to the core protein via a serine residue. During linker formation, xylose (Xyl) is first attached by a xylosyltransferase (XT); galactosyl-transferases I and II (GT I and II) then transfer two galactoses (Gal) from UDP-Gal to xylose, and finally, glucoronic acid (GlcA) is transferred from UDP-GlcA by glucuronosyl-transferase I (GlcAT). After the formation of the linker, GAG biosynthesis takes different routes for CS/DS or heparin/HS synthesis. Synthesis of CS/DS chains first involves the transfer of N-acetylgalactosamine (GalNAc) by GalNAc transferase to the terminal GlcA, while that of heparin/HS chains depends on the transfer of N-acetylglucosamine (GlcNAc) by GlcNAc transferase. The GAG chains are then elongated by sequential addition of repeated disaccharide units composed of an amino sugar, [N-acetylgalactosamine (GalNAc) or N-acetylgalactosamine (GalNAc)], and a uronic acid [glucoronic acid (GlcA) or iduronic acid (IdoA)]. In HS and heparin, the repeated disaccharide unit comprises N-acetylglucosamine and glucoronic acid residues. EXT enzymes mediate HS/heparin chain elongation (Kreuger and Kjellén, 2012).
After their synthesis, GAG chains are modified in the Golgi apparatus by different enzymes. All HS chain modifier enzymes have been identified in zebrafish (Bink et al., 2003; Chen et al., 2005; Ghiselli and Farber, 2005; Cadwallader and Yost, 2006a,b, 2007; Filipek-Górniok et al., 2015). Major modifications on the HS chains are sulfations, which are mediated by two classes of sulfotransferases in a hierarchical manner. The expression patterns of these enzymes are characterized in zebrafish through early developmental stages (0–48 hpf). First, the N-deacetylase/N-sulfotransferases (NDSTs) replace the acetyl group in GlcNAc by a sulfate group. Five NDST genes (ndst1a, ndst1b, ndst2a, ndst2b, and ndst3) have been identified in zebrafish. All isoforms are expressed in various brain regions during early zebrafish development, while a distinct isoform (ndst3) is expressed in the spinal cord (Filipek-Górniok et al., 2015). HS chains can be further modified through epimerization of GlcA into iduronic acid (IdoA) by C-5 epimerase. The zebrafish C-5 epimerases Glce-A and Glce-B are expressed in brain through early embryonic development (Cadwallader and Yost, 2007).
The second class of sulfotransferases comprises the HS O-sulfotransferases (OSTs) 2-OST, 6-OST, and 3-OST. Zebrafish 2-OST or HS2ST is expressed in all brain regions in early developmental stages. Of the four zebrafish 6-OSTs or HS6STs (6-OST-1a, 6-OST-1b, 6-OST-2, and 6-OST-3) all are expressed in the brain in early development, while only 6-OST-1a and 6-OST-1b isoforms are expressed in the spinal cord (Cadwallader and Yost, 2006b). Comparison of HS structure between adult zebrafish and porcine intestine revealed similar global structures. Differences were documented as higher 2-O-sulfated iduronic acid (IdoA2S) content and lower levels of GlcA (Zhang et al., 2009).
It has been suggested that the evolutionary origin of the Hs3sts goes back to a common ancestor of bilaterians and cnidarians in the early eumetazoan evolution. Trichoplax, a placozoan identified as a sister clade to bilaterians and cnidarians, has no Hs3st homologs, but expresses all the other HS sulfotransferases. Invertebrates express fewer Hs3st isozymes than vertebrates. Hydra expresses one Hs3st, whereas Nematostella (sea anemone), belonging to the Cnidarian phylum, expresses two Hs3sts. The Hs3st enzymes in Hydra and Nematostella share near 50% amino acid sequence identity with human Hs3sts. Strongylocentrotus (sea urchin) and Planaria (flatworm) express a single Hs3st, whereas Drosophila (fruit fly) and Caenorhabditis (nematode) have two Hs3st. Vertebrates express a greater number of Hs3sts. Homo and other mammals express 7 Hs3sts, while Danio (zebrafish) expresses 8 (Thacker et al., 2014). 3-O-Sulfated disaccharides and tetrasaccharides have been identified in HS from zebrafish embryos, which were diminished by morpholinos directed against specific 3-ost genes. During early cleavage stages in the zebrafish, 3-OST enzymes are encoded by maternally deposited mRNAs, which are evenly distributed throughout the embryos. By contrast, during later stages, and particularly in the developing brain, the zebrafish Hs3st genes display a complex combinatorial expression profile with each gene showing a unique and cell-type specific expression pattern. The 3ost1 transcripts are ubiquitously accumulated to high levels during early cleavage stages prior to becoming restricted to head and anterior somites by 24 hpf. In 48 hpf embryos, 3ost1 expression is restricted to head, gut and pectoral fin. Expression of the 3ost2 gene is detected from 24 hpf onward in the brain and otic vesicle. At 48 hpf, 3ost2 is expressed in all major brain areas, as well as in the olfactory region. Zebrafish 3ost3X and -3Z are the orthologs of the mammalian 3-OST-3a and -3b genes, respectively. 3ost3X transcripts are evenly distributed during early cleavage stages and are restricted to hindbrain and spinal cord from 36 hpf onward. By contrast, while expression of the 3ost3Z gene is not detected during early cleavage stages, by 48 hpf it is transcribed in telencephalon, tectal regions, hindbrain, and spinal cord. 3ost4 RNAs are maternally expressed and ubiquitously accumulated in early cleavage stage embryos, before being specifically transcribed in midbrain, hindbrain, olfactory epithelium and otic vesicle by 48 hpf onward. Similarly, ubiquitous accumulation of maternal 3ost5 transcripts is detected in early cleavage and embryonic stages, while from mid-somitogenesis onward, these RNAs are specifically transcribed in forebrain, midbrain and spinal cord neurons. Distribution of 3ost6 transcripts is detected during early cleavage stages, while from 36 hpf onward these RNAs are expressed in the hindbrain. Finally, the 3ost7 gene is expressed during early cleavage stages and becomes restricted to brain neurons by 48 hpf (Cadwallader and Yost, 2006a). The zebrafish 3ost2 and 3ost3 generate receptors for herpes simplex virus 1 (HSV1) entry, highlighting the functionality and similarity of zebrafish neuronal 3-O-sulfated motifs to those found in humans (Antoine et al., 2014).
Heparan sulfate PGs (HSPGs) comprise one or more HS-sulfated GAG chains covalently linked to a core protein. 17 distinct types of HSPGs have been identified, which are classified into three groups based on their locations: (i) syndecans and glypicans (membrane HSPGs), (ii) agrin, perlecan, and type-XVIII collagen (secreted extracellular matrix HSPGs), and (iii) serglycin (secretory vesicle HSPG) (Lindahl et al., 2017). Agrin also occurs as a transmembrane proteoglycan resulting from alternative splicing (Burgess et al., 2000; Neumann et al., 2001). The structure and function of zebrafish syndecan-4, required for neural crest migration, closely resembles that found in higher vertebrates (Whiteford et al., 2008). Using brains of BACE-1 deficient zebrafish, glypican-1 is identified as a substrate for the enzyme. BACE-1 is implicated in pathogenesis of AD by contributing to generation of Aβ peptide (Hogl et al., 2013).
Heparan sulfate chains of HSPGs can also be modified post-assembly by several enzymes. In particular, the shortening and degradation of HS chains involve heparanase (HPSE), and the removal of sulfate motifs by SULFs (a family of plasma membrane endosulfatases) entails finer modifications. A single gene orthologous to human heparanase is found in zebrafish (Wei and Liu, 2014). Orthologs to mammalian SULFs have been identified in zebrafish; they consist of three genes, all expressed in the central nervous system (CNS) (Gorsi et al., 2010). PG degradation is mediated by their internalization, which induces endolytic cleavage of the core protein by proteases and degradation of heparan sulfate chains by heparanase and endo-β-glucuronidase. Complete degradation of the remaining small HS chains takes place in lysosomes and involves a series of exoglycosidases and sulfatases (Lamanna et al., 2008; Lindahl et al., 2017). The ortholog of the human iduronate-2-sulfatase in zebrafish, whose defects are responsible for mucopolysaccharidosis type II (MPS II), has been identified (Moro et al., 2010). The identification of HSs in zebrafish, together with an array of orthologs for HS biosynthetic and chain modifier enzymes in the nervous system, makes zebrafish a model organism well-suited for the study of HS contribution to tauopathies.
HS-Protein Interactions
Heparan sulfates can bind non-covalently to a variety of proteins. An array of heparin-binding proteins have been identified, many of which interact with HSs under physiological conditions and modulate their biological activities (Bishop et al., 2007; Ori, 2008; Xu and Esko, 2014). Protein-HS interactions promote protein presentation, protection and stabilization, as well as conformational changes and oligomerization (Thacker et al., 2014). The conformation of GAG chains and the negative charges provided by the sulfate groups and uronic acid epimers constitute the binding site for positively charged amino acids of heparin-binding proteins (Lindahl and Li, 2009; Sarrazin et al., 2011). The ligand-binding sites of HSPGs rely on the orientation of carboxyl groups and organization of sulfate groups (Sarrazin et al., 2011). In particular, the sulfation pattern of HSPGs, which is more diverse than other GAG chain modifications, plays an essential role in the specificity of the binding sites of HSPGs to heparin-binding proteins (Aviezer et al., 1994; Sanderson et al., 1994; Herndon et al., 1999). Moreover, it has been shown that different patterns of sulfation in HSPGs with similar core proteins lend them different binding specificities (Kato et al., 1994; Sanderson et al., 1994). Hence the concerted action of enzymes confers highly variable patterns of sulfation to HS chains, thus driving their complexity and micro-heterogeneity (Shriver et al., 2012; Thacker et al., 2014). Importantly, data suggest that GAG chain sulfation does not go to completion on sulfated polysaccharide chains, yielding alternate highly sulfated domains (NS domains) and regions showing low or no sulfation (NA domains) (Sarrazin et al., 2011).
The sulfation pattern of most HSPGs changes during development, thus modifying their binding specificities (Nurcombe et al., 1993; Brickman et al., 1998; Yabe et al., 2005), a process playing a critical role in nervous system development (Irie et al., 2002; Bülow and Hobert, 2004). HS modifications dynamically evolve during zebrafish development (Cadwallader and Yost, 2006a,b, 2007; Zhang et al., 2009). Studies in zebrafish have demonstrated a critical requirement for HS in axon pathfinding during development (Lee et al., 2004; Kim et al., 2007; Kastenhuber et al., 2009; Wang et al., 2012; Poulain and Chien, 2013). Chemical disruption of HS and CS biosynthesis in early zebrafish development leads to brain disorganization (Beahm et al., 2014). However, the specific binding motifs for many heparin-binding proteins remain to be defined (Thacker et al., 2014). Full knowledge of HS-protein interactions has important therapeutic implications. Surfen, a non-specific heparan sulfate antagonist interferes with numerous biological processes, showing potential as a therapeutic agent. The properties of surfen ranges from anti-inflammatory (Warford et al., 2018) and immuomodulatory (Warford et al., 2014) to suppression of stem cell differentiation (Huang et al., 2018), inhibition of HIV type 1 infection (Roan et al., 2010) and rescue of tauopathy in a zebrafish model (Alavi Naini et al., 2018). However, non-specificity limits the beneficial effects of surfen. Surfen is reported to decrease neuroinflammation through HSPGs while increasing both the beneficial and harmful CS proteoglycans involved in remyelination (Warford et al., 2018). Thus complete knowledge of the PG interactome promotes development of specific therapies targeting harmful intractions and/or enhancing beneficial ones.
Tau and Tauopathies
Tauopathies are neurodegenerative disorders characterized by microtubule-associated protein tau (MAPT) hyperphosphorylation and aggregation into paired helical filaments (PHFs) or straight filaments (SFs), forming neurofibrillary tangles (NFTs) in brain. Unlike amyloid-beta (Aβ) aggregates, which are specifically detected in AD patients, tau tangles are found in many neurodegenerative disorders such as corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), Pick’s disease, dementia pugilistica, frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), and many others including AD (Alavi Naini and Soussi-Yanicostas, 2015). MAPT gene mutations are identified in a number of tauopathies. The mutations alter the amino acid sequence of tau, disrupt splicing or both (Alavi Naini and Soussi-Yanicostas, 2015). AD and other dementias are currently major challenges for health care systems, and major public health priorities worldwide, for which there is an unmet need for disease-modifying treatments (Frankish and Horton, 2017).
The tau protein is identified as a microtubule-associated protein (MAP) with a wide range of potential functions. In the adult human CNS six different tau isoforms are expressed (Goedert et al., 1989b). They are generated via alternative splicing of a single MAPT gene located on chromosome 17q21.31 and comprising 16 exons (Neve et al., 1986). Tau isoforms range in length from 352 to 441 amino acids, with a molecular weight of 45–65 kDa. The exons E1, E4, E5, E7, E9, E11, E12 and E13, are included in all tau isoforms. Tau isoforms are differentiated by three (3R) or four (4R) carboxy-terminal tandem repeats of 31 amino acids, which are designated as microtubule-binding domains (MBDs). These repeated domains, which show strong sequence conservation, are encoded by exons E9, E10, E11, and E12, the exclusion or inclusion of exon 10 generates the 3R or 4R tau isoforms, respectively (Goedert et al., 1989a,b; Andreadis et al., 1992).
The main functions recognized for tau are microtubule stabilization and polymerization. Microtubules are part of the eukaryotic cytoskeletal framework and are primarily composed of α- and β-tubulin, forming tubular polymers. Microtubules are essential for cytoskeletal maintenance and intracellular transport (Nogales, 2001). Tau mutations alter their affinity for interaction and binding with microtubules (Alavi Naini and Soussi-Yanicostas, 2015). Tau is able to bind to the outside and probably to the inside of microtubules, while the N- and C-terminal regions project outward (Kar et al., 2003; Santarella et al., 2004). The tandem repeat sequences in the (MBD) provide a net positive charge that interacts directly with the negatively charged residues of tubulin (Kar et al., 2003; Jho et al., 2010). The 4R-tau isoforms have higher affinity for binding to microtubules than 3R-tau and are more efficient at promoting microtubule assembly, likely due to the presence of the inter-repeat sequences between the first and second microtubule-binding repeats, which possess over twice the binding affinity of any individual microtubule-binding repeat (Goedert and Jakes, 1990; Goode and Feinstein, 1994).
Tau alterations as biomarkers for dementia have mainly been studied in AD. The amount of total tau (T-tau) in CSF correlates with the intensity of neurodegeneration (Gao et al., 2018) and hyperphosphorylated tau (P-tau) levels in CSF correlate with hippocampal atrophy in prodromal AD (De Leon et al., 2006; Fagan and Holtzman, 2010). Interestingly, Aβ peptide accumulation does not correlate with neurodegeneration in prodromal AD (Iaccarino et al., 2018). Moreover the mean content of abnormally phosphorylated tau from several brain regions in individual AD patients closely correlates with disease severity (Holzer et al., 1994). These observations along with tau mutations responsible for a number of tauopathies, highlight the importance of tau alterations as therapeutic targets in tauopathies.
Role of Heparan Sulfate (HS) in Tauopathies
In 1855 Virchow reported the presence of polysaccharides in amyloid deposits in brain (Virchow, 1855). In 1942 Hass characterized the polysaccharides in amyloid lesions as sulfated amino-sugar based polysaccharides, later called heparin-like glycosaminoglycans (GAGs), and then heparan sulfate proteoglycans (HSPGs) (Hass, 1942). Tauopathies are amyloid disorders (Iadanza et al., 2018). Amyloid disorders are a diverse group of protein-misfolding disorders (PMDs) characterized by disease-specific protein aggregates containing heparan sulfate proteoglycans (HSPGs) in most cases (Ancsin, 2003; Nishitsuji and Uchimura, 2017).
The association of GAGs with protein aggregates in Alzheimer’s disease (AD) and other tauopathies was described decades ago in the literature (Kato et al., 1991; Su et al., 1992; DeWitt et al., 1993, 1994). Further investigations demonstrated that sulfated GAGs played a critical role in tau hyperphosphorylation and aggregation, but also amyloid precursor protein (APP) cleavage and the resulting Aβ peptide fibrillization (Fraser et al., 1992; Brunden et al., 1993; Goedert et al., 1996; Hasegawa et al., 1997; Scholefield et al., 2003; Beckman et al., 2006). However, the nature of the sulfated GAGs involved in these processes remained unclear. In particular, GAG-tau interactions have received little attention as potential therapeutic targets.
Heparan sulfate proteoglycans are associated with both amyloid-beta (Aβ) plaques and neurofibrillary tangles (NFTs) in Alzheimer’s disease (AD) (Snow et al., 1990; Kato et al., 1991; Perlmutter et al., 1991; Perry et al., 1991; Su et al., 1992), the most frequent form of tauopathy (Hernández et al., 2018). While the principal HSPGs accumulating in the AD-associated aggregates are membrane-associated HSPGs, the presence of perlecan (a non-membrane-associated HSPG) in Aβ deposits is debated (Van Gool et al., 1993; Snow et al., 1994; Van Horssen et al., 2002). Specifically, the initially reported association of perlecan with Aβ plaques and neurofibrillary tangles in AD was later challenged because of the cross-reactivity of the antibodies used (Verbeek et al., 1999). Perlecan is increased in AD brains (Liu et al., 2016). However, a study of the perlecan mRNA levels in the hippocampus of AD patients and age-matched controls has shown similar expression levels (Maresh et al., 1996), and mice overexpressing the perlecan core protein did not develop plaques or tangles (Hart et al., 2001). Agrin is strongly expressed in the hippocampus and amygdala (Bowe and Fallon, 1995; Donahue et al., 1999), two areas strongly affected in AD (Braak and Braak, 1991). Agrin is the major HSPG associated with both Aβ plaques and tau-containing NFTs in AD (Verbeek et al., 1999; Cole and Liu, 2006). Last, heparanase (HPSE) is overexpressed in brain areas showing degeneration in AD as measured by RT-PCR (García et al., 2017), suggested be a protective mechanim. Thus membrane-associated HSPGs provide a link between the two major hallmarks of AD, Aβ plaques and NFTs. This suggests that the membrane-associated HSPGs might be involved in processes upstream of AD lesions.
In many neuropathies, abnormal HSPG accumulations are often observed at early stages of the disease. HSPGs accumulate in neuronal cell bodies in Down syndrome patients years before the first clinical symptoms of dementia occur. HSPGs are also associated with Aβ deposits in the early stages of AD. Specifically, HSPGs are associated with both immature diffuse Aβ plaques (Snow et al., 1990, 1994; Su et al., 1992; Cotman et al., 2000) and hyperphosphorylated tau aggregates, during the earliest stages of neurofibrillary pathology (Spillantini et al., 1999). Interestingly, in this situation HSPG staining is more intense than that of tau, suggesting that HSPGs play a role in initiation of disease.
In 1996 in vitro biochemical investigations demonstrated that interactions between heparin and tau protein led to the assembly of tau into NFT-like filaments (Goedert et al., 1996). Moreover, sulfated motifs on the GAG chain are critical for tau aggregation, as the non-sulfated hyaluronic acid does not enhance tau polymerization (Arrasate et al., 1997). Aside from structural changes, heparin was also shown to markedly stimulate the phosphorylation of tau by different protein kinases, leading to tau hyperphosphorylation, a key feature of tauopathies. In particular, tau phosphorylation by cdc28, cAMP-dependent protein kinase, GSK3b and several stress-activated protein kinases (SAP kinases) are markedly stimulated in vitro by heparin (Mawal-Dewan et al., 1992; Brandt et al., 1994; Yang et al., 1994; Goedert et al., 1997; Hasegawa et al., 1997). Heparin also prevents tau binding to taxol-stabilized microtubules and promotes disassembly of microtubules formed by tau and tubulin, suggesting that highly sulfated GAGs compete with microtubules for tau binding (Goedert et al., 1996). However, further research showed that the effect of heparin on tau was mimicked by several GAGs and depended on their degree of sulfation: highly sulfated dextran sulfate, pentosan polysulfate and heparin exerted a marked effect, while a number of intermediately sulfated GAGs such as HS, CS and DS had a moderate effect, and non-sulfated dextran and hyaloronic acid had no effect (Hasegawa et al., 1997). Interestingly, the concentration of sulfated GAGs required to stimulate tau phosphorylation was lower than that required for tau assembly and aggregation (Hasegawa et al., 1997), suggesting that tau phosphorylation may precede its assembly into NFTs. The increased phosphorylation and aggregation of tau observed in tauopathies may involve a substrate modulator effect of GAGs on tau conformation, as previously shown in other biological contexts (Faucher et al., 1988; Abdel Ghany et al., 1990).
Association of HSPGs with proteins can lead to conformational changes in the bound proteins, as demonstrated by the allosteric effect of heparin on anti-thrombin. Heparin induces a conformational change in tau (Paudel and Li, 1999; Sibille et al., 2006; Elbaum-Garfinkle and Rhoades, 2012), and heparin-induced conformational changes have been shown to expose previously masked tau phosphorylation sites (Zheng-Fischhöfer et al., 1998; Paudel and Li, 1999; Yoshida and Goedert, 2012). Electron microscopy data has provided evidence that sulfated GAGs present in tau aggregates affect paired helical filament (PHF) conformation: treatment of PHF-tau extracted from AD patients with heparinase or chondroitinase resulted in untwisting of PHF filaments (Arrasate et al., 1997). Furthermore, treatment of PHF-tau tangles with heparinase alters their immunodecoration properties, suggesting conformational changes upon heparinase treatment (Hernández et al., 2002). Specifically, heparin interacts with the second (R2) and third (R3) repeat regions of the tau microtubule-binding domain (Sibille et al., 2006). More recently, a small domain located in the N-terminal region of the R2 repeat was identified as critical for binding (Mukrasch et al., 2005; Zhao et al., 2017). Interestingly, Hasegawa et al. (1997), have suggested that sequence differences among moderately sulfated GAGs likely play a role in their interaction with tau (Hasegawa et al., 1997), as the overall level of sulfation cannot account for the observed effects. This further suggests that specific sulfated sequences on GAGs are required for interaction with tau.
Most importantly, cell surface HSs also act as receptors for Aβ (Kanekiyo et al., 2011) and extracellular tau aggregates, likely mediating a prion-like propagation of the pathology (Holmes et al., 2013), as reflected by the stages defined by Braak and Braak (1991). Membrane-associated heparan sulfate is involved in cell surface binding, uptake and internalization of tau seeds (Holmes et al., 2013) suggesting that propagation of tau seeds can also be blocked by pharmacological targeting of cell surface HSPGs. This approach may slow the progression of tauopathies (Holmes and Diamond, 2014). Alteration of heparan sulfate structure with aging may also be implicated in neurodegeneration. In particular, HS is important for adult neurogenesis by modulating FGF signaling; age-related alteration of HS binding properties in the hippocampus (Huynh et al., 2012), significantly compromises local regenerative capacities (Yamada et al., 2017). Membrane-bound HS, such as syndecans and glypicans are critical for regulation of neurogenesis (Yu et al., 2017).
Heparan sulfate has emerged as a potential multiplayer in tau pathology. HS is potentially implicated in the initiation and propagation of tau pathology and may play a role in limiting regenerative potential in dementia-susceptible regions. Further studies are needed to elucidate the role of different HSPGs in tauopathies. Memebrane-bound HS, mostly syndecan-3, syndecan-4 and glypican-3, are increased in AD brain (Liu et al., 2016). Particular attention should be paid to processes defining regional susceptibility to tauopathy.
Zebrafish for Drug Discovery in Tauopathies
The zebrafish (Danio rerio), is a well-established model for the study of developmental biology, gene function and human diseases. As a vertebrate, it is closer to humans than invertebrate models, such as nematodes and fruit flies, and offers many advantages over invertebrate and other alternative vertebrate models.
The zebrafish is particularly well-suited for forward and reverse genetic and high-throughput functional studies. Major advantages of zebrafish include a precisely characterized genome with genes showing 50–80% sequence similarities with their human orthologs. Orthologous counterparts of many genes relevant to tauopathies or specifically to Alzheimer’s disease are identified in zebrafish. The zebrafish has undergone an ancient genome duplication followed by loss of many duplicated genes. Two paralogs of the human MAPT gene, mapta and maptb, have been identified in zebrafish. Interestingly, in early developmental stages mapta transcripts contain predominantly 4-6 Rs, while most maptb transcripts contain 3Rs (Chen et al., 2009). In embryonic stages, the expression of mapta and maptb show a positive correlation with sex-linked ubiquitin-specific peptidase 9 (USP9) (Köglsberger et al., 2017). In the adult zebrafish brain, hypoxia is reported to shift splicing of mapta and maptb transcripts toward 4-6 R isoforms (Moussavi Nik et al., 2014). Orthologs to human APOE (apoea and apoeb), APP (appa and appb) and the APP cleaving complex are also identified in zebrafish (Newman et al., 2014).
Further advantages of the zebrafish include low cost, high fecundity, short generation time, external fertilization, external embryonic development and embryo transparency allowing easy manipulation and visualization. This small fish is particularly advantageous for the study of the nervous system as its small brain size facilitates imaging. Despite differences in CNS development, anatomy and chemistry (Panula et al., 2010), overall brain organization (Rupp et al., 1996) with neuroanatomical (Rink and Wullimann, 2004; Mueller and Wullimann, 2009) and neurochemical (Mueller et al., 2004) pathways show striking similarities in zebrafish and humans. The adult zebrafish also displays high-order behaviors such as memory, social contacts and conditioned responses (Guo, 2004). In addition, the transparency and small size of its brain allow recording of neuronal activity by the recently developed molecular sensors such as genetically encoded calcium indicators or voltage sensors (Lin and Schnitzer, 2016).
Microinjection of antisense morpholino oligonucleotides (MOs) into one-cell stage zebrafish embryos allows rapid, simple and specific reverse genetic studies by inhibition of mRNA splicing or translation (Nasevicius and Ekker, 2000; Eisen and Smith, 2008). Efficient transgenesis methods and large-scale mutagenesis screens, mRNA injection, and genomic editing tools such as the CRISPR/cas9 approach are developed in zebrafish (Prykhozhij et al., 2018). Zebrafish embryos are also suitable for large-scale chemical screening to identify novel therapeutic targets and compounds. Whole cell, tissue or organism assays are twice as often successful in drug discovery efforts than target-based assays, and the ideal assay settings are whole organism assays. Phenotypic screens, particularly in CNS drug discovery, have been markedly more efficient in identifying novel therapeutic entities than target-based assays in recent pre-clinical studies (1999–2008) (Swinney and Anthony, 2011). Zebrafish embryos have also been used to study the potential toxicity of an imaging probe for NFTs (Anumala et al., 2013) and a neuroprotective agent for AD (Torres et al., 2014).
Recent successes in drug discovery in zebrafish rely on conserved physiological pathways and drug metabolism. Here we cite two examples of such discoveries. Clemizole, approved by the FDA, has been found to inhibit seizures in a zebrafish phenotypic screen for antiepileptic drugs (Baraban et al., 2013). Zebrafish larval assays have also revealed an FDA-approved substance that protects against the ototoxicity of aminoglycoside, which enters a phase I clinical trial this year (Smuga-Otto, 2018).
The analogy of brain structure and neuronal subtypes between zebrafish and humans, together with expression of key tau phosphorylating kinases, makes the zebrafish an attractive model organism for investigating tau pathology (Santana et al., 2012). Several transgenic zebrafish models of tauopathy expressing either the wild-type or mutant human tau protein are generated. These models represent the proof of concept that zebrafish models of tauopathy recapitulate key aspects of tau pathology. First, a zebrafish model transiently expressing a mutant human tau protein fused to GFP reproduced the cytoskeletal pathology in the brain including the accumulation of tau in NFT-like aggregates and the presence of hyperphosphorylated tau (Tomasiewicz et al., 2002). The hybrid protein was under the control of the pan-neuronal gata2 promoter, which is expressed in the brain, retina and spinal cord and shows mosaic expression. This study demonstrated the conservation of pathways involved in tauopathy in zebrafish, making it possible to robustly model the pathology in this species.
More recently, a stable zebrafish transgenic line was produced that expresses the human 4-repeat tau under the control of the zebrafish enolase-2 (eno2) promoter. The utility of the eno2 promoter lies in its expression, which starts in differentiated neurons and persists into adulthood. Tau deposits resembling NFTs were found within neuronal cell bodies and proximal axons in brain regions relevant to PSP (Bai et al., 2007; Bai and Burton, 2011). Later, a stable zebrafish transgenic line expressing the human tau protein carrying the tauP301L mutation linked to frontotemporal dementia (FTD) under the control of the zebrafish pan-neuronal HuC promoter was generated (Paquet et al., 2009). In this model, the Tg[HuC::hTauP301L/DsRed], the vector system allows expression of the mutated human tau P301L along with simultaneous expression of DsRed fluorescent reporter, facilitating the identification of the mutated tau expressing neurons. Tg[HuC::hTauP301L/DsRed] develops spinal motoneuron axon extention and elongation defects and neurodegeneration in early developmental stages and as a consequence deficits in the escape response reflex. Similar defects were also observed in zebrafish larvae expressing the mutant A152T tau protein (Lopez et al., 2017). The chronic expression of the P301L mutant tau into adulthood has also been recently examined in zebrafish (Cosacak et al., 2017). Focusing on the adult zebrafish brain, Cosacak et al reported tau hyperphosphorylation in the telencephalon. However, the authors did not find tauopathy-related symptoms in the telencephalon, such as formation of neurofibrillary tangles. This is in contrasts with another report (Bai and Burton, 2011) on tau mislocalization and tau-laden neurofibrillary like structures in tau transgenic adult zebrafish brain.
The observed discrepancies may be due to the specific brain regions studied, telencephalon (Cosacak et al) vs. optic tectum and brainstem (Bai and Burton), or different transgenic systems or labeling methologies used. However, the possible resistance of adult zebrafish telencephalon to tauopathy is an interesting avenue of investigation that may reveal potential protective mechanisms. Moreover, HS plays a role in nervous system regeneration (Fuller-Carter et al., 2015; Yamada et al., 2017). High regenerative capacities in zebrafish (Gemberling et al., 2013) makes the organism a suitable model to study the implication of HS in regenerative processes.
A transient expression system is also established that expresses either full-length or truncated zebrafish 3R or human 4R tau, fused to GFP, under the control of HuC promoter (Wu et al., 2016). The expression of all constructs was found to be toxic, causing significant neurodegeneration. Using this experimental system enhancing an anti-apoptotic, anti-oxidative or neurotrophic mechanisms, was found to be protective against neurodegeneration.
The zebrafish offers numerous, diverse advantages as an animal model for the study of nervous system pathology. Numerous genes orthologous to those involved in tauopathies have been identified in the zebrafish. Experiments on zebrafish have confirmed the conservation of essential pathways between humans and the zebrafish that enable it to model tau pathology. Specific brain regions in the zebrafish are reported to bear resistance to NFT formation, possibly due to high regenerative capacities. Elucidation of these mechanisms, in particular the role of HS in the pathological vs. protective or regenerative processes in zebrafish will help identify potential pathways and targets to prevent or treat tau pathology.
Therapeutic Approaches Targeting HSPGS in Tauopathies
A better knowledge of HS-protein interactions has huge therapeutic implications for human pathologies (Capila and Linhardt, 2002; Lindahl, 2007). The primary therapeutic approach based on targeting PGs/GAGs in tauopathies consists of treatment with exogenous GAGs or GAG mimetics. Administration of exogenous GAGs is believed to competitively inhibit the harmful processes mediated by endogenous GAGs (Wang and Ding, 2014). A summary of therapeutic approaches involving exogenous GAGs and GAG mimetics is presented in Table 1. Indeed, LMW heparin oligomers efficiently inhibit the stimulatory effect of heparin on APP secretion, and inhibit the binding of heparin to Aβ1-28 peptide in vitro (Leveugle et al., 1998). However, large sulfated GAGs such as heparin and DS inhibit the binding of HSPGs to APP in vitro (Narindrasorasak et al., 1991). Several polysulfated GAGs and synthetic sulfate-containing compounds such as the Aβ-binding sulfonated dye Congo Red, along with several other sulfonated dyes, have been found to attenuate the neurotoxic effects of Aβ25-35 and Aβ1-40 in vitro (Pollack et al., 1995a,b). Moreover, several small anionic substances with sulfonate and sulfate residues efficiently decrease Aβ deposition (Kisilevsky et al., 1995). Accordingly, arrays of LMW GAGs are developed as competitive inhibitors of PG interactions (Gervais et al., 2001). Neuroparin (C3), an LMW GAG derivative of heparin, has been shown to have neuroprotective and neuroreparative properties in several animal models of AD (Dudas et al., 2008). Interestingly, neuroparin is reported to attenuate the abnormal tau immunoreactivity (representative of an AD-related conformational alteration of tau) in rat hippocampus following unilateral injection of Aβ25-35 into the amygdala (Dudas et al., 2002). In addition, this abnormal Aβ25-35 induced tau immunoreactivity was fully prevented following chronic subcutaneous injections of the LMW GAG ceroparin (Walzer et al., 2002). Heparin-like oligosaccharides reduce uptake of tau oligomers limiting their infectivity (Wang et al., 2018).
Table 1.
Classic strategies targeting HSPGs in tauopathies.
| Category | Names | Targets | Settings/Organisms | Outcomes | References | |
|---|---|---|---|---|---|---|
| Sulfated glycosaminoglycans (GAGs) | Conventional GAGs | Heparin, dextran sulfate and other GAGs | APP/Aβ | In vitro, PC12 cells | Inhibition of HSPG binding to APP | Narindrasorasak et al., 1991; Leveugle et al., 1994 |
| Heparin analogs | BACE1 | In vitro | BACE1 inhibition | Patey et al., 2006, 2008 | ||
| Low molecular weight (LMW) heparins | Heparin oligosaccharides | APP/Aβ | In vitro, Human | Inhibition of the stimulatory effect of heparin on APP secretion and heparin binding to Aβ1-28 | Leveugle et al., 1998 | |
| Tau | Neuroblastoma culture | Reduction in cellular uptake and cytotoxicity of tau oligomers | Wang et al., 2018 | |||
| Enoxaparin/Dalteparin | Aβ | In vitro | Disassembley of Aβ40 fibrils | Zhu et al., 2001 | ||
| Certoparin/Neuroparin (C3) | Aβ/Tau | Rat | Reduction in tau abnormal conformation in hippocampus, stimulated by Aβ25-35 intra-amygdaloid injection | Walzer et al., 2002/ Dudas et al., 2002, 2008 | ||
| Enoxaparin | Aβ | APP23 mice | Decrease in Aβ brain concentration, deposition and reactive astrocytosis | Bergamaschini et al., 2004 | ||
| APPswe/PS1dE9 mice | Improvement of spatial memory, decrease in Aβ deposition if treated in early stages of Aβ accumulation | Timmer et al., 2010 | ||||
| Low molecular weight anionic sulfonate or sulfate compounds | Congo red and analogs | Aβ | PC12/Hela cells | Attenuation of Aβ25-35 and Aβ40 toxicity | Pollack et al., 1995b,a | |
| Sodium 1,3-propanediol disulphate | Aβ | In vitro | Disassembly of Aβ40 fibrils | Kisilevsky et al., 1995 | ||
| Tramiprosate (3-amino-1-propanesulfonic acid; 3APS; homotaurine; AlzhemedTM) | Aβ | TgCRND8 mice | Significant reduction of Aβ in brain and plasma, reduction of Aβ plaques in brain | Gervais et al., 2007 | ||
| Humans: Phase II trial in probable mild to moderate AD | Satisfactory safety and tolerability, significant decrease of CSF Aβ42 levels, no cognitive benefits | |||||
| Phase III trial in mild to moderate AD | No cognitive benefits, post-hoc analyses: decrease in loss of hippocampal volume | Aisen et al., 2006 | ||||
| Cognitive benefits in APOE4/4 patients | Abushakra et al., 2017 | |||||
Another ionic compound, tramiprosate (3-amino-1-propanesulfonic acid; 3APS; AlzhemedTM), has been shown to bind preferentially the soluble Aβ peptide, maintain Aβ peptide in a soluble conformation, and reduce Aβ burden in TgCRND8 mice, a transgenic AD model that develops early-onset and aggressive brain Aβ amyloidosis (Gervais et al., 2007). However, in November 2007, tramiprosate was rejected for further pharmaceutical development because of a lack of proven efficacy in a phase III human clinical trial (Carter et al., 2010). However, while clinical trials of tramiprosate in mild-to-moderate AD have not shown therapeutic efficacy, post-hoc analyses have shown some significantly positive outcomes on secondary endpoints and in some particular subgroups of patients (Aisen et al., 2011; Caltagirone et al., 2012). In particular, the disease-modifying potential is highlighted in APOE4 allele carrier patients by the observed cognitive benefits (Caltagirone et al., 2012; Abushakra et al., 2017).
The classic strategies aimed at HSPGs are mostly tested in AD where benefits are observed in distinct patient subgroups. In these strategies, APP and Aβ (not tau) are mostly studied as targets. An alternative approach consists in targeting HS sulfation. Recently, overexpression of several HS biosynthetic enzymes such as 3-O-sulfotransferase-2 (3ost2 or Hs3st2) was demonstrated in the hippocampus of AD patients. Inactivation of the zebrafish ortholog of the HS chain modifier enzyme Hs3st2 significantly decreased tau hyperphosphorylation and tau-related neuropathology in vitro and in vivo in the Tg[HuC::hTauP301L/DsRed] zebrafish model of tauopathy (Sepulveda Diaz et al., 2015). An additional approach consists of treatment with heparan sulfate antagonist molecules. In a recent proof of concept study a rescue of neuronal abnormalities upon treatment with surfen and oxalyl surfen was observed in the Tg[HuC::hTauP301L/DsRed] zebrafish model (Alavi Naini et al., 2018). Figure 1 summarizes the two approaches in Tg[HuC::hTauP301L/DsRed] zebrafish.
FIGURE 1.
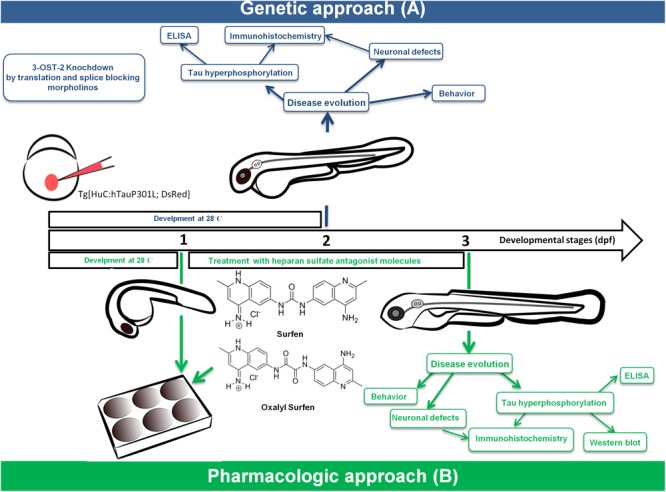
Combination of genetic (A) and pharmacological (B) approaches identified novel therapeutics for tauopathies using a zebrafish transgenic model of tau pathology.
3-O-Sulfation in HS chains forms binding sites for HSPG binding proteins. However, biochemical characterization of this rare modification among an array of other sulfated groups requires large sample quantities, and shortage of material for studying 3-O-sulfation has so far hampered work on these sulfated motifs. So far six HSPG-binding proteins have been shown to require 3-O-sulfation for their association with GAG chains (Thacker et al., 2014).
The addition of a 3-O-sulfate group is one of the last chain modifications in HS biosynthesis (Zhang et al., 2001a,b). This modification is a rare and discrete modification confined to a limited number of chains when it occurs at all, and its prevalence among naturally occurring HSs is largely unknown (Thacker et al., 2014). The 3-O-sulfate group was initially identified while searching for sulfate group removing enzymes on heparin (Leder, 1980), and later confirmed with chemical, NMR and mass spectrometry analyses (Meyer et al., 1981; Yamada et al., 1993).
The best-studied case of 3-O-sulfate group-dependent binding is the association of anti-thrombin (AT) to heparin and HS. Binding of heparin to AT induces conformational changes in the protein that radically increase its catalytic activity (Rosenberg and Damus, 1973; Huntington et al., 1996). The activated AT then inactivates several proteases, including thrombin, factor IXa, and factor Xa, which are involved in blood coagulation. This is why heparin is used as a routine anticoagulant agent in clinical practice. The characterization of AT binding sites on the sulfated sugar chains is facilitated by their enrichment in heparin, which is readily available commercially (Yamada et al., 1993). The minimum AT binding site on heparin is a pentasaccharide sequence with a critical 3-O-sulfated motif on the middle N-sulfo-glucosamine residue (Thunberg et al., 1982; Choay et al., 1983). In the absence of this 3-O-sulfate group, the affinity of heparin to AT was markedly decreased, along with its inhibitory effect on factor Xa (Atha et al., 1985, 1987).
3-O-Sulfated HSs at the cell surface also serve as entry receptors for the herpes simplex envelope glycoprotein, glycoprotein D (gD) (Shukla et al., 1999), and the binding domain has been identified as an octosaccharide containing a 3-O-sulfated motif (Liu et al., 2002). In a similar manner, fibroblast growth factor 7 (FGF7) and fibroblast growth factor receptors (FGFR) are thought to rely on 3-O-sulfated motifs for binding to HS and heparin (McKeehan et al., 1999; Ye et al., 2001; Luo et al., 2006). Moreover, 3-O-sulfation on heparin/HS is suggested to mediate the binding of “cyclophilin B” and “stabilin” (Vanpouille et al., 2007; Pempe et al., 2012). Cyclophilin B promotes lymphocyte adhesion and migration following binding to HS located at the cell surface (Allain et al., 2002). Stabilins (1, 2) mediate heparin clearance in hepatic sinusoidal endothelial cells (Hansen et al., 2005; Harris et al., 2008).
Combination of genetic (A) (morpholino gene-KO) (Sepulveda-Diaz et al., 2015) and pharmacological (B) (small molecule) (Alavi Naini et al., 2018) approaches in the zebrafish Tg[HuC::hTauP301L; DsRed] tauopathy model identified 3-OS-sulfated HS as a therapeutic target for tauopathies and the small molecules surfen and oxalyl surfen as novel therapeutic candidates.
Alterations in the fine structure of GAGs have previously been described in several pathological situations. The sulfate moieties on GAG chains have been shown to contribute to amylin fibrillization in islet amyloidosis (Castillo et al., 1998). Furthermore, alteration of the O-sulfation pattern and specific disaccharide compositions has been observed in amyloid-laden liver and spleen tissues (Lindahl and Lindahl, 1997). An analysis of the HSs in the cerebral cortex in AD and control subjects demonstrated an alteration in N-sulfated residue distribution in AD brain, with an increase in GlcNSO3 residues (Lindahl et al., 1995). However, in studies of cerebral tissue the abnormal HSs may have been diluted with normal HSs, as analyses were performed on whole brains. In particular, alterations in specific HSPG populations or modifications of small domains in HSPG chains may have been hidden. Moreover, in AD, analysis of skin fibroblasts from patients revealed alterations in HS sulfation. Zebrower and Kieras reported that cultured skin fibroblasts from AD patients produced 30% more GlcA-GlcNSO3, and 40% less GlcA-GlcN-6-OSO3 compared to samples from age-matched healthy individuals (Zebrower et al., 1992). Authors suggested that an alteration in HS-sulfotransferase activity was responsible for their observations (Zebrower and Kieras, 1993). Importantly, a critical requirement for the presence of 6-O-sulfated disaccharide-containing HS in internalization and spreading of infectious tau species has recently been demonstrated (Rauch et al., 2018; Stopschinski et al., 2018). 6-O-Sulfation was also identified as critical for heparin-tau interaction by surface plasmon resonance (SPR) and nuclear magnetic resonance (NMR) spectroscopy (Zhao et al., 2017), while the requirement for 3-O-sulfation was not investigated in these recent studies. These studies together with studies on 3-O-sulfation (Sepulveda-Diaz et al., 2015) support the hypothesis that specific GAG chain sulfation mediated by a combination of 2-, 3- and 6-O-sulfotransferases play a key role in the physiopathology of tauopathies. Interestingly, hierarchical sulfation of disaccharide residues allows 3-O-sulfated motifs to be generated only on 6-O-sulfated domains of GAG chains.
Aside from hyperphosphorylation and aggregation of the tau protein, the sulfated motifs on GAG chains have also been shown to play a role in Aβ fibrillization and aggregation. This was first suggested by Fraser and coworkers, based on X-ray diffraction studies (Fraser et al., 1992). A later in vitro study showed that the sulfated motifs on GAG chains directly bind fibrillar Aβ (Gupta-Bansal et al., 1995). In addition, while removal of N-sulfated motifs on heparin GAG chains slightly reduced the heparin-induced Aβ fibril formation (Castillo et al., 1999), removal of O-sulfates led to a significant loss of heparin-enhanced Aβ fibrillization. Heparin enhancement of Aβ fibrillization was significantly greater than that induced by HS, consistent with the lower sulfate content in HS. By contrast, inorganic sulfate was found to have no effect (Castillo et al., 1999).
In turn, Aβ peptides have been suggested to influence GAG composition and localization (Miller et al., 1997; Timmer et al., 2009). In particular, Aβ42 carrying the Dutch mutation has been reported to induce cellular relocalization and production of agrin and glypican-1 in vitro, together with altered glycosylation and increased sulfate incorporation (Timmer et al., 2009). Interestingly, the treatment of human brain pericytes (HBPs) with Aβ42Dutch leads to the association of Aβ fibrils to cell surface glypican-1 prior to their internalization (Timmer et al., 2009). TGF-β enhancement of Aβ fibril formation is also suggested to involve an increase in HSPG synthesis or chain modification (Castillo et al., 1999). Aβ degradation takes place in the lysosomal compartment (Li et al., 2012), the intracellular compartment where HSPG degradation also occurs (Brauker and Wang, 1987; Yanagishita and Hascall, 1992; Bame, 1993). Interestingly, Aβ has been shown to inhibit the heparanase-mediated degradation of GAGs and HSPGs in vitro (Bame et al., 1997), suggesting that a similar process may hamper HSPGs degradation in vivo in tauopathies, creating a scenario similar to that observed in patients with mucopolysaccharidoses.
Apolipoprotein E (whose E4 allele is a major risk factor in sporadic AD) promotes GAG sulfation in cultured neuroblastoma cells, and this effect is greater for ApoE4 than ApoE3 (Bonay and Avila, 2001). The interaction between HSPGs and apolipoprotein is necessary for the uptake of apolipoprotein E-containing lipoprotein by low-density lipoprotein (LDL) receptor related protein (LRP) by hepatocytes in vitro. Lipoprotein uptake in neurons is also mediated by the HSPG/LRP pathway (Mahley, 1996). Interestingly, apoE has been shown to interact with HSPGs through a binding site, which is structurally complementary to HS rich in N- and O-sulfate groups (Libeu et al., 2001). Moreover, the inhibitory effect of ApoE4 (not observed for ApoE3) on neurite outgrowth in vitro is likely mediated by the HSPG/LRP pathway (Bellosta et al., 1995). Moreover, a synthetic peptide that contain the amino acid residues 141-155, together with a 22 kDa N-terminal thrombin-cleavage fragment of apoE display significant toxicity to neurites from embryonic chick neurons in vitro, with apoE4 fragments exhibiting greater toxicity than apoE3. The toxic effect is likely mediated via the HSPG/LRP pathway, and treatments antagonizing HS prevent neurite degeneration (Crutcher et al., 1994; Marques et al., 1996). HSPG acts jointly with LRP1 for cell surface binding and uptake of Aβ (Kanekiyo et al., 2011). Finally, ApoE (Li et al., 2012) and LRP (Fuentealba et al., 2010) promote lysosomal trafficking of Aβ. ApoE acts in an isoform-dependent manner, with apoE3 enhancing Aβ trafficking and degradation more efficiently than apoE4 (Li et al., 2012). These observations support the hypothesis that a defect in the metabolism of HS orchestrated by Aβ and apoE may play a role in the physiopathology of tauopathies.
The internalization of HS in the intracellular space has been described in several pathological situations (Cole and Liu, 2006). The intracellular accumulation of HS long before detection of tau pathology in neurons in AD and in Down syndrome was described decades ago (Snow et al., 1990; Goedert et al., 1996). However, their involvement in the series of events leading to the abnormal tau phosphorylation and neurodegeneration has been largely disregarded, possibly because of the paradigm confining the biological roles of HSPGs to the extracellular space (Shriver et al., 2012).
The association of sulfated GAGs with tau in neurons has long been puzzling and was seen to result from leakage of newly synthesized sulfated GAGs from intracellular organelles such as Golgi apparatus, endosomes and lysosomes (Díaz-Nido et al., 2002). Interestingly, under two distinct pathological situations in neuroblastoma cultures, expression of human tauP301L protein and exposure to oxidative stress, HS was localized in intracellular space. An intense intracellular uptake of membrane-associated HS and the concomitant hyperphosphorylation of the tau protein were observed (Sepulveda Diaz et al., 2015). These findings point to a possible altered metabolism and distribution of sulfated HS in tau-related neuropathologies. Regarding the molecular mechanisms linking 3-O-sulfated HSs and tau abnormal phosphorylation, in vitro data suggest that the sulfated GAGs bind to tau and not to the GSK3β tau-kinase (Sepulveda Diaz et al., 2015). Accordingly, in the presence of heparin (a highly sulfated HS), tau rapidly undergoes conformational changes that allow kinase access to epitopes otherwise inaccessible for phosphorylation (Hasegawa et al., 1997; Zheng-Fischhöfer et al., 1998; Paudel and Li, 1999; Sibille et al., 2006; Yoshida and Goedert, 2012). The substrate modulator effect of 3-O-sulfated HS motifs further supports the hypothesis that these sulfated polysaccharides act as molecular chaperones able to induce key conformational changes on tau protein and triggering its phosphorylation at newly unmasked tau residues. The hypothesis is further supported by the observation of an increase in hippocampal 3-O-sulfation with aging (Huynh et al., 2012). Recently, a GAG sulfotransferase inhibitor has been identified (Cheung et al., 2017) that sets a basic scaffold for the future development of highly specific and potent sulfotransferase inhibitors, which are promising candidates as potential therapeutics for tauopathies.
Conclusion
Heparan sulfate-protein interactions are emerging as potential therapeutic targets in tauopathies. Previous work suggest a multifactorial role played by GAGs in the pathogenesis of tauopathies. Increased biosynthesis, internalization and/or decreased degradation of HS GAGs, or alterations in HS chain modifications likely contribute to tauopathies. These observations raise the question of whether specific motifs on HS chains are involved in binding to tau and induction of misfolded states. The zebrafish has recently emerged as an attractive animal model for Alzheimer’s disease research and provides an ideal tool for drug screening prior to clinical testing in mammals. Thanks to this small fish, HS is targeted in tau pathology by two different approaches. A critical requirement for 3-O-sulfated HS in tau pathology was discovered and HS antagonists (surfen and oxalyl surfen) were identified as efficient therapeutic candidates. 6-O-sulfated HS motifs have recently been identified as critical for tau seed internalization. 3-O-sulfation, a fine chain modification, is only generated in 6-O-sulfated regions. Together, these recent findings strengthen the hypothesis of distinct HS sequences implicated in tau misfolding, hyperphosphorylation, aggregation and propagation. These findings are steps toward defining of specific HS targets in tauopathies.
Author Contributions
Both authors contributed to writing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abdel Ghany M., El Gendy K., Zhang S., Racker E. (1990). Control of src kinase activity by activators, inhibitors, and substrate chaperones. Proc. Natl. Acad. Sci. U.S.A. 87 7061–7065. 10.1073/pnas.87.18.7061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abushakra S., Porsteinsson A., Scheltens P., Sadowsky C., Vellas B., Cummings J., et al. (2017). Clinical effects of tramiprosate in APOE4/4 homozygous patients with mild Alzheimer’s disease suggest disease modification potential. J. Prev. Alzheimer’s Dis. 4 149–156. 10.14283/jpad.2017.26 [DOI] [PubMed] [Google Scholar]
- Aisen P. S., Gauthier S., Ferris S. H., Saumier D., Haine D., Garceau D., et al. (2011). Tramiprosate in mild-to-moderate Alzheimer’s disease - A randomized, double-blind, placebo-controlled, multi-centre study (the alphase study). Arch. Med. Sci. 7 102–111. 10.5114/aoms.2011.20612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aisen P. S., Saumier D., Briand R., Laurin J., Gervais F., Tremblay P., et al. (2006). A Phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology 67 1757–1763. 10.1212/01.wnl.0000244346.08950.64 [DOI] [PubMed] [Google Scholar]
- Alavi Naini S. M., Soussi-Yanicostas N. (2015). Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell. Longev. 2015:17. 10.1155/2015/151979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavi Naini S. M., Yanicostas C., Hassan-Abdi R., Blondeel S., Bennis M., Weiss R. J., et al. (2018). Surfen and oxalyl surfen decrease tau hyperphosphorylation and mitigate neuron deficits in vivo in a zebrafish model of tauopathy. Transl. Neurodegener. 7:6. 10.1186/s40035-018-0111-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allain F., Vanpouille C., Carpentier M., Slomianny M.-C., Durieux S., Spik G. (2002). Interaction with glycosaminoglycans is required for cyclophilin B to trigger integrin-mediated adhesion of peripheral blood T lymphocytes to extracellular matrix. Proc. Natl. Acad. Sci. U.S.A. 99 2714–2719. 10.1073/pnas.052284899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancsin J. B. (2003). Amyloidogenesis: historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid 10 67–79. 10.3109/13506120309041728 [DOI] [PubMed] [Google Scholar]
- Andreadis A., Brown W. M., Kosik K. S. (1992). Structure and novel exons of the human tau gene. Biochemistry 31 10626–10633. 10.1021/bi00158a027 [DOI] [PubMed] [Google Scholar]
- Antoine T. E., Jones K. S., Dale R. M., Shukla D., Tiwari V. (2014). Zebrafish: modeling for herpes simplex virus infections. Zebrafish 11 17–25. 10.1089/zeb.2013.0920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anumala U. R., Gu J., Lo Monte F., Kramer T., Heyny-Von Haußen R., Hölzer J., et al. (2013). Fluorescent rhodanine-3-acetic acids visualize neurofibrillary tangles in Alzheimer’s disease brains. Bioorganic Med. Chem. 21 5139–5144. 10.1016/j.bmc.2013.06.039 [DOI] [PubMed] [Google Scholar]
- Arrasate M., Pérez M., Valpuesta J. M., Avila J. (1997). Role of glycosaminoglycans in determining the helicity of paired helical filaments. Am. J. Pathol. 151 1115–1122. [PMC free article] [PubMed] [Google Scholar]
- Atha D. H., Lormeau J. C., Petitou M., Choay J., Rosenberg R. D. (1985). Contribution of monosaccharide residues in heparin binding to antithrombin III. Biochemistry 24 6723–6729. 10.1021/bi00344a063 [DOI] [PubMed] [Google Scholar]
- Atha D. H., Lormeau J. C., Petitou M., Rosenberg R. D., Choay J. (1987). Contribution of 3-O- and 6-O-sulfated glucosamine residues in the heparin-induced conformational change in antithrombin III. Biochemistry 26 6454–6461. 10.1021/bi00394a024 [DOI] [PubMed] [Google Scholar]
- Aviezer D., Levy E., Safran M., Svahn C., Buddecke E., Schmidt A., et al. (1994). Differential structural requirements of heparin and heparan sulfate proteoglycans that promote binding of basic fibroblast growth factor to its receptor. J. Biol. Chem. 269 114–121. [PubMed] [Google Scholar]
- Bai Q., Burton E. A. (2011). Zebrafish models of tauopathy. Biochim. Biophys. Acta – Mol. Basis Dis. 12 353–363. 10.1016/j.bbadis.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Q., Garver J. A., Hukriede N. A., Burton E. A. (2007). Generation of a transgenic zebrafish model of tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 35 6501–6516. 10.1093/nar/gkm608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bame K. J. (1993). Release of heparan sulfate glycosaminoglycans from proteoglycans in Chinese hamster ovary cells does not require proteolysis of the core protein. J. Biol. Chem. 268 19956–19964. [PubMed] [Google Scholar]
- Bame K. J., Danda J., Hassall A., Tumova S. (1997). Abeta(1-40) prevents heparanase-catalyzed degradation of heparan sulfate glycosaminoglycans and proteoglycans in vitro. A role for heparan sulfate proteoglycan turnover in Alzheimer’s disease. J. Biol. Chem. 272 17005–17011. 10.1074/jbc.272.27.17005 [DOI] [PubMed] [Google Scholar]
- Baraban S. C., Dinday M. T., Hortopan G. A. (2013). Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 4:2410. 10.1038/ncomms3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beahm B. J., Dehnert K. W., Derr N. L., Kuhn J., Eberhart J. K., Spillmann D., et al. (2014). A visualizable chain-terminating inhibitor of glycosaminoglycan biosynthesis in developing zebrafish. Angew. Chemie – Int. Ed. 53 3347–3352. 10.1002/anie.201310569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman M., Holsinger R. M. D., Small D. H. (2006). Heparin activates β-secretase (BACE1) of Alzheimer’s disease and increases autocatalysis of the enzyme. Biochemistry 45 6703–6714. 10.1021/bi052498t [DOI] [PubMed] [Google Scholar]
- Bergamaschini L., Rossi E., Storini C., Pizzimenti S., Distaso M., Perego C., et al. (2004). Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J. Neurosci. 24 4181–4186. 10.1523/JNEUROSCI.0550-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta S., Nathan B. P., Orth M., Dong L. M., Mahley R. W., Pitas R. E. (1995). Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J. Biol. Chem. 270 27063–27071. 10.1074/jbc.270.45.27063 [DOI] [PubMed] [Google Scholar]
- Bink R. J., Habuchi H., Lele Z., Dolk E., Joore J., Rauch G. J., et al. (2003). Heparan sulfate 6-O-sulfotransferase is essential for muscle development in zebrafish. J. Biol. Chem. 278 31118–31127. 10.1074/jbc.M213124200 [DOI] [PubMed] [Google Scholar]
- Bishop J. R., Schuksz M., Esko J. D. (2007). Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446 1030–1037. 10.1038/nature05817 [DOI] [PubMed] [Google Scholar]
- Bonay P., Avila J. (2001). Apolipoprotein E4 stimulates sulfation of glycosaminoglycans in neural cells. Biochim. Biophys. Acta Mol. Basis Dis. 1535 217–220. 10.1016/S0925-4439(00)00096-X [DOI] [PubMed] [Google Scholar]
- Bowe M. A., Fallon J. R. (1995). The role of agrin in synapse formation. Annu. Rev. Neurosci. 18 443–462. 10.1146/annurev.ne.18.030195.002303 [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82 239–259. 10.1007/BF00308809 [DOI] [PubMed] [Google Scholar]
- Brandt R., Lee G., Teplow D. B., Shalloway D., Abdel-Ghany M. (1994). Differential effect of phosphorylation and substrate modulation on tau’s ability to promote microtubule growth and nucleation. J. Biol. Chem. 269 11776–11782. [PubMed] [Google Scholar]
- Brauker J. H., Wang J. L. (1987). Nonlysosomal processing of cell-surface heparan sulfate proteoglycans. Studies of I-cells and NH4Cl-treated normal cells. J. Biol. Chem. 262 13093–13101. [PubMed] [Google Scholar]
- Brickman Y. G., Ford M. D., Gallagher J. T., Nurcombe V., Bartlett P. F., Turnbvill J. E. (1998). Structural modification of fibroblast growth factor-binding heparan sulfate at a determinative stage of neural development. J. Biol. Chem. 273 4350–4359. 10.1074/jbc.273.8.4350 [DOI] [PubMed] [Google Scholar]
- Brunden K. R., Richter-Cook N. J., Chaturvedi N., Frederickson R. C. A. (1993). pH-dependent binding of synthetic β-amyloid peptides to glycosaminoglycans. J. Neurochem. 61 2147–2154. 10.1111/j.1471-4159.1993.tb07453.x [DOI] [PubMed] [Google Scholar]
- Bülow H. E., Hobert O. (2004). Differential sulfations and epimerization define heparan sulfate specificity in nervous system development. Neuron 41 723–736. 10.1016/S0896-6273(04)00084-4 [DOI] [PubMed] [Google Scholar]
- Burgess R. W., Skarnes W. C., Sanes J. R. (2000). Agrin isoforms with distinct amino termini: differential expression, localization, and function. J. Cell Biol. 151 41–52. 10.1083/jcb.151.1.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwallader A. B., Yost H. J. (2006a). Combinatorial expression patterns of heparan sulfate sulfotransferases in zebrafish: I, The 3-O-sulfotransferase family. Dev. Dyn. 235 3423–3431. 10.1002/dvdy.20991 [DOI] [PubMed] [Google Scholar]
- Cadwallader A. B., Yost H. J. (2006b). Combinatorial expression patterns of heparan sulfate sulfotransferases in zebrafish: II, the 6-O-sulfotransferase family. Dev. Dyn. 235 3432–3437. 10.1002/dvdy.20990 [DOI] [PubMed] [Google Scholar]
- Cadwallader A. B., Yost H. J. (2007). Combinatorial expression patterns of heparan sulfate sulfotransferases in zebrafish: III. 2-O-sulfotransferase and C5-epimerases. Dev. Dyn. 236 581–586. 10.1002/dvdy.21051 [DOI] [PubMed] [Google Scholar]
- Caltagirone C., Ferrannini L., Marchionni N., Nappi G., Scapagnini G., Trabucchi M. (2012). The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: a review. Aging Clin. Exp. Res. 24 580–587. 10.3275/8585 [DOI] [PubMed] [Google Scholar]
- Capila I., Linhardt R. J. (2002). Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 41 391–412. [DOI] [PubMed] [Google Scholar]
- Carter M. D., Simms G. A., Weaver D. F. (2010). The development of new therapeutics for Alzheimer’s disease. Clin. Pharmacol. Ther. 88 475–486. 10.1038/clpt.2010.165 [DOI] [PubMed] [Google Scholar]
- Castillo G. M., Cummings J. A., Yang W., Judge M. E., Sheardown M. J., Rimvall K., et al. (1998). Sulfate content and specific glycosaminoglycan backbone of perlecan are critical for perlecan’s enhancement of islet amyloid polypeptide (amylin) fibril formation. Diabetes Metab. Res. Rev. 47 612–620. 10.2337/diabetes.47.4.612 [DOI] [PubMed] [Google Scholar]
- Castillo G. M., Lukito W., Wight T. N., Snow A. D. (1999). The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J. Neurochem. 72 1681–1687. 10.1046/j.1471-4159.1999.721681.x [DOI] [PubMed] [Google Scholar]
- Crutcher K. A., Clay M. A., Scott S. A., Tian X., Tolar M., Harmony J. A. K. (1994). Neurite degeneration elicited by apolipoprotein E peptides. Exp. Neurol. 130 120–126. 10.1006/exnr.1994.1191 [DOI] [PubMed] [Google Scholar]
- Chen E., Stringer S. E., Rusch M. A., Selleck S. B., Ekker S. C. (2005). A unique role for 6-O sulfation modification in zebrafish vascular development. Dev. Biol. 284 364–376. 10.1016/j.ydbio.2005.05.032 [DOI] [PubMed] [Google Scholar]
- Chen M., Martins R. N., Lardelli M. (2009). Complex splicing and neural expression of duplicated tau genes in zebrafish embryos. J. Alzheimer’s Dis. 18 305–317. 10.3233/JAD-2009-1145 [DOI] [PubMed] [Google Scholar]
- Cheung S. T., Miller M. S., Pacoma R., Roland J., Liu J., Schumacher A. M., et al. (2017). Discovery of a small-molecule modulator of glycosaminoglycan sulfation. ACS Chem. Biol. 12 3126–3133. 10.1021/acschembio.7b00885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choay J., Petitou M., Lormeau J. C., Sinaÿ P., Casu B., Gatti G. (1983). Structure-activity relationship in heparin: a synthetic pentasaccharide with high affinity for antithrombin III and eliciting high anti-factor Xa activity. Biochem. Biophys. Res. Commun. 116 492–499. 10.1016/0006-291X(83)90550-8 [DOI] [PubMed] [Google Scholar]
- Cole G. J., Liu I.-H. (2006). “Glycosaminoglycans, proteoglycans, and conformational disorders,” in Protein Misfolding, Aggregation and Conformational Diseases, Protein Aggregation and Conformational Diseases Vol. 1 eds Uversky V., Fink A. L. (New York, NY: Kluwer Academic/Plenum Publishers; ) 10.1007/0-387-25919-8_5 [DOI] [Google Scholar]
- Cosacak M. I., Bhattarai P., Bocova L., Dzewas T., Mashkaryan V., Papadimitriou C., et al. (2017). Human TAUP301L overexpression results in TAU hyperphosphorylation without neurofibrillary tangles in adult zebrafish brain. Sci. Rep. 7:12959. 10.1038/s41598-017-13311-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman S. L., Halfter W., Cole G. J. (2000). Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer’s disease brain. Mol. Cell. Neurosci. 15 183–198. 10.1006/mcne.1999.0816 [DOI] [PubMed] [Google Scholar]
- Couchman J. R., Pataki C. A. (2012). An introduction to proteoglycans and their localization. J. Histochem. Cytochem. 60 885–897. 10.1369/0022155412464638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon M. J., DeSanti S., Zinkowski R., Mehta P. D., Pratico D., Segal S., et al. (2006). Longitudinal CSF and MRI biomarkers improve the diagnosis of mild cognitive impairment. Neurobiol. Aging 27 394–401. 10.1016/j.neurobiolaging.2005.07.003 [DOI] [PubMed] [Google Scholar]
- DeWitt D. A., Richey P. L., Praprotnik D., Silver J., Perry G. (1994). Chondroitin sulfate proteoglycans are a common component of neuronal inclusions and astrocytic reaction in neurodegenerative diseases. Brain Res. 656 205–209. 10.1016/0006-8993(94)91386-2 [DOI] [PubMed] [Google Scholar]
- DeWitt D. A., Silver J., Canning D. R., Perry G. (1993). Chondroitin sulfate proteoglycans are associated with the lesions of alzheimer’s disease. Exp. Neurol. 121 149–152. 10.1006/exnr.1993.1081 [DOI] [PubMed] [Google Scholar]
- Díaz-Nido J., Wandosell F., Avila J. (2002). Glycosaminoglycans and β-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 23 1323–1332. 10.1016/S0196-9781(02)00068-2 [DOI] [PubMed] [Google Scholar]
- Donahue J. E., Berzin T. M., Rafii M. S., Glass D. J., Yancopoulos G. D., Fallon J. R., et al. (1999). Agrin in Alzheimer’s disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proc. Natl. Acad. Sci. U.S.A. 96 6468–6472. 10.1073/pnas.96.11.6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas B., Cornelli U., Lee J. M., Hejna M. J., Walzer M., Lorens S. A., et al. (2002). Oral and subcutaneous administration of the glycosaminoglycan C3 attenuates Aβ(25-35)-induced abnormal tau protein immunoreactivity in rat brain. Neurobiol. Aging 23 97–104. 10.1016/S0197-4580(01)00255-X [DOI] [PubMed] [Google Scholar]
- Dudas B., Rose M., Cornelli U., Pavlovich A., Hanin I. (2008). Neuroprotective properties of glycosaminoglycans: potential treatment for neurodegenerative disorders. Neurodegen. Dis. 5 200–205. 10.1159/000113702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen J. S., Smith J. C. (2008). Controlling morpholino experiments: don’t stop making antisense. Development 135 1735–1743. 10.1242/dev.001115 [DOI] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S., Rhoades E. (2012). Identification of an aggregation-prone structure of tau. J. Am. Chem. Soc. 134 16607–16613. 10.1021/ja305206m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan A. A., Holtzman D. D. (2010). Cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark. Med. 4 51–63. 10.2217/BMM.09.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucher M., Gironès N., Hannun Y. A., Bell R. M., Davis R. J. (1988). Regulation of the epidermal growth factor receptor phosphorylation state by sphingosine in A431 human epidermoid carcinoma cells. J. Biol. Chem. 263 5319–5327. [PubMed] [Google Scholar]
- Filipek-Górniok B., Carlsson P., Haitina T., Habicher J., Ledin J., Kjellén L. (2015). The NDST gene family in zebrafish: role of Ndst1b in pharyngeal arch formation. PLoS One 10:e0119040. 10.1371/journal.pone.0119040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish H., Horton R. (2017). Prevention and management of dementia: a priority for public health. Lancet 390 2614–2615. 10.1016/S0140-6736(17)31756-7 [DOI] [PubMed] [Google Scholar]
- Fraser P. E., Nguyen J. T., Chin D. T., Kirschner D. A. (1992). Effects of sulfate ions on alzheimer β/A4 peptide assemblies: implications for amyloid fibril-proteoglycan interactions. J. Neurochem. 59 1531–1540. 10.1111/j.1471-4159.1992.tb08470.x [DOI] [PubMed] [Google Scholar]
- Fuentealba R. A., Liu Q., Zhang J., Kanekiyo T., Hu X., Lee J. M., et al. (2010). Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Aβ42 uptake and lysosomal trafficking. PLoS One 5:e11884. 10.1371/journal.pone.0011884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller-Carter P. I., Carter K. W., Anderson D., Harvey A. R., Giles K. M., Rodger J. (2015). Integrated analyses of zebrafish miRNA and mRNA expression profiles identify miR-29b and miR-223 as potential regulators of optic nerve regeneration. BMC Genomics 16:591. 10.1186/s12864-015-1772-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.-L., Wang N., Sun F.-R., Cao X.-P., Zhang W., Yu J.-T. (2018). Tau in neurodegenerative disease. Ann. Transl. Med. 6:175. 10.21037/atm.2018.04.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García B., Martín C., García-Suárez O., Muñiz-Alonso B., Ordiales H., Fernández-Menéndez S., et al. (2017). Upregulated expression of heparanase and heparanase 2 in the brains of Alzheimer’s disease. J. Alzheimer’s Dis. 58 185–192. 10.3233/JAD-161298 [DOI] [PubMed] [Google Scholar]
- Gemberling M., Bailey T. J., Hyde D. R., Poss K. D. (2013). The zebrafish as a model for complex tissue regeneration. Trends Genet. 29 611–620. 10.1016/j.tig.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais F., Chalifour R., Garceau D., Kong X., Laurin J., Mclaughlin R., et al. (2001). Glycosaminoglycan mimetics: a therapeutic approach to cerebral amyloid angiopathy. Amyloid 8 28–35. [PubMed] [Google Scholar]
- Gervais F., Paquette J., Morissette C., Krzywkowski P., Yu M., Azzi M., et al. (2007). Targeting soluble Aβ peptide with tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 28 537–547. 10.1016/j.neurobiolaging.2006.02.015 [DOI] [PubMed] [Google Scholar]
- Ghiselli G., Farber S. A. (2005). D-glucuronyl C5-epimerase acts in dorso-ventral axis formation in zebrafish. BMC Dev. Biol. 12:19. 10.1186/1471-213X-5-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Hasegawa M., Jakes R., Lawler S., Cuenda A., Cohen P. (1997). Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 409 57–62. 10.1016/S0014-5793(97)00483-3 [DOI] [PubMed] [Google Scholar]
- Goedert M., Jakes R. (1990). Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 9 4225–4230. 10.1002/J.1460-2075.1990.TB07870.X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., Crowther R. A. (1996). Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383 550–553. 10.1038/383550a0 [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Jakes R., Rutherford D., Crowther R. A. (1989a). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3 519–526. 10.1016/0896-6273(89)90210-9 [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Potier M. C., Ulrich J., Crowther R. A. (1989b). Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 8 393–399. 10.1002/j.1460-2075.1989.tb03390.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode B. L., Feinstein S. C. (1994). Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J. Cell Biol. 124 769–782. 10.1083/jcb.124.5.769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsi B., Whelan S., Stringer S. E. (2010). Dynamic expression patterns of 6-O endosulfatases during zebrafish development suggest a subfunctionalisation event for sulf2. Dev. Dyn. 239 3312–3323. 10.1002/dvdy.22456 [DOI] [PubMed] [Google Scholar]
- Guo S. (2004). Linking genes to brain, behavior and neurological diseases: what can we learn from zebrafish? Genes Brain Behav. 3 63–74. 10.1046/j.1601-183X.2003.00053.x [DOI] [PubMed] [Google Scholar]
- Gupta-Bansal R., Frederickson R. C. A., Brunden K. R. (1995). Proteoglycan-mediated inhibition of Aβ proteolysis. A potential cause of senile plaque accumulation. J. Biol. Chem. 270 18666–18671. 10.1074/jbc.270.31.18666 [DOI] [PubMed] [Google Scholar]
- Hansen B., Longati P., Elvevold K., Nedredal G. I., Schledzewski K., Olsen R., et al. (2005). Stabilin-1 and stabilin-2 are both directed into the early endocytic pathway in hepatic sinusoidal endothelium via interactions with clathrin/AP-2, independent of ligand binding. Exp. Cell Res. 303 160–173. 10.1016/j.yexcr.2004.09.017 [DOI] [PubMed] [Google Scholar]
- Harris E. N., Weigel J. A., Weigel P. H. (2008). The human hyaluronan receptor for endocytosis (HARE/stabilin-2) is a systemic clearance receptor for heparin. J. Biol. Chem. 283 17341–17350. 10.1074/jbc.M710360200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart M., Li L., Tokunaga T., Hassell J. R., Snow A. D., Ki F., et al. (2001). Overproduction of perlecan core protein in cultured cells and transgenic mice. J. Pathol. 194 262–9. [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Crowther R. A., Jakes R., Goedert M. (1997). Alzheimer-like changes in microtubule-associated protein tau induced by sulfated glycosaminoglycans. Inhibition of microtubule binding stimulation of phosphorylation filament assembly depend on the degree of sulfation. J. Biol. Chem. 272 33118–33124. 10.1074/jbc.272.52.33118 [DOI] [PubMed] [Google Scholar]
- Hass G. (1942). Studies of amyloid II: the isolation of a polysaccharide from amyloid-bearing tissues. Arch. Path 34 92–105. [Google Scholar]
- Hernández F., Llorens-Martín M., Bolós M., Pérez M., Cuadros R., Pallas-Bazarra N., et al. (2018). New beginnings in Alzheimer’s disease: the most prevalent tauopathy. J. Alzheimer’s Dis. 64(s1) S529–S534. 10.3233/JAD-179916 [DOI] [PubMed] [Google Scholar]
- Hernández F., Pérez M., Lucas J. J., Avila J. (2002). Sulfo-glycosaminoglycan content affects PHF-tau solubility and allows the identification of different types of PHFs. Brain Res. 935 65–72. 10.1016/S0006-8993(02)02455-1 [DOI] [PubMed] [Google Scholar]
- Herndon M. E., Stipp C. S., Lander A. D. (1999). Interactions of neural glycosaminoglycans and proteoglycans with protein ligands: assessment of selectivity, heterogeneity and the participation of core proteins in binding. Glycobiology 9 143–155. 10.1093/glycob/9.2.143 [DOI] [PubMed] [Google Scholar]
- Hogl S., Van Bebber F., Dislich B., Kuhn P. H., Haass C., Schmid B., et al. (2013). Label-free quantitative analysis of the membrane proteome of Bace1 protease knock-out zebrafish brains. Proteomics 13 1519–1527. 10.1002/pmic.201200582 [DOI] [PubMed] [Google Scholar]
- Holmes B. B., DeVos S. L., Kfoury N., Li M., Jacks R., Yanamandra K., et al. (2013). Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110 E3138–E3147. 10.1073/pnas.1301440110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B. B., Diamond M. I. (2014). Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 289 19855–19861. 10.1074/jbc.R114.549295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer M., Holzapfel H. P., Zedlick D., Brückner M. K., Arendt T. (1994). Abnormally phosphorylated tau protein in Alzheimer’s disease: heterogeneity of individual regional distribution and relationship to clinical severity. Neuroscience 63 499–516. 10.1016/0306-4522(94)90546-0 [DOI] [PubMed] [Google Scholar]
- Huang M. L., Michalak A. L., Fisher C. J., Christy M., Smith R. A. A., Godula K. (2018). Small molecule antagonist of cell surface glycosaminoglycans restricts mouse embryonic stem cells in a pluripotent state. Stem Cells 36 45–54. 10.1002/stem.2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington J. A., Olson S. T., Fan B., Gettins P. G. W. (1996). Mechanism of heparin activation of antithrombin. Evidence for reactive center loop preinsertion with expulsion upon heparin binding. Biochemistry 35 8495–8503. 10.1021/bi9604643 [DOI] [PubMed] [Google Scholar]
- Huynh M. B., Villares J., Sepúlveda Díaz J. E., Christiaans S., Carpentier G., Ouidja M. O., et al. (2012). Glycosaminoglycans from aged human hippocampus have altered capacities to regulate trophic factors activities but not Aβ42 peptide toxicity. Neurobiol. Aging 33 1005.e11–1005.e22. 10.1016/j.neurobiolaging.2011.09.030 [DOI] [PubMed] [Google Scholar]
- Iaccarino L., Tammewar G., Ayakta N., Baker S. L., Bejanin A., Boxer A. L., et al. (2018). Local and distant relationships between amyloid, tau and neurodegeneration in Alzheimer’s disease. NeuroImage Clin. 17 452–464. 10.1016/j.nicl.2017.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadanza M. G., Jackson M. P., Hewitt E. W., Ranson N. A., Radford S. E. (2018). A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 19 755–773. 10.1038/s41580-018-0060-8 [DOI] [PubMed] [Google Scholar]
- Irie A., Yates E. A., Turnbull J. E., Holt C. E. (2002). Specific heparan sulfate structures involved in retinal axon targeting. Development 129 61–70. [DOI] [PubMed] [Google Scholar]
- Jho Y. S., Zhulina E. B., Kim M. W., Pincus P. A. (2010). Monte carlo simulations of tau proteins: effect of phosphorylation. Biophys. J. 99 2387–2397. 10.1016/j.bpj.2010.06.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T., Zhang J., Liu Q., Liu C.-C., Zhang L., Bu G. (2011). Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid- uptake. J. Neurosci. 31 1644–1651. 10.1523/JNEUROSCI.5491-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S., Fan J., Smith M. J., Goedert M., Amos L. A. (2003). Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 22 70–77. 10.1093/emboj/cdg001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber E., Kern U., Bonkowsky J. L., Chien C.-B., Driever W., Schweitzer J. (2009). Netrin-DCC, robo-slit, and heparan sulfate proteoglycans coordinate lateral positioning of longitudinal dopaminergic diencephalospinal axons. J. Neurosci. 29 8914–8926. 10.1523/JNEUROSCI.0568-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Wang H., Bernfield M., Gallagher J. T., Turnbull J. E. (1994). Cell surface syndecan-1 on distinct cell types differs in fine structure and ligand binding of its heparan sulfate chains. J. Biol. Chem. 269 18881–18890. [PubMed] [Google Scholar]
- Kato T., Sasaki H., Katagiri T., Sasaki H., Koiwai K., Youki H., et al. (1991). The binding of basic fibroblast growth factor to Alzheimer’s neurofibrillary tangles and senile plaques. Neurosci. Lett. 122 33–36. 10.1016/0304-3940(91)90186-W [DOI] [PubMed] [Google Scholar]
- Kim M. J., Liu I. H., Song Y., Lee J. A., Halfter W., Balice-Gordon R. J., et al. (2007). Agrin is required for posterior development and motor axon outgrowth and branching in embryonic zebrafish. Glycobiology 17 231–247. 10.1093/glycob/cwl069 [DOI] [PubMed] [Google Scholar]
- Kisilevsky R., Lemieux L. J., Fraser P. E., Kong X., Hultin P. G., Szarek W. A. (1995). Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer’s disease. Nat. Med. 1 143–148. 10.1038/nm0295-143 [DOI] [PubMed] [Google Scholar]
- Köglsberger S., Cordero-Maldonado M. L., Antony P., Forster J. I., Garcia P., Buttini M., et al. (2017). Gender-specific expression of ubiquitin-specific peptidase 9 modulates tau expression and phosphorylation: possible implications for tauopathies. Mol. Neurobiol. 54 7979–7993. 10.1007/s12035-016-0299-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuger J., Kjellén L. (2012). Heparan sulfate biosynthesis: regulation and variability. J. Histochem. Cytochem. 60 898–907. 10.1369/0022155412464972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamanna W. C., Frese M. A., Balleininger M., Dierks T. (2008). Sulf loss influences N-, 2-O-, and 6-O-sulfation of multiple heparan sulfate proteoglycans and modulates fibroblast growth factor signaling. J. Biol. Chem. 283 27724–27735. 10.1074/jbc.M802130200 [DOI] [PubMed] [Google Scholar]
- Leder I. G. (1980). A novel 3-0 sulfatase from human urine acting on methyl-2-deoxy-2-sulfamino-α-D-glucopyranoside 3-sulfate. Biochem. Biophys. Res. Commun. 94 1183–1189. 10.1016/0006-291X(80)90544-6 [DOI] [PubMed] [Google Scholar]
- Lee J. S., Von Der Hardt S., Rusch M. A., Stringer S. E., Stickney H. L., Talbot W. S., et al. (2004). Axon sorting in the optic tract requires HSPG synthesis by ext2 (dackel) and extl3 (boxer). Neuron 44 947–960. 10.1016/j.neuron.2004.11.029 [DOI] [PubMed] [Google Scholar]
- Leveugle B., Ding W., Laurence F., Dehouck M. P., Scanameo A., Cecchelli R., et al. (1998). Heparin oligosaccharides that pass the blood-brain barrier inhibit beta-amyloid precursor protein secretion and heparin binding to beta-amyloid peptide. J. Neurochem. 70 736–744. 10.1046/j.1471-4159.1998.70020736.x [DOI] [PubMed] [Google Scholar]
- Leveugle B., Scanameo A., Ding W., Fillit H. (1994). Binding of heparan sulfate glycosaminoglycan to β-amyloid peptide – Inhibition by potentially therapeutic polysulfated compounds. Neuroreport 5 1389–1392. 10.1097/00001756-199406270-00024 [DOI] [PubMed] [Google Scholar]
- Li J., Kanekiyo T., Shinohara M., Zhang Y., LaDu M. J., Xu H., et al. (2012). Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 287 44593–44601. 10.1074/jbc.M112.420224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libeu C. P., Lund-Katz S., Phillips M. C., Wehrli S., Hernáiz M. J., Capila I., et al. (2001). New insights into the heparan sulfate proteoglycan-binding activity of apolipoprotein E. J. Biol. Chem. 276 39138–39144. 10.1074/jbc.M104746200 [DOI] [PubMed] [Google Scholar]
- Lin M. Z., Schnitzer M. J. (2016). Genetically encoded indicators of neuronal activity. Nat. Neurosci. 19 1142–1153. 10.1038/nn.4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl B., Eriksson L., Lindahl U. (1995). Structure of heparan sulphate from human brain, with special regard to Alzheimer’s disease. Biochem. J. 306 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl B., Lindahl U. (1997). Amyloid-specific heparan sulfate from human liver and spleen. J. Biol. Chem. 272 26091–26094. [DOI] [PubMed] [Google Scholar]
- Lindahl U. (2007). Heparan sulfate-protein interactions – A concept for drug design? Thromb. Haemost. 98 109–115. 10.1160/TH07-04-0310 [DOI] [PubMed] [Google Scholar]
- Lindahl U., Couchman J., Kimata K., Esko J. D. (2017). Proteoglycans and Sulfated Glycosaminoglycans. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. 10.1101/GLYCOBIOLOGY.3E.017 [DOI] [PubMed] [Google Scholar]
- Lindahl U., Li J. (2009). Interactions between heparan sulfate and proteins—Design and functional implications. Int. Rev. Cell Mol. Biol. 276 105–159. 10.1016/S1937-6448(09)76003-4 [DOI] [PubMed] [Google Scholar]
- Liu C. C., Zhao N., Yamaguchi Y., Cirrito J. R., Kanekiyo T., Holtzman D. M., et al. (2016). Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med. 8:332ra44. 10.1126/scitranslmed.aad3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Shriver Z., Pope R. M., Thorp S. C., Duncan M. B., Copeland R. J., et al. (2002). Characterization of a heparan sulfate octasaccharide that binds to herpes simplex virus type 1 glycoprotein D. J. Biol. Chem. 277 33456–33467. 10.1074/jbc.M202034200 [DOI] [PubMed] [Google Scholar]
- Lopez A., Lee S. E., Wojta K., Ramos E. M., Klein E., Chen J., et al. (2017). A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 140 1128–1146. 10.1093/brain/awx005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Ye S., Kan M., McKeehan W. L. (2006). Structural specificity in a FGF7-affinity purified heparin octasaccharide required for formation of a complex with FGF7 and FGFR2IIIb. J. Cell. Biochem. 97 1241–1258. 10.1002/jcb.20724 [DOI] [PubMed] [Google Scholar]
- Mahley R. W. (1996). Heparan sulfate proteoglycan/low density lipoprotein receptor-related protein pathway involved in type III hyperlipoproteinemia and Alzheimer’s disease. Isr. J. Med. Sci. 32 414–429. [PubMed] [Google Scholar]
- Maresh G. A., Erezyilmaz D., Murry C. E., Nochlin D., Snow A. D. (1996). Detection and quantitation of perlecan mrna levels in Alzheimers disease and normal aged hippocampus by competitive reverse transcription polymerase chain reaction. J. Neurochem. 67 1132–1144. 10.1046/j.1471-4159.1996.67031132.x [DOI] [PubMed] [Google Scholar]
- Marques M. A., Tolar M., Harmony J. A. K., Crutcher K. A. (1996). A thrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity. Neuroreport 7 2529–2532. 10.1097/00001756-199611040-00025 [DOI] [PubMed] [Google Scholar]
- Mawal-Dewan M., Sen P. C., Abdel-Ghany M., Shalloway D., Racker E. (1992). Phosphorylation of tau protein by purified p34cdc28 and a related protein kinase from neurofilaments. J. Biol. Chem. 267 19705–19709. [PubMed] [Google Scholar]
- McKeehan W. L., Wu X., Kan M. (1999). Requirement for anticoagulant heparan sulfate in the fibroblast growth factor receptor complex. J. Biol. Chem. 274 21511–21514. 10.1074/JBC.274.31.21511 [DOI] [PubMed] [Google Scholar]
- Meyer B., Thunberg L., Lindahl U., Larm O., Leder I. G. (1981). The antithrombin-binding sequence of heparin studied by n.m.r. spectroscopy. Carbohydr. Res. 88 C1–C4. 10.1016/S0008-6215(00)84615-7 [DOI] [PubMed] [Google Scholar]
- Miller J. D., Cummings J., Maresh G. A., Walker D. G., Castillo G. M., Ngo C., et al. (1997). Localization of perlecan (or a perlecan-related macromolecule) to isolated microglia in vitro and to microglia/macrophages following infusion of beta-amyloid protein into rodent hippocampus. Glia 21 228–243. [DOI] [PubMed] [Google Scholar]
- Moro E., Tomanin R., Friso A., Modena N., Tiso N., Scarpa M., et al. (2010). A novel functional role of iduronate-2-sulfatase in zebrafish early development. Matrix Biol. 29 43–50. 10.1016/j.matbio.2009.09.001 [DOI] [PubMed] [Google Scholar]
- Moussavi Nik S. H., Newman M., Ganesan S., Chen M., Martins R., Verdile G., et al. (2014). Hypoxia alters expression of zebrafish microtubule-associated protein tau (mapta, maptb) gene transcripts. BMC Res. Notes 7:767. 10.1186/1756-0500-7-767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T., Vernier P., Wullimann M. F. (2004). The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res. 1011 156–169. 10.1016/j.brainres.2004.02.073 [DOI] [PubMed] [Google Scholar]
- Mueller T., Wullimann M. F. (2009). An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav. Evol. 74 30–42. 10.1159/000229011 [DOI] [PubMed] [Google Scholar]
- Mukrasch M. D., Biernat J., Von Bergen M., Griesinger C., Mandelkow E., Zweckstetter M. (2005). Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 280 24978–24986. 10.1074/jbc.M501565200 [DOI] [PubMed] [Google Scholar]
- Narindrasorasak S., Lowery D., Gonzalez-DeWhitt P., Poorman R. A., Greenberg B., Kisilevsky R. (1991). High affinity interactions between the Alzheimer’s beta-amyloid precursor proteins and the basement membrane form of heparan sulfate proteoglycan. J. Biol. Chem. 266 12878–12883. [PubMed] [Google Scholar]
- Nasevicius A., Ekker S. C. (2000). Effective targeted gene “knockdown” in zebrafish. Nat. Genet. 26 216–220. 10.1038/79951 [DOI] [PubMed] [Google Scholar]
- Neumann F. R., Bittcher G., Annies M., Schumacher B., Kröger S., Ruegg M. A. (2001). An alternative amino-terminus expressed in the central nervous system converts agrin to a type II transmembrane protein. Mol. Cell. Neurosci. 17 208–225. 10.1006/mcne.2000.0932 [DOI] [PubMed] [Google Scholar]
- Neve R. L., Harris P., Kosik K. S., Kurnit D. M., Donlon T. A. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol. Brain Res. 387 271–280. 10.1016/0169-328X(86)90033-1 [DOI] [PubMed] [Google Scholar]
- Newman M., Ebrahimie E., Lardelli M. (2014). Using the zebrafish model for Alzheimer’s disease research. Front. Genet. 5:189 10.3389/fgene.2014.00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitsuji K., Uchimura K. (2017). Sulfated glycosaminoglycans in protein aggregation diseases. Glycoconj. J. 34 453–466. 10.1007/s10719-017-9769-4 [DOI] [PubMed] [Google Scholar]
- Nogales E. (2001). Structural insights into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 69 277–302. 10.1146/annurev.biophys.30.1.397 [DOI] [PubMed] [Google Scholar]
- Nurcombe V., Ford M. D., Wildschut J. A., Bartlett P. F. (1993). Developmental regulation of neural response to FGF-1 and FGF-2 by heparan sulfate proteoglycan. Science (80) 260 103–106. 10.1126/science.7682010 [DOI] [PubMed] [Google Scholar]
- Ori A. (2008). The heparanome and regulation of cell function: structures, functions and challenges. Front. Biosci. 13:4309–4338. 10.2741/3007 [DOI] [PubMed] [Google Scholar]
- Panula P., Chen Y. C., Priyadarshini M., Kudo H., Semenova S., Sundvik M., et al. (2010). The comparative neuroanatomy and neurochemistry of zebrafish CNS systems of relevance to human neuropsychiatric diseases. Neurobiol. Dis. 40 46–57. 10.1016/j.nbd.2010.05.010 [DOI] [PubMed] [Google Scholar]
- Paquet D., Bhat R., Sydow A., Mandelkow E., Berg S., Hellberg S., et al. (2009). A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J. Clin. Invest. 119 1382–1395. 10.1172/JCI37537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patey S. J., Edwards E. A., Yates E. A., Turnbull J. E. (2006). Heparin derivatives as inhibitors of BACE-1, the Alzheimer’s β-secretase, with reduced activity against factor Xa and other proteases. J. Med. Chem. 49 6129–6132. 10.1021/jm051221o [DOI] [PubMed] [Google Scholar]
- Patey S. J., Edwards E. A., Yates E. A., Turnbull J. E. (2008). Engineered heparins: novel β-secretase inhibitors as potential Alzheimer’s disease therapeutics. Neurodegen. Dis. 5 197–199. 10.1159/000113701 [DOI] [PubMed] [Google Scholar]
- Paudel H. K., Li W. (1999). Heparin-induced conformational change in microtubule-associated protein tau as detected by chemical cross-linking and phosphopeptide mapping. J. Biol. Chem. 274 8029–8038. 10.1074/jbc.274.12.8029 [DOI] [PubMed] [Google Scholar]
- Pempe E. H., Xu Y., Gopalakrishnan S., Liu J., Harris E. N. (2012). Probing structural selectivity of synthetic heparin binding to stabilin protein receptors. J. Biol. Chem. 287 20774–20783. 10.1074/jbc.M111.320069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter L. S., Barrón E., Saperia D., Chui H. C. (1991). Association between vascular basement membrane components and the lesions of Alzheimer’s disease. J. Neurosci. Res. 30 673–681. 10.1002/jnr.490300411 [DOI] [PubMed] [Google Scholar]
- Perry G., Siedlak S. L., Richey P., Kawai M., Cras P., Kalaria R. N., et al. (1991). Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci. 11 3679–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack S. J., Sadler I. I., Hawtin S. R., Tailor V. J., Shearman M. S. (1995a). Sulfated glycosaminoglycans and dyes attenuate the neurotoxic effects of beta-amyloid in rat PC12 cells. Neurosci. Lett. 184 113–116. 10.1016/0304-3940(94)11182-I [DOI] [PubMed] [Google Scholar]
- Pollack S. J., Sadler I. I., Hawtin S. R., Tailor V. J., Shearman M. S. (1995b). Sulfonated dyes attenuate the toxic effects of β-amyloid in a structure-specific fashion. Neurosci. Lett. 197 211–214. 10.1016/0304-3940(95)11939-T [DOI] [PubMed] [Google Scholar]
- Poulain F. E., Chien C. B. (2013). Proteoglycan-mediated axon degeneration corrects pretarget topographic sorting errors. Neuron 78 49–56. 10.1016/j.neuron.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prykhozhij S. V., Caceres L., Berman J. N. (2018). New developments in CRISPR/Cas-based functional genomics and their implications for research using zebrafish. Curr. Gene Ther. 17 286–300. 10.2174/1566523217666171121164132 [DOI] [PubMed] [Google Scholar]
- Rauch J. N., Chen J. J., Sorum A. W., Miller G. M., Sharf T., See S. K., et al. (2018). Tau Internalization is regulated by 6-O sulfation on heparan sulfate proteoglycans (HSPGs). Sci. Rep. 8:6382. 10.1038/s41598-018-24904-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink E., Wullimann M. F. (2004). Connections of the ventral telencephalon (subpallium) in the zebrafish (Danio rerio). Brain Res. 1011 206–220. 10.1016/j.brainres.2004.03.027 [DOI] [PubMed] [Google Scholar]
- Roan N. R., Sowinski S., Münch J., Kirchhoff F., Greene W. C. (2010). Aminoquinoline surfen inhibits the action of SEVI (semen-derived enhancer of viral infection). J. Biol. Chem. 285 1861–1869. 10.1074/jbc.M109.066167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg R. D., Damus P. S. (1973). The purification and mechanism of action of human antithrombin-heparin cofactor. J. Biol. Chem. 248 6490–6505. [PubMed] [Google Scholar]
- Rupp B., Wullimann M. F., Reichert H. (1996). The zebrafish brain: a neuroanatomical comparison with the goldfish. Anat. Embryol. (Berl). 194 187–203. 10.1007/BF00195012 [DOI] [PubMed] [Google Scholar]
- Sanderson R. D., Turnbull J. E., Gallagher J. T., Lander A. D. (1994). Fine structure of heparan sulfate regulates syndecan-1 function and cell behavior. J. Biol. Chem. 269 13100–13106. [PubMed] [Google Scholar]
- Santana S., Rico E. P., Burgos J. S. (2012). Can zebrafish be used as animal model to study Alzheimer’s disease? Am. J. Neurodegener. Dis. 1 32–48. [PMC free article] [PubMed] [Google Scholar]
- Santarella R. A., Skiniotis G., Goldie K. N., Tittmann P., Gross H., Mandelkow E. M., et al. (2004). Surface-decoration of microtubules by human tau. J. Mol. Biol. 339 539–553. 10.1016/j.jmb.2004.04.008 [DOI] [PubMed] [Google Scholar]
- Sarrazin S., Lamanna W. C., Esko J. D. (2011). Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 3:a004952. 10.1101/cshperspect.a004952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholefield Z., Yates E. A., Wayne G., Amour A., McDowell W., Turnbull J. E. (2003). Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer’s beta-secretase. J. Cell Biol. 163 97–107. 10.1083/jcb.200303059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda-Diaz J. E., Alavi Naini S. M., Huynh M. B., Ouidja M. O., Yanicostas C., Chantepie S., et al. (2015). HS3ST2 expression is critical for the abnormal phosphorylation of tau in Alzheimer’s disease-related tau pathology. Brain 138 1339–1354. 10.1093/brain/awv056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver Z., Capila I., Venkataraman G., Sasisekharan R. (2012). Heparin and heparan sulfate: analyzing structure and microheterogeneity. Handb. Exp. Pharmacol. 207 159–176. 10.1007/978-3-642-23056-1_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D., Liu J., Blaiklock P., Shworak N. W., Bai X., Esko J. D., et al. (1999). A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99 13–22. 10.1016/S0092-8674(00)80058-6 [DOI] [PubMed] [Google Scholar]
- Sibille N., Sillen A., Leroy A., Wieruszeski J. M., Mulloy B., Landrieu I., et al. (2006). Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy. Biochemistry 45 12560–12572. 10.1021/bi060964o [DOI] [PubMed] [Google Scholar]
- Smuga-Otto K. (2018). Inner Workings: zebrafish assay forges new approach to drug discovery. Proc. Natl. Acad. Sci. U.S.A. 115 5306–5308. 10.1073/pnas.1806440115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow A. D., Mar H., Nochlin D., Sekiguchi R. T., Kimata K., Koike Y., et al. (1990). Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am. J. Pathol. 137 1253–1270. [PMC free article] [PubMed] [Google Scholar]
- Snow A. D., Sekiguchi R. T., Nochlin D., Kalaria R. N., Kimata K. (1994). Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer’s disease brain. Am. J. Pathol. 144 337–347. [PMC free article] [PubMed] [Google Scholar]
- Spillantini M. G., Tolnay M., Love S., Goedert M. (1999). Microtubule-associated protein tau, heparan sulphate and alpha-synuclein in several neurodegenerative diseases with dementia. Acta Neuropathol. 97 585–594. 10.1007/s004010051034 [DOI] [PubMed] [Google Scholar]
- Stopschinski B. E., Holmes B. B., Miller G. M., Manon V. A., Vaquer-Alicea J., Prueitt W. L., et al. (2018). Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus -synuclein and -amyloid aggregates. J. Biol. Chem. 293 10826–10840. 10.1074/jbc.RA117.000378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J. H., Cummings B. J., Cotman C. W. (1992). Localization of heparan sulfate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer’s disease. Neuroscience 51 801–813. 10.1016/0306-4522(92)90521-3 [DOI] [PubMed] [Google Scholar]
- Swinney D. C., Anthony J. (2011). How were new medicines discovered? Nat. Rev. Drug Discov. 10 507–519. 10.1038/nrd3480 [DOI] [PubMed] [Google Scholar]
- Thacker B. E., Xu D., Lawrence R., Esko J. D. (2014). Heparan sulfate 3-O-sulfation: a rare modification in search of a function. Matrix Biol. 35 60–72. 10.1016/j.matbio.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunberg L., Bäckström G., Lindahl U. (1982). Further characterization of the antithrombin-binding sequence in heparin. Carbohydr. Res. 100 393–410. 10.1016/S0008-6215(00)81050-2 [DOI] [PubMed] [Google Scholar]
- Timmer N. M., van Horssen J., Otte-Holler I., Wilhelmus M. M. M., David G., van Beers J., et al. (2009). Amyloid β induces cellular relocalization and production of agrin and glypican-1. Brain Res. 1260 38–46. 10.1016/j.brainres.2008.12.063 [DOI] [PubMed] [Google Scholar]
- Timmer N. M., van Dijk L., van der Zee C. E., Kiliaan A., de Waal R. M. W., Verbeek M. M. (2010). Enoxaparin treatment administered at both early and late stages of amyloid β deposition improves cognition of APPswe/PS1dE9 mice with differential effects on brain Aβ levels. Neurobiol. Dis. 40 340–347. 10.1016/j.nbd.2010.06.008 [DOI] [PubMed] [Google Scholar]
- Tomasiewicz H. G., Flaherty D. B., Soria J. P., Wood J. G. (2002). Transgenic zebrafish model of neurodegeneration. J. Neurosci. Res. 70 734–745. 10.1002/jnr.10451 [DOI] [PubMed] [Google Scholar]
- Torres M., Price S. L., Fiol-Deroque M. A., Marcilla-Etxenike A., Ahyayauch H., Barceló-Coblijn G., et al. (2014). Membrane lipid modifications and therapeutic effects mediated by hydroxydocosahexaenoic acid on Alzheimer’s disease. Biochim. Biophys. Acta – Biomembr. 1838 1680–1692. 10.1016/j.bbamem.2013.12.016 [DOI] [PubMed] [Google Scholar]
- Van Gool D., David G., Lammens M., Baro F., Dom R. (1993). Heparan sulfate expression patterns in the amyloid deposits of patients with Alzheimer’s and Lewy body type dementia. Dementia 4 308–314. [DOI] [PubMed] [Google Scholar]
- Van Horssen J., Kleinnijenhuis J., Maass C. N., Rensink A. A. M., Otte-Höller I., David G., et al. (2002). Accumulation of heparan sulfate proteoglycans in cerebellar senile plaques. Neurobiol. Aging 23 537–545. 10.1016/S0197-4580(02)00010-6 [DOI] [PubMed] [Google Scholar]
- Vanpouille C., Deligny A., Delehedde M., Denys A., Melchior A., Liénard X., et al. (2007). The heparin/heparan sulfate sequence that interacts with cyclophilin B contains a 3-O-sulfated N-unsubstituted glucosamine residue. J. Biol. Chem. 282 24416–24429. 10.1074/jbc.M701835200 [DOI] [PubMed] [Google Scholar]
- Verbeek M. M., Otte-Höller I., Van Den Born J., Van Den Heuvel L. P. W. J., David G., Wesseling P., et al. (1999). Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer’s disease brain. Am. J. Pathol. 155 2115–2125. 10.1016/S0002-9440(10)65529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virchow R. (1855). Zur cellulose-frage. Arch. Pathol. Anat. Physiol. Klin. Med. 8 140–144. 10.1007/BF01935322 [DOI] [Google Scholar]
- Walzer M., Lorens S., Hejna M., Fareed J., Hanin I., Cornelli U., et al. (2002). Low molecular weight glycosaminoglycan blockade of beta-amyloid induced neuropathology. Eur. J. Pharmacol. 445 211–220. 10.1016/S0014-2999(02)01759-4 [DOI] [PubMed] [Google Scholar]
- Wang F., Wolfson S. N., Gharib A., Sagasti A. (2012). LAR receptor tyrosine phosphatases and HSPGs guide peripheral sensory axons to the skin. Curr. Biol. 22 373–382. 10.1016/j.cub.2012.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Ding K. (2014). Proteoglycans and glycosaminoglycans in misfolded proteins formation in Alzheimer’s disease. Protein Pept. Lett. 21 1048–1056. 10.2174/0929866521666140626095145 [DOI] [PubMed] [Google Scholar]
- Wang P., Lo Cascio F., Gao J., Kayed R., Huang X. (2018). Binding and neurotoxicity mitigation of toxic tau oligomers by synthetic heparin like oligosaccharides. Chem. Commun. 6:72. 10.1039/C8CC05072D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warford J., Doucette C. D., Hoskin D. W., Easton A. S. (2014). Murine T cell activation is regulated by surfen (bis-2-methyl-4-amino-quinolyl-6-carbamide). Biochem. Biophys. Res. Commun. 443 524–530. 10.1016/j.bbrc.2013.11.119 [DOI] [PubMed] [Google Scholar]
- Warford J. R., Lamport A.-C., Clements D. R., Malone A., Kennedy B. E., Kim Y., et al. (2018). Surfen, a proteoglycan binding agent, reduces inflammation but inhibits remyelination in murine models of Multiple Sclerosis. Acta Neuropathol. Commun. 6 1–21. 10.1186/s40478-017-0506-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K. H., Liu I. H. (2014). Heparan sulfate glycosaminoglycans modulate migration and survival in zebrafish primordial germ cells. Theriogenology 81 1275–1285. 10.1016/j.theriogenology.2014.02.009 [DOI] [PubMed] [Google Scholar]
- Whiteford J. R., Ko S., Lee W., Couchman J. R. (2008). Structural and cell adhesion properties of zebrafish syndecan-4 are shared with higher vertebrates. J. Biol. Chem. 283 29322–29330. 10.1074/jbc.M803505200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B. K., Yuan R. Y., Lien H. W., Hung C. C., Hwang P. P., Chen R. P. Y., et al. (2016). Multiple signaling factors and drugs alleviate neuronal death induced by expression of human and zebrafish tau proteins in vivo. J. Biomed. Sci. 23:25. 10.1186/s12929-016-0237-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D., Esko J. D. (2014). Demystifying heparan sulfate-protein Interactions. Annu. Rev. Biochem. 83 129–157. 10.1146/annurev-biochem-060713-035314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabe T., Hata T., He J., Maeda N. (2005). Developmental and regional expression of heparan sulfate sulfotransferase genes in the mouse brain. Glycobiology 15 982–993. 10.1093/glycob/cwi090 [DOI] [PubMed] [Google Scholar]
- Yamada S., Yoshida K., Sugiura M., Sugahara K., Khoo K. H., Morris H. R., et al. (1993). Structural studies on the bacterial lyase-resistant tetrasaccharides derived from the antithrombin III-binding site of porcine intestinal heparin. J. Biol. Chem. 268 4780–4787. [PubMed] [Google Scholar]
- Yamada T., Kerever A., Yoshimura Y., Suzuki Y., Nonaka R., Higashi K., et al. (2017). Heparan sulfate alterations in extracellular matrix structures and fibroblast growth factor-2 signaling impairment in the aged neurogenic niche. J. Neurochem. 142 534–544. 10.1111/jnc.14081 [DOI] [PubMed] [Google Scholar]
- Yanagishita M., Hascall V. C. (1992). Cell surface heparan sulfate proteoglycans. J. Biol. Chem. 267 9451–9454. [PubMed] [Google Scholar]
- Yang S.-D., Yu J. S., Shiah S.-G., Huang J.-J. (1994). Protein kinase FA/glycogen synthase kinase-3α after heparin potentiation phosphorylates τ on sites abnormally phosphorylated in Alzheimer’s disease brain. J. Neurochem. 63 1416–1425. 10.1046/j.1471-4159.1994.63041416.x [DOI] [PubMed] [Google Scholar]
- Ye S., Luo Y., Lu W., Jones R. B., Linhardt R. J., Capila I., et al. (2001). Structural basis for interaction of FGF-1, FGF-2, and FGF-7 with different heparan sulfate motifs. Biochemistry 40 14429–14439. 10.1021/bi011000u [DOI] [PubMed] [Google Scholar]
- Yoshida H., Goedert M. (2012). Phosphorylation of microtubule-associated protein tau by AMPK-related kinases. J. Neurochem. 120 165–176. 10.1111/j.1471-4159.2011.07523.x [DOI] [PubMed] [Google Scholar]
- Yu C., Griffiths L. R., Haupt L. M. (2017). Exploiting heparan sulfate proteoglycans in human neurogenesis-controlling lineage specification and fate. Front. Integr. Neurosci. 11:28. 10.3389/fnint.2017.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebrower M., Beeber C., Kieras F. J. (1992). Characterization of proteoglycans in alzheimer’s disease fibroblasts. Biochem. Biophys. Res. Commun. 184 1293–1300. 10.1016/S0006-291X(05)80023-3 [DOI] [PubMed] [Google Scholar]
- Zebrower M., Kieras F. J. (1993). Are heparan sulphate (HS) sulphotransferases implicated in the pathogenesis of alzheimer’s disease? Glycobiology 3 3–5. 10.1093/glycob/3.1.3 [DOI] [PubMed] [Google Scholar]
- Zhang F., Zhang Z., Thistle R., McKeen L., Hosoyama S., Toida T., et al. (2009). Structural characterization of glycosaminoglycans from zebrafish in different ages. Glycoconj. J. 26 211–218. 10.1007/s10719-008-9177-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Beeler D. L., Lawrence R., Lech M., Liu J., Davis J. C., et al. (2001a). 6-O-sulfotransferase-1 represents a critical enzyme in the anticoagulant heparan sulfate biosynthetic pathway. J. Biol. Chem. 276 42311–42321. 10.1074/jbc.M101441200 [DOI] [PubMed] [Google Scholar]
- Zhang L., Lawrence R., Schwartz J. J., Bai X., Wei G., Esko J. D., et al. (2001b). The effect of precursor structures on the action of glucosaminyl 3-O-sulfotransferase-1 and the biosynthesis of anticoagulant heparan sulfate. J. Biol. Chem. 276 28806–28813. 10.1074/jbc.M100204200 [DOI] [PubMed] [Google Scholar]
- Zhao J., Huvent I., Lippens G., Eliezer D., Zhang A., Li Q., et al. (2017). Glycan determinants of heparin-tau interaction. Biophys. J. 112 921–932. 10.1016/j.bpj.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng-Fischhöfer Q., Biernat J., Mandelkow E. M., Illenberger S., Godemann R., Mandelkow E. (1998). Sequential phosphorylation of tau by glycogen synthase kinase-3β and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 252 542–552. 10.1046/j.1432-1327.1998.2520542.x [DOI] [PubMed] [Google Scholar]
- Zhu H., Yu J., Kindy M. S. (2001). Inhibition of amyloidosis using low-molecular-weight heparins. Mol. Med. 7 517–522. 10.1007/BF03401857 [DOI] [PMC free article] [PubMed] [Google Scholar]