

Abstract
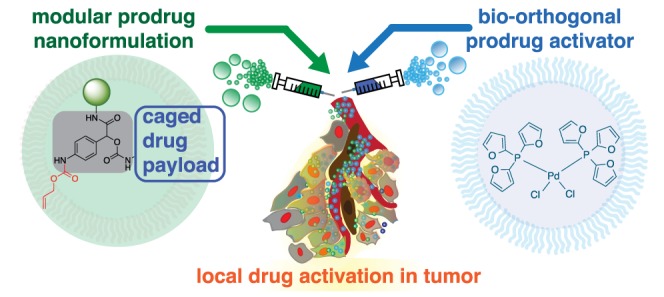
Prodrug strategies that facilitate localized and controlled activity of small-molecule therapeutics can reduce systemic exposure and improve pharmacokinetics, yet limitations in activation chemistry have made it difficult to assign tunable multifunctionality to prodrugs. Here, we present the design and application of a modular small-molecule caging strategy that couples bioorthogonal cleavage with a self-immolative linker and an aliphatic anchor. This strategy leverages recently discovered in vivo catalysis by a nanoencapsulated palladium compound (Pd-NP), which mediates alloxylcarbamate cleavage and triggers release of the activated drug. The aliphatic anchor enables >90% nanoencapsulation efficiency of the prodrug, while also allowing >104-fold increased cytotoxicity upon prodrug activation. We apply the strategy to a prodrug formulation of monomethyl auristatin E (MMAE), demonstrating its ability to target microtubules and kill cancer cells only after selective activation by Pd-NP. Computational pharmacokinetic modeling provides a mechanistic basis for the observation that the nanotherapeutic prodrug strategy can lead to more selective activation in the tumor, yet in a manner that is more sensitive to variable enhanced permeability and retention (EPR) effects. Combination treatment with the nanoencapsulated MMAE prodrug and Pd-NP safely blocks tumor growth, especially when combined with a local radiation therapy regimen that is known to improve EPR effects, and represents a conceptual step forward in prodrug design.
Keywords: translational nanomedicine, neo-adjuvant tumor priming, systems pharmacology, drug delivery, doxorubicin, mononuclear phagocyte system, macrophage
Bioorthogonal chemical reactions are increasingly developed as tools for the controlled delivery and activation of therapeutics, molecular imaging or detection agents, and as synthetic biology reagents.1 “Ligation” reactions, for example, based on stepwise administration of a pretargeting agent and a complementary imaging2 or drug3 ligand, have demonstrated the potential of using bioorthogonal approaches such as inverse electron-demand Diels–Alder reaction (IEDDA) to enhance selective targeting in models of cancer. Alternatively, bioorthogonal bond cleavage reactions1 may employ a variety of strategies, including (i) photoinduced decaging using nitroaromatics,4 (ii) deallylation using palladium5 and ruthenium catalysts,6 (iii) depropargylation using palladium7 or gold8 particles, (iv) IEDDA-induced “click-to-release”,9 and (v) cleavage induced by strain-promoted alkene–azide cycloaddition.10 These strategies have matured to the point of becoming promising tools for activating caged prodrugs.
Prodrugs are a proven route to limiting exposure in off-target tissues, especially for cytotoxic anticancer compounds such as microtubule-targeting agents. Drug caging, typically by protecting a primary amine critical for drug activity, can be used to restrict drug action until the prodrug is activated by a spatiotemporally controlled deprotection reaction. Nanoparticle formulation can achieve the same goals, and it is appealing, in principle, to combine the two approaches for controlling exposure in target vs off-target tissues. Sufficient drug activation at the target disease site is often a limiting factor for both prodrug and nanoparticle (NP) strategies. Reactants are required to maintain chemical stability until they reach the target, at which point they must sufficiently react to achieve therapeutically active drug concentrations. Compared to click-chemistry approaches, bioorthogonal catalysts have been attractive for their diverse reactivity and efficient activation of caged compounds. Unfortunately, most strategies with transition-metal catalysts have relied on microparticles,8 elemental powders, resins,11 and for traditional synthetic chemistry applications, simple salts or phenylphosphines, all of which have issues with biocompatibility, stability, toxicity, and systemic bioavailability. As a result, in vivo demonstration of bioorthogonal catalysis has been limited, especially as applied to nanoformulated drugs.
Recently, a nanoencapsulated palladium catalyst (Pd-NP) was reported to overcome these issues using bis[tri(2-furyl)phosphine] palladium(II) dichloride, PdCl2(TFP)2, in a biocompatible poly(lactic-co-glycolic acid)-b-polyethylene glycol platform.5 The 60 nm Pd-NP formulation demonstrated stability in biological solutions and exploited the “enhanced permeability and retention” (EPR) effect to passively accumulate in solid tumors following systemic administration. The EPR effect is further exploited by co-administering a separate nanoformulation of prodrug, which cooperatively reduces off-target drug activation and toxicity.5 Pd-NP has been both safe and effective at locally activating prodrugs in xenograft tumors, and its first demonstration relied on model allyloxycarbonyl (Alloc) and propargyloxycarbonyl (Poc) derivatives of the DNA-damaging chemotherapeutic, doxorubicin (DOX). Although effective, this amphiphilic prodrug was nanoencapsulated with relatively poor efficiency (22%), and its relatively low potency in cell killing required high doses.5 Furthermore, other drugs may be completely incompatible for nanoencapsulation or activity caging with this approach. Therefore, a need exists to expand the repertoire of bioorthogonal cleavage functionality, thereby facilitating improved prodrug engineering—in this case, to improve nanoencapsulation efficiency and cytotoxic turn-on.
This work presents the design of a bioorthogonal cleavage strategy based on coupling palladium-catalyzed deallylation with a self-immolative linker, which has been functionalized with an aliphatic anchor for efficient nanoencapsulation and blockage of prodrug action. This strategy provides a modular platform for adding functionality to prodrugs. In this application, prodrug nanoencapsulation efficiency is increased to >90% and >104 fold-increase in cytotoxicity is realized upon prodrug activation. As a proof-of-principle, we design a prodrug of the microtubule targeting agent, monomethyl auristatin E (MMAE). MMAE is typical of a drug that is too toxic for systemic delivery, but that has met with success using tumor-selective targeting mostly in antibody-drug conjugates (ADCs). However, ADCs are very limited in the on-target exposure that they can achieve. We therefore formulated caged MMAE as a NP using alkyl chain immobilization, thus combining two principles for improving tumor vs whole body exposure. Upon activation by Pd-NP, caged MMAE (C16proMMAE) disrupts microtubule activity in live cells and becomes cytotoxic at <100pM. C16proMMAE safely blocks tumor growth in mouse models of cancer when combined with Pd-NP. Computational modeling suggests that the dual NP strategy can limit systemic exposure of toxic activated drugs relative to what is achieved in the tumor, but especially relies on the EPR effect. To exploit this reliance, we used single low-dose radiation to enhance EPR, which led to synergistic tumor shrinkage when combined with the prodrug strategy. Overall, these results advance bioorthogonal catalysis toward clinical applicability and expand the possibilities for prodrug design.
Results/Discussion
Nanoparticulate Prodrug Design, Optimization, and Characterization
The overall design for prodrug multifunctionality lies in a central three-branched self-immolative linker (SIL) based on 4-aminomandelic acid (Figure 1). The NH2-trigger is protected by the palladium-reactive Alloc group, while the benzylic position and the carboxyl group accommodate both the drug and an additional functional moiety, respectively, which in this case was used to tune hydrophobicity based on the particular application. Upon palladium-mediated cleavage of Alloc, self-immolation of the linker leads to drug release via rapid 1,6-elimination (Figure 1).
Figure 1.

Overview of modular prodrug design strategy. A self-immolative linker (gray) bridges three modular functional aspects of an inactive nontoxic prodrug: a bioorthogonally cleavable protective group (shown here in red as allyloxycarbonyl, alloc) that is removed upon exposure to a triggering agent, a nanoencapsulation anchor (shown here in green using an aliphatic C16 chain), and the caged drug payload (blue). A bioorthogonal activating agent, here using Pd-NP based on the polymeric micellar encapsulation of PdCl2(TFP)2, leads to drug uncaging and activation in a spatiotemporally controlled manner.
As a first test, we designed a fluorogenic probe to screen reactivity against a panel of palladium compounds with organic ligands that were discovered from previous studies to be active in physiological solutions.5 The central SIL and Alloc groups were used to cage 4-methyl-7-aminocoumarin (AMC), with addition of a polyethylene glycol (PEG) side chain to improve solubility, yielding Alloc-SIL-PEG4-AMC (Figure S1a). Bioorthogonal activation, monitored by fluorescence turn-on (Figure S1b), was screened in Hank’s buffered saline solution (HBSS) and minimal essential medium (MEM), two physiologically relevant aqueous solutions that are ubiquitous in mammalian cell culture modeling. Consistent with previous studies of Alloc- and Poc-deprotection,5 PdCl2(TFP)2 was most efficient, achieving >75% yield in HBSS (Figure S1c).
Using PdCl2(TFP)2, we measured cleavage kinetics of the Alloc-SIL caging strategy and compared them to bis-alloc-rhodamine-110 (Alloc2R110) deprotection, as a previously used model prodrug substrate.5 Encouragingly, Pd-NP uncaged Alloc-SIL-PEG4-AMC approximately 1.5-fold faster than Alloc2R110 (based on calculated second-order rate constants, Figure S1d). Thus, the Alloc-SIL strategy enables multifunctionality without compromising reaction kinetics.
We next applied the prodrug strategy to two model anticancer therapies, MMAE and DOX. The Alloc-SIL group was employed as above, but using a C16 aliphatic anchor rather than PEG in order to facilitate efficient nanoencapsulation into a clinically relevant polymeric micellar formulation containing a hydrophobic PLGA-based core. The resulting lipophilic prodrugs Alloc-SIL-C16-MMAE (“C16proMMAE”, Figure 2a) and Alloc-SIL-C16-DOX (“C16proDOX”, Figure 2b) were both nanoencapsulated with >90% efficiency with acceptable size and polydispersity, shown by transmission electron microscopy (TEM, Figure 2c–d) and dynamic light scattering (Figure S2a). Without the C16 anchor, the amphiphilic DOX (cLogP = −0.7) exhibited only moderate encapsulation (22%),5 and encapsulation of parent MMAE was undetectable using the same nanoprecipitation strategy. The C16 anchored prodrug nanoformulations were stable, with no increase in size or polydispersity after 72 h at 37 °C in PBS; size was uniform with PDI of 0.11–0.13 throughout (Figure S2b). Over 72 h at 37 °C in PBS, release of the prodrug payload from the nanoformulation was 20% ± 6% and 9% ± 1% for C16proDOX and C16proMMAE, respectively (n = 3).
Figure 2.

Caged MMAE and DOX encapsulate into nanoparticles and are selectively cytotoxic in the presence of the Pd-NP bioorthogonal trigger. (a–b) Chemical structure of caged MMAE (a) and DOX (b), color-coded according to the scheme in Figure 1. (c–d) TEM imaging of C16proMMAE (c) and C16proDOX (d) encapsulated in a formulation of PLGA-PEG polymeric micelles. Particle diameters were quantified according to their distribution (black bars) and Gaussian fit (red curve), with representative images shown at right (scale bar, 100 nm). (e–f) Cytotoxicity was determined for caged MMAE (e) and DOX (f) at the indicated concentration (x-axis), in the presence of varying amounts of Pd-NP bioorthogonal trigger, over 72 h treatment using HT1080 fibrosarcoma cells (n = 2, data are means ± s.e.m.). Parent noncaged compounds were also tested as controls (black curves).
Bioorthogonally Triggered in Vitro Cytotoxicity of the Prodrug
In addition to enhancing nanoencapsulation efficiency, the SIL strategy further reduced cytotoxicity of DOX in its caged form, tested using HT1080 fibrosarcoma cancer cells (a model extensively characterized for its in vitro and in vivo responsiveness to prodrug nanoformulations). Alloc caging increased the concentration at which 50% of cells died in a resazurin-based cytotoxicity assay (IC50) from 0.1 μM to 18 μM; however, the IC50 was not reached at concentrations up to 50 μM for the C16proDOX compound. The concentration at which 20% of cells died (IC20) was roughly 15-fold higher than for the previously described prodrug Alloc-DOX5 and 1900-fold higher than for uncaged DOX (Figure S3). For both prodrug formulations, co-incubation of cells with Pd-NP restored drug cytotoxicity (Figure S3c).
Compared to the DOX prodrugs, C16proMMAE exhibited even greater potency upon Pd-NP activation, showing a >104-fold increase in IC50 to 15 pM, which is comparable to cytotoxicity of the uncaged parent compound, MMAE (Figure 2e–f). C16proMMAE shows >10-fold enhanced cytotoxicity when incubated with sub-micromolar Pd-NP, which is promising considering 5–7.5 μM tumoral Pd-NP concentrations have been safely achieved in xenograft tumor models.5 Based on cytotoxicity, the level of C16proMMAE activation approaches >90% as Pd-NP concentration is increased to 50 μM. Although this concentration is high for typical small-molecule therapeutics, past studies have shown it to be relatively nontoxic (typical IC50 > 100 μM),5 and it is modest when considering that many transition-metal bioorthogonal catalysts, including those based on Pd, are used as heterogeneous resins and implants that often employ much larger doses of metal by mass.12
Mechanisms of Cellular NP Uptake and Activity
We next investigated the mechanism of cellular prodrug uptake. HT1080 cells were made to transgenically express Rab7a-RFP and Lamp1-RFP fluorescent fusion proteins, which localize to the late endosome and lysosome, respectively. After 24 h treatment with fluorescent NPs based on the C16 prodrug formulation, co-localization was quantified between the NPs and Rab7a or Lamp1 positive vesicles (Figures 3a and S4a). Fluorescence microscopy revealed that NPs accumulated at high levels in vesicle-sized puncta within cells (Figure S4a). Approximately 1/3 of these NP puncta were located in Rab7a-RFP+ vesicles, while roughly 2/3 were associated with Lamp1-RFP+ vesicles, suggesting NP uptake through endosomal/lysosomal pathways. Co-treatment with inhibitors of macropinocytosis (5-(N-ethyl-N-isopropyl)amiloride, EIPA) and actin-dependent processes including macropinocytosis/endocytosis (latrunculin A and cytochalasin D as inhibitors of actin polymerization) acutely reduced the uptake of NPs (Figure 3b). Broad inhibition of kinase signaling (via staurosporine), which is thought to be especially important for macropinocytic uptake in RAS mutant cancer cells such as HT1080,13 also decreased NP accumulation. These results collectively suggest that uptake of PLGA-PEG NPs, similar in structure to Pd-NP and C16 prodrug NPs, occurs through actin-dependent endocytic/macropinocytic processes that lead to predominant lysosomal accumulation.
Figure 3.

Imaging reveals in vitro pathways of cell uptake and C16proDOX activation. (a) HT1080 tumor cells expressing either Rab7a-RFP or Lamp1-RFP fusion proteins were incubated with a fluorescently labeled NP based on the prodrug formulation (PLGA-PEG + PLGA-BODIPY630) and imaged 24 h later. Co-localization was determined by tabulating the fraction of NP-positive puncta (vesicles) in cells that contained high RFP expression or not (labeled red and gray, respectively; n > 10 cells, bars are mean ± std. dev.). (b) Following 30 min pretreatment with the indicated compounds, HT1080 cells were treated with the same NP as in (a) and imaged 3 h later (n > 6 biological replicates; mean ± s.e.m.). (c–d) HT1080 tumor cells were treated with C16proDOX, DOX, or Pd-NP with C16proDOX for 24 h and then imaged by fluorescence microscopy (representative images shown in (c)) to evaluate subcellular drug accumulation based on endogenous anthracycline fluorescence (scale bar, 20 μm). Yellow lines (c) denote representative examples of how fluorescence intensity profiles were quantified from line-scans through cells (d), shown as means (thick line) ± s.e.m. (shading; n > 10), for hoechst (green) and anthracycline fluorescence (red).
Past reports have delved into mechanisms of intracellular hydrolytic and enzymatic PLGA degradation for controlled drug release.13 While environmentally sensitive polymers have been successfully used in the past to facilitate endosomal escape, here these strategies did not appear necessary for prodrug activation and cytotoxic action. Despite endosomal/lysosomal NP accumulation, Pd-NP co-treatment was still able to restore a large fraction of the prodrug’s cytotoxicity, such that its potency approaches that achieved with the parent drug (especially for C16proMMAE, Figure 2e). Prodrug activation can occur both extracellularly and intracellularly depending on relative pharmacokinetics and rates of prodrug release from the NP vehicle. In the models used here, we infer that the majority of C16prodrug activation occurs intracellularly, given its slow extracellular release rate from the NP vehicle compared with time scales of pharmacokinetic clearance; <10% of C16proDOX is activated when Pd-NP and prodrug-NPs are co-incubated in 37 °C PBS (measured by HPLC as in Figure S5), and by 72 h, the majority of NPs have been systemically cleared and taken up by cells, at least in the xenograft models used. Previous studies indicate Pd-mediated prodrug activation can occur extracellularly, including in the interstitium of tumor xenografts.5 However, the prodrug in those cases did not contain a C16 anchor, therefore leading to roughly 90% in vitro prodrug release from the NP vehicle into solution by 72 h at 37 °C in PBS. In contrast, <10% of C16proMMAE is released in vitro into solution after 72 h (Figure S2), which corresponds to a 20-fold slower kinetic release rate. Furthermore, past work with C16-anchored fluorescent prodrugs encapsulated in similar PLGA-PEG NPs has shown direct cellular NP uptake to be a dominant component of delivery, including in tumor xenografts and in the HT1080 model.14
Several recent studies have implicated lysosomal pH as a contributing factor in payload release, using chloroquine (CQ) as an inhibitor of endosomal acidification and of autophagosome fusion with lysosomes.15,16 We co-treated HT1080 cells with CQ, Pd-NP, and C16proDOX to understand whether CQ could block catalytic prodrug activation. In agreement with previous studies, we found that CQ treatment led to the enhanced accumulation of NPs in distinctly large vesicles consistent with the induction of LC3+ autophagosome enrichment (Figure S4b).15,16 Despite this morphological change, only modest impact was observed in the cytotoxicity of combined Pd-NP and C16proDOX treatment, suggesting at least in this in vitro model that CQ effects, including its impact on lysosomal pH, are not substantial enough to completely block Pd-mediated prodrug activation (Figure S4c). Moreover, our past work has indicated that low pH is not required for release and activity of Pd from its NP vehicle.5 While further investigation of the subcellular mechanisms by which Pd-NP activates prodrugs extends beyond the scope of this manuscript, data at hand suggest a model of endosomal/lysosomal accumulation and gradual liberation of the prodrug and catalyst from the NP vehicles, allowing them to react with each other. Controlled prodrug bioorthogonal cleavage then leads to the formation of an active compound that is able to escape sequestration and act on its intracellular target. Past work using fluorescent C16-anchored prodrugs within PLGA-PEG nanoformulations has highlighted how drug payloads can freely escape lysosomal sequestration and even act on neighboring cells (including in HT1080 tumors), even though their polymeric vehicle remains intracellularly confined.14
Imaging the Molecular Action of Pd-Activated Prodrugs
The intrinsic fluorescence of DOX enabled us to track where the prodrug actually accumulated in cells with and without activation by Pd-NP. Uncaged parent DOX is capable of covalently binding DNA via the 3′-NH2 group of the daunosamine sugar moiety, thus leading to topoisomerase II disruption and DNA damage. Consequently, covalently reacting and DNA-intercalating DOX primarily accumulates in the nuclei of cancer cells. However, fluorescence microscopy in HT1080 cancer cells showed that lipophilic C16proDOX accumulated primarily in cytoplasmic/perinuclear cellular compartments consistent with late endosomal and lysosomal uptake but not in the nucleus itself (Figure S5a). Encouragingly, co-incubation of cells with Pd-NP led to co-localization of the catalyst with its prodrug substrate in the perinuclear cellular compartment (Figure S5a–b) and caused a detectable increase of drug in the nucleus compared to the cytoplasm. This suggests that Pd-mediated prodrug activation leads to enhanced DNA association of the prodrug (Figure 3c). HPLC fluorescence detection in lysate of treated cells confirmed a decrease in intracellular prodrug concentration when cells were co-treated with Pd-NP (Figure S5c–d). Residual cytoplasmic/lysosomal accumulation of C16proDOX was observed despite Pd-NP co-treatment, although this was also observed to some degree for DOX treatment as well (Figure 3, S5). These results suggest successful intracellular activation of C16proDOX by Pd-NP.
We next studied how C16proMMAE and its subsequent activation compares to the biological behavior of the parent compound MMAE, a well understood antimitotic agent that blocks the polymerization of tubulin and consequently inhibits cell division. Across a panel of four cancer cell lines, C16proMMAE exhibited no detectable impact on cell growth at concentrations ≤10 μM (Figure S6a). However, its co-treatment with Pd-NP led to cytotoxic responses that closely mirrored those of the parent compound in terms of both the concentration at which 50% of the effect was observed (IC50) and the maximum inhibitory effect that was achieved (Emax; Figure 4a–c). Thus, at the cellular level, C16proMMAE behaves nearly identically to MMAE once activated by Pd-NP (R2 > 0.95, Figure 4b–c). Of note, Pd-NP by itself did not detectably impact cell growth at concentrations up to 50 μM for three of the four tested cell lines (Figure S6b), consistent with past reports and its known safety profile.5
Figure 4.

Caged MMAE only disrupts microtubule dynamics in the presence of Pd-NP in live cancer cells. (a–c) Cytotoxic drug effects were quantified by a resazurin-based cell viability assay, 72 h post-treatment. A representative dose–response curve using the ES2 cell line and MMAE illustrate Emax and IC50 calculations (a; n = 2, mean ± s.e.m.), which were then compared across multiple cell lines and between treatments with MMAE vs the combination of 50 μM Pd-NP and C16proMMAE (b–c; two-tailed t test). See Figure S6 for full data. (d) HT1080 cells transgenically expressing EB3-mApple were confocally imaged over time to monitor the dynamics of microtubule plus-end tips in live cells (left). EB3 microtubule “comets” were automatically detected and computationally tracked (center; pseudocolored according to comet speed) following drug treatment. Scale bar, 20 μm. See Movie S1 for representative movies of all conditions. (e–f) Neither the average number of tracks detected (e) nor the speed of those tracks moving in cells (f) substantially changed with either 1 μM C16proMMAE or 35 μM Pd-NP treatment alone, but both treatments together eliminated all EB3 comets in a manner similar to the parent uncaged drug (median ± i.q.r.; 24 h treatment).
To examine C16proMMAE effects at the molecular level, we used confocal microscopy of growing microtubule ends as a biomarker of drug actions in live cells. We engineered the HT1080 cancer cell line to transgenically express a red fluorescent fusion protein, EB3-mApple. The end-binding protein 3 (EB3) is also known as microtubule-associated protein RP/EB family member 3 (MAPRE3) and binds to the plus-end of growing microtubules (Figure 4d). As a result, EB3-fluorescent protein fusions are widely used for studying microtubule dynamics in live cells, with plus-ends visible as microtubule “comets” transiting the cell.17 HT1080-EB3-mApple cells displayed microtubule comets that were not substantially perturbed by individual treatment with either Pd-NP or C16proMMAE, either according to microtubule comet prevalence in cells (Figure 4e) or their growth speed (Figure 4f). However, the combination of both led to complete elimination of visible comets, just as observed with the parent compound MMAE. LC/MS analysis confirmed Pd-NP causes generation of MMAE from the prodrug (Figure S5). Together, these results suggest that MMAE caging was effective at preventing microtubule perturbation and that activation by Pd-NP restored its microtubule disruption capacity (Figure 4; Movie S1).
Safety and Efficacy Using Human Xenograft and Syngeneic Mouse Models of Cancer
Given the success of Pd-NP in activating C16proMMAE, we next tested its ability to safely and effectively treat tumors in two complementary mouse models of cancer: subcutaneous HT1080 xenograft tumors in nu/nu mice, and MC38 murine colon adenocarcinoma tumors grown intradermally in immunocompetent C57BL/6 mice. Upon palpable tumor formation, HT1080-bearing animals were treated with doses of either Pd-NP, C16proMMAE NP, or the combination of the two by intravenous injection. It was previously determined that staggering the administration of Pd-NP and a subsequent prodrug NP administration by several hours (as opposed to co-injecting both NPs together) could lead to more selective tumoral accumulation of activated prodrug.5 Indeed, although Pd accumulates in the clearance organs (liver, spleen, and kidney) in mice bearing HT1080 tumors, staggered administration of the prodrug NP leads to comparably lower off-target accumulation of the activated prodrug, especially relative to levels of prodrug activation in the tumor (Figure S7a).5 Using the prescribed 5 h dose staggering scheme, the dual treatment of Pd-NP and C16proMMAE NP successfully blocked tumor growth (Figure 5a), while animals receiving either Pd-NP or C16proMMAE NP as single-treatments saw no change in their tumor growth compared to the untreated cohort (Figure 5a; P > 0.05; also see ref (5). Similarly, combined Pd-NP and C16proMMAE treatment was effective at slowing tumor growth in the MC38 tumor model (Figure 5b). In fact, in both models tumor growth was completely blocked at 2 days following the second round of Pd-NP and C16proMMAE treatment (Figure 5c–d). However, the treatment was not curative, and a fraction of tumors eventually resumed growth (individual tumors growth curves are shown in Figure S7b–c).
Figure 5.

Dual Pd-NP and C16proMMAE treatment safely blocks tumor growth in multiple tumor models. (a–b) HT1080 (a) and MC38 (b) tumors were treated with Pd-NP, C16proMMAE, or the combination of the two at the indicated time points (red arrows) following tumor formation. Tumor volumes were monitored over time by caliper (n ≥ 6, means ± s.e.m.). (c–d) Changes in individual HT1080 (c) and MC38 (d) tumor volumes were quantified and compared at day 7 (see (a); n ≥ 6, means ± s.e.m; two-tailed Mann–Whitney test). (e–f) Body weight was monitored in animals bearing HT1080 (e) or MC38 (f) tumors following treatment with the combination of Pd-NP and C16proMMAE, showing no significant loss compared to the vehicle control group (n ≥ 4, means ± s.e.m.). (g) At the end of the study end point, plasma of MC38 tumor-bearing mice was analyzed for signs of toxicity, and no significant changes were observed (n ≥ 4, means ± s.e.m.; two-tailed t test at α = 0.05 significance level).
In vitro tests had demonstrated C16proMMAE to be a much more potent drug once activated by Pd-NP, compared to DOX-based formulations (Figure 2). When compared to previously published results, the dual treatment with Pd-NP and C16proMMAE performed at least as well as the combination of Pd-NP and Alloc-DOX NP, even at <2% the relative molar dose (0.8 μmol kg–1 compared to 48 μmol kg–1 of prodrug).5 Control experiments using equivalent doses of parent MMAE were not performed, as it is known to be severely toxic and above the maximum tolerated dose in mouse models (e.g., see ref (18)). For these reasons, MMAE itself is not a cancer drug candidate and is only used as an antibody-drug conjugate or nanotherapeutic. In contrast, animals treated with dual Pd-NP and C16proMMAE did not show drug-induced weight loss (Figure 5e–f), and blood chemistry analysis of treated animals showed no signs of drug-induced liver or kidney toxicity (Figure 5g). Thus, these results suggest that Pd-mediated activation of C16proMMAE can safely and effectively block tumor growth.
Modeling in Vivo Mechanisms of Dual Nanotherapy Action
Data reported here and elsewhere indicate that dual Pd-NP and prodrug-NP strategies can lead to more selective activation in the tumor compared to traditional solvent- and nanoformulations of the parent drug. In this work, the dual Pd-NP and C16proMMAE treatment was effective and well-tolerated. Previously, we found that dual Pd-NP and Alloc-DOX treatment was similarly safe and effective (albeit requiring much higher prodrug doses for efficacy). In past head-to-head experiments using HT1080 xenografts, traditional solvent- and nanoformulations of active DOX both exhibited myelotoxicity at equimolar doses, while the dual Pd-NP and Alloc-DOX strategy did not.5 Others have likewise reported that traditional DOX nanoencapsulation can fail to prevent myelotoxicity in mouse models of cancer.19 What mechanisms enable dual Pd-NP and prodrug treatment to more selectively activate in the tumor, particularly compared to other traditional nanoformulations?
To address these questions, we developed a computational multicompartment model of pharmacokinetics and prodrug activation (Figure 6a). As with all such models, simplifications were made for both practical implementation and manageable interpretation, and we roughly based our study on prior models used for pharmacokinetic analysis of biologics and nanomaterials.20−23 The model consists of 27 parameters (12 of which were optimized, 15 fixed from prior experimental data; Table S1), 30 ordinary differential equations (Table S2), and 4 primary organ-level compartments (Figure 6a): the central compartment (plasma), peripheral tissue (simplified here as the heart, as a representative example with known drug toxicity concerns), the mononuclear phagocyte system (simplified here as the liver, which clears a large fraction of nanomaterials), and the tumor. Following distribution from vessels into tissue, NPs are taken up by phagocytes in the liver (Kupffer cells) and tumor (tumor-associated macrophages, TAMs) along with tumor cells themselves. Importantly, we modeled myeloid phagocytic capacity as saturable, based on prior data using Pd-NPs5 as well as a host of studies examining the effects of nanomaterial “loading doses” on the phagocytic clearance of subsequently administered nanomaterials.24−27 While TAMs were also modeled as saturable, tumor cells were not, on the basis of published experimental data indicating the latter accumulate lower NP levels on a per-cell basis, at a slower rate, and well below their saturation levels achieved in vitro compared to TAMs and Kupffer cells (Table S3 for data and references). Despite differences in particular nanomaterial properties used in the “loading dose” studies, their overall findings are relatively consistent (see Table S3). Once taken up into cells, catalytic and prodrug NPs react to yield active drug. For simplification, degradation was modeled as gradual transport of NPs from endocytic/lysosomal compartments into “downstream compartments” that were much less conducive to drug activation (for instance, as if the prodrug or Pd compound were metabolized into degradation products).
Figure 6.

A multicompartment pharmacokinetic model accurately reflects the benefits of the prodrug strategy. (a) Schematic depicting the computational pharmacokinetic model, with particle colors corresponding to the legend at right (Tables S1–S2 contain full equations and parameters). (b) Average fit of the computational model according to objective pharmacokinetic and biodistribution parameters that were experimentally measured (see Table S3). (c) Model simulation showing biodistribution of the catalyst, the prodrug (administered 5 h after the catalyst), and the activated prodrug over 48 h. Thick line and shading denote mean and std. dev. of simulations across n = 24 optimizations. (d) From data in (c), the modeled ratio of tumor: liver accumulation (n = 24) was compared to the experimentally observed ratio of tumor:clearance organs (liver, spleen, kidney; see Figure S7a for details; n = 3). Data are means ± s.e.m. (*paired two-tailed t tests).
Parameters in the model were iteratively fit to 12 features derived from experimental data, most using the same HT1080 xenograft model and PLGA-PEG-based nanoformulations described here. In particular, the model was guided by (i) time-lapse intravital microscopy of nanomaterial systemic clearance, extravasation, and cellular uptake in HT1080 tumors; (ii) organ-level biodistribution measured by fluorescence (for fluorochrome-labeled NPs) and mass spectrometry (e.g., for Pd-NP), including in the HT1080 model; (iii) cell-level biodistribution of fluorescent PLGA-PEG NPs measured by flow cytometry; and (iv) the relative ratios of catalytic NPs, prodrug NPs, and activated drug determined by fluorescence and/or mass spectrometry in the HT1080 models and loading dose studies (see Table S3). Overall, an ensemble of model results were compiled that showed a reasonable fit to the experimental data (Figure 6b), with a median parameter error (21%) roughly within the biological uncertainty observed across the experimental data (average standard error 23%, Table S3). Pharmacokinetics and drug activation in the model accurately captured the time-staggered dosing of the catalyst- and prodrug-NPs (Figure 6c). We performed a parametric sensitivity analysis to gauge how changing individual model parameters can influence overall system behavior (Figure S8). From this analysis and an examination of the simulation results, an important trend was captured: through the combination of saturable phagocytic clearance in the liver (amplified through staggered NP administration), and compounded EPR effect in the tumor, drug activation was observed to be more selective in the tumor compared to distribution of the catalyst NP or its prodrug NP substrate (Figure 6d). This trend was matched by experimental biodistribution data in the HT1080 model (Figure 6d).
Exploiting the Enhanced Reliance on EPR for Dual Nanotherapy Action
Perhaps intuitively, the parametric sensitivity analysis of the computational model (Figure S8) revealed that the bioorthogonal prodrug strategy relies to an especially high degree on factors related to the tumor EPR effect. As prime examples, the model suggests that changes in vascular permeability (Pt) and overall tumor vascularization (St) influence tumoral accumulation of active drug to a greater degree than accumulation of either the catalytic or prodrug NPs individually (highlighted in Figure S8). Further experimental data underscores the strategy’s susceptibility to variable EPR effects. Our recent studies have highlighted how the EPR effect is highly variable in the HT1080 tumor xenograft model.28,29 Closer examination of the tumor growth responses reveals that the dual Pd-NP and C16proMMAE treatment leads to the most variable response of any other treatment, including to all controls, traditional single-nanotherapeutic treatments, and solvent-based treatments (Figure S7d).
To overcome this variability, previous reports have shown that single low-dose tumor irradiation can increase NP accumulation via enhanced vascular permeability, TAM recruitment, and other physiological effects, and therefore considerably enhance EPR effects and NP efficacy in solid tumors.30 To further improve efficacy, we tested whether dual Pd-NP and C16proMMAE NP treatment could benefit from such an approach. Based on measured impacts of such irradiation on vascular permeability (Table S3), the computational model predicted an especially responsive enhancement in drug activation, above the expected ∼2-fold increase in NP accumulation (Figure 7a). Indeed, HT1080 tumors exhibited greater accumulation of PLGA-PEG NP in the tumor following conformal 5 Gy γ irradiation (the curative radiation dose is ∼10× higher), as measured by confocal fluorescence microscopy (Figure 7b) and flow cytometry (Figure 7c). Although local tumor irradiation itself did not durably control tumor growth (as reported previously, ref (30)), it caused tumors to dramatically shrink when combined with the Pd-NP and C16proMMAE NP treatment regimen (Figure 7d-e), with no observed weight loss from irradiation (Figure S7e). Notably, when radiation was added to the Pd-NP and C16proMMAE treatment, the tumor responses no longer exhibited the same degree of heterogeneity that was seen without radiation (all tumors shrank, Figure S7d). Overall, these results indicate that Pd-mediated C16proMMAE activation is even more effective when tumors are conditioned by RT to accumulate greater levels of NPs.
Figure 7.

Single low-dose irradiation enhances delivery and efficacy of dual Pd-NP and C16proMMAE treatment. (a) Based on published experimental data (see Table S3), computational modeling predicts how local 5 Gy irradiation, performed 72 h prior to NP injection, impacts tumoral NP accumulation and prodrug activation (n = 24 optimizations; means ± s.e.m.). (b–c) HT1080 tumor cells and the accumulation of PLGA-PEG NP were compared with or without irradiation as modeled in (a), shown by confocal fluorescence microscopy in live tumors (b; n ≥ 3, scale bar 100 μm), and flow cytometry of excised and digested tumors (c; n ≥ 4), 24 h post-treatment with NP. (d–e) HT1080 tumor xenografts were treated as in Figure 6a, but with addition of a single-dose of 5 Gy irradiation locally to the tumor site (un-irradiated groups are re-shown for reference; n ≥ 5, means ± s.e.m). Corresponding changes in the volumes of individual tumors were quantified at day 7 (n ≥ 5, means ± s.e.m; two-tailed t tests).
Comparison to Prior Prodrug Designs
This report advances bioorthogonal chemistry applications by enabling multifunctional prodrug engineering for more efficient in vivo drug action delivered as NPs. We present a modular design based on a multifunctional self-immolative linker that allows sterically bulky substituents (here, the aliphatic C16 anchor) to be designed and utilized for tuning prodrug properties, while catalytic removal by a bioorthogonal Pd-NP catalyst (using the Alloc protecting group) efficiently restores drug activity. Compared to initial efforts in developing Pd-mediated prodrugs, this strategy led to development of C16proMMAE, a NP-formulated prodrug with improvements including (i) 100-fold greater turn-on capacity in cytotoxicity compared to Alloc-DOX (>70,000× turn-on for C16proMMAE compared to ∼700× for Alloc-DOX); (ii) a roughly 100-fold greater in vitro potency compared to Alloc-DOX in the presence of Pd-NP; (iii) a 50-fold reduction in the prodrug dose required for controlling tumor growth in vivo; and (iv) highly efficient and stable nanoencapsulation properties. The approach is also able to take advantage of recent discoveries into how local radiation therapy can improve tumoral NP delivery and action, leading to synergistic response.30 These results, and their comparison with traditional solvent- and nanoformulations of active drugs, are summarized in Table S4.
Prodrugs based on MMAE have a successful history in the clinic, largely based on their implementation in antibody drug conjugates (ADC) such as brentixumab vedotin (Adcetris), the anti-CD30 ADC used to treat refractory Hodgkin lymphoma among other indications. Other MMAE-based ADCs are undergoing clinical trials, while alternative microtubule-disrupting drugs are used in clinically approved ADCs such as trastuzumab emtansine (Kadcyla). Although ADCs have demonstrated an ability to extend survival in patients, they typically exhibit several drawbacks, primarily dose-limiting toxicities and incomplete tumor penetration. ADC resistance is still common, especially in advanced malignancies. Toxicity arises from off-target ADC uptake; instability of the antibody-drug conjugation, especially when disulfide and hydrazone bonds are used, leading to systemic drug exposure; limited drug loading on antibodies; and poor ability to tightly control and decouple prodrug activation from antibody behavior. Limited efficacy may be due to low exposure of target cells, which is inevitable given the high molecular weight of antibodies. The bioorthogonal prodrug strategy presented here helps address some of these issues. In general, nanoencapsulation supports far greater prodrug loading capacity compared to antibodies. Use of a catalyst for prodrug activation offers the possibility of substoichiometric reactions, while use of a distinct bioorthogonal trigger (here, Pd-NP) can provide greater control over when and where the prodrug becomes activated, especially compared to prodrugs that rely on endogenous and often widely expressed enzymatic reactions, such as cathepsin proteases relied on by many ADCs.
Future Directions
The prodrug design concept presented in this work is especially suited for combinatorial NP delivery strategies, although other bioconjugation strategies (for instance, to antibodies or targeted small molecules) may be feasible. In principle, the C16 anchor could be replaced with other nanoencapsulation anchors, linkers to different types of molecular targeting entities, or other therapeutic payloads. In the context of Pd-NP bioorthogonal catalysis, nanoencapsulation has been shown to improve the stability, solubility, and selective in vivo delivery;5 future studies may use the technology developed here to explore bioorthogonal catalysis with more advanced nanoformulations, such as those which are molecularly targeted, environmentally responsive, based on implanted biomaterial scaffolds, or incorporated into adoptively transferred cell therapies.
Given that our strategy requires accumulation of two different NPs in tumors, it is highly dependent on tumor uptake via the EPR effect, and this is underscored by the computational pharmacokinetic modeling results. Patients vary in how well their tumors accumulate NPs, but personalization of therapy may be feasible using biomarkers of NP uptake.31 For instance, recent work has highlighted how an FDA-approved magnetic NP, ferumoxytol (Feraheme), can be used as an MRI contrast agent and companion diagnostic to identify tumors with high EPR and to predict corresponding nanomedicine response.28 This approach has been especially promising in predicting the clinical activity of a liposomal formulation of the prodrug irinotecan (Onivyde), which was recently approved by the FDA for treatment of refractory pancreatic cancer.32 Along these lines, local low-dose tumor irradiation, as used here, can enhance the EPR effect in a manner detectable by ferumoxytol imaging and leads to synergistic responses.29
Conclusions
Here we present DOX and MMAE as two proof-of-principle drugs with which to apply the prodrug strategy, both containing primary amines that are important for their function. Their broad and proven clinical activity in treating cancer, combined with well understood mechanisms of action and dose-limiting systemic toxicities, motivated their use in this application. Furthermore, the endogenous fluorescence of DOX provided some insight into its intracellular distribution as a prodrug, its co-localization with Pd-NP, and its subsequently enhanced association with DNA once activated. In future work, the prodrug design has the potential to extend beyond antimitotic drugs and apply to a variety of therapeutics, including immunomodulatory agents, targeted inhibitors, and radiopharmaceuticals. Our data indicated that the C16 nanoencapsulation anchor was important in blocking biological (i.e., cytotoxic) effects of the prodrug and influenced subcellular distribution. The modular design strategy of the approach theoretically allows for further optimization and modification of the C16 anchor, especially for application to different drug payloads acting in different subcellular compartments.
Methods
Full methods and characterization describing chemical syntheses can be found in the online Supplementary Methods.
Cell Lines and Animal Models
All animal research was performed in accordance with guidelines from the Institutional Subcommittee on Research Animal Care. HT1080 xenografts were generated by 2 × 106 cells implanted subcutaneously in flanks of 6–8 week old female nu/nu mice (Cox7/MGH) in 50 μL PBS. MC38 syngeneic tumors were formed by intradermal implantation of 2 × 106 cells in 50 μL PBS, in the flanks of 7–12 week old female C57BL/6 mice (JAX), following previously described inoculation protocols.33 Four blinded caliper measurements per tumor were used to measure tumor size according to the formula V = (width)2(length)/2; animals were randomly assigned to treatment groups. Experiments were built on prior imaging, biodistribution, and longitudinal tumor growth data that likewise guided determination of experimental sample sizes.5,30 Treatment began approximately 3 weeks post-implantation once tumors reached an average diameter of 5.5 ± 1.8 mm in the HT1080 model (mean ± std. dev.; n = 72) and roughly 2 weeks post-implantation in the MC38 model once tumors reached an average diameter of 5.45 ± 1.2 mm (mean ± std. dev.; n = 14). Study results were pooled across independent cohorts (n ≥ 2), therefore sample sizes of some control groups are over-represented. Drug-free NP vehicle controls were used throughout, and nanoformulations were freshly prepared prior to injection. Treatments were 0.8 μmol kg–1 C16proMMAE, 50 mg kg–1 Pd-NP, or a combination of the two staggered by 5 h on the same day, all by tail-vein injection in 50 μL PBS, on the indicated days. Following pre-established criteria, mice were sacrificed when tumor burden reached more than 1 cm in diameter, or 2 cm in diameter if only one tumor was present, or according to a body condition score of 2. Drug-induced weight loss did not exceed 10% in any treatment group. Blood chemistry readouts were measured from plasma collected in heparinized tubes by terminal cardiac puncture under vaporized isoflurane anesthesia, using the MGH Veterinary Clinical Pathology lab and the automated DriChem blood chemistry analyzer (Heska). The HT1080 cell line was obtained directly from ATCC, was cultured according to the provider’s guidelines, was not independently verified, and underwent routine mycoplasma testing. Transgenic cell lines were generated as described previously.5 EB3-mApple cells were generated by transfection and repeated rounds of sorting by FACS. The construct mApple-EB3-7 was a gift from Michael Davidson (Addgene plasmid # 54892). For all procedures, mice were anesthetized with an isoflurane vaporizer on a heated stage, euthanasia was performed by CO2 chamber when necessary, and all treatment groups underwent procedures and monitoring consecutively on the same day when possible, but in a randomized order.
Nanoformulation and Characterization
C16proMMAE and C16proDOX nanoformulations were synthesized by nanoprecipitation by first combining 0.1 mg prodrug, 5 mg PLGA(75:25 lactide:glycolide)8.3 kDa-PEG5.5 kDa (Advanced Polymer Materials, Inc.; by manufacturer, 70% functionality by 1H NMR, PI 1.38 according to GPC), and 1 mg PLGA(50:50 lactide:glycolide)30–60 kDa (Sigma) in a 212 μL mixture of 1:1 dimethylformamide (DMF):acetonitrile (MeCN), then added dropwise to 20 mL H2O under stirring at room temperature for 4 h, then filtered through a 0.45 μm cellulose acetate syringe filter (Cole-Parmer), and concentrated in Amicon 100 kDa molecular-weight-cutoff centrifugal filters (Millipore) spun at 3000g for 30 min. Initial experiments determined up to 1 mg of C16 prodrug could be used with the same polymer composition with no detectable loss in loading efficiency. For fluorescence-based imaging and flow-cytometric detection of NP uptake, PLGA-BODIPY630 was used instead of PLGA (described previously).5 NP drug and Pd compound loadings were determined by absorbance (Nanodrop spectrophotometer), interpolation from a standard curve (R2 > 0.99) after 1:10 dilution in DMF. Size and ζ potential measurements were performed using dynamic light scattering (Malvern Zetasizer). Prodrug loading efficiency is defined as the fraction of initial drug used in the nanoprecipitation reaction that was successfully encapsulated and recovered in final NP product.
TEM was performed at the Microscopy Core of the Center for Systems Biology/Program in Membrane Biology (MGH). A JEOL 1011 electron microscope was used for TEM, with sample preparation by deposition of 20 μL NP (1.0 mg mL–1) onto a carbon-coated copper grid. Excess solution was blotted, grids were stained with phosphotungstic acid, and then blotted, dried, and imaged. In vitro NP prodrug release was performed by incubating in PBS at 37 °C, separating NPs using a 30 kDa molecular weight cutoff filter (Millipore Amicon) after 72 h, and measuring flow-through for drug content by absorbance (Nanodrop). NP was dissolved in DMF and also measured for drug content by absorbance. Pd-NP, Alloc2R110 and its nanoformulation, and all Pd compounds were synthesized and characterized as previously described.5
In Vitro NP Characterization
For cytotoxicity assessment, 5000 cells per well were added to 96-well plates; cells were treated after overnight seeding with compound or the appropriate buffer control (drug-free PLGA-PEG NP) and assessed for viability 72 h later using PrestoBlue (Life Technologies) following the manufacturer’s protocol.
For in vitro C16proDOX and DOX quantification, 15 cm confluent plates of HT1080 cells were washed 3× in PBS, lysed using 100 μL lysis buffer (150 mM NaCl, 1% Triton X-100, 50 mM Tris, Roche complete protease inhibitor, pH 8.0), and drug was extracted as described.34 Concentrations were fit from integrated fluorescence chromatography as before, using linear approximation as deemed appropriate from reference standards of purified DOX and C16 solutions, correcting for fluorescence efficiencies between C16proDOX and DOX.5 Cells were treated with 1 μM DOX or C16proDOX and/or 70 μM Pd-NP for 24 h. C16proMMAE activation was detected by incubation of 20 μM C16proMMAE with 60 μM Pd-NP in DMF for 24 h at 37 °C.
Microtubule Imaging
EB3 tracking was performed using an FV1000 confocal laser scanning microscope on a 37 °C heated stage, with XLUMPLFLN 20× (NA 1.0) or LUMFLN 60× (NA 1.1) water-immersion objectives, 1–10× digital zoom, 559 nm diode laser, and BA575–620 emission filter (all Olympus America). Cells were treated with 1 μM MMAE, C16proMMAE, and/or 35 μM Pd-NP for 24 h prior to imaging. EB3 tracks were automatically identified and analyzed from time-lapse data sets using u-track software,35 with tuning of maximum gap number and minimum frame number across data sets to account for differences in image quality; ambiguous and spurious tracks and artifacts were excluded according to requirements for directional continuity (directional persistence as net displacement/path length >0.8) and movement (value >3 pixels). Speed was determined between frames in each track and averaged for each track by computing the mean of the middle 80% of between-frame values. Track overlay figures were produced using a custom python script. Averaged track speeds excluded outliers falling more than 1.5× the interquartile range for each biological replicate.
In Vitro NP Uptake Imaging
To quantify subcellular localization of NPs in HT1080 cells, Rab7a-RFP and Lamp1-RFP fusion constructs were expressed using a commercial baculovirus platform (CellLight BacMam 2.0, Invitrogen), following manufacturing guidelines. Pharmacological modulation of NP uptake was performed using the following: staurosporine (1 μM; LC laboratories), latrunculin B (1 μM; Tocris), cytochalasin D (1 μM; Sigma), chloroquine (50 μM; Sigma), and ethylisopropyl amiloride EIPA (100 μM; Tocris). Cells were rinsed in fresh media immediately prior to imaging; only adherent cells were quantified. Chloroquine dose–response measurements were normalized to the C16proDOX control (rather than the chloroquine-free control), in order to compare relative effects on the potency of Pd-mediated C16proDOX activation itself. 50 μM chloroquine alone caused a 15–30% decrease in cell viability.
Flow Cytometry and Confocal Tumor Imaging
Subcutaneous HT1080 tumors were harvested 3 weeks post-implantation and 24 h post-treatment with 3 mg kg–1 PLGA-PEG therapeutic NPs14 encapsulated with PLGA-BODIPY630 as a validated near-infrared label of NP uptake, administered by tail-vein injection in 50 μL PBS. Flow cytometry and confocal imaging are described previously.30 Single-cell quantification of NP uptake was performed in bilateral subcutaneous tumors, matched such that one tumor received 5 Gy local γ irradiation 72 h prior to NP administration. Following animal sacrifice at 24 h post-injection, single-cell suspensions of resected tumors were stained for tumor cells (CD45-hCD29+) using CD45 (BD 30-F11) and hCD29 (BD MAR4) antibodies, and single-cell NP uptake was measured by the mean fluorescence intensity of gated cells on an LSRII flow cytometer. Confocal tumor imaging was performed on mice bearing subcutaneous HT1080 tumors under a dorsal window chamber as previously described;36 tumor cells were subcutaneously injected 30 min after surgical chamber implantation, and imaged and irradiated 2 weeks later.
Radiation Therapy
Dual source 137Cs Gammacell 40 Exactor (Best Theratronics) with a custom-built lead shield was used for conformal tumor irradiation, using a setup described previously.30 Immediately prior to RT, mice were anesthetized via 87.5 mg/kg ketamine and 12.5 mg/kg xylazine i.p., immobilized in the lead shielding chamber, and irradiated individually according to the calibrated dose rate of 0.6 Gy min–1.
Biodistribution
Accumulation of palladium and prodrug NP in tissues was assessed as previously described, shown here as a combined ratiometric analysis presented relative to tumor concentrations. All measurements were determined 24 h post-treatment with Pd-NP in HT1080 subcutaneous tumor-bearing mice, as used throughout. Palladium biodistribution was determined by an Agilant 7500 Series ICP-MS, fitting to a 9-point standard curve and using Pd ICP standard solution. Biodistribution of prodrug NP vehicle and activation was quantified by Olympus OV110 fluorescence reflectance imaging of freshly excised tissue that had been rinsed in PBS. For a model prodrug NP, PLGA-PEG nanoformulation encapsulating the near-infrared NP label PLGA-BODIPY630 and the model Pd-NP substrate Alloc2R110 were used, all as described and characterized previously.5 Fluorescence intensity values were calculated from regions of interest defined manually in ImageJ, after correcting for background autofluorescence. Measurements were averaged according to the mean across n = 3 replicates, and calculated concentrations were divided by the average concentrations observed in the tumor. To compare tumor levels to concentrations seen in clearance organs, tumor levels were divided by the pooled average concentrations observed across the liver, spleen, and kidney.
Computational Pharmacokinetic Modeling
The multicompartment model was developed based on custom Matlab scripts and was simulated using the ordinary differential equation solver ode15s. Parameters were optimized according to iterative and stochastically sampled rounds of bounded optimization. The cost function consisted of experimentally measured features of biodistribution and pharmacokinetics (Table S3), and n = 24 optimization runs were computed based on stochastic initial parameter values and cost functions with slightly different weights on each of these features. Average modeling results were then tabulated from the 24 optimizations. The final model was generated after iterative rounds of increasing the model complexity and compartments (e.g., through the use of saturable phagocytic uptake) to better fit the experimental data, until yielding sufficiently accurate results.
Statistical Analysis
Statistical analyses were performed using Prism (GraphPad), MATLAB (Mathworks), and Excel (Microsoft). Measurement statistics and error bars are described in the figure legends. Two-tailed tests were used with false-positive thresholds of α = 0.05.
Acknowledgments
Part of this work was supported by NIH/NCI grants R00CA207744, R01CA206890, U01CA206997, and T32CA079443 and the Austrian Science Fund (FWF, J 3586-B23 to H.M.). We thank Javier Piñeda (HMS) and Jonathan Carlson (MGH) for helpful discussions. We also thank Diane Capen and Dennis Brown (MGH) for TEM assistance. Data and materials availability: All cell lines were obtained through material transfer agreements. Requests for collaboration involving materials used in this research will be fulfilled provided that a written agreement is executed in advance between Massachusetts General Hospital and the requesting parties.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b07954.
Supplementary Figure S1. A model fluorogenic uncaging reaction reveals the kinetics and efficiency of Pd-mediated deprotection. Supplementary Figure S2. Nanoformulated prodrug size distribution and stability. Supplementary Figure S3. Improved prodrug caging increases maximum nontoxic dose in cells. Supplementary Figure S4. Monitoring in vitro prodrug activation. Supplementary Figure S5. Microscopic evaluation of intracellular NP localization. Supplementary Figure S6. Dose–response of PdNP and prodrugs across multiple cancer cell lines. Supplementary Figure S7. Analysis of tumor growth data. Supplementary Figure S8. Pharmacokinetic model sensitivity analysis. Supplementary Table S1. Pharmacokinetic computational model parameters. Supplementary Table S2. Pharmacokinetic model equations. Supplementary Table S3. Pharmacokinetic model optimization parameters. Supplementary Table S4. Overview of the prodrug strategy efficacy and safety (PDF)
Supplementary methods: Compiled chemical synthesis methods and characterization (PDF)
Movie S1: Time-lapse microscopy of microtubule comets. (AVI)
Author Contributions
# These authors contributed equally. M.A.M., H.M., and R.W. developed the concept. All authors designed the experiments. M.A.M., H.M., and R.W. wrote the paper. All authors analyzed the results and edited the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Li J.; Chen P. R. Development and Application of Bond Cleavage Reactions in Bioorthogonal Chemistry. Nat. Chem. Biol. 2016, 12, 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Wang B.; Zhao N.; Tian Z.; Dai Y.; Nie Y.; Tian J.; Wang Z.; Chen X. Improved Tumor Targeting and Longer Retention Time of NIR Fluorescent Probes Using Bioorthogonal Chemistry. Theranostics 2017, 7, 3794–3802. 10.7150/thno.20912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hapuarachchige S.; Kato Y.; Artemov D. Bioorthogonal Two-Component Drug Delivery in HER2(+) Breast Cancer Mouse Models. Sci. Rep. 2016, 6, 24298. 10.1038/srep24298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Askevold B.; Mikula H.; Kohler R. H.; Pirovich D.; Weissleder R. Nano-Palladium is a Cellular Catalyst for In Vivo Chemistry. Nat. Commun. 2017, 8, 15906. 10.1038/ncomms15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völker T.; Meggers E. Chemical Activation in Blood Serum and Human Cell Culture: Improved Ruthenium Complex for Catalytic Uncaging of Alloc-Protected Amines. ChemBioChem 2017, 18, 1083–1086. 10.1002/cbic.201700168. [DOI] [PubMed] [Google Scholar]
- Li J.; Yu J.; Zhao J.; Wang J.; Zheng S.; Lin S.; Chen L.; Yang M.; Jia S.; Zhang X.; Chen P. R. Palladium-Triggered Deprotection Chemistry for Protein Activation in Living Cells. Nat. Chem. 2014, 6, 352–361. 10.1038/nchem.1887. [DOI] [PubMed] [Google Scholar]
- Pérez-López A. M.; Rubio-Ruiz B.; Sebastián V.; Hamilton L.; Adam C.; Bray T. L.; Irusta S.; Brennan P. M.; Lloyd-Jones G. C.; Sieger D.; Santamaría J.; Unciti-Broceta A. Gold-Triggered Uncaging Chemistry in Living Systems. Angew. Chem., Int. Ed. 2017, 56, 12548–12552. 10.1002/anie.201705609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to Release: Instantaneous Doxorubicin Elimination Upon Tetrazine Ligation. Angew. Chem., Int. Ed. 2013, 52, 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Matikonda S. S.; Orsi D. L.; Staudacher V.; Jenkins I. A.; Fiedler F.; Chen J.; Gamble A. B. Bioorthogonal Prodrug Activation Driven By a Strain-Promoted 1, 3-Dipolar Cycloaddition. Chem. Sci. 2015, 6, 1212–1218. 10.1039/C4SC02574A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. T.; Dawson J. C.; Macleod K. G.; Rybski W.; Fraser C.; Torres-Sánchez C.; Patton E. E.; Bradley M.; Carragher N. O.; Unciti-Broceta A. Extracellular Palladium-Catalysed Dealkylation of 5-Fluoro-1-Propargyl-Uracil as a Bioorthogonally Activated Prodrug Approach. Nat. Commun. 2014, 5, 3277. 10.1038/ncomms4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Ruiz B.; Weiss J. T.; Unciti-Broceta A. Efficient Palladium-Triggered Release of Vorinostat From a Bioorthogonal Precursor. J. Med. Chem. 2016, 59, 9974–9980. 10.1021/acs.jmedchem.6b01426. [DOI] [PubMed] [Google Scholar]
- Yameen B.; Choi W. I.; Vilos C.; Swami A.; Shi J.; Farokhzad O. C. Insight Into Nanoparticle Cellular Uptake and Intracellular Targeting. J. Controlled Release 2014, 190, 485–499. 10.1016/j.jconrel.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Zheng Y. R.; Gadde S.; Pfirschke C.; Zope H.; Engblom C.; Kohler R. H.; Iwamoto Y.; Yang K. S.; Askevold B.; Kolishetti N.; Pittet M.; Lippard S. J.; Farokhzad O. C.; Weissleder R. Tumour-Associated Macrophages Act as a Slow-Release Reservoir of Nano-Therapeutic Pt(IV) Pro-Drug. Nat. Commun. 2015, 6, 8692. 10.1038/ncomms9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zeng X.; Liang X.; Yang Y.; Li X.; Chen H.; Huang L.; Mei L.; Feng S. S. The Chemotherapeutic Potential of PEG-B-PLGA Copolymer Micelles That Combine Chloroquine as Autophagy Inhibitor and Docetaxel as an Anti-Cancer Drug. Biomaterials 2014, 35, 9144–9154. 10.1016/j.biomaterials.2014.07.028. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chang D.; Yang Y.; Zhang X.; Tao W.; Jiang L.; Liang X.; Tsai H.; Huang L.; Mei L. Systematic Investigation on the Intracellular Trafficking Network of Polymeric Nanoparticles. Nanoscale 2017, 9, 3269–3282. 10.1039/C7NR00532F. [DOI] [PubMed] [Google Scholar]
- Stepanova T.; Slemmer J.; Hoogenraad C. C.; Lansbergen G.; Dortland B.; De Zeeuw C. I.; Grosveld F.; van Cappellen G.; Akhmanova A.; Galjart N. Visualization of Microtubule Growth in Cultured Neurons Via the Use of Eb3-Gfp (End-Binding Protein 3-Green Fluorescent Protein). J. Neurosci. 2003, 23, 2655–2664. 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legigan T.; Clarhaut J.; Tranoy-Opalinski I.; Monvoisin A.; Renoux B.; Thomas M.; Le Pape A.; Lerondel S.; Papot S. The First Generation of Β-Galactosidase-Responsive Prodrugs Designed for the Selective Treatment of Solid Tumors in Prodrug Monotherapy. Angew. Chem., Int. Ed. 2012, 51, 11606–11610. 10.1002/anie.201204935. [DOI] [PubMed] [Google Scholar]
- Xu R.; Zhang G.; Mai J.; Deng X.; Segura-Ibarra V.; Wu S.; Shen J.; Liu H.; Hu Z.; Chen L.; Huang Y.; Koay E.; Huang Y.; Liu J.; Ensor J. E.; Blanco E.; Liu X.; Ferrari M.; Shen H. An Injectable Nanoparticle Generator Enhances Delivery of Cancer Therapeutics. Nat. Biotechnol. 2016, 34, 414–418. 10.1038/nbt.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter L. T.; Zhu H.; Mackensen D. G.; Jain R. K. Physiologically Based Pharmacokinetic Model for Specific and Nonspecific Monoclonal Antibodies and Fragments in Normal Tissues and Human Tumor Xenografts in Nude Mice. Cancer Res. 1994, 54, 1517–1528. [PubMed] [Google Scholar]
- Schluep T.; Hwang J.; Hildebrandt I. J.; Czernin J.; Choi C. H.; Alabi C. A.; Mack B. C.; Davis M. E. Pharmacokinetics and Tumor Dynamics of the Nanoparticle IT-101 From Pet Imaging and Tumor Histological Measurements. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 11394–11399. 10.1073/pnas.0905487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks B. S.; Reynolds J. G.; Klinz S. G.; Geretti E.; Lee H.; Leonard S. C.; Gaddy D. F.; Espelin C. W.; Nielsen U. B.; Wickham T. J. Multiscale Kinetic Modeling of Liposomal Doxorubicin Delivery Quantifies the Role of Tumor and Drug-Specific Parameters in Local Delivery to Tumors. CPT: Pharmacometrics Syst. Pharmacol. 2012, 1, e15. 10.1038/psp.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Moss M. L.; Powell G.; Petrovich R.; Edwards L.; Meyer A. S.; Griffith L. G.; Lauffenburger D. A. Targeting Autocrine HB-EGF Signaling With Specific ADAM12 Inhibition Using Recombinant ADAM12 Prodomain. Sci. Rep. 2015, 5, 15150. 10.1038/srep15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Yan X.; Jacobson O.; Sun W.; Wang Z.; Tong X.; Xia Y.; Ling D.; Chen X. Improved Tumor Uptake By Optimizing Liposome Based Res Blockade Strategy. Theranostics 2017, 7, 319–328. 10.7150/thno.18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang D. J.; Moon C.; Oh E. Improved Tumor Targeting and Antitumor Activity of Camptothecin Loaded Solid Lipid Nanoparticles By Preinjection of Blank Solid Lipid Nanoparticles. Biomed. Pharmacother. 2016, 80, 162–172. 10.1016/j.biopha.2016.03.018. [DOI] [PubMed] [Google Scholar]
- Liu L.; Hitchens T. K.; Ye Q.; Wu Y.; Barbe B.; Prior D. E.; Li W. F.; Yeh F. C.; Foley L. M.; Bain D. J.; Ho C. Decreased Reticuloendothelial System Clearance and Increased Blood Half-Life and Immune Cell Labeling for Nano- and Micron-Sized Superparamagnetic Iron-Oxide Particles Upon Pre-Treatment With Intralipid. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 3447–3453. 10.1016/j.bbagen.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Ye Q.; Lu M.; Lo Y. C.; Hsu Y. H.; Wei M. C.; Chen Y. H.; Lo S. C.; Wang S. J.; Bain D. J.; Ho C. A. New Approach to Reduce Toxicities and to Improve Bioavailabilities of Platinum-Containing Anti-Cancer Nanodrugs. Sci. Rep. 2015, 5, 10881. 10.1038/srep10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Gadde S.; Pfirschke C.; Engblom C.; Sprachman M. M.; Kohler R. H.; Yang K. S.; Laughney A. M.; Wojtkiewicz G.; Kamaly N.; Bhonagiri S.; Pittet M. J.; Farokhzad O. C.; Weissleder R. Predicting Therapeutic Nanomedicine Efficacy Using a Companion Magnetic Resonance Imaging Nanoparticle. Sci. Transl. Med. 2015, 7, 314ra183. 10.1126/scitranslmed.aac6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Arlauckas S.; Weissleder R. Prediction of Anti-Cancer Nanotherapy Efficacy By Imaging. Nanotheranostics 2017, 1, 296–312. 10.7150/ntno.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Chandra R.; Cuccarese M. F.; Pfirschke C.; Engblom C.; Stapleton S.; Adhikary U.; Kohler R. H.; Mohan J. F.; Pittet M. J.; Weissleder R. Radiation Therapy Primes Tumors for Nanotherapeutic Delivery Via Macrophage-Mediated Vascular Bursts. Sci. Transl. Med. 2017, 9, eaal0225 10.1126/scitranslmed.aal0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Kantoff P. W.; Wooster R.; Farokhzad O. C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan R. K.; Korn R. L.; Raghunand N.; Sachdev J. C.; Newbold R. G.; Jameson G.; Fetterly G. J.; Prey J.; Klinz S. G.; Kim J.; Cain J.; Hendriks B. S.; Drummond D. C.; Bayever E.; Fitzgerald J. B. Correlation Between Ferumoxytol Uptake in Tumor Lesions By MRI and Response to Nanoliposomal Irinotecan in Patients With Advanced Solid Tumors: A Pilot Study. Clin. Cancer Res. 2017, 23, 3638–3648. 10.1158/1078-0432.CCR-16-1990. [DOI] [PubMed] [Google Scholar]
- Arlauckas S. P.; Garris C. S.; Kohler R. H.; Kitaoka M.; Cuccarese M. F.; Yang K. S.; Miller M. A.; Carlson J. C.; Freeman G. J.; Anthony R. M.; Weissleder R.; Pittet M. J. In Vivo Imaging Reveals a Tumor-Associated Macrophage-Mediated Resistance Pathway in Anti-PD-1 Therapy. Sci. Transl. Med. 2017, 9, eaal3604 10.1126/scitranslmed.aal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright C. F.; Graciani N.; Han W.; Yue E.; Stein R.; Lai Z.; Diamond M.; Dowling R.; Grimminger L.; Zhang S. Y.; Behrens D.; Musselman A.; Bruckner R.; Zhang M.; Jiang X.; Hu D.; Higley A.; Dimeo S.; Rafalski M.; Mandlekar S.; et al. Matrix Metalloproteinase-Activated Doxorubicin Prodrugs Inhibit HT1080 Xenograft Growth Better Than Doxorubicin With Less Toxicity. Mol. Cancer Ther. 2005, 4, 751–760. 10.1158/1535-7163.MCT-05-0006. [DOI] [PubMed] [Google Scholar]
- Applegate K. T.; Besson S.; Matov A.; Bagonis M. H.; Jaqaman K.; Danuser G. Plustiptracker: Quantitative Image Analysis Software for the Measurement of Microtubule Dynamics. J. Struct. Biol. 2011, 176, 168–184. 10.1016/j.jsb.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Askevold B.; Yang K. S.; Kohler R. H.; Weissleder R. Platinum Compounds for High-Resolution In Vivo Cancer Imaging. ChemMedChem 2014, 9, 1131–1135. 10.1002/cmdc.201300502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.