

Abstract
Myocardial infarction is a leading cause of mortality worldwide, while myocardial ischemia and timely reperfusion contribute to myocardial injury. The mitochondria are involved in the injury and mediate the apoptosis of cardiomyocytes. In order to develop novel therapeutic approaches for myocardial infarction, the present study evaluated the myocardial protective effects of eriodictyol and investigated relevant mechanisms in H9c2 cardiomyocytes. As a result, eriodictyol was observed to improve the H9c2 cardiomyocyte viability and block the leakage of cytosolic lactate dehydrogenase under hypoxia/reoxygenation. In addition, the dysfunction of mitochondria induced by hypoxia/reoxygenation was ameliorated by eriodictyol through suppressing the overload of intracellular Ca2+, preventing overproduction of reactive oxygen species, blocking mitochondrial permeability transition pore opening, increasing mitochondrial membrane potential level and decreasing ATP depletion. Finally, the apoptosis of H9c2 cardiomyocyte induced by hypoxia/reoxygenation was prevented by eriodictyol through upregulation of the expression of B-cell lymphoma-2 (Bcl-2) and downregulation of the expression levels of Bcl-2-associated X protein and caspase-3. These results provided evidence for further investigation on myocardial protection and the treatment of myocardial infarction using eriodictyol.
Keywords: eriodictyol, H9c2 cardiomyocyte, hypoxia/reoxygenation, mitochondrial dysfunction, myocardial protection
Introduction
Myocardial infarction is a leading cause of mortality in humans worldwide, which results from myocardial ischemia (1,2). Timely reperfusion is considered as an effective therapy to limit the infarction size, although reperfusion of the ischemic myocardium will increase the number of patients with heart failure due to the cardiomyocyte death (3). The mitochondria are involved in the reperfusion injury, and the opening of mitochondrial permeability transition pore (MPTP) contributes to the infarction (4,5). In addition, intracellular Ca2+ overload and oxidative stress in mitochondria are also associated with myocardial infarction (6). In the development of effective therapeutic approaches for myocardial infarction, natural compounds serve an important role, including berberine (7), tanshinone IIA (8) and lycopene (9).
Eriodictyol (Fig. 1) is a flavonoid identified in numerous medicinal plants, such as Bauhinia ungulata (10), Arcytophyllum thymifolium (11), Elsholtzia bodinieri (12) and Clinopodium chinense (13). Pharmacological investigations have revealed that eriodictyol possesses several bioactivities, including neuroprotection (14–16), renoprotection (17) and lung protection (18), exerted via anti-inflammation and anti-oxidation. In addition, its anti-inflammatory and anti-oxidative capacities have drawn attention to its therapeutic potential (19–23).
Figure 1.
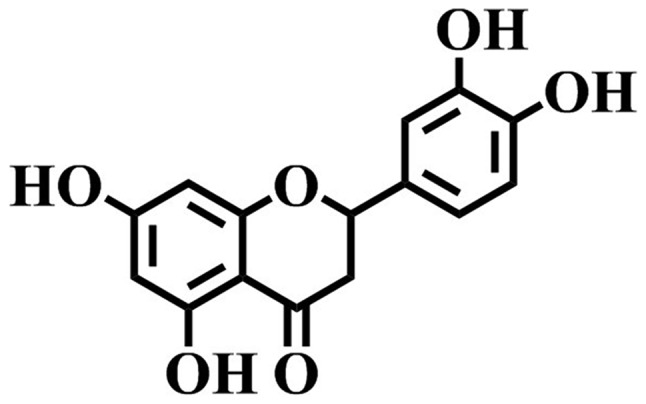
Chemical structure of eriodictyol.
With the aim to investigate bioactive phytochemicals for the treatment of myocardial infarction, the protective effects of eriodictyol on H9c2 cardiomyocyte injury induced by hypoxia/reoxygenation are investigated in the present study. The protective effect of eriodictyol is reported in vitro.
Materials and methods
Chemicals and reagents
Eriodictyol was purchased from YuanYe Biotechnology Co., Ltd. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS) and calcein acetoxymethyl (calcein-AM) were obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Dimethyl sulfoxide (DMSO) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and were purchased from Sigma-Aldrich (Merck AG, Darmstadt, Germany). Reactive oxygen species (ROS), lactate dehydrogenase (LDH) and bicinchoninic acid (BCA) assay kits were supplied by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). JC-1, Fluo-3 AM, ATP detection kit and caspase-3 assay kit, as well as cleaved caspase-3 (cat. no. AC033), B-cell lymphoma-2 (Bcl-2; cat. no. AB112), Bcl-2-associated X protein (Bax; cat. no. AB026) and actin (cat. no. AA128) antibodies (all 1:1,000) were purchased from Beyotime Institute of Biotechnology (Nantong, China).
Cell culture and treatment
The H9c2 cardiomyocytes, a rat embryonic cardiac myoblast line, were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China) and cultured as previously described (9). Briefly, H9c2 cells were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin under humid condition with 5% CO2 and 95% air at 37°C. Next, cells at the logarithmic phase were incubated in 96-well plates at a density of 1×105/ml. The cells were then divided into the control group (CG), model group (MG) and three eriodictyol groups, which were pretreated with 1, 10 and 50 µM eriodictyol in DMSO for 4 h. In order to establish the hypoxia/reoxygenation model, H9c2 cells in the MG and eriodictyol groups were subjected to an atmosphere with 95% N2 and 5% O2 at 37°C for 4 h, and then cultured under a condition of 95% air and 5% O2 at 37°C for a further 4 h. The CG cells were cultured under normoxic conditions.
Cell viability assay
To evaluate the protective effect of eriodictyol on the H9c2 cardiomyocyte injury induced by hypoxia/reoxygenation, an MTT assay was conducted. Following the aforementioned treatments, the cells were incubated with 0.2 ml MTT for 4 h at 37°C. Subsequently, 200 µl DMSO was added into each well to dissolve the formazan crystals, and the absorbance was recorded on an iMark microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 490 nm. The experiments were repeated three times.
Determination of LDH activity
The extracellular LDH activity was determined using the LDH assay kit, according to the manufacturer's instructions. Following treatment and incubation, the H9c2 cardiomyocyte culture medium was centrifuged at 400 × g and room temperature for 5 min, and then 20 µl supernatant was mixed with 20 µl 2,4-dinitrophenylhydrazine. The mixture was incubated at 37°C for 15 min. Next, 250 µl NaOH (0.4 M) was added into the reaction system and incubated for a further 15 min at 37°C. Subsequent to keeping at room temperature for 5 min, the absorbance was recorded on a microplate reader at 450 nm. The activity of LDH was calculated based on the absorbance as previously reported (24) and is expressed as U/l.
Detection of intracellular Ca2+
In order to monitor the cytosolic Ca2+ content in H9c2 cardiomyocytes, the Fluo-3 AM molecular fluorescence probe was employed. Following treatment as described earlier, H9c2 cardiomyocytes were incubated with Fluo-3 AM at 37°C for 30 min and washed twice with phosphate-buffered saline (PBS) to remove any extracellular dye. Subsequent to incubation for a further 30 min, the fluorescence intensity was measured on a SpectraMax M5 microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA) at an excitation wavelength of 488 nm and emission wavelength of 525 nm.
Measurement of ROS generation
The production of intracellular ROS was detected by a fluorescence method (25) with the ROS assay kit, according to the manufacturer's protocol. Following treatment, the medium was replaced and the cells were loaded with 10 µM 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA). After incubation at 37°C for 30 min, the cells were rinsed with PBS and the fluorescence intensity was recorded on a fluorescence microplate reader at an excitation wavelength of 480 nm and emission wavelength of 525 nm.
Assessment of mitochondrial membrane potential (MMP)
The fluorescent probe JC-1 was used to detect the MMP in H9c2 cardiomyocytes. In normal mitochondria, the JC-1 monomer aggregates in the matrix, whereas JC-1 maintains its monomeric form in mitochondria with reduced MMP. The fluorescence of JC-1 aggregates is measured at an excitation wavelength of 530 nm and emission wavelength of 590 nm. In the current investigation, cardiomyocytes were loaded with JC-1 (100 µM) at 37°C for 20 min and then washed with PBS. The fluorescence intensity of the JC-1 aggregate was recorded on a fluorescence microplate reader, and the MMP was determined as the ratio of the JC-1 fluorescence intensity to that of the control group.
Opening of MPTP
The opening of MPTP was evaluated by determining the release of mitochondrial calcein, as previously described (26). Briefly, cardiomyocytes were incubated with 2 µM calcein-AM and 1 mM CoCl2 at room temperature for 30 min. Next, free calcein-AM and CoCl2 were washed away with Hank's balanced salt solution, and cells were incubated with CoCl2 for a further 20 min at 37°C in order to quench the fluorescence of free cytosolic calcein. The fluorescence of mitochondrial calcein in the cardiomyocytes was recorded on a fluorescence microplate reader at 490 nm for excitation and 515 nm for emission. The quenching of fluorescence in H9c2 cardiomyocytes indicated the opening of MPTP.
Level of intracellular ATP
The intercellular ATP in H9c2 cardiomyocytes was determined by the firefly luciferase method (25) with the ATP detection kit according to the manufacturer's protocol. Luciferin generates fluorescence under the catalysis of firefly luciferase, and the process consumes ATP quantitatively. The treated H9c2 cardiomyocytes were lysed on ice with 200 µl lysis reagent from the assay kit. The lysed cells were then centrifuged at 12,000 × g for 4 min at 4°C, and 100 µl supernatant was mixed with 100 µl ATP monitoring reagent. Subsequently, the luminescence was detected on a microplate reader, and the level of intracellular ATP was derived from the standard curve.
Caspase-3 activity
The activity of caspase-3 was quantified through a colorimetric detection kit, following the manufacturer's instructions. Briefly, subsequent to the aforementioned treatments, the H9c2 cardiomyocytes were lysed and centrifuged at 16,000 × g for 10 min at 4°C. The supernatant was then incubated with the substrate Ac-DEVD-pNA at 37°C for 2 h, and the absorbance was measured on a microplate reader at 405 nm. The relative caspase-3 activity was expressed as a percentage of the control group, as previous described (27).
Western blot analysis
The protein expression levels of caspase-3, Bcl-2 and Bax in pretreated H9c2 cardiomyocytes were analyzed by western blot analysis. In brief, the cells were lysed with lysis reagent containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100 and 1 mM phenylmethylsulfonyl fluoride on ice for 30 min. Next, the lysate was centrifuged at 12,000 × g for 15 min at 4°C, and the supernatant was collected for the analysis of cleaved caspase-3, Bcl-2 and Bax levels. The total protein concentration in the samples was determined by BCA assay kit. Subsequently, the protein was separated by electrophoresis on a 15% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes. Following blocking with 5% skimmed milk at room temperature for 1 h, the membranes were incubated overnight at 4°C with primary antibodies against cleaved caspase-3, Bcl-2, Bax and actin. The membranes were was with TBST three times, treated with the respective secondary antibodies conjugated to horseradish peroxidase (cat. no. LDAN0310; 1:1,000; Shanghai Lengton Bioscience Co., Ltd., Shanghai, China) at room temperature for 1 h and detected by an Enhanced ECL Chemiluminescent Substrate kit (cat. no. 36222ES60; Shanghai Yeasen Biotechnology Co., Ltd., Shanghai, China). Actin was used as the internal control.
Statistical analysis
All results are expressed as the means ± standard deviation. GraphPad Prism (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA) was employed to analyze the results. Statistical differences between different groups were compared by one-way analysis of variance followed by Dunnett's test for multiple comparisons and Student's t-test for single comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of eriodictyol on H9c2 cardiomyocyte viability
As shown in Fig. 2, the MTT assay demonstrated that the viability of H9c2 cardiomyocytes decreased when subjected to the hypoxia/reoxygenation (P<0.001). Upon treatment with different dosages of eriodictyol, the survival of H9c2 cardiomyocytes was significantly improved and the cells viability was 77.75±7.06% of the cell viability of CG when cells were treated with 50 µM eriodictyol (P<0.001). The viability in H9c2 cardiomyocytes treated with 50 µM eriodictyol (77.75±7.06%) was significantly higher than the cells treated with 10 µM eriodictyol (63.56±2.75%; P<0.001) and the latter was significantly increased compared with the group treated with 1 µM eriodictyol (58.16±3.17%; P<0.05). These results indicate the potential cardioprotective effect of eriodictyol on H9c2 cells in a dose-dependent manner at the range of 1–50 µM eriodictyol.
Figure 2.
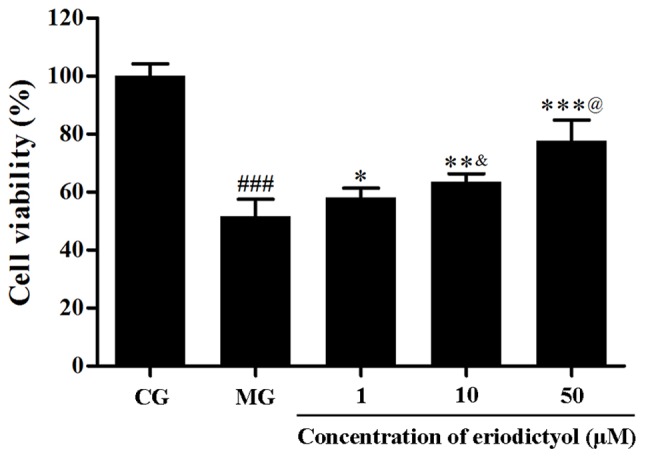
Effect of eriodictyol treatment (1, 10 and 50 µM) on H9c2 cardiomyocyte viability. Cell viability was determined by MTT assay (n=6). ###P<0.001 vs. the CG; *P<0.05, **P<0.01 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. CG, control group; MG, model group.
Effect of eriodictyol on LDH activity
The leakage of LDH from the cytoplasm is associated with cell death. In this investigation, the activity of LDH in the culture medium of the MG (301.01±28.81%) was significantly higher as compared with that in the CG (P<0.001), which further demonstrated that the viability of H9c2 cells was affected by hypoxia/reoxygenation. When pretreated with eriodictyol, the activity of LDH in the 10 µM group with was reduced to 256.81±13.75%, which was significantly higher than the 50 µM group (191.56±13.75%; P<0.001) and lower than the 1 µM group (272.94±8.44%; P<0.05). The results indicated the release of LDH from the cytosol of H9c2 cardiomyocytes was evidently decreased in a dosage-dependent manner (Fig. 3).
Figure 3.
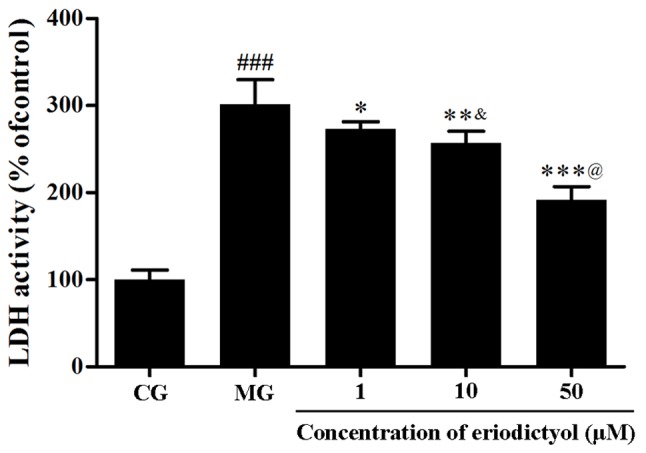
Effect of eriodictyol treatment (1, 10 and 50 µM) on the activity of extracellular LDH in H9c2 cardiomyocytes. n=6. ###P<0.001 vs. the CG; *P<0.05, **P<0.01 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. LDH, lactate dehydrogenase; CG, control group; MG, model group.
Effect of eriodictyol on intracellular Ca2+
To assess the intracellular Ca2+ content, the fluorescence probe Fluo-3 AM was used. Following the induction of hypoxia/reoxygenation, the level of intracellular Ca2+ was markedly elevated to 155.28±6.13% as compared with the CG (P<0.001; Fig. 4). However, in comparison with the model group, eriodictyol reduced the overload of intracellular Ca2+ to 145.68±8.45, 122.41±7.64 and 102.39±8.17% upon pretreatment with 1, 10 and 50 µM, respectively (Fig. 4). These results provided evidence that eriodictyol was able to reduce the overload of intracellular Ca2+ in the hypoxia/reoxygenation cell model.
Figure 4.
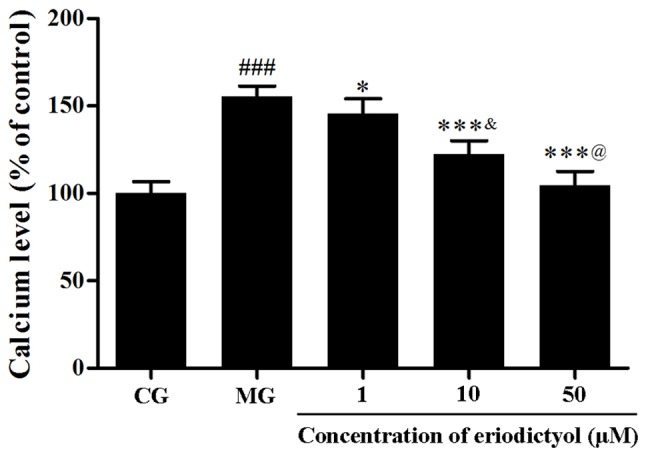
Effect of eriodictyol treatment (1, 10 and 50 µM) on intracellular Ca2+. n=6. ###P<0.001 vs. the CG; *P<0.05 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. CG, control group; MG, model group.
Effect of eriodictyol on ROS generation
The intracellular ROS generation was determined through the fluorescence intensity of DCFH-DA. The results indicated that the relative fluorescence intensity in the MG (148.84±5.16%) was significantly higher compared with that in the CG (P<0.001). By contrast, when cells were treated with eriodictyol, the fluorescence intensity was significantly decreased to 139.25±8.68% (P<0.05), 111.89±9.10 and 101.33±4.57% (both P<0.001) compared with the MG group (Fig. 5). The fluorescence intensity of 10 µM (111.89±9.10%) was lower compared with 1 µM (139.25±8.68%; P<0.05) and higher compared with 50 µM (101.33±4.57%; P<0.05). This indicated that eriodictyol was able to downregulate the generation of intracellular ROS.
Figure 5.
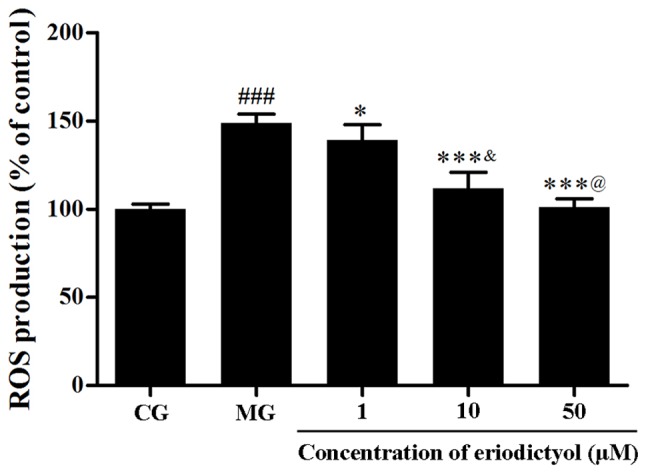
Effect of eriodictyol treatment (1, 10 and 50 µM) on ROS production. n=6. ###P<0.001 vs. the CG; *P<0.05 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. ROS, reactive oxygen species; CG, control group; MG, model group.
Effect of eriodictyol on MMP
In order to determine the MMP in H9c2 cardiomyocytes, the fluorescent probe JC-1 was used. Compared with the CG, the fluorescence intensity in the MG (41.89±3.75%) was significantly decreased (P<0.001), which indicated the collapse of MMP in H9c2 cardiomyocytes following hypoxia/reoxygenation. However, when pretreated with 1, 10 and 50 µM eriodictyol, the fluorescence intensity was significantly elevated to 48.00±4.74% (P<0.05), 62.92±8.56 and 79.89±6.24% (both P<0.001), respectively, compared with the MG group (Fig. 6). Meanwhile, among these groups treated with eriodictyol, the 10 µM group was significantly higher than the 1 µM group lower than the 50 µM (both P<0.05). These findings revealed that the collapse of MMP in hypoxia/reoxygenation-treated H9c2 cardiomyocytes was attenuated by eriodictyol.
Figure 6.
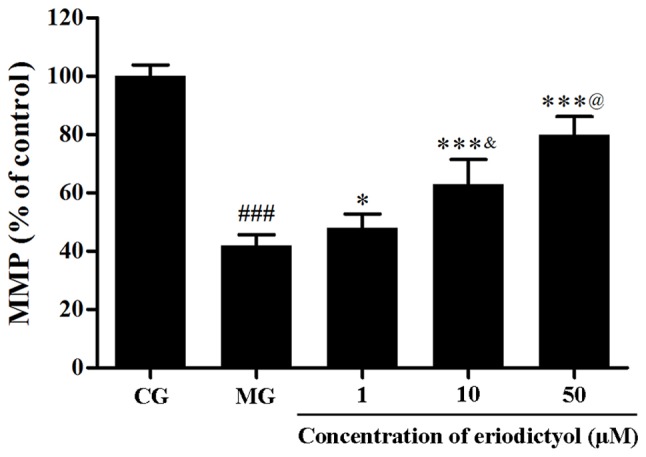
Effect of eriodictyol treatment (1, 10 and 50 µM) on the MMP in H9c2 cardiomyocytes. n=6. ###P<0.001 vs. the CG; *P<0.05 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. MMP, mitochondrial membrane potential; CG, control group; MG, model group.
Effect of eriodictyol on the opening of MPTP
The MPTP opening was evaluated through the fluorescence intensity of free calcein in mitochondria. As shown in Fig. 7, under hypoxia/reoxygenation, the fluorescence intensity of mitochondrial calcein was approximately half that of the CG (50.11±6.00%; P<0.001), which demonstrated that the MPTP opened. Upon treatment with 1, 10 and 50 µM eriodictyol, the fluorescence intensity increased significantly to 58.83±6.84% (P<0.05), 63.75±8.00% (P<0.01) and 80.40±5.92% (P<0.001), respectively, compared to the MG group (Fig. 7). Compared with the CG group (100±11.83%), the ATP level in the MG group (52.62±5.87%) was significantly decreased (P<0.001). Eriodictyol (1, 10 and 50 µM) pre-treatement increased ATP levels to 59.69±4.95% (P<0.05), 69.23±3.27% (P<0.01) and 80.52±9.52% (P<0.001), respectively, compared with the MG group. These results implied that eriodictyol inhibited the MPTP opening.
Figure 7.
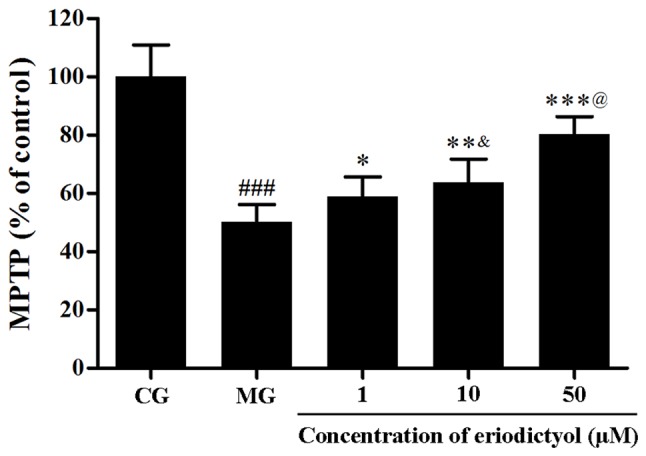
Effect of eriodictyol treatment (1, 10 and 50 µM) on MPTP opening. n=6. ###P<0.001 vs. the CG; *P<0.05, **P<0.01 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. MPTP, mitochondrial permeability transition pore; CG, control group; MG, model group.
Effect of eriodictyol on ATP depletion
The level of intracellular ATP represents the function of mitochondria. To detect the depletion of intracellular ATP, the firefly luciferase method was used. Hypoxia/reoxygenation in H9c2 cardiomyocytes resulted in the decline of the intracellular ATP level, whereas treatment with eriodictyol enhanced the ATP level. The ATP level in the 10 µM eriodictyol group was significantly higher than that in the 1 µM group as well as lower than 50 µM group (both P<0.05; Fig. 8). Compared with the CG group (100±11.83%), the ATP level in the MG group (52.62±5.87%) was significantly decreased (P<0.001). Pretreated with eriodictyol (1, 10 and 50 µM), compared with the MG group, the ATP levels were increased to 59.69±4.95% (P<0.05), 69.23±3.27% (P<0.001) and 80.52±9.52% (P<0.001), respectively. These results indicated that eriodictyol treatment was able to improve the depletion of intracellular ATP in the cardiomyocytes.
Figure 8.
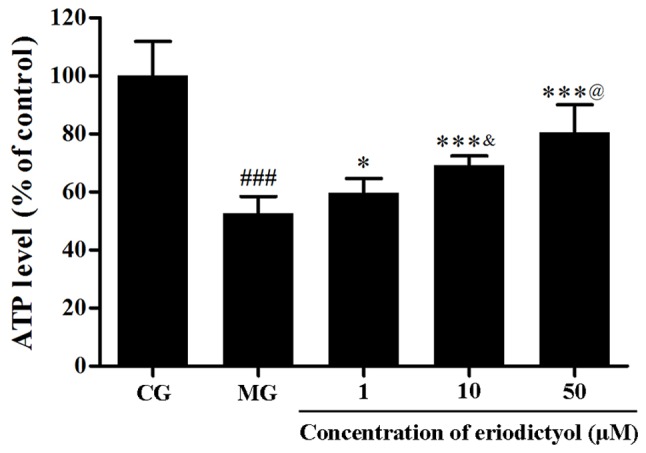
Effect of eriodictyol treatment (1, 10 and 50 µM) on the depletion of ATP. n=6. ###P<0.001 vs. the CG; *P<0.05 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. CG, control group; MG, model group.
Effect of eriodictyol on caspase-3 activity and expression levels of caspase-3, Bcl-2 and Bax
As a member of the cysteinyl aspartate specific protease family, caspase-3 serves a pivotal role in apoptosis through hydrolyzed cleavage (28). Western blot analysis revealed that hypoxia/reoxygenation promoted the expression of caspase-3 in H9c2 cardiomyocytes, while eriodictyol reduced this expression by different extents (Fig. 9A). In addition, colorimetric detection further confirmed the activity of caspase-3 quantitatively. Compared with the control group, the caspase-3 activity was markedly elevated in H9c2 cardiomyocytes treated by hypoxia/reoxygenation. However, in the presence of eriodictyol, the increased activity of caspase-3 was inhibited accordingly. Similarly, the activity of caspase-3 in the 10 µM eriodictyol group was significantly lower than that in 1 µM group as well as higher than 50 µM group (both P<0.05; Fig. 9B). The activity of caspase-3 in the MG group (253.38±17.80%) was significantly decreased compared with the CG group (100.00±24.74%; P<0.001). In contrast to the MG group, caspase-3 activity in the 1, 10 and 50 µM eriodictyol-treated groups was significantly decreased to 230.80±15.03% (P<0.05), 179.40±18.85% (P<0.001) and 147.09±17.41% (P<0.001), respectively.
Figure 9.
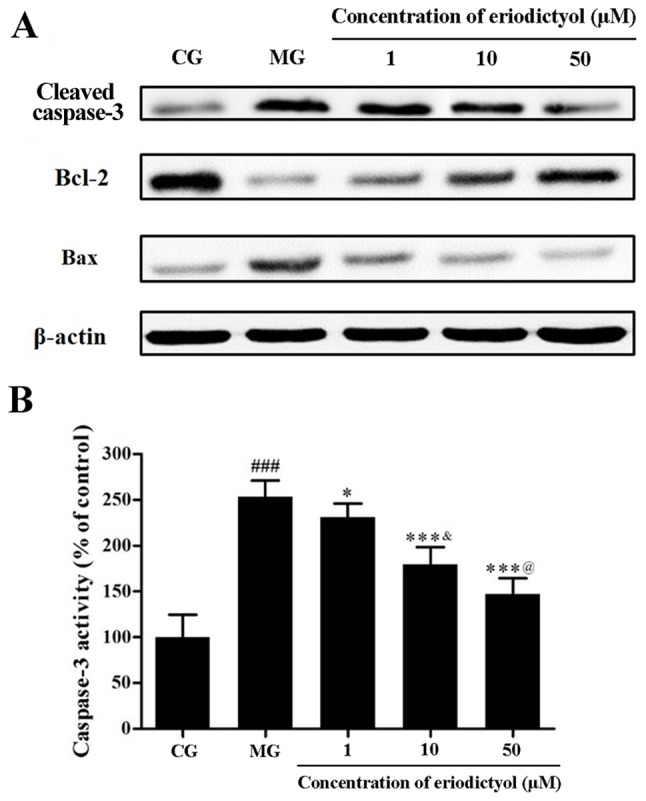
Effect of eriodictyol on the apoptosis of H9c2 cardiomyocytes induced by hypoxia/reoxygenation. (A) Western blot analysis of the protein expression levels of cleaved caspase-3, Bcl-2 and Bax. (B) Activity of caspase-3 in H9c2 cardiomyocytes (n=6). ###P<0.001 vs. the CG; *P<0.05 and ***P<0.001 vs. the MG; &P<0.05 vs. the 1 µM eriodictyol; @P<0.05 vs. the 10 µM eriodictyol. Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated X protein; CG, control group; MG, model group.
Bcl-2 and Bax are the major members of the Bcl-2 protein family, which are involved in mitochondrion-mediated apoptosis. The former demonstrates an anti-apoptotic effect, whereas the latter exhibits a pro-apoptotic effect (29). In the present study, the expression of Bcl-2 was downregulated by hypoxia/reoxygenation in contrast to that in the control group. However, treatment with eriodictyol was observed to upregulate Bcl-2 expression. Accordingly, hypoxia/reoxygenation upregulated the expression of Bax, while eriodictyol treatment suppressed this increased expression (Fig. 9B).
Discussion
Myocardial ischemia and subsequent reperfusion is the major cause of myocardial infarction. The oxygen deprivation will lead to the breakdown of redox homeostasis and ROS overproduction. As the major site of ROS production, mitochondria serve an important role in the injury of myocardial ischemia and reperfusion (30). In addition to the overproduction of ROS, overload of intracellular Ca2+ also affects the function of the mitochondria (31). As the key determinant of mitochondrial dysfunction, the MPTP will open (32). The electrochemical gradient across the inner mitochondrial membrane (MMP) is necessary for mitochondrial function (33). Following the opening of MPTP, free solutes and proteins can be distributed across the inner mitochondrial membrane and result in the collapse of the MMP (34). The dysfunction of mitochondria also leads to the depletion of ATP due to MPTP opening (35), and finally results in the cardiomyocyte apoptosis (36).
Caspases are cysteinyl aspartate specific proteases with a central role in apoptosis, and their activation occurs through cleavage at specific sites (37). As an effector enzyme, caspase-3 is the key mediator responsible for promoting cell apoptosis (38). In addition, Bcl-2 and Bax are members of the Bcl-2 protein family that participate in mitochondrion-mediated apoptosis. Bcl-2 prevents apoptosis and blocks the activation of caspase-3, while Bax promotes cell apoptosis (39).
In the present study, mitochondrial dysfunction and cell injury induced by hypoxia/reoxygenation were observed. Pretreatment with eriodictyol increased the cell survival and blocked the leakage of LDH from the cytosol, which indicates the potential cardioprotective effect of eriodictyol. Further experiments revealed that eriodictyol improved the dysfunction of mitochondria through suppressing the overload of intracellular Ca2+, preventing the overproduction of ROS, blocking the opening of MPTP, increasing the MMP level and decreasing ATP depletion. As important intracellular signaling molecules, there is interplay between Ca2+ and ROS production. Ca2+ may increase ROS production by enhancing metabolism and ROS regulates Ca2+ homeostasis through reciprocal redox (40). Meanwhile, the interplay between ROS and Ca2+ triggers the opening of MPTP opening, which leads to the collapse of MMP (41). In addition, as the main source of ATP, mitochondria may fail to synthesize enough ATP to maintain cellular function due to the dysfunction of MMP (42).
Furthermore, eriodictyol inhibited the apoptosis of H9c2 cardiomyocytes through upregulating the expression of Bcl-2 and downregulating the expression levels of Bax and caspase-3, as well as reducing the activity of caspase-3. As a bioactive flavonoid, eriodictyol showed many protective effects through the inhibition of oxidative stress (14,15,17,18). At the same time, eriodictyol and its glucoside can protect against cerebral ischemia injury in vitro and in vivo (16,43). To the best of our knowledge, the present study is the first to demonstrate the protective effects of eriodictyol on cardiomyocytes injured by hypoxia/reoxygenation, which indicates its potential application in the prevention of myocardial ischemia and reperfusion injury.
In conclusion, the results of the present study demonstrated the myocardial protective effects of eriodictyol and relevant mechanisms in vitro. Eriodictyol can enhance the survival of H9c2 cardiomyocytes injured by hypoxia/reoxygenation. The mechanisms involve the improvement of mitochondrial dysfunction and inhibition of apoptosis via the mitochondria-mediated signaling pathway, including the upregulation of Bcl-2, downregulation of Bax and inhibition of caspase-3. These results provided evidence for further evaluations in vivo for the development of novel therapeutic approaches for myocardial infarction.
References
- 1.Yellon DM, Hausenloy DJ. Realizing the clinical potential of ischemic preconditioning and postconditioning. Nat Clin Pract Cardiovasc Med. 2005;2:568–575. doi: 10.1038/ncpcardio0346. [DOI] [PubMed] [Google Scholar]
- 2.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. New Eng J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 3.Hausenloy DJ, Yellon DM. Targeting myocardial reperfusion injury-the search continues. New Eng J Med. 2015;373:1073–1075. doi: 10.1056/NEJMe1509718. [DOI] [PubMed] [Google Scholar]
- 4.Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 2015;78:23–34. doi: 10.1016/j.yjmcc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Hausenloya DJ, Duchenb MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 6.Pagliaro P, Moro F, Tullio F, Perrelli MG, Penna C. Cardioprotective pathways during reperfusion: Focus on redox signaling and other modalities of cell signaling. Antioxid Redox Signal. 2011;14:833–850. doi: 10.1089/ars.2010.3245. [DOI] [PubMed] [Google Scholar]
- 7.Zhao GL, Yu LM, Gao WL, Duan WX, Jiang B, Liu XD, Zhang B, Liu ZH, Zhai ME, Jin ZX, et al. Berberine protects rat heart from ischemia/reperfusion injury via activating JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress. Acta Pharmacol Sin. 2016;37:354–367. doi: 10.1038/aps.2015.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Shen L, Wang Z, Jiang HP, Liu LX. Tanshinone IIA protects against myocardial ischemia reperfusion injury by activating the PI3K/Akt/mTOR signaling pathway. Biomed Pharmacother. 2016;84:106–114. doi: 10.1016/j.biopha.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 9.Gao Y, Jia P, Shu W, Jia D. The protective effect of lycopene on hypoxia/reoxygenation-induced endoplasmic reticulum stress in H9C2 cardiomyocytes. Eur J Pharmacol. 2016;774:71–79. doi: 10.1016/j.ejphar.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 10.de Sousa LM, de Carvalho JL, da Silva HC, Lemos TL, Arriaga AM, Braz-Filho R, Militão GC, Silva TD, Ribeiro PR, Santiago GM. New cytotoxic bibenzyl and other constituents from Bauhinia ungulata L. (Fabaceae) Chem Biodivers. 2016;13:1630–1635. doi: 10.1002/cbdv.201600058. [DOI] [PubMed] [Google Scholar]
- 11.Milella L, Milazzo S, De Leo M, Vera Saltos MB, Faraone I, Tuccinardi T, Lapillo M, De Tommasi N, Braca A. α-Glucosidase and α-amylase inhibitors from arcytophyllum thymifolium. J Nat Prod. 2016;79:2104–2112. doi: 10.1021/acs.jnatprod.6b00484. [DOI] [PubMed] [Google Scholar]
- 12.Zhong JD, Feng Y, Li HM, Xia XS, Li RT. A new flavonoid glycoside from Elsholtzia bodinieri. Nat Prod Res. 2016;30:2278–2284. doi: 10.1080/14786419.2016.1164698. [DOI] [PubMed] [Google Scholar]
- 13.Zeng B, Chen K, Du P, Wang SS, Ren B, Ren YL, Yan HS, Liang Y, Wu FH. Phenolic compounds from Clinopodium chinense (Benth.) O. Kuntze and their inhibitory effects on α-Glucosidase and vascular endothelial cells injury. Chem Biodivers. 2016;13:596–601. doi: 10.1002/cbdv.201500187. [DOI] [PubMed] [Google Scholar]
- 14.Lou H, Jing X, Ren D, Wei X, Zhang X. Eriodictyol protects against H2O2-induced neuron-like PC12 cell death through activation of Nrf2/ARE signaling pathway. Neurochem Int. 2012;61:251–257. doi: 10.1016/j.neuint.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Jing X, Shi H, Zhu X, Wei X, Ren M, Han M, Ren D, Lou H. Eriodictyol attenuates β-amyloid 25-35 peptide-induced oxidative cell death in primary cultured neurons by activation of Nrf2. Neurochem Res. 2015;40:1463–1471. doi: 10.1007/s11064-015-1616-z. [DOI] [PubMed] [Google Scholar]
- 16.Ferreira Ede O, Fernandes MY, Lima NM, Neves KR, Carmo MR, Lima FA, Fonteles AA, Menezes AP, Andrade GM. Neuroinflammatory response to experimental stroke is inhibited by eriodictyol. Behav Brain Res. 2016;312:321–332. doi: 10.1016/j.bbr.2016.06.046. [DOI] [PubMed] [Google Scholar]
- 17.Li CZ, Jin HH, Sun HX, Zhang ZZ, Zheng JX, Li SH, Han SH. Eriodictyol attenuates cisplatin-induced kidney injury by inhibiting oxidative stress and inflammation. Eur J Pharmacol. 2016;772:124–130. doi: 10.1016/j.ejphar.2015.12.042. [DOI] [PubMed] [Google Scholar]
- 18.Zhu GF, Guo HJ, Huang Y, Wu CT, Zhang XF. Eriodictyol, a plant flavonoid, attenuates LPS-induced acute lung injury through its antioxidative and anti-inflammatory activity. Exp Ther Med. 2015;10:2259–2266. doi: 10.3892/etm.2015.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JK. Anti-inflammatory effects of eriodictyol in lipopolysaccharide-stimulated Raw 264.7 murine macrophages. Arch Pharm Res. 2011;34:671–679. doi: 10.1007/s12272-011-0418-3. [DOI] [PubMed] [Google Scholar]
- 20.Rossato MF, Trevisan G, Walker CI, Klafke JZ, de Oliveira AP, Villarinho JG, Zanon RB, Royes LF, Athayde ML, Gomez MV, Ferreira J. Eriodictyol: A flavonoid antagonist of the TRPV1 receptor with antioxidant activity. Biochem Pharmacol. 2011;81:544–551. doi: 10.1016/j.bcp.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Habtemariam S, Dagne E. Comparative antioxidant, prooxidant and cytotoxic activity of sigmoidin A and eriodictyol. Planta Med. 2010;76:589–594. doi: 10.1055/s-0029-1240604. [DOI] [PubMed] [Google Scholar]
- 22.Walker J, Reichelt KV, Obst K, Widder S, Hans J, Krammer GE, Ley JP, Somoza V. Identification of an anti-inflammatory potential of Eriodictyon angustifolium compounds in human gingival fibroblasts. Food Funct. 2016;7:3046–3055. doi: 10.1039/C6FO00482B. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira PS, Spolidorio LC, Manthey JA, Cesar TB. Citrus flavanones prevent systemic inflammation and ameliorate oxidative stress in C57BL/6J mice fed high-fat diet. Food Funct. 2016;7:2675–2681. doi: 10.1039/C5FO01541C. [DOI] [PubMed] [Google Scholar]
- 24.Chen C, He H, Luo Y, Zhou M, Yin D, He M. Involvement of Bcl-2 signal pathway in the protective effects of apigenin on anoxia/reoxygenation-induced myocardium injury. J Cardiovasc Pharmacol. 2016;67:152–163. doi: 10.1097/FJC.0000000000000331. [DOI] [PubMed] [Google Scholar]
- 25.Ji HJ, Wang DM, Hu JF, Sun MN, Li G, Li ZP, Wu DH, Liu G, Chen NH. IMM-H004, a novel courmarin derivative, protects against oxygen-and glucose-deprivation/restoration-induced apoptosis in PC12 cells. Eur J Pharmacol. 2014;723:259–266. doi: 10.1016/j.ejphar.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, Sun GB, Zhang JY, Luo Y, Yu YL, Xu XD, Meng XB, Zhang MD, Lin WB, Sun XB. Elatoside C protects the heart from ischaemia/reperfusion injury through the modulation of oxidative stress and intracellular Ca2+ homeostasis. Int J Cardiol. 2015;185:167–176. doi: 10.1016/j.ijcard.2015.03.140. [DOI] [PubMed] [Google Scholar]
- 27.Li JZ, Yu SY, Wu JH, Shao QR, Dong XM. Paeoniflorin protects myocardial cell from doxorubicin-induced apoptosis through inhibition of NADPH oxidase. Can J Physiol Pharmacol. 2012;90:1569–1575. doi: 10.1139/y2012-140. [DOI] [PubMed] [Google Scholar]
- 28.Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–539. doi: 10.1038/cdd.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding H, Han R, Chen X, Fang W, Liu M, Wang X, Wei Q, Kodithuwakku ND, Li Y. Clematichinenoside (AR) attenuates hypoxia/reoxygenation-induced H9c2 cardiomyocyte apoptosis via a mitochondria-mediated signaling pathway. Molecules. 2016;21(pii):E683. doi: 10.3390/molecules21060683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madungwe NB, Zilberstein NF, Feng Y, Bopassa JC. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. Am J Cardiovasc Dis. 2016;6:93–108. [PMC free article] [PubMed] [Google Scholar]
- 31.Hurst S, Hoek J, Sheu SS. Mitochondrial Ca2+ and regulation of the permeability transition pore. J Bioenerg Biomembr. 2017;49:27–47. doi: 10.1007/s10863-016-9672-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 33.Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kroemer G. Mitochondrial control of apoptosis: An overview. Biochem Soc Symp. 1999;66:1–15. doi: 10.1042/bss0660001. [DOI] [PubMed] [Google Scholar]
- 35.Li YY, Xiao L, Qiu LY, Yan YF, Wang H, Duan GL, Liao ZP, Chen HP. Sasanquasaponin-induced cardioprotection involves inhibition of mPTP opening via attenuating intracellular chloride accumulation. Fitoterapia. 2017;116:1–9. doi: 10.1016/j.fitote.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 37.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 38.Uchiyama T, Otani H, Okada T, Ninomiya H, Kido M, Imamura H, Nogi S, Kobayashi Y. Nitric oxide induces caspase-dependent apoptosis and necrosis in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2002;34:1049–1061. doi: 10.1006/jmcc.2002.2045. [DOI] [PubMed] [Google Scholar]
- 39.Youle RJ, Strasser A. The Bcl-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 40.Yan Y, Wei CL, Zhang WR, Cheng HP, Liu J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol Sin. 2006;27:821–826. doi: 10.1111/j.1745-7254.2006.00390.x. [DOI] [PubMed] [Google Scholar]
- 41.Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jašová M, Kancirová I, Waczulíková I, Ferko M. Mitochondria as a target of cardioprotection in models of preconditioning. J Bioenerg Biomembr. 2017;49:357–368. doi: 10.1007/s10863-017-9720-1. [DOI] [PubMed] [Google Scholar]
- 43.Jing X, Ren D, Wei X, Shi H, Zhang X, Perez RG, Lou H, Lou H. Toxicol Appl Pharmacol. 2013;273:672–679. doi: 10.1016/j.taap.2013.10.018. [DOI] [PubMed] [Google Scholar]