

Abstract
Hypertrophic cardiomyopathy (HCM) is the most common genetic cardiovascular disease. Although some genes and miRNAs related with HCM have been studied, the molecular regulatory mechanisms between miRNAs and transcription factors (TFs) in HCM have not been systematically elucidated. In this study, we proposed a novel method for identifying dysregulated miRNA‐TF feed‐forward loops (FFLs) by integrating sample matched miRNA and gene expression profiles and experimentally verified interactions of TF‐target gene and miRNA‐target gene. We identified 316 dysregulated miRNA‐TF FFLs in HCM, which were confirmed to be closely related with HCM from various perspectives. Subpathway enrichment analysis demonstrated that the method was outperformed by the existing method. Furthermore, we systematically analysed the global architecture and feature of gene regulation by miRNAs and TFs in HCM, and the FFL composed of hsa‐miR‐17‐5p, FASN and STAT3 was inferred to play critical roles in HCM. Additionally, we identified two panels of biomarkers defined by three TFs (CEBPB, HIF1A, and STAT3) and four miRNAs (hsa‐miR‐155‐5p, hsa‐miR‐17‐5p, hsa‐miR‐20a‐5p, and hsa‐miR‐181a‐5p) in a discovery cohort of 126 samples, which could differentiate HCM patients from healthy controls with better performance. Our work provides HCM‐related dysregulated miRNA‐TF FFLs for further experimental study, and provides candidate biomarkers for HCM diagnosis and treatment.
Keywords: feed‐forward loop, miRNA, transcription factor
1. INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most common genetic cardiovascular disease and is a leading cause of disability and death in patients of all ages, especially sudden and unexpected cardiac death in young people.1 From encoding protein RNAs to non‐coding RNAs, our understanding about HCM has improved dramatically both clinically and pathophysiologically. However, the potential molecular mechanisms underlying the pathology of HCM have not been fully understood.
Amongst many genetic factors, miRNAs and transcription factors (TFs) are two types of key gene regulators, and they both participate in many important cellular processes, including cell differentiation, proliferation, and apoptosis.2 MiRNAs mainly regulate gene expression at the post‐transcriptional level, while TFs modulate gene transcription at the transcriptional level. Researchers have demonstrated that miRNAs and TFs may synergistically regulate the same target genes, and they may mutually regulate one another; hence forming feed‐forward loops (FFLs), which have been reported to form recurrent network motifs and play important roles in the mammalian gene regulatory network.2, 3 Thus, dysregulated miRNA‐TF FFLs will lead to a series of diseases, and deciphering the interplay between miRNAs and TFs by means of FFLs will yield new mechanistic insights into specific biological events.
Currently available biological databases have integrated different types of molecular interactions, which made it possible to identify miRNA‐TF FFLs. For instance, TarBase,4 miRTarBase,5 and miRecords6 collected experimentally verified miRNA‐gene regulatory relationships for different organisms, while TargetScan7 predicted biological targets for miRNAs. TRED8 and Transfac9 offered experimentally confirmed TF‐gene regulatory relationships, while JASPAR10 predicted TF targets. TransmiR database11 recruited experimentally verified TF‐miRNA regulatory pairs. Additionally, large amount of genome‐wide data such as microarray data and next‐generation sequencing data also provide us more valuable information to investigate dysregulated FFLs implicated with specific cellular processes and diseases.
Much efforts have been devoted to detect miRNA‐TF FFLs, which were used to dissect potential regulatory mechanisms underlying human diseases.12, 13 On the one hand, started from disease‐related molecules and different regulatory relationships amongst miRNAs, genes and TFs, Ye et al14 revealed that miR‐19 inhibited CYLD through the identified disrupted FFLs in T‐cell acute lymphoblastic leukaemia. Qin et al15 constructed gene regulatory networks involved in miRNA‐TF FFLs for three subtypes of breast cancer, and investigated their distinct and common characteristics. In addition, 4‐node FFLs were proposed and identified by Sun et al16 in glioblastoma, from which critical miRNAs and subnetworks were detected. Recently, Zhang et al12 and Arora et al13 have reviewed miRNA and TF FFLs involved in various biological processes and diseases. On the other hand, computational methods have been developed for identifying dysregulated miRNA and TF FFLs based on sample matched mRNA and miRNA expression profiles.17, 18 For example, Yan et al17 proposed a method, dChip‐GemiNI, for identifying significant miRNA‐TF FFLs associated with five cancers. Subsequently, Jiang et al18 developed an algorithm to dissect dysregulated miRNA‐TF FFLs across 13 tumor types, and identified 26 pan‐cancer FFLs. Therefore, using FFLs to decipher the pathological and physiological mechanisms underlying diseases will provide new clues for understanding disease initiation and progression.
In this study, we presented a method for systematically identifying dysregulated miRNA‐TF FFLs in HCM, and 316 dysregulated FFLs were obtained. We found that these dysregulated FFLs were significantly enriched in significantly differentially expressed molecules and the known HCM‐related molecules. Functional analysis also demonstrated that the FFLs were closely associated with HCM. We further investigated the global architecture and feature of regulation between miRNAs and TFs in HCM by constructing a dysregulated miRNA‐TF regulatory network, from which a FFL (hsa‐miR‐17‐5p, STAT3 and FASN) might play important roles in HCM and two panels of diagnostic biomarkers defined by three TFs and four miRNAs were identified.
2. MATERIALS AND METHODS
2.1. Obtaining and preprocessing of miRNA, gene and TF expression profiles
The HCM related sample‐mathed gene expression profiles and miRNA expression profiles (GSE36961 and GSE36946) were downloaded from the Gene Expression Omnibus database. This expression profiles were obtained by measuring human cardiac tissues from 106 HCM patients and 20 control donors. Protein‐coding genes were retained and miRNA names were manually mapped to standard mature miRNA names based on miRBase database (release 21). If multiple probes corresponded to one gene, the expression values were averaged. A list of human TFs were acquired from a previous study19 and TF expression profiles were extracted from gene expression data. Finally, 23 601 mRNAs, 805 miRNAs, 1369 TFs and their corresponding expression profiles were obtained.
2.2. Regulatory relationships amongst miRNAs, genes, and TFs
Experimentally verified regulatory relationships amongst miRNAs, genes, and TFs were used. Firstly, experimentally confirmed miRNA‐gene interactions were collected from TarBase (version 6.0),4 miRTarBase (version7.0),5 and miRecords (version 4)6 databases. Secondly, using TFs obtained above, the miRNA‐TF regulatory relationships were determined from miRNA‐gene interactions. Thirdly, experimentally confirmed TF‐gene interactions were collected from TRED8 and Transfac,9 and experimentally verified TF‐miRNA regulatory relations were retrieved from TransmiR (version 1.2)11 database.
2.3. Candidate miRNA‐TF FFLs
In this study, we focused on three‐node miRNA‐TF FFLs, which included a gene, a miRNA, and a TF. According to the main regulator, miRNA‐TF FFLs can be typically classified into three types12, 13: miRNA‐FFL, TF‐FFL, and composite FFL (Figure S1). In a miRNA‐FFL, miRNA is the main regulator, which controls the expression of a TF and their common target gene, while in a TF‐FFL, TF is the primary regulator. In a composite‐FFL, a miRNA regulates the expression of a TF and a target gene. Simultaneously, the TF dominates the expression of the miRNA and the target gene. By integrating expression profile data and the interactions between miRNAs, TFs, and genes, we identified 6,809 candidate miRNA‐TF FFLs (Figure 1A), which comprised of 5549 miRNA‐FFLs (81.49%), 851 TF‐FFLs (12.50%) and 409 composite FFLs (6.01%). These candidate FFLs included 434 miRNAs, 1253 genes, and 223 TFs.
Figure 1.
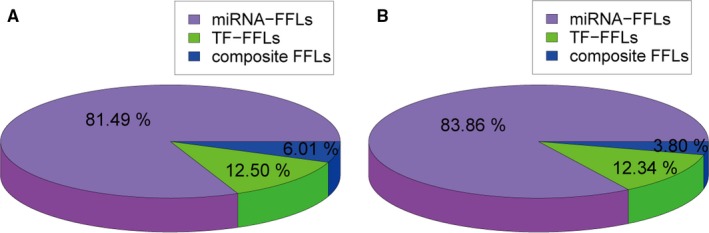
Statistical result of miRNA‐TF FFLs in HCM. (A) The distribution of three types of FFLs in candidate miRNA‐TF FFLs. (B) The distribution of three types of FFLs in dysregulated miRNA‐TF FFLs
2.4. Identification of dysregulated miRNA‐TF FFLs in HCM
The dysregulated miRNA‐TF FFLs in HCM were identified using sample matched expression profile data. Firstly, each node was scored according to the extent of differential expression18, 20 using the formulas (1) and (2):
| (1) |
| (2) |
where φ−1 is the inverse normal cumulative distribution function. p is the P‐value which indicates the significance of differential expression computed by the R “limma” package. FC is the fold change of this gene expression.
Secondly, each edge was scored according to the change of gene co‐expression between HCM case samples and control samples using the following equations18, 21, 22:
| (3) |
| (4) |
| (5) |
where r case and r control are the Spearman correlation coefficient of gene expression in case and control samples, respectively. While n case and n control represent the samples size, respectively. Function of F is Fisher transformation, which have been shown that applying it could improve the power of identifying differentially rewired genes.23
Subsequently, the score of a candidate miRNA‐TF FFL was calculated by combining the node score and the edge score as follows:
| (6) |
where n node and n edge denote the number of nodes and edges in the miRNA‐TF FFL. The parameter α∈(0,1) is used to control the weight of node score and edge score. Here, we considered the node and edge score were weighted equally, and selected α as 0.5.
Finally, a P‐value was computed to reflect the significance of a miRNA‐TF FFL. We constructed a random miRNA‐TF FFL by randomly selecting a miRNA, a gene, and a TF, and then calculated the score of this FFL through the above procedure. This process repeated 10 000 times. The empirical P‐value was defined as the proportion of randomly obtained FFL scores larger than the real FFL score as below:
| (7) |
In this article, the FFL with P‐value <0.01 were selected as dysregulated FFLs.
2.5. Collection of genes and miRNAs related to HCM
Genes related to HCM were compiled from a comprehensive human gene‐disease association database, DisGeNET (V5.0),24 which integrated many currently widely used gene‐disease databases, such as the Online Mendelian Inheritance in Man database,25 the Comparative Toxicogenomics Database,26 the Genetic Association Database,27 the Mouse Genome Database,28 PubMed and Uniprot.29 After removing repeating gene‐disease entries, 300 unique genes associated with HCM were obtained.
HCM‐related miRNAs were collected by performing a comprehensive literature review. We searched three manually curated and experimentally confirmed human miRNA‐disease association databases: HMDD (version 2.0),30 miR2Disease,31 and PhenomiR (February 2011),32 and found that none of these databases contained miRNAs related to HCM, although HMDD and miR2Disease included miRNAs associated with cardiac hypertrophy. Therefore, we inquired PubMed using the phrase “hypertrophic cardiomyopathy AND microRNA.” Each article was manually searched for miRNAs with aberrant expression in HCM. The miRNAs were then mapped to standard mature miRNA names from miRBase database (release 21),33 and 54 unique mature miRNAs were ultimately selected.
2.6. Subpathway enrichment analysis
Existing researches have shown that type‐specific biological functions tend to be located in local areas of the pathway (subpathway) instead of the entire pathway, and thus subpathway may provide more detailed explanations for pathogenesis.34, 35 KEGG subpathway (local area of the entire biological pathway) enrichment analysis was performed by the R “SubpathwayMiner” package.34 Significantly enriched subpathways were identified with a P < 0.05. To show the results more clearly, we retained the subpathway with the minimum P‐value if multiple significantly enriched subpathways corresponded to an entire pathway.
2.7. Identification of potential diagnostic biomarkers in HCM
Potential biomarkers for distinguishing HCM patients from controls were identified using a classification model based on support vector machine (SVM), which was performed using the R “e1071” package. The performance was evaluated by classification accuracy and the area under the receiving operating curve (AUC) using 5‐fold cross‐validation.
The optimal biomarkers in HCM diagnosis were selected using Li et al's method.36 We computed classification accuracy for all combinations by applying SVM, and the optimal biomarkers were selected considering a balance between classification accuracy and the number of biomarkers.
3. RESULTS
3.1. Dysregulated miRNA‐TF FFLs in HCM
We identified 316 dysregulated miRNA‐TF FFLs in HCM using our method (Figure 1B and Table S1). These dysregulated FFLs included 265 miRNA‐FFLs (83.86%), 39 TF‐FFLs (12.34%), and 12 composite FFLs (3.80%). Merging these FFLs 118 miRNAs, 102 genes and 53 TFs were obtained. The number of nodes and links in the FFLs was shown in Table 1.
Table 1.
Summary of three types of dysregulated miRNA‐TF FFLs in HCM
| Motif | Number of FFLs | Number of nodes | Number of links | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genes | miRNAs | TFs | Total | miRNA‐gene | miRNA‐TF | TF‐gene | TF‐miRNA | Total | ||
| miRNA‐FFL | 265 | 98 | 101 | 53 | 252 | 232 | 181 | 151 | – | 564 |
| TF‐FFL | 39 | 17 | 29 | 11 | 57 | 38 | – | 22 | 30 | 90 |
| Composite‐FFL | 12 | 9 | 5 | 5 | 19 | 10 | 7 | 11 | 7 | 35 |
| Total | 316 | 102 | 118 | 53 | 273 | 268 | 188 | 171 | 37 | 664 |
3.2. Validation of dysregulated miRNA‐TF FFLs for their roles in HCM
We searched for experimentally confirmed miRNA‐TF FFLs in HCM by performing a comprehensive literature review, but no result obtained. Therefore, we validated the dysregulated FFLs from several other perspectives in the absence of gold standard set of FFLs in HCM and compared the method with Jiang et al's method.18
We investigated significantly differentially expressed (SDE) miRNAs, genes and TFs in dysregulated FFLs. The R “limma” package with P < 0.01 and fold change >1.2 were used to select SDE molecules. As shown in Figure S2, The proportion of SDE molecules was significantly higher than that of candidate FFLs (hypergeometric test, P < 0.001). Amongst these SDE molecules, most were down‐regulated. Meanwhile, the proportion of HCM‐related molecules in dysregulated FFLs was significantly higher than candidate FFLs (hypergeometric test, P < 0.001). Additionally, SDE molecules and HCM‐related molecules in top 5%, 10%, 20%, 30%, 40%, 50% dysregulated FFLs were examined (Figure 2A and B, Table S2). We observed that the top 5% dysregulated FFLs contained the largest proportion of molecules that were SDE (62.50%) and related to HCM (21.88%). By comparison, the FFLs obtained by Jiang et al's method didn't show this phenomenon.
Figure 2.
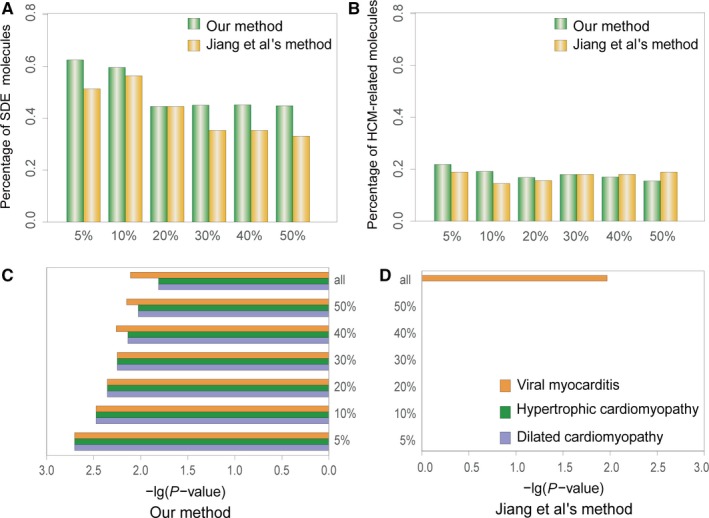
Comparison of our method and Jiang et al.'s method in top 5%, 10%, 20%, 30%, 40%, 50% dysregulated FFLs. (A) The distribution of SDE molecules. (B) The distribution of HCM‐related molecules. (C and D) The distribution of significantly enriched subpathways
Biological pathways implicated with dysregulated FFLs were also studied. SubpathwayMiner34 was performed to identify significantly enriched KEGG subpathways, and 89 significant subpathways were obtained (P < 0.05, Table S3). It is noteworthy that current version of KEGG included five pathways in cardiovascular diseases, and three pathways were significantly enriched: viral myocarditis (P = 7.76 × 10−3), hypertrophic cardiomyopathy (P = 1.55 × 10−2), and dilated cardiomyopathy (P = 1.55 × 10−2). Simultaneously, several other pathways closely related with HCM were identified, including signal transduction‐related PI3K‐Akt and MAPK signalling pathway, inflammation, and immune‐related leucocyte transendothelial migration and T cell receptor signalling pathway, heart function signalling‐related focal adhesion, and axon guidance. We also explored significantly enriched subpathways of top 5%, 10%, 20%, 30%, 40%, 50% dysregulated FFLs. Consequently, they all significantly enriched in the above three cardiovascular diseases pathways, and P‐values of the top 5% dysregulated FFLs were the smallest (Figure 2C, Table S3). Compared to Jiang et al's method,18 we found that they were not significantly enriched in any cardiovascular diseases pathway, while all the dysregulated FFLs were only enriched in viral myocarditis (Figure 2D, Table S4). All the results demonstrated that the dysregulated FFLs we identified played key roles in HCM, and the more significant the dysregulated FFLs, the more valuable information they will provide for HCM. Additionally, when we limited miRNAs, genes and TFs to be SDE, 19 dysregulated FFLs were obtained. But none of cardiovascular disease pathways was significantly enriched (Table S5), suggesting that the initiation and progression of diseases was caused by not only the expression change of one single gene, but more importantly the inter‐gene interaction.
We paid close attention to the top 5% dysregulated FFLs (Figure 3A). These FFLs included 32 molecules, of which 20 were SDE, seven were the known HCM‐related molecules and the remaining 25 molecules were all verified to be implicated with cardiac development or cardiovascular disorders (Table 2 and Table S6). The subpathways enrichment analysis showed that the three cardiovascular disease pathways were all ranked at the top positions (Figure 3B). GO analysis revealed that the genes in top 5% dysregulated FFLs tended to be significantly enriched in three GO terms (Biological Process, P < 0.05) functional clusters using Cytoscape plug‐in ClueGO, and these clusters mainly included apoptotic. The known researches showed that apoptosis was involved in the development of myocardial fibrosis in familiar HCM37 and apoptosis constituted a major biological phenomenon in the development of HCM.38 Interestingly, the FFL formed by hsa‐miR‐17‐5p, MCL1, and STAT3 has been confirmed in a recent report.39 Kumar et al showed that miR‐17‐5p regulated autophagy in murine mycobacterium tuberculosis‐infected macrophages by targeting MCL1 and STAT3, and STAT3 silencing suppressed MCL1 levels.39 Simultaneously, Xiang et al reported that STAT3 up‐regulated expression of anti‐apoptotic MCL1 in neonatal rat cardiomyocytes.40
Figure 3.
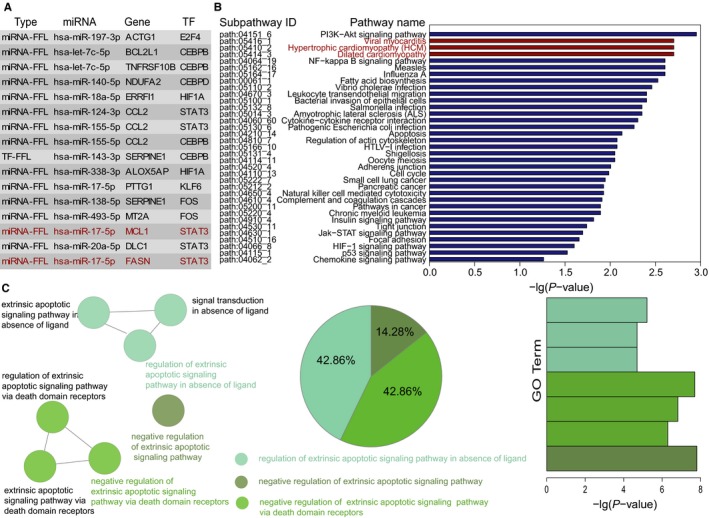
The top 5% dysregulated FFLs and their functional analysis. (A) The top 5% dysregulated FFLs. The red coloured FFLs denote the FFLs mentioned later. (B) Significantly enriched KEGG subpathways. The red coloured pathways denote that they belong to cardiovascular disease pathways in KEGG. (C) Significantly enriched GO terms. The similar GO terms are labelled in the same color
Table 2.
Literature validation of nodes in top 5% dysregulated FFLs associated with cardiac development and cardiovascular disorders
| Type | Molecule | Known research | Year | PMID |
|---|---|---|---|---|
| miRNA | hsa‐let‐7c‐5p | (1) Cardiac development | (1) 2017 | (1) 29057256 |
| (2) Cardiac development | (2) 2014 | (2) 24365598 | ||
| (3) Heart failure | (3) 2016 | (3) 27072074 | ||
| miRNA | hsa‐miR‐124‐3p | (1) Cardiomyocyte hypertrophy | (1) 2017 | (1) 28478799 |
| (2) Vascular smooth muscle cells proliferation and migration | (2) 2017 | (2) 29042195 | ||
| (3) Atherosclerosis | (3) 2017 | (3) 28457624 | ||
| miRNA | hsa‐miR‐138‐5p | (1) Congenital Heart Disease | (1) 2018 | (1) 29298094 |
| (2) Alcoholic cardiomyopathy | (2) 2015 | (2) 25791397 | ||
| (3) Cardiac development | (3) 2008 | (3) 19004786 | ||
| miRNA | hsa‐miR‐140‐5p | (1) Cardiac development | (1) 2015 | (1) 26465880 |
| (2) Heart failure | (2) 2016 | (2) 27072074 | ||
| (3) Cardiotoxicity | (3) 2017 | (3) 29304479 | ||
| miRNA | hsa‐miR‐143‐3p | (1) Dilated cardiomyopathy | (1) 2018 | (1) 29335596 |
| (2) Insulin action in cardiomyocytes | (2) 2013 | (2) 23812417 | ||
| (3) Coronary heart disease | (3) 2017 | (3) 29321799 | ||
| miRNA | hsa‐miR‐155‐5p | (1) Cardiac hypertrophy | (1) 2015 | (1) 26086795 |
| (2) Dilated cardiomyopathy | (2) 2015 | (2) 25840506 | ||
| miRNA | hsa‐miR‐197‐3p | (1) Cardiometabolic | (1) 2017 | (1) 28178938 |
| (2) Cardiovascular death | (2) 2015 | (2) 26720041 | ||
| miRNA | hsa‐miR‐338‐3p | (1)Diabetic cardiomyopathy | (1) 2014 | (1) 23797610 |
| (2) Autophagy in cardiomyocytes | (2) 2017 | (2) 29247537 | ||
| miRNA | hsa‐miR‐493‐5p | (1) Coronary microembolisation | (1) 2017 | (1) 28968594 |
| Gene | ACTG1 | (1) Myocardial injury | (1) 2018 | (1) 29068691 |
| Gene | ALOX5AP | (1) Familial hypercholesterolemia | (1) 2009 | (1) 19361804 |
| (2) Coronary heart disease | (2) 2010 | (2) 21199733 | ||
| Gene | BCL2L1 | (1) Cardiac dysfunction | (1) 2008 | (1) 18313710 |
| Gene | CCL2 | (1) Cardiomyopathy | (1) 2014 | (1) 24980781 |
| (2) Ischaemic cardiomyopathy | (2) 2007 | (2) 17692033 | ||
| (3) Cardiac fibrosis | (3) 2009 | (3) 19482709 | ||
| Gene | DLC1 | (1) Congenital heart disease | (1) 2014 | (1) 24587289 |
| Gene | ERRFI1 | (1) Metabolic syndrome | (1) 2016 | (1) 27778020 |
| Gene | MCL1 | (1) Survival of cardimyocytes during oxidative stress | (1) 2016 | (1) 27220418 |
| (2) Myocardial homoeostasis and autophagy | (1) 2013 | (1) 24165322 | ||
| Gene | MT2A | (1) Cardiomyopathy | (1) 2016 | (1) 27477335 |
| (2) Coronary heart disease | (1) 2014 | (2) 25555862 | ||
| Gene | NDUFA2 | (1) Cardiomyocytes oxidative stress | (2) 2013 | (1) 23891692 |
| Gene | SERPINE1 | (1) Hypertrophic cardiomyopathy | (1) 2013 | (1) 23756156 |
| (2) Heart failure | (2) 2016 | (2) 27284354 | ||
| Gene | TNFRSF10B | (1) Plasma fatty acid distribution | (1) 2010 | (1) 20410100 |
| TF | CEBPB | (1) Cardiovascular disease | (1) 2010 | (1) 20460359 |
| (2) Cardiac fibroblast senescence | (2) 2015 | (2) 25472717 | ||
| TF | CEBPD | (1) Ischaemic cardiomyopathy | (1) 2015 | (1) 25884818 |
| TF | E2F4 | (1) Cardiomyocyte proliferation | (1) 2010 | (1) 19955219 |
| (2) Cardiomyoycte cell proliferation | (2) 2006 | (2) 17102628 | ||
| TF | KLF6 | (1) Cardiac fibrosis | (1) 2015 | (1) 25987545 |
| (2) Cardiac fibrosis | (2) 2013 | (2) 23724005 | ||
| TF | STAT3 | (1) Cardiac Hypertrophy and Fatty Heart | (1) 2015 | (1) 26161779 |
| (2) Familial hypertrophic cardiomyopathy | (2) 2008 | (2) 18362229 | ||
| (3) Cardiomyocyte apoptosis | (3) 2014 | (3) 25200830 |
3.3. The global architecture and feature of regulation between miRNAs and TFs in HCM
To investigate the global architecture and feature of gene regulation by miRNAs and TFs in HCM, we constructed dysregulated miRNA‐TF regulatory network (DmiR_TF_Net) via merging three types of dysregulated FFLs identified above. As shown in Figure 4A, the network included 273 nodes (118 miRNAs, 102 genes and 53 TFs) and 664 edges. Amongst these edges, 268 belonged to miRNA‐gene pairs, 188 belonged to miRNA‐TF pairs, 171 belonged to TF‐gene pairs, and 37 belonged to TF‐miRNA pairs.
Figure 4.
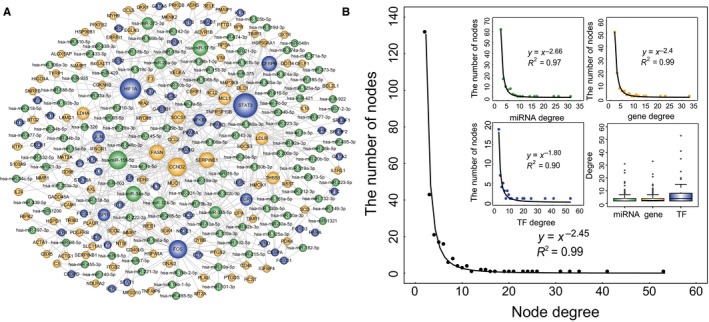
DmiR_TF_Net and its structural features. (A) Global view of the DmiR_TF_Net. The DmiR_TF_Net consists of 664 edges between 118 miRNAs (green circles), 102 genes (yellow circles), and 53 TFs (blue circles). The node size is proportional to the node degree in the network. (B) Degree distribution of all nodes in the DmiR_TF_Net, and degree distribution of miRNAs, genes, and TFs
To examine the global view of the DmiR_TF_Net, we calculated degree and their distribution. Like many other biological networks, degree distribution of this network displayed a power law with a slope of −2.45 and an R 2 of 0.99, indicating that the network was scale‐free (Figure 4B). Therefore, we observed that most nodes degree was low and only a few nodes highly connected with other nodes. Additionally, the individual degree distribution of miRNAs, genes and TFs also demonstrated the same features. The average node degree of miRNAs, genes, and TFs was 4.18 (range 2‐31), 4.30 (range 2‐33), and 7.47 (range 2‐53), respectively (Figure 4B).
We further investigated degree and betweenness centrality (BC) of the known HCM‐related nodes and nodes in top 5% dysregulated FFLs. As shown in Figure 5A‐D, HCM‐related nodes and top 5% dysregulated FFLs nodes have significantly higher degree and BC than other nodes (Wilcoxon rank sum test). The nodes with highly connected features (hubs) and high BC are usually considered to play critical roles in maintaining the overall connectivity of the network. We selected hub nodes using the method proposed by Yu et al,41 and four hub miRNAs (hsa‐miR‐155‐5p, hsa‐miR‐17‐5p, hsa‐miR‐34a‐5p, and hsa‐miR‐20a‐5p), four hub genes (CCND2, FASN, SERPINE1, LDLR) and four hub TFs (STAT3, HIF1A, FOS, and CEBPB) were identified (Table 3). The top 5% miRNAs and TFs with the highest BC were six miRNAs (hsa‐miR‐182‐5p, hsa‐miR‐155‐3p, hsa‐miR‐124‐3p, hsa‐miR‐34a‐5p, hsa‐miR‐17‐5p, and hsa‐miR‐181a‐5p), five genes (PDK4, CCND2, FASN, SERPINE1, and SCD) and three TFs (STAT3, HIF1A, and FOS) (Table 4). Additionally, we observed that the hub nodes and the nodes with high BC were both significantly enriched in nodes related with HCM and nodes within top 5% dysregulated FFLs (Hypergeometric test, Figure 5E and F). All the observations demonstrated that the nodes with high degree and BC might play critical roles in HCM.
Figure 5.
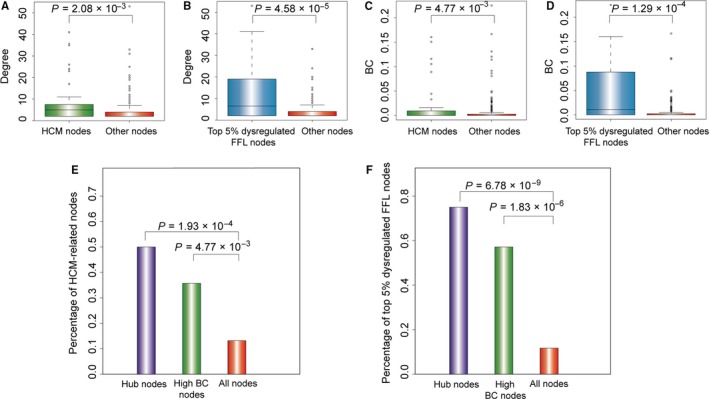
HCM‐related nodes and nodes in top 5% dysregulated FFLs tend to be hubs and high BC nodes. (A and B) The degree of HCM‐related nodes and nodes in top 5% dysregulated FFLs were significantly higher than that of other nodes. (C and D) The BC of HCM‐related nodes and nodes in top 5% dysregulated FFLs was significantly higher than that of other nodes. (E) The percentage of HCM‐related nodes in hub nodes, high BC nodes, and all nodes. (F) The percentage of top 5% dysregulated FFL nodes in hub nodes, high BC nodes, and all nodes
Table 3.
Hub miRNAs, hub genes, and hub TFs in the DmiR_TF_Net in HCM
| miRNAs | Degree | Genes | Degree | TFs | Degree |
|---|---|---|---|---|---|
| hsa‐miR‐155‐5p∆ | 31 | CCND2 | 33 | STAT3∆ | 53 |
| hsa‐let‐17‐5p#∆ | 26 | FASN#∆ | 23 | HIF1A#∆ | 41 |
| hsa‐miR‐34a‐5p# | 24 | SERPINE1∆ | 21 | FOS#∆ | 35 |
| hsa‐miR‐20a‐5p#∆ | 17 | LDLR | 15 | CEBPB∆ | 25 |
miRNAs, genes and TFs with “∆” denote the miRNAs, genes and TFs are within top 5% dysregulated FFLs, and those with “#” indicate the known miRNAs, genes and TFs associated with HCM.
Table 4.
MiRNAs, genes, and TFs with the highest (top 5%) betweenness centrality (BC) in the DmiR_TF_Net in HCM
| miRNAs | BC | Genes | BC | TFs | BC |
|---|---|---|---|---|---|
| hsa‐miR‐182‐5p | 0.1667 | PDK4 | 0.1667 | STAT3∆ | 0.2252 |
| hsa‐miR‐155‐5p∆ | 0.1311 | CCND2 | 0.1144 | HIF1A#∆ | 0.1605 |
| hsa‐miR‐124‐3p∆ | 0.1205 | FASN#∆ | 0.1053 | FOS#∆ | 0.1508 |
| hsa‐miR‐34a‐5p# | 0.1161 | SERPINE1∆ | 0.0916 | ||
| hsa‐miR‐17‐5p#∆ | 0.0891 | SCD | 0.0369 | ||
| hsa‐miR‐181a‐5p | 0.0496 |
miRNAs and TFs with “∆” denote the miRNAs, genes, and TFs are within top 5% dysregulated FFLs, and those with “#” indicate the known miRNAs, genes, and TFs associated with HCM.
Amongst these hub nodes and high BC nodes, we found that hsa‐miR‐17‐5p, FASN, and STAT3 formed a FFL, and this FFL was within the top 5% dysregulated FFLs (Figure 3). Hsa‐miR‐17‐5p and FASN were both known HCM‐related genes. While increased levels of phosphorylated STAT3 were observed in a double‐mutation mouse model of familial HCM, and corresponded with the occurrence of disease.42 Additionally, it has been reported that by targeting STAT3, miR‐17‐5p regulated mouse cardiomyocyte apoptosis in response to ischaemia followed by reperfusion43 and induced protective autophagy and anti‐apoptosis in vascular smooth muscle cells.44
3.4. Potential diagnostic biomarkers in HCM
Based on the above observations, we focused on the 12 regulators (five TFs and seven miRNAs) with high degree and BC in the DmiR_TF_Net, which might be associated with HCM occurrence. There were 25 − 1 = 31 and 27 − 1 = 127 combinations of these TFs and miRNAs, respectively. We calculated classification accuracies for all the combinations by applying SVM classification model, and the optimal biomarkers were achieved. Finally, two panel biomarkers defined by three TFs (CEBPB, HIF1A and STAT3) and four miRNAs (hsa‐miR‐155‐5p, hsa‐miR‐17‐5p, hsa‐miR‐20a‐5p, and hsa‐miR‐181a‐5p) with the highest classification accuracy were identified. For the signature of three TFs, an accuracy of 0.937 and an AUC value of 0.927 were obtained in the training set based on 5‐fold cross‐validation (Figure 6A). We further examined this signature in an independent test set (GSE68316) including seven HCM patients and five healthy individuals, and an accuracy of 0.833 and an AUC value of 0.800 were obtained (Figure 6B). Similarly, for the signature of four miRNAs, we achieved an accuracy of 0.889 and an AUC value of 0.883 in the training set (Figure 6C). Hierarchical clustering analysis was performed using expression data of the two panel biomarkers and two major sample clusters with clear differences were found (Figure 6D‐F). For three TFs, the rates of HCM patients in the predicted HCM group were 100% both in training set (106/106) and test set (7/7), whereas the corresponding rates in the predicted healthy group were 45% (9/20) and 80% (4/5), respectively. For four miRNAs, the rates of HCM patients in the predicted HCM group were 92.45% (98/106) in training set, whereas the corresponding rates in the predicted healthy group were 10% (2/20). All these results indicated that the signatures we identified had better performance in distinguishing HCM patients from controls.
Figure 6.
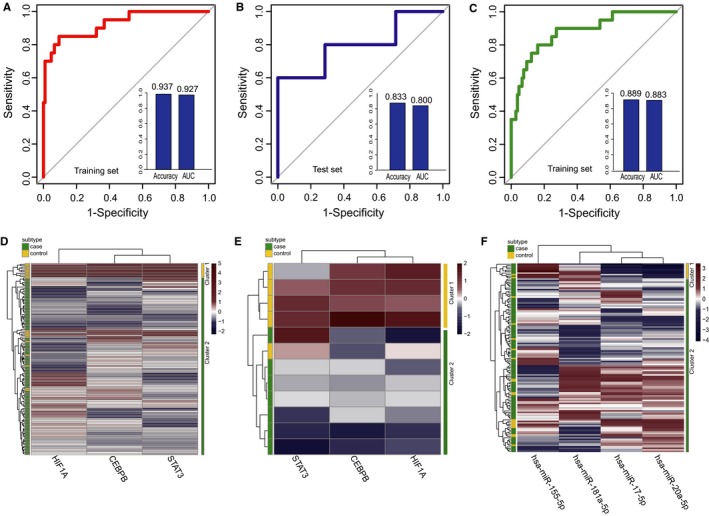
Classification performance of the identified two panel diagnostic biomarkers defined by TFs and miRNAs in HCM based on 5‐fold cross‐validation analysis. (A and B) Performance evaluation of three TFs diagnostic biomarkers in the training set and test set, respectively. (C) Performance evaluation of four miRNAs diagnostic biomarkers in training set. (D and F) The hierarchical clustering heat map of 126 samples based on expression profiles of three TFs and four miRNAs in training set, respectively. (E) Hierarchical clustering heat map of 12 samples based on expression profiles of three TFs in test set
4. DISCUSSION
TFs and miRNAs are two key regulators that mainly regulate gene expression at the transcriptional and post‐transcriptional level. Accumulating evidence has demonstrated the important roles of their combinational regulation acting as FFLs in various cellular processes and diseases. In this study, we introduced a novel analysis approach for identifying dysregulated miRNA‐TF FFLs between two biological states such as health and disease. We then identified dysregulated miRNA‐TF FFLs associated with HCM, which were confirmed to be closely related with HCM from different perspectives. We investigated the global architecture and feature of dysregulated FFLs in HCM, from which a FFL (hsa‐miR‐17‐5p, STAT3 and FASN) might play important roles in HCM and two panels of diagnostic biomarkers defined by three TFs and four miRNAs were identified.
The main idea of the computational method we proposed is differential co‐expression in normal and disease states. For a miRNA‐TF FFL, we evaluated the strength of its dysregulation by integrating differential expression of the nodes and differential co‐expression of the edges in the FFL. Therefore, differential co‐expression analysis can not only reflect dynamic changes of a single gene, but also capture the connections between genes. This approach was originated from Jiang's method,18 and we improved it. Compared with Jiang's method, we observed that the more significant the FFLs we identified, the more HCM‐related biological information they contained. The investigation of biological pathway also revealed that the dysregulated FFLs we identified were significantly enriched in three cardiovascular disease pathways including HCM pathway, while Jiang's method18 only enriched in viral myocarditis.
More importantly, the dysregulated FFLs we identified provide important clues for further experimentally validation and studying combinational regulation of miRNAs and TFs in HCM. The dysregulated FFLs were based on experimentally verified regulatory relationships among miRNAs, genes, and TFs. Here, we focused more on the accuracy than the coverage, and thus the computational predicted interactions were not used. However, it is neither complete nor unbiased for experimentally confirmed data, and many regulatory relationships among miRNAs, genes, and TFs were not verified in HCM‐related cells or tissues. Therefore, further experimental confirmation was warranted. With an improvement of the quantity and quality of these data, the dysregulated miRNA‐TF FFLs we identified will be more accurate and comprehensive.
In summary, we systematically identified dysregulated miRNA‐TF FFLs in HCM, and confirmed their important roles in HCM. Our results provide important clues for investigation of gene regulation by miRNAs and TFs in HCM, and shed new light on deciphering the pathogenesis of HCM at the transcriptional and post‐transcriptional levels.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
ACKNOWLEDGEMENTS
This work was supported in part by the National Natural Science Foundation of China (grant nos. 31500675), the Innovation Special Fund of Harbin Science and Technology Bureau of Heilongjiang Province (2017RAQXJ203), the Postdoctoral Foundation of Heilongjiang Province (LBH‐Q17133) and the Scientific Research Project of Heilongjiang Province Health and Family Planning Commission (grant nos. 2016‐131).
Shi H, Li J, Song Q, et al. Systematic identification and analysis of dysregulated miRNA and transcription factor feed‐forward loops in hypertrophic cardiomyopathy. J Cell Mol Med. 2019;23:306–316. 10.1111/jcmm.13928
Contributor Information
Hongbo Shi, Email: shihongbo@ems.hrbmu.edu.cn.
Zhenzhen Wang, Email: wangzz@ems.hrbmu.edu.cn.
Guangde Zhang, Email: zhangguangde@ems.hrbmu.edu.cn.
REFERENCES
- 1. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242‐255. [DOI] [PubMed] [Google Scholar]
- 2. Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319:1785‐1786. [DOI] [PubMed] [Google Scholar]
- 3. Tsang J, Zhu J, van Oudenaarden A. MicroRNA‐mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell. 2007;26:753‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vergoulis T, Vlachos IS, Alexiou P, et al. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2011;40:D222‐D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hsu SD, Tseng YT, Shrestha S, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA‐target interactions. Nucleic Acids Res. 2014;42:D78‐D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiao F, Zuo Z, Cai G, et al. miRecords: an integrated resource for microRNA‐target interactions. Nucleic Acids Res. 2009;37:D105‐D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Friedman RC, Farh KK, Burge CB, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang C, Xuan Z, Zhao F, et al. TRED: a transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. 2007;35:D137‐D140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matys V, Fricke E, Geffers R, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mathelier A, Zhao X, Zhang AW, et al. JASPAR 2014: an extensively expanded and updated open‐access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142‐D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang J, Lu M, Qiu C, et al. TransmiR: a transcription factor‐microRNA regulation database. Nucleic Acids Res. 2010;38:D119‐D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang HM, Kuang S, Xiong X, et al. Transcription factor and microRNA co‐regulatory loops: important regulatory motifs in biological processes and diseases. Brief Bioinform. 2015;16:45‐58. [DOI] [PubMed] [Google Scholar]
- 13. Arora S, Rana R, Chhabra A, et al. miRNA‐transcription factor interactions: a combinatorial regulation of gene expression. Mol Genet Genomics. 2013;288:77‐87. [DOI] [PubMed] [Google Scholar]
- 14. Ye H, Liu X, Lv M, et al. MicroRNA and transcription factor co‐regulatory network analysis reveals miR‐19 inhibits CYLD in T‐cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012;40:5201‐5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qin S, Ma F, Chen L. Gene regulatory networks by transcription factors and microRNAs in breast cancer. Bioinformatics. 2015;31:76‐83. [DOI] [PubMed] [Google Scholar]
- 16. Sun J, Gong X, Purow B, et al. Uncovering microRNA and transcription factor mediated regulatory networks in glioblastoma. PLoS Comput Biol. 2012;8:e1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yan Z, Shah PK, Amin SB, et al. Integrative analysis of gene and miRNA expression profiles with transcription factor‐miRNA feed‐forward loops identifies regulators in human cancers. Nucleic Acids Res. 2012;40:e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang W, Mitra R, Lin CC, et al. Systematic dissection of dysregulated transcription factor‐miRNA feed‐forward loops across tumor types. Brief Bioinform. 2016;17:996‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang G, Shi H, Wang L, et al. MicroRNA and transcription factor mediated regulatory network analysis reveals critical regulators and regulatory modules in myocardial infarction. PLoS ONE. 2015;10:e0135339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiao Y, Hsiao TH, Suresh U, et al. A novel significance score for gene selection and ranking. Bioinformatics. 2014;30:801‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Q, Yu H, Zhao Z, et al. EW_dmGWAS: edge‐weighted dense module search for genome‐wide association studies and gene expression profiles. Bioinformatics. 2015;31:2591‐2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McKenzie AT, Katsyv I, Song WM, et al. DGCA: a comprehensive R package for differential gene correlation analysis. BMC Syst Biol. 2016;10:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hou L, Chen M, Zhang CK, et al. Guilt by rewiring: gene prioritization through network rewiring in genome wide association studies. Hum Mol Genet. 2014;23:2780‐2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bauer‐Mehren A, Rautschka M, Sanz F, et al. DisGeNET: a Cytoscape plugin to visualize, integrate, search and analyze gene‐disease networks. Bioinformatics. 2010;26:2924‐2926. [DOI] [PubMed] [Google Scholar]
- 25. Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514‐D517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davis AP, Murphy CG, Johnson R, et al. The Comparative Toxicogenomics Database: update 2013. Nucleic Acids Res. 2013;41:D1104‐D1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Becker KG, Barnes KC, Bright TJ, et al. The Genetic Association Database. Nat Genet. 2004;36:431‐432. [DOI] [PubMed] [Google Scholar]
- 28. Eppig JT, Blake JA, Bult CJ, et al. The Mouse Genome Database (MGD): comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res. 2012;40:D881‐D886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. UniProt C. Activities at the universal protein resource (UniProt). Nucleic Acids Res. 2014;42:D191‐D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y, Qiu C, Tu J, et al. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42:D1070‐D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang Q, Wang Y, Hao Y, et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37:D98‐D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruepp A, Kowarsch A, Schmidl D, et al. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010;11:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Griffiths‐Jones S, Grocock RJ, van Dongen S, et al. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140‐D144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li C, Li X, Miao Y, et al. SubpathwayMiner: a software package for flexible identification of pathways. Nucleic Acids Res. 2009;37:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li X, Jiang W, Li W, et al. Dissection of human MiRNA regulatory influence to subpathway. Brief Bioinform. 2012;13:175‐186. [DOI] [PubMed] [Google Scholar]
- 36. Li J, Chen Z, Tian L, et al. LncRNA profile study reveals a three‐lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut. 2014;63:1700‐1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ino T, Nishimoto K, Okubo M, et al. Apoptosis as a possible cause of wall thinning in end‐stage hypertrophic cardiomyopathy. Am J Cardiol. 1997;79:1137‐1141. [DOI] [PubMed] [Google Scholar]
- 38. Kavantzas NG, Lazaris AC, Agapitos EV, et al. Histological assessment of apoptotic cell death in cardiomyopathies. Pathology. 2000;32:176‐180. [PubMed] [Google Scholar]
- 39. Kumar R, Sahu SK, Kumar M, et al. MicroRNA 17‐5p regulates autophagy in Mycobacterium tuberculosis‐infected macrophages by targeting Mcl‐1 and STAT3. Cell Microbiol. 2016;18:679‐691. [DOI] [PubMed] [Google Scholar]
- 40. Xiang Y, Liao XH, Li JP, et al. Myocardin and Stat3 act synergistically to inhibit cardiomyocyte apoptosis. Oncotarget. 2017;8:99612‐99623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu H, Greenbaum D, Xin LuH, et al. Genomic analysis of essentiality within protein networks. Trends Genet. 2004;20:227‐231. [DOI] [PubMed] [Google Scholar]
- 42. Tsoutsman T, Kelly M, Ng DC, et al. Severe heart failure and early mortality in a double‐mutation mouse model of familial hypertrophic cardiomyopathy. Circulation. 2008;117:1820‐1831. [DOI] [PubMed] [Google Scholar]
- 43. Du W, Pan Z, Chen X, et al. By targeting Stat3 microRNA‐17‐5p promotes cardiomyocyte apoptosis in response to ischemia followed by reperfusion. Cell Physiol Biochem. 2014;34:955‐965. [DOI] [PubMed] [Google Scholar]
- 44. Hao MX, Wang X, Jiao KL. MicroRNA‐17‐5p mediates hypoxia‐induced autophagy and inhibits apoptosis by targeting signal transducer and activator of transcription 3 in vascular smooth muscle cells. Exp Ther Med. 2017;13:935‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials