

Abstract
Key regulators of the Wnt signalling, DVL1, DVL2 and DVL3, in astrocytomas of different malignancy grades were investigated. Markers for DVL1,DVL2 and DVL3 were used to detect microsatellite instability (MSI) and gross deletions (LOH), while immunohistochemistry and immunoreactivity score were used to determine the signal strengths of the three DVL proteins and transcription factors of the pathway, TCF1 and LEF1. Our findings demonstrated that MSI at all three DVL loci was constantly found across tumour grades with the highest number in grade II (P = 0.008). Collectively, LOHs were more frequent in high‐grade tumours than in low grade ones. LOHs of DVL3 gene were significantly associated with grade IV tumours (P = 0.007). The results on protein expressions indicated that high‐grade tumours expressed less DVL1 protein as compared with low grade ones. A significant negative correlation was established between DVL1 expression and malignancy grades (P < 0.001). The expression of DVL2 protein was found similar across grades, while DVL3 expression significantly increased with malignancy grades (P < 0.001). The signal strengths of expressed DVL1 and DVL3 were negatively correlated (P = 0.002). However, TCF1 and LEF1 were both significantly upregulated and increasing with astrocytoma grades (P = 0.001). A positive correlation was established between DVL3 and both TCF1 (P = 0.020) and LEF1 (P = 0.006) suggesting their joint involvement in malignant progression. Our findings suggest that DVL1 and DVL2 may be involved during early stages of the disease, while DVL3 may have a role in later phases and together with TCF1 and LEF1 promotes the activation of Wnt signalling.
Keywords: astrocytic brain tumours, Dishevelled, DVL1, DVL2, DVL3, transcription factors TCF1 & LEF1, Wnt signalling
1. INTRODUCTION
Astrocytomas are the most common and deadliest form of primary brain tumours.1 According to the World Health Organization (WHO) classification, there are four grades of astrocytic brain tumours, considering their histology, molecular characteristics and prognosis.2 The least aggressive are pilocytic astrocytomas corresponding to WHO grade I, while diffuse (grade II) and anaplastic astrocytomas (grade III) are malignant types with intrinsic ability to progress to higher grades of malignancy. Of these, glioblastoma multiforme (grade IV) is the most aggressive, fastest‐growing and highly invasive tumour with survival times of about a year.3 Despite recent advances in understanding the molecular basis of astrocytoma development and progression, additional research is required to develop more effective therapies.
By its biological characteristics astrocytomas are genetically and pathohistologically a very heterogeneous group of tumours. Complex mechanism of gliomagenesis is the outcome of overlapping between altered signalling pathways. A long‐scale study conducted by The Cancer Genome Atlas (TCGA) revealed numerous data on specific genetic and epigenetic alterations underlying gliomas.4 Although it is still not possible to define the exact number and chronology of changes during gliomagenesis, they found that key genes responsible for the formation and progression of astrocytic brain tumours are most frequently involved into deregulated oncogenic pathways: RTK/RAS/PI3K, TP53 and RB.4, 5 Recent studies have shown that altered signal transduction in Wnt signalling pathway is also involved in the molecular pathogenesis of glial tumours.6, 7, 8, 9
The Wnt signalling is an evolutionarily conserved pathway that plays very important roles during embryonic development and tumourigenesis.10 In the absence of Wnt ligand, cytoplasmic β‐catenin protein is constantly being degraded by the action of the destruction complex composed of Axin, APC and 2 phosphokinases: casein kinase 1 (CK1) and glycogen synthase kinase 3 (GSK3). Casein kinase 1 and GSK3 phosphorylate β‐catenin, which leads to its proteasomal degradation. Continuous elimination of β‐catenin from the cell prevents β‐catenin from reaching the nucleus, thereby repressing transcription of Wnt target genes.11, 12
Binding of Wnt ligand to a Frizzled (Fz) receptor and its co‐receptor LRP5/6 activates Wnt/β‐catenin pathway. In this stage the concentration of DVL protein in cytoplasm increases, resulting in recruitment of components of destruction complex to the cell membrane. These events lead to inhibition of β‐catenin phosphorylation, its stabilization and accumulation in the cytoplasm. β‐catenin transfers to the nucleus where it forms complexes with T cell factor 1 (TCF1) and lymphoid‐enhancer factor 1 (LEF1) and in such a fashion activates Wnt target gene transcription.13 DVL is the central component of Wnt signalling and key cytoplasmic regulator that rescues β‐catenin from degradation. Three homologous Dishevelled genes (DVL1, DVL2 and DVL3), that show a high degree of similarity, have been found in humans. Knock‐out mouse models have shown that each of the DVL proteins can act individually but also in the combination with other family members.14 Dishevelled gene proteins function as branching points for the differential interpretation of distinct Wnt ligands.15 They are activated by phosphorylation in response to Wnt signals; whereas the ubiquitination of DVL proteins leads to an effective inhibition of the β‐catenin destruction complex.13 It has been postulated that DVL overexpression may play a role in the progression of several cancers.16, 17, 18, 19, 20 As a result it represents a potential target for cancer therapy.
In the present investigation, we searched for changes of all three human DVL genes and proteins and tried to define their involvement in specific astrocytoma grade. We were also interested about their effect on Wnt signalling activation and examined transcription factors TCF1 and LEF1.
2. METHODS
2.1. Tumour specimens
Eighty‐three astrocytoma samples together with corresponding autologous blood tissue were collected with patients’ consents from the Departments of Neurosurgery and Departments of Pathology University Hospital Centers “Zagreb” and “Sisters of Charity”, Croatia and Brain Tumour Tissue Banks from Croatia and Canada. All tumours were studied by certified neuropathologist and classified according to the WHO criteria.2 The patients had no family history of brain tumours and did not undergo any cancer treatment (chemotherapy or radiotherapy) prior to surgery. The sample consisted of 17 pilocytic astrocytomas (grade I), 14 diffuse (grade II), 17 anaplastic (grade III) and 35 glioblastomas (grade IV). Thirty‐six patients were female and 47 male. The age of patients varied from 2 to 77 (mean age = 45, median = 44 years). The mean age of diagnosis for females was 49 (median 54.5) and for males 42 years (median 41).
Ethical approval was received from the Ethics Committees School of Medicine University of Zagreb (Case number: 380‐59‐10106‐14‐55/147; Class: 641‐01/14‐02/01) and University Hospital Centers “Sisters of Mercy” (number EP‐7426/14‐9) and “Zagreb” (number 02/21/JG, class: 8.1.‐14/54‐2). Patients gave their informed consent.
2.2. DNA extraction
For the genomic DNA extraction, approximately 0.5 g of tumour tissue was homogenized with 1 mL extraction buffer (10 mmol L−1 Tris–HCl, pH 8.0; 0.1 M EDTA, pH 8.0; 0.5% sodium dodecyl sulfate) and incubated with proteinase K (100 μg/mL; Sigma‐Aldrich, St. Louis, MO, USA) overnight at 37°C. Phenol chloroform extraction and ethanol precipitation followed. Blood was used to extract DNA from leukocyte. Five millilitre of blood was lysed with 15 mL RCLB (Red Blood Cell Lysis Buffer) (0.16 M NH4Cl; 10 mmol L−1 KHCO3; 10 mmol L−1 EDTA; pH 7.6) and centrifuged (15 min/5000 g). After overnight incubation with 2 mL SE buffer (Sodium‐EDTA; 75 mmol L−1 NaCl; 25 mmol L−1 Na2EDTA; pH 8), 200 μL 10% SDS (Sodium‐dodecyl sulphate) and 15 μL proteinase K (Sigma, Germany) (20 mg/mL) at 37°C, salting‐out method by isopropanol precipitation followed.
2.3. Polymerase chain reaction, multiplex PCR, microsatellite instability, loss of heterozygosity
Genetic changes were tested using PCR amplification of polymorphic microsatellite markers for DVL1 (D1S468 and D1S243), DVL2 (D17S960) and DVL3 (D3S1262) genes to detect microsatellite instability (MSI) and loss of heterozygosity (LOH).21 All markers were amplified in a total volume of 25 μL: 5X PCR buffer, 1.5 mmol L−1 MgCl2, 2.5 mmol L−1 of each dNTP, each primer 0.4 μmol, 100 ng DNA, 1 U Taq polymerase (Promega, Fitchburg, WI, USA). PCR conditions and primers for D1S468, D1S243, D17S960 and D3S1262 markers are shown in Table 1.
Table 1.
PCR conditions for microsatellite markers D1S468, D1S243, D17S960 and D3S1262
| Marker | Initial denaturation | Cycles | Final extension | Primers | ||
|---|---|---|---|---|---|---|
| D1S468 | 94°C 5 min | 94°C 30 s | 60°C 30 s | 72°C 30 s | 72°C 10 min | 5′ TTAACCGTTTTGGTCCTACC 3′ 5′ CTCTGACCAGCATTAAAGATTC 3′ |
| D1S243 | 95°C 5 min | 95°C 30 s | 56°C 30 s | 72°C 30 s | 72°C 10 min | 5′ CAC ACA GGC TCA CAT GCC 3′ 5′ GCT CCA GCG TCA TGG ACT 3′ |
| D17S960 | 95°C 5 min | 95°C 30 s | 58°C 30 s | 72°C 30 s | 72°C 30 s | 5′ CAA TGG AAC CAA ATG TGG TC 3′ 5′ AGT CCG ATA ATG CCA GGA TG 3′ |
| D3S1262 | 94°C 5 min | 94°C 30 s | 56°C 35 s | 72°C 30 s | 72°C 10 min | 5′ CGG CCC TAG GAT ATT TTC AA 3′ 5′ CCA GTT TTT ATG GAC GGG GT 3′ |
Marker D3S1262 was also used for multiplex PCR. It was amplified in the same reaction together with markers for genes SHGC‐68373 (222 bp) 22 and Apex1 (321 bp) 23 that were used as references. Data from the literature indicated that genetic regions where reference genes are located are not changed in glioblastoma and therefore could be used as reference to study oncogenic amplifications.
Genetic alterations were visualized on Spreadex EL 400 Mini gels (Elchrom Scientific, ALLabortechnik, AL‐Diagnostic GmbH, Amstetten, Austria) and stained with SYBR Gold (Invitrogen, Molecular Probes, Eugene, OR, USA). Samples were considered positive for MSI if additional DNA bands appeared in tumour sample, or if the existing DNA bands have changed position in comparison to bands of autologous blood tissue. Absence or significant decrease in the intensity of one polymorphic allele in tumour as compared to the DNA from autologous blood was considered as LOH.
2.4. Microsatellite genotyping analysis
Loss of heterozygosity was confirmed by capillary electrophoresis performed on instrument 3730XL (Applied Biosystems Inc.). The 5′‐end of the forward primer was fluorescently labelled with a 6‐FAM dye, and PCR amplification was performed with the AmpliTaq Gold master‐mix (360) (Applied Biosystems Inc.). The labelled PCR products were separated using capillary electrophoresis. Alleles were sized relative to an internal size standard (500 LIZ; Thermo Fisher Scientific, Carlsbad, CA, USA). Raw data and graphical representation of LOH samples were reviewed using GeneMapper 5 and Peak Scanner (Thermo Fisher Scientific).
2.5. Immunohistochemistry
In order to establish the levels of expression and cellular localization of DVL1, DVL2, DVL3 gene products immunohistochemistry was used, as well as for testing the upregulation of transcription factors, TCF1 and LEF1. The samples were fixed in formalin, embedded in paraffin, sliced into 4‐μm thick sections, and then fixed onto capillary gap microscope slides (DakoCytomation, Glostrup, Denmark). Briefly, sections were deparaffinized in xylene, rehydrated in decreasing concentrations of ethanol and then microwaved twice for 7 minutes at 400 W in citrate buffer and three times for 5 minutes at 700 W to unmask epitopes. To block endogenous peroxidase activity, cells were fixed in dark with 3% H2O2 for 10 minutes. Non‐specific binding was blocked by incubating samples with protein block serum‐free ready‐to‐use (Dako, Carpinteria, CA, USA) for 30 minutes at 4°C. Next, the primary antibodies, mouse monoclonal anti‐human for DVL1, TCF1 and LEF1 (1:50; Santa Cruz Biotechnology Inc., Dallas, TX, USA) and rabbit polyclonal anti‐human for DVL2 and DVL3 (1:500; 1:50, Abcam, Cambridge, UK) were applied overnight at 4°C. Slides were then washed three times in phosphate‐buffered saline (PBS), and secondary LINK antibody was applied for 30 min at 4°C. Slides were again washed three times in PBS and incubated with substrate chromogen solution (EnVision™, Dako REAL™) for 30 seconds. Sections were immunostained using streptavidin horseradish peroxidase/DAB (3,3‐diaminobenzidine) (Dako REAL™ EnVision™, Glostrup, Denmark).
The level of expression of DVL1, DVL2 and DVL3 proteins in the healthy brain was determined by using cerebral cortex of human brain (Amsbio, Oxfordshire, UK). We have found that the levels of immunoreactivity of all homologues of the Dishevelled protein family in the healthy brain tissue were very low, and the signal was present only in the cytoplasm.
Negative controls underwent the same staining procedure but without incubating samples with primary antibodies. The frontal cortex of a healthy human brain, placenta, liver tissue and normal bronchial epithelia all served as positive controls. Antibody labelling was analysed by three independent and blinded observers using an Olympus BH‐2 microscope and a digital scanner (NanoZoomer 2.0‑RS; Hamamatsu Photonics, Hamamatsu, Japan). ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to determine the cell number and the intensity of protein expression. We counted 200 cells in tumour hot spot area and performed semi‐quantitative analysis introducing immunoreactivity score (IRS) in order to determine the signal strength. Immunoreactivity score is a factor that best correlates with computational photo analysis and was calculated by multiplying the percentage of cells with a positive signal in the sample (PP score) with staining intensity (SI score). PP score was determined as follows: no immunopositivity in tumour cells = score 0; 1%‐25% positive cells = score 1; >25%‐50% = score 2; >50%‐85% = score 3; >85% = score 4. SI score was assessed in three categories mirroring staining intensities, no staining or weak = score 1, moderate staining = score 2 and strong staining = score 3. Immunoreactivity score in our study ranged from 0 to 12. For statistical analysis needs, IRS values were converted to numbers/symbols: 1 (0/+) (IRS = 0‐4) no expression or very weak expression; 2 (++) (IRS = 6‐8) moderate expression; 3 (+++) (IRS = 9‐12) strong expression.
2.6. Statistical analysis
The statistical evaluations for all the obtained data including tumour intracranial localization, pathohistological type and grade, age, sex, genetic findings and protein expression findings were performed with the SPSS v.19.0.1 (SPSS, Chicago, IL, USA) statistical program, with the statistical significance set at P < 0.05.
The normality of the distribution of individual parameters within a group was tested using a Kolmogorov‐Smirnov test for normality where low significance (P < 0.05) indicates that the distribution significantly differs from normal. Depending on the results of the analysis and the number of patients per group, parametric and nonparametric statistical tests were further used. The differences in the frequency of the chosen parameters were analysed by Pearson's χ2 test using Yates's correlation when appropriate. Differences in the values between the four grades were tested by a one‐way variance analysis (ANOVA) in the case of normal distribution. When the assumptions of one‐way ANOVA are not met, Kruskal‐Wallis test was applied. In the case of normal distribution, differences between the two groups were tested by Student's t test, and in case of a deviation from normality by the Mann‐Whitney test. When the distribution between individual parameters was normal, we used Pearson correlation, while values derived from Spearman correlation were taken into account if distribution deviated from normality.
2.7. Analysis of public databases cBioPortal, COSMIC and GEPIA
In order to test the compatibility of our results we investigated data from publicly available databases. Genetic changes publicly reported on DVL genes were assessed from cBioPortal and COSMIC databases,24, 25 while survival plots were performed from GEPIA database.26
3. RESULTS
3.1. MSI at all three DVL loci was constant across tumour grades while LOHs were associated to higher grades
Our study explored DVL1, DVL2 and DVL3 gene alterations in association with tumour grade. The presence of LOH and MSI, two aspects of the genomic instability of tumour cells, was detected in astrocytoma of different malignancy grades.
For DVL1 gene analysis, two polymorphic microsatellite markers located in the close proximity to DVL1 gene in chromosomal region 1p36 were selected. Marker D1S468 showed a substantial rate of microsatellite instability (MSI) across all astrocytoma grades, pilocytic (grade I) showed 21.4%, diffuse (grade II) 53.8%, anaplastic (grade III) 36.4% and glioblastoma (grade IV) 17.1%. Contrary LOH was detected only in the highest grade—glioblastomas, in 8.6% of patients (Table 2). Statistical analysis confirmed that the distribution regarding tumour grades was significant. Genetic changes were significantly more distributed to glioblastoma group (P = 0.020). Furthermore, our results indicated that MSI was significantly associated to diffuse astrocytomas (grade II) as compared with other astrocytoma grades (P = 0.008).
Table 2.
Polymorphic status of microsatellite markers used and the observed genetic changes of DVL1, DVL2 and DVL3 genes
| Patient no | Grade | DVL1 | DVL2 | DVL3 |
|---|---|---|---|---|
| D1S468 & D1S243 | D17S960 | D3S1262 | ||
| 1 | I | HETERO | HOMO | HETERO |
| 2 | I | HETERO | HOMO | HETERO |
| 3 | I | HETERO | HETERO | HETERO |
| 4 | I | HETERO | HETERO | HETERO |
| 5 | I | HETERO | HETERO | HOMO |
| 6 | I | MSI | LOH | MSI |
| 7 | I | HETERO | HOMO | MSI |
| 8 | I | MSI | MSI | MSI |
| 9 | I | HETERO | HOMO | HETERO |
| 10 | I | MSI | HOMO | HETERO |
| 11 | I | MSI | HETERO | HETERO |
| 12 | I | HETERO | HOMO | HETERO |
| 13 | I | HETERO | HETERO | HETERO |
| 14 | I | HETERO | HETERO | HETERO |
| 15 | II | HETERO | HETERO | HETERO |
| 16 | II | HETERO | LOH | HETERO |
| 17 | II | MSIa | LOH | MSI |
| 18 | II | MSI | HETERO | HETERO |
| 19 | II | MSI | MSI | HETERO |
| 20 | II | MSIa | HOMO | HETERO |
| 21 | II | HETERO | HOMO | HETERO |
| 22 | II | HETERO | HOMO | HETERO |
| 23 | II | MSI | HETERO | HETERO |
| 24 | II | HETERO | HETERO | HOMO |
| 25 | II | MSI | MSI | MSI |
| 26 | II | MSI | LOH | HOMO |
| 27 | II | MSI | HETERO | HETERO |
| 28 | III | HETERO | HOMO | HETERO |
| 29 | III | MSI | HETERO | LOH |
| 30 | III | LOH | LOH | MSI |
| 31 | III | MSI | HOMO | HETERO |
| 32 | III | HETERO | HETERO | MSI |
| 33 | III | HETERO | HOMO | HETERO |
| 34 | III | MSI | HOMO | MSI |
| 35 | III | MSI&LOH | HETERO | HETERO |
| 36 | III | MSI | HETERO | HETERO |
| 37 | III | MSI | LOH | HETERO |
| 38 | III | HETERO | HOMO | HETERO |
| 39 | IV | HETERO | HOMO | LOH |
| 40 | IV | MSI&LOH | HETERO | HETERO |
| 41 | IV | HETERO | HETERO | MSI |
| 42 | IV | MSI | HOMO | HETERO |
| 43 | IV | HETERO | HOMO | HETERO |
| 44 | IV | MSI | HOMO | HOMO |
| 45 | IV | LOH | HETERO | HETERO |
| 46 | IV | MSI | HETERO | LOH |
| 47 | IV | HETERO | HOMO | LOH |
| 48 | IV | MSI | HETERO | HETERO |
| 49 | IV | LOH | LOH | HETERO |
| 50 | IV | HETERO | HETERO | HETERO |
| 51 | IV | MSI | HETERO | LOH |
| 52 | IV | HETERO | HETERO | LOH |
| 53 | IV | HETERO | HOMO | MSI |
| 54 | IV | HETERO | HETERO | HETERO |
| 55 | IV | MSI | MSI | LOH |
| 56 | IV | MSI | HOMO | LOH |
| 57 | IV | MSI | HOMO | HETERO |
| 58 | IV | MSI | HETERO | LOH |
| 59 | IV | LOH&MSI | HETERO | HETERO |
| 60 | IV | MSI | MSI | LOH |
| 61 | IV | HETERO | HETERO | HOMO |
| 62 | IV | HETERO | HETERO | MSI |
| 63 | IV | LOH&MSI | LOH | HETERO |
| 64 | IV | HETERO | LOH | HETERO |
| 65 | IV | MSI | HETERO | HETERO |
| 66 | IV | HETERO | HETERO | MSI |
| 67 | IV | HETERO | HOMO | LOH |
| 68 | IV | HETERO | LOH | HETERO |
| 69 | IV | MSI | MSI | MSI |
| 70 | IV | HETERO | HOMO | HETERO |
| 71 | IV | LOH&MSI | LOH | HETERO |
| 72 | IV | HETERO | HETERO | HETERO |
| 73 | IV | HETERO | HETERO | HOMO |
HETERO, heterozygous; HOMO, homozygous; MSI, microsatellite instability; LOH, loss of heterozygosity.
MSI found by both markers.
The second marker, D1S243, confirmed the constant rates of MSI across all grades: grade I with 7.1%, grade II with 23.1%, grade III with 18.2% and grade IV with 28.6% of MSI, whereas this marker revealed LOH in grades III (18.2%) and IV (9.1%) tumours.
Collective results for both markers for DVL1 showed that MSI was detected in 28.6% of pilocytic, 61.5% diffuse, 45.5% anaplastic astrocytomas and in 34.3% of glioblastomas, while LOHs were found in 18.2% of anaplastic and 17.2% of glioblastomas (Figure 1A).
Figure 1.
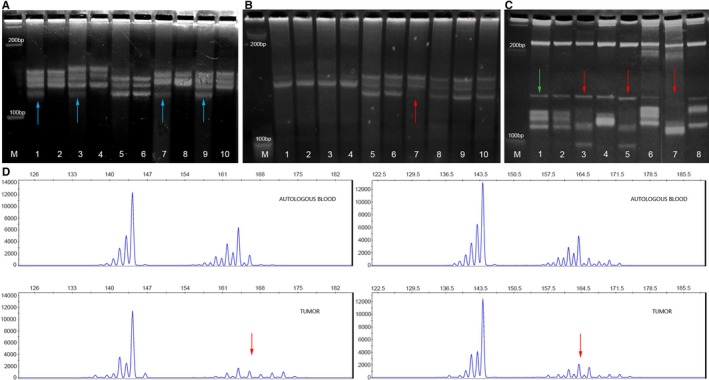
Microsatellite instability (MSI), loss of heterozygosity (LOH) and amplification of A) DVL1, B) DVL2 and C) DVL3 gene in astrocyoma samples on Spreadex gels. Lane M—100 bp DNA standard; odd lanes show astrocytoma samples; even lanes show corresponding blood samples. Samples demonstrating MSI are indicated with blue arrow; samples demonstrating LOH are indicated with red arrow; samples demonstrating amplification are indicated with green arrow. D) Confirmation of LOH samples by microsatellite genotyping analysis (capillary electrophoresis). Upper figures show DNA samples isolated from the blood, while bottom figures show DNA samples isolated from tumour tissue of the same patient
For DVL2 gene analysis marker D17S960 was selected and it showed a lower rate of MSI in all tumour grades, 7.1% in grade I, 15.4% in grade II, 0% in grade III and 8.6% in grade VI. Again grade II tumours (diffuse) harboured the highest frequency of MSI. However, gross deletions of DVL2 were detected in all grades (Figure 1B). Thus, grade II and III each harboured 33.3% of LOHs, followed by grade IV with 21.7% and grade I with the lowest frequency of 12.5% (Table 2). The distribution of genetic changes was not significantly associated to any specific grade (P = 0.843).
DVL3 gene was analysed with D3S1262 marker (Figure 1C). Microsatellite instability rate was again relatively constant across different grades (grade I with 21.4%; grade II with 15.4%; grade III with 14.3% and grade IV with 14.3%) while gross deletions were associated only to higher malignancy grades (grade III with 18.2% and grade IV with 31.2%) (Table 2). Statistical analysis revealed that LOH of DVL3 gene was significantly more frequent in glioblastomas as compared with astrocytomas of lower grades (P = 0.007).
To ascertain results on observed genetic changes, genotyping by capillary electrophoresis was additionally performed and LOHs were confirmed as shown in Figure 1D. Taking both approaches together our findings were additionally confirmed.
Besides gross deletions, DVL3 gene also showed amplifications in 8.6% of glioblastoma patients (Figure 1C). Polymorphic status of used microsatellite markers and observed genetic changes of DVL1, DVL2 and DVL3 genes are presented in Table 2.
3.2. Low rate of somatic mutations and deletions of DVL genes is reported on cBioPortal and COSMIC databases
We tested public datasets downloaded from the Cancer Genome Atlas Database, an open access database that is publicly available at http://www.cbioportal.org and found that the numbers differed to the results of our experiments. Combined collective public datasets from eight studies that are examining low‐grade gliomas and glioblastomas encompassing 4216 samples demonstrate the following distribution: somatic mutations were found in 8/4216 (0.19%) cases for DVL1, 12/4216 (0.28%) for DVL2 and 5/4216 (0.12%) for DVL3. Amplifications are reported mainly for DVL3 (53 cases showed amplification). Nevertheless, low number of cases with amplified DVL1 (0.19%) were also reported. Furthermore, deletions of DVL1 were found in seven glioblastomas and deletions of DVL2 in two glioblastomas, while deletions of DVL3 were not reported. A low rate of fusion mutations was also reported. The reported mutations are predominantly missense somatic mutations. It is also obvious that mutational burden is much more frequent in glioblastomas than low‐grade tumours and those amplifications are particularly associated to DVL3 gene and to glioblastomas. The publicly available data for each DVL gene are shown in Figure 2.
Figure 2.
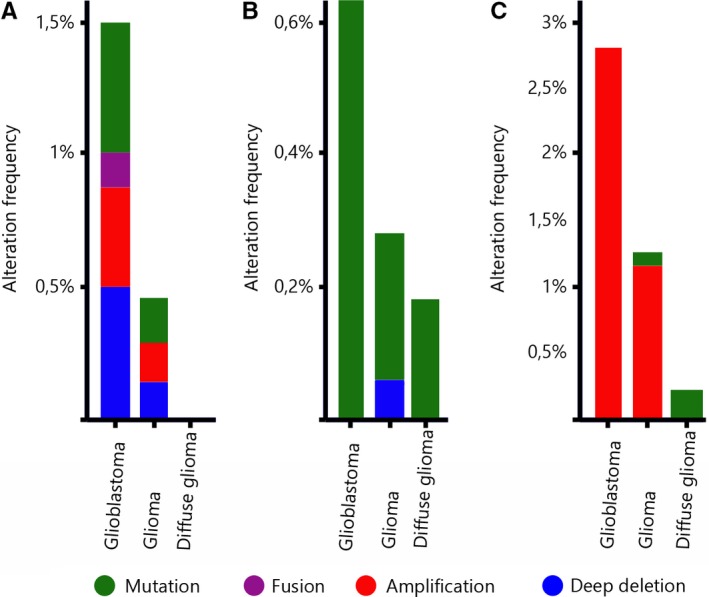
cBioPortal data sets querying 4238 samples in nine studies for each DVL gene. Percentages of alteration frequencies of the DVL genes are shown. A) DVL1; B) DVL2; C) DVL3
3.3. High grade tumours expressed less DVL1 protein as compared with low grade ones, the expression of DVL2 protein was similar across grades, while DVL3 expression increased with malignancy grades
The effect of genetic changes on the protein expressions levels was investigated in the same patients. The expression of DVL1 protein greatly differed among different tumour grades. The highest number of samples with moderate and strong signal was present in the anaplastic astrocytoma group (92.9%) followed by pilocytic (85.7%) and diffuse (70%), while glioblastomas showed low or lack of DVL1 expression in 67.6% of cases. Cell count analysis of DVL1 signal strength showed statistically significant differences in the number of cells with low, moderate and strong expression among different tumour grades (P < 0.001). Glioblastomas had significantly more cells with low expression as compared with pilocytic (P = 0.001) and anaplastic astrocytomas (P = 0.001). Accordingly, glioblastomas had a lower number of cells with strong DVL1 expression as compared with pilocytic (P = 0.001) and anaplastic astrocytomas (P = 0.001). Moreover, anaplastic astrocytomas showed significantly lower number of cells with low expression levels when compared with diffuse astrocytomas (P = 0.008). Negative correlation between DVL1 protein expression and malignancy grades (rs = −0.479, df = 70, P < 0.001) was established (Figure 3A). Of note, in tumours with low levels of DVL1 expression, the signal was predominantly localized in the cytoplasm or in the cytoplasm and cell membrane. In tumours with moderate and strong expression levels of the DVL1 protein, the signal was significantly more frequently localized in cytoplasm and nucleus (P < 0.001).
Figure 3.
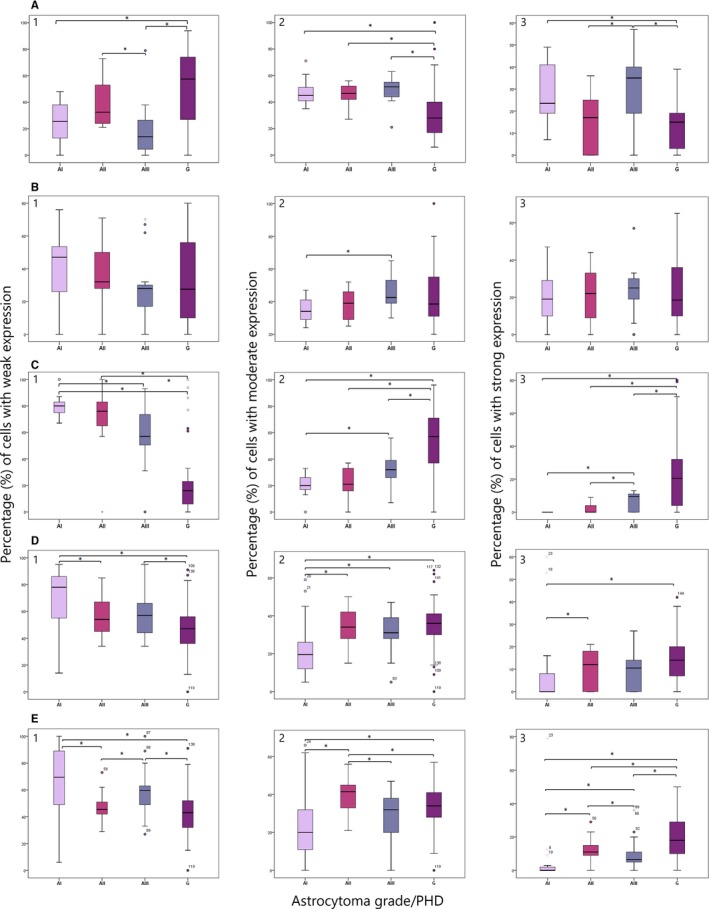
Number of cells with (1) low, (2) moderate and (3) high levels of expression of A) DVL1, B) DVL2, C) DVL3, D) TCF1 and E) LEF1 protein across different malignancy grades. Asterisks (*) denotes statistical significance (P < 0.05)
The results of our study demonstrated increased DVL2 expression in all tumour grades when compared with the control tissue. The highest number of samples with moderate and strong signal was present in the group of anaplastic astrocytoma (77.6%) followed by diffuse astrocytoma (70%), glioblastoma (61.7%) and pilocytic astrocytoma (57.2%). As the expression of DVL2 protein was closely the same in all tumour grades, cell count analysis with respect to DVL2 signal strengths did not show significant differences among malignancy grades (Figure 3B). Once again, tumours with the highest proportion of cells with moderate and strong signal had protein located in the cytoplasm and nuclei (P < 0.001).
Unlike the two previously analysed proteins, the DVL3 expression increase with malignancy grade was very pronounced. Glioblastoma had highest proportion of cells with moderate and strong expression (82.4%) (Figure 3C), while astrocytomas grade I and II were characterized with weak DVL3 expression, as none of the analysed samples showed more than 50% of positive cells in the tumour hot spot area. Anaplastic astrocytomas had 7.2% samples with moderate expression levels. Accordingly, cell count analysis regarding DVL3 signal strength showed significant differences between tumour grades (P < 0.001). Glioblastomas had the lowest number of cells showing lack of expression and the highest number of cells showing moderate and strong expression as compared to astrocytoma grades I to III (P 1 < 0.001; P 2 = 0.001; P 3 = 0.013, respectively). Tumours with low signal intensities had predominant cytoplasmic localization, while samples with moderate and strong expression had signal localized in both the cytoplasm and the nucleus (P = 0.004) (Figure 3C). This observation suggests that the increased DVL3 expression in glioblastoma leads to more frequent transfer of this protein into the nucleus (Figure 4).
Figure 4.
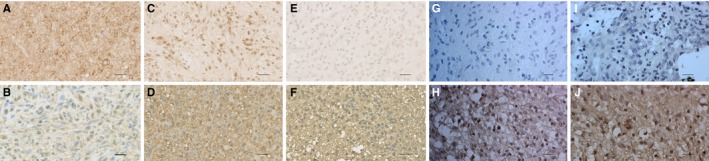
Characteristic immunohistochemical staining of DVL1, DVL2, DVL3, TCF1 and LEF1 proteins in astrocytoma. A) astrocytic brain tumour grade I showing strong cytoplasmic and nuclear staining of DVL1, B) astrocytic brain tumour grade IV showing weak cytoplasmic staining of DVL1, C) astrocytic brain tumour grade I showing strong nuclear staining of DVL2, D) astrocytic brain tumour grade IV showing strong cytoplasmic staining of DVL2, E) astrocytic brain tumour grade I showing lack of expression of DVL3, F) astrocytic brain tumour grade IV showing strong membranous, cytoplasmic and nuclear staining of DVL3, G) astrocytic brain tumour grade I showing weak nuclear expression of TCF1, H) astrocytic brain tumour grade IV showing moderate and strong expression of TCF1, I) astrocytic brain tumour grade I showing weak nuclear expression of LEF1, J) astrocytic brain tumour grade IV showing moderate and strong nuclear expression of LEF1. Scale bar 50 μm
3.4. TCF1 and LEF1 protein expression levels increased with astrocytoma grades
Next we asked how the Dishevelleds’ expression is impacting Wnt signalling activation. For this we analysed the expression levels of two transcriptional factors located at the end of the Wnt signalling cascade, TCF1 and LEF1, whose elevated expression indicates the activity of the pathway. Both factors were found to be expressed in our total sample where the levels of both TCF1 and LEF1 expression increased with astrocytoma grades. Accordingly, 61.1% of pilocytic astrocytomas showed the lowest levels of TCF1 expression, while weak or lack of TCF1 expression was confined to 50% of diffuse astrocytomas. The highest expression levels were found in anaplastic and glioblastomas cases, 77.8% and 80% respectively. The signal was present exclusively in the nuclei of tumour cells (Figure 3D).
Statistical analysis for TCF1 protein showed that pilocytic had significantly more cells with low expression levels as compared with diffuse astrocytomas (P = 0.040) and glioblastomas (P = 0.003). Furthermore, diffuse (P = 0.047), anaplastic (P = 0.022) astrocytomas and glioblastomas (P = 0.001) had significantly higher number of cells with strong TCF1 expression as compared with pilocytic (Figure 3D).
Similar results were obtained for LEF1 protein expression. Low or lack of LEF1 expression was found in 61.1% of pilocytic astrocytomas, while 70% of glioblastomas showed strong LEF1 expression. Almost the same proportions of samples with moderate and strong expression were present in the group of diffuse astrocytoma (88.9%) and glioblastoma (90%), while in anaplastic cases the observed rate was lower (66.6%).
The cell count analysis confirmed the previous observations. Thus, pilocytic astrocytomas had significantly more cells with low LEF1 expression than both diffuse astrocytomas (P = 0.006) and glioblastomas (P = 0.001) (Figure 3E). The highest number of cells with strong LEF1 expression was in glioblastomas comparing to pilocytic (P < 0.001), diffuse (P = 0.032) and anaplastic types (P = 0.003). Diffuse astrocytomas had significantly more cells with strong LEF1 expression as compared with pilocytic (P < 0.001) and anaplastic (P = 0.008), while anaplastic had significantly more cells with strong LEF1 expression than pilocytic (P = 0.008) (Figure 3E). The results on transcription factors suggest that Wnt activation is accompanying the progression of the disease.
3.5. The correlations of molecular findings and clinical parameters
Immunoreactivity score values for DVL1, DVL2 and DVL3 were calculated in order to determine the potential correlations between the expression levels of the proteins.
There was no significant correlation between the expression levels of DVL1 and DVL2 proteins (P = 0.799) (Figure 5A). However, DVL1 and DVL3 were significantly negatively correlated in our total sample (P = 0.002) (Figure 5B). Lower tumour grades (I and II) showed significantly higher levels of DVL1 expression than high grades (III and IV), while strong DVL3 expression was significantly associated to high‐grade tumours. Comparing the IRS values for DVL2 and DVL3, statistically significant negative correlation was established only for pilocytic astrocytomas (r = −0.699, t = −3.388, P = 0.005), while anaplastic astrocytomas showed a trend of negative correlation for the two proteins (r = −0.488, t = 0.236, P = 0.077) (Figure 5C). Correlation of IRS values for TCF1 and LEF1 revealed significant positive correlation between the two proteins across all tumour grades (r s = 0.474, df = 92, P < 0.001) (Figure 5D).
Figure 5.
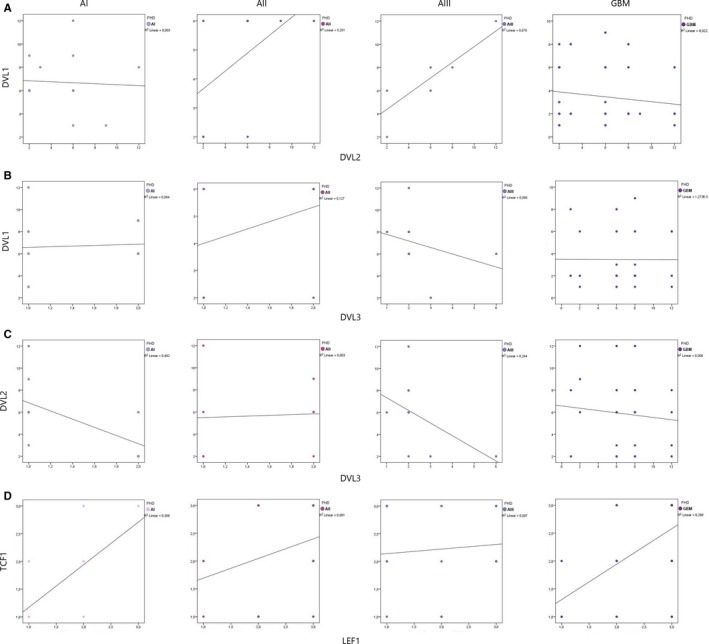
Correlation between A) DVL1 and DVL2, B) DVL1 and DVL3, C) DVL2 and DVL3, D) TCF1 and LEF1 in astrocytic brain tumours grades I‐IV
Bivariate correlation of all analysed proteins revealed a statistically significant positive correlation between DVL3 and TCF1 (r s = 0.410, df = 30, P = 0.020) and DVL3 and LEF1 (r s = 0.475, df = 30, P = 0.006). Correlation of DVL1 and TCF1; DVL1 and LEF1; DVL2 and TCF1; DVL2 and LEF1 showed no statistically significant association between analysed proteins.
Finally we have found that two molecular parameters were significantly associated with aging. Younger patients had a stronger DVL1 expression than older ones (r s = −0.226, df = 70, P = 0.057). On the other hand, older patients had significantly higher DVL3 expression comparing to the younger ones (r s = 0.395, df = 70, P = 0.001). No statistical significance was found in relation to the sex of the patients.
Kaplan‐Meier survival curves were generated from GEPIA database for each DVL member. The curves showed differences among DVL genes expressions and overall and disease‐free survivals. The plots are shown in Figure 6. The plots demonstrated that for low‐grade astrocytomas (Figure 6B,D,F), the low expression levels of DVL1 and DVL2 are correlated to better both overall and disease‐free survival. The same applies for the disease‐free plot of DVL3, while the overall survival plot shows similar survival for both low and high DVL3 levels. Glioblastoma group (Figure 6A,C,E) shows differences. Curves demonstrate similar survival for both high and low DVL1 expression levels, however, disease‐free survival plots for DVL2 and DVL3 show that higher levels of the proteins predict better survival (Figure 6C,E).
Figure 6.
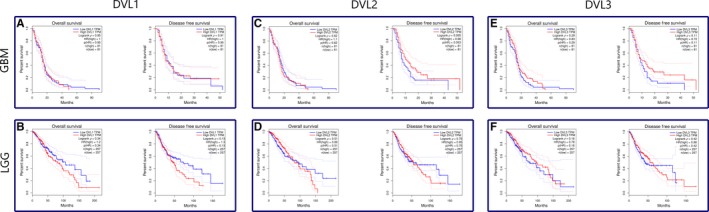
Kaplan‐Meier survival curves generated from GEPIA database for each DVL member. A,B) represent DVL1; C,D) DVL2; E,F) DVL3. A,C,E) demonstrate glioblastoma, B,D,F) low‐grade gliomas
4. DISCUSSION
Despite advances in molecular and clinical oncology, in the case of astrocytoma, and in particular glioblastoma, no significant progress in the increase of the survival rates has been made over the last decades. Therefore, the identification of novel molecular targets responsible for tumour progression and biomarkers for prediction of disease outcome and response to treatment is extremely requisite. Several novel studies indicate the involvement of Wnt signalling in the molecular pathogenesis of glial tumours.7, 8, 9, 20 However, very little is known about the possible association of the proteins involved in the Wnt pathway with tumour malignancy. Especially rare are studies about the roles of DVL1, DVL2 and DVL3 in human astrocytic brain tumours.27, 28, 29 The results of the present study showed that genetic and protein changes of all three Dishevelleds have distinct roles in the process of astrocytoma formation and progression.
We have found the constant rate of MSI across all astrocytoma grades which indicates that genomic instability is part of genetic blueprint of those tumours. Microsatellite instability at DVL1 locus was detected in 28.6% of pilocytic, 61.5% diffuse, 45.5% anaplastic astrocytomas and 34.3% of glioblastomas. Diffuse astrocytomas displayed significantly higher rate of DVL1 MSI than other astrocytoma grades (P = 0.008). Although at a bit lower frequencies, MSI for DVL2 was also constantly detected with the exception of anaplastic astrocytomas. Nevertheless, diffuse type again showed the highest number of MSI. For DVL3 gene MSI was constantly present across different grades.
The lowest frequencies of DVL1 and DVL2 MSI were observed for pilocytic astrocytomas which are benign tumours and not prone to malignant progression. However, the highest MSI frequencies were confined to diffuse cases suggesting that the increased genomic instability could be characteristic for astrocytomas that are able to progress to higher grades. It has been known that the increase of mutation accumulation results in an acceleration of the tumour cell evolution.30 But besides having effect on cell proliferation, the increased rate of mutations within tumour cells could also lead to cell death. Therefore, only those cells in which the number of mutations does not exceed a certain threshold will have the potential to proliferate. The lower frequencies of MSI found in glioblastomas for all three DVL homologs could be explained by the inability of tumour cells to proceed into much desired apoptosis.31
As MSI results from impaired DNA mismatch repair (MMR), several groups32, 33, 34, 35, 36 analysed MMR genes in astrocytoma of different grades but reported low rate of mutations. Nevertheless, the observed presence of MSI may suggest that yet another mechanism of MMR gene inactivation besides mutations is causative of this molecular phenomenon in astrocytoma. Microsatellites are generally found in non‐coding regions and their role in various malignant diseases is still unexplained.37, 38 Establishing a pattern of characteristic microsatellite loci in tumours could have predictive potential for tumour behaviour.
The results on gross deletions of the three human DVL genes indicate that LOHs of DVL1 and DVL3 genes were confined only to the high‐grade malignant group (grades III and IV) while LOHs of DVL2 were appearing across all grades. Collectively for all DVL genes the number of LOHs was much more frequent in higher‐grade tumours (23 LOHs) than in low grade ones (4 LOHs). Statistical analysis pointed out that deletions of DVL3 gene were significantly associated to the highest grade (P = 0.007) and could be connected to tumour progression.
The chromosomal area on the chromosome 17 where DVL2 is located was often deleted in our study. This is consistent with several earlier studies investigating aberrations of chromosome 17 in various tumour types including lung, breast, ovarian and brain tumours.39, 40, 41, 42, 43 Lee et al39 found LOH on locus 17p in 50% of anaplastic astrocytomas and glioblastomas, but did not detect it in tumours of lower grades. We too, have found LOH to be more frequent in malignant types (grades III and IV) or those types prone to malignancy (grade II).
Genetic changes of DVL3 gene suggest its involvement in the process of glioma progression. We noticed that the percentage of LOH increased with the malignancy grade (grade III with 18.2% and grade IV with 31.2%), while the presence of MSI was slightly decreasing in higher grades (grade I with 21.4%; grade II with 15.4%; grade III with 14.3% and grade IV with 14.3%). Statistical analysis revealed that LOH of DVL3 was significantly more frequent in glioblastomas than in lower‐grade tumours (P = 0.007). We also found the amplification of DVL3 gene in 8.6% of glioblastoma which could indicate different cellular roles for this gene. Other research groups showed similar results. Hu et al44 detected 30% of glioblastomas with LOH in the chromosomal region 3q where DVL3 is located. On the other hand, Hui et al45 investigated amplifications in the glioblastoma genome using the aCGH, and found that the region 3q was amplified in 14.3% of the tumour.
Data from cBioPortal and COSMIC databases24, 25 report on low rate of somatic mutations, fusions and deep deletions of all DVL genes. However, the mutations that have been found are much more frequent in glioblastomas than low‐grade tumours. The reported rate of amplifications of DVL3 is similar to the ones we found.
Taken together, we showed that MSI occurred constantly across all four tumour grades, with more frequent incidence in lower astrocytoma grades, suggesting its association with tumour formation. Loss of heterozygosity was found to be often present in anaplastic astrocytoma and glioblastoma and therefore could be involved in the process of tumour progression as a background mechanism for inactivation of tumour suppressor genes. All things considered, it is clear that LOH and MSI contribute to the genomic profile of astrocytoma.46, 47, 48, 49The levels of DVL1, DVL2 and DVL3 protein expressions were studied as the functional consequences of each Dishevelled homologue on astrocytoma tumour formation are still not fully clarified, and the data from the literature are contradictory. Most studies report on gene amplifications and high expression of DVL proteins in general,17, 50 but others describe large deletions of these genes as well.51, 52 Although our results generally showed higher DVL1 expression levels when compared to the control tissue, when dividing the sample to malignancy grade groups it became obvious that the expression levels of DVL1 differed significantly across tumour grades (P < 0.001). Glioblastomas displayed predominant low DVL1 expression, while anaplastic, pilocytic and diffuse astrocytomas showed predominantly moderate and strong signals. Our results indicate that expression levels of DVL1 dropped in the highest grade. Glioblastomas had significantly more cells with low expression and significantly less cells with strong expression as compared with pilocytic (P < 0.001) and anaplastic astrocytomas (P < 0.001). Moreover, diffuse astrocytomas had significantly more cells with low expression when compared to anaplastic cases (P = 0.008). Negative correlation between DVL1 protein expression and malignancy grades (r s = −0.479, df = 70, P < 0.001) was established indicating that elevated protein expression could be attributed to early events. Also younger patients had a stronger DVL1 expression than older ones (r s = −0.226, df = 70, P = 0.057). We also noticed that in tumours with low DVL1 expression, the signal was predominantly localized in the cytoplasm or in the cytoplasm and cell membrane, while in those with moderate and strong expression, the signal was confined to cytoplasm and nucleus (P < 0.001). Several authors showed that DVL1 gene amplification and elevated protein expression are involved in the development of breast and prostate cancer16, 53, 54 accompanied with positive correlation between DVL1 and β‐catenin location in the nucleus.53
DVL2 was also upregulated in all tumour grades in comparison to control samples. The highest number of samples with moderate and strong signal was present in anaplastic astrocytoma (77.6%) followed by diffuse astrocytoma (70%), glioblastoma (61.7%) and pilocytic astrocytoma (57.2%). As the expression of DVL2 protein was quite similar across tumour grades, cell count analysis of signal strengths did not show significant differences among malignancy grades. Once again, tumours with the highest proportion of cells with moderate and strong signal had protein located in the cytoplasm and nuclei (P < 0.001). A study by Pulvirenti et al55 report on increased DVL2 expression in 70% of glioblastomas. The authors confirmed the role of DVL2 in human glioblastoma by DVL2 gene attenuation using RNAi. Furthermore, when DVL2 activity was decreased in the glioblastoma cells it prevented tumour development in the immunodeficiency mice. There are also studies showing that DVL2 is involved in the progression of colon cancer.56
The clear picture on the increase of the expression levels with the malignancy grade was obtained for DVL3. In all grade I and II astrocytomas samples, DVL3 expression was characterized as weak, while 82.4% of glioblastomas showed moderate and strong expression levels. Accordingly, cell count analysis regarding DVL3 signal strength showed significant differences among tumour grades (P < 0.001). Glioblastomas had the lowest number of cells showing lack of expression and the highest number of cells showing moderate and strong expression as compared to other grades (P 1 < 0.001; P 2 = 0.001; P 3 = 0.013 respectively). Unlike the two previously analysed proteins, the DVL3 expression increase with malignancy grade was very pronounced. Of note is that older patients had significantly higher DVL3 expression comparing to the younger ones (r s = 0.395, df = 70, P = 0.001). There was no significant correlation between the expression levels of DVL1 and DVL2 proteins (P = 0.799). However, DVL1 and DVL3 were significantly negatively correlated in our total sample (P = 0.002). Stronger DVL1 expression was associated to low‐grade tumours and strong DVL3 expression to high‐grade tumours. As the correlation between the decreased protein expression of DVL3 and the presence of gross deletions has not been established, there is a possibility that the observed LOHs are the reflection of tumour heterogeneity occurring only in certain glioblastoma subtypes.
With regard to subcellular localization of DVL3 we noticed that tumours with low signal intensities showed predominant cytoplasmic localization, while moderate and strong expression was usually localized in both the cytoplasm and the nucleus (P = 0.004). This observation suggests that the increased DVL3 expression in glioblastoma leads to more frequent transfer of this protein into the nucleus. The data from the literature report on two cellular pools of DVL: one in the cytoplasm and another in the nucleus. Gan et al57 found that DVL3 was accumulated in the nucleus in 36% of the analysed colon cancer samples. It seems that DVL migrates from one cell compartment to another after its concentration reaches a level necessary for signal transduction. As our results of the bivariate correlation between DVL3 and transcription factors TCF1 and LEF1 showed that the proteins were significantly positively correlated, we can assume that the increased DVL3 expression could ultimately lead to enhanced transcriptional activity of target genes.
Similar to us, two studies17, 27 also showed different behaviour of DVL members in human tumours. Sareddy et al27 in astrocytoma showed that the expression of DVL3 mRNA was elevated in all three malignancy grades, while the expression of DVL1 and DVL2 mRNAs was not detected. Authors also noticed a strong positive correlation between DVL3, β‐catenin and LEF1 at the mRNA level, indicating increased activity of the Wnt signalling pathway. Uematsu et al17 observed increased DVL3 expression in non‐small cell lung cancer, while expression of DVL1 and DVL2 was not detected. Other papers studied Dishevelled expression29, 58 and found that it increased with the histopathological grade. Dishevelled was positively correlated with the proliferation index Ki‐67, tumour invasion index MMP‐9 and β‐catenin, and transferred to the nucleus which indicated its involvement in the proliferation and invasion processes. Our group previously analysed DVL1 and DVL3 proteins in metastases from lung to the brain. The results showed elevated DVL1 (87.1%) and DVL3 (90.3%) expression and cytoplasmic and nuclear localization of the signal.59 It is important to discuss the potential roles that each DVL member plays in signalling circumstances. Such studies are rare and how DVLs conduct activity still remains poorly understood. Firstly, it has been proposed that because of the high similarity of DVL genes in mice and humans, members play equal roles and there may be redundancy in how they work. However, Sharma et al,60 based on mouse knockout models, indicated that each DVL member may have a unique role to play. To elucidate the specific roles of DVL genes, rescue studies on double knockout mice were conducted. Double knockout of DVL1 and DVL3 did not display neural tube defects which indicate that each DVL has a unique role to play. Another point in favour of different roles is the aberrant expression of DVL genes linked to different human disorders. DVL1 has been reported to be a candidate gene for human development disorder Schwartz‐Jampel syndrome, Charcot‐Marie‐Tooth disease type 2A and DiGeorge Syndrome, while DVL3 have been linked to Hirschsprung's disease.
Interestingly, Sharma et al60 performed a TCGA analysis of DVL1, DVL2 and DVL3 expression across of four different types of cancer including glioblastoma and found that DVL RNA expression in majority of those cancers did not differ as compared to adjacent normal tissues except for DVL1. The authors report on downregulation of DVL1 RNA in glioblastoma and upregulation of DVL3 RNA in lung cancer. This is very similar to our results and may suggest the importance of the post‐transcriptional regulation of DVL proteins. The activation of Wnt signalling is primarily mediated by transcription factors from the TCF/LEF family. It is well known that the TCF1 and LEF1 proteins can have truly diverse effects in mediating signalling. Their impact on activation or repression of the target genes depends on the protein partner they interact with. Numerous studies investigated TCF1 and LEF1 transcription factors in various tumour types including colon, kidney, breast, adenocarcinoma, melanoma, several different types of leukaemia and lymphoma60, 61, 62 and concluded that their expression is important in maintaining the physiological functions of cells, while their overexpression causes invasion of tumour cells60 and promotes epithelial‐to‐mesenchymal transition.63
The results of our study showed that both TCF1 and LEF1 showed elevated expression and this upregulation indicates that the Wnt pathway was activated. Moreover, the levels of both TCF1 and LEF1 expression increased with astrocytoma grades. Astrocytomas of higher grades had significantly higher number of cells with strong TCF1 expression as compared with lower grades (P = 0.022 anaplastic and P = 0.001 glioblastoma). Similarly, the highest number of cells with strong LEF1 expression was confined to glioblastomas as compared with pilocytic (P < 0.001), diffuse (P = 0.032) and anaplastic types (P = 0.003). Correlation of IRS values for TCF1 and LEF1 revealed significant positive correlation between the two proteins across all tumour grades (r s = 0.474, df = 92, P < 0.001).
Positive correlation was established between DVL3 and both TCF1 (r s = 0.410, df = 30, P = 0.020) and LEF1 (r s = 0.475, df = 30, P = 0.006) indicating once again DVL3 link to progression.
There are several studies investigating the role of Wnt signalling in human astrocytoma.27, 64 Sareddy et al65 demonstrated strong expression of TCF4 and LEF1 in diffuse, anaplastic astrocytomas and glioblastomas. They showed that the activation of the transcription factors was induced by β‐catenin migration into the nucleus. A positive correlation was also found between the expression level of TCF4 and LEF1 compared to the astrocytoma grade. Although we analysed another member of TCF family, the findings are still consistent to our results.
Our previous study66 on beta‐catenin expression in neuroepithelial brain tumours of different grades strengthens the claim on Wnt signalling pathway's activity. We have shown that beta‐catenin was upregulated in 53.1% of samples. Moreover, in 21.4% of glioblastomas beta‐catenin was accumulated exclusively in the nucleus, while in 38.1% in both the cytoplasm and nucleus.
5. CONCLUSIONS
Together the results of this study show that important regulatory components of the Wnt signalling pathway, DVLs, have different roles in astrocytomas. Our study revealed that LOH and MSI contribute to the genomic profile of astrocytoma. Furthermore, a noticeable difference in the expression levels of DVL proteins in different tumour grades was established. We have found that DVL1 and DVL2 in astrocytic brain tumours act in one fashion and may be involved during early stages of the disease, while DVL3 behaves differently being involved in the progression while having activating effect on TCF1 and LEF1 transcription factors of Wnt pathway.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was conducted in compliance with the declaration of Helsinki regarding ethical principles for medical research involving human volunteers. This research was approved by Ethics Committees of the School of Medicine University of Zagreb and University Hospital Centers “Sisters of Charity” and “Zagreb”.
ACKNOWLEDGEMENT
We thank Brain Tumour Foundation of Canada for donating brain tumour samples.
AUTHOR CONTRIBUTION
AK contributed to data acquisition and analysis, performed experimental work, wrote the manuscript and revised it for important intellectual content. MB performed experimental work, participated in data collection, interpretation and analysis. DT participated in tumour sample analysis and results interpretation and revised the manuscript for important intellectual content; KŽ participated in tumour sample analysis and revised the manuscript for important intellectual content; AB contributed to the data interpretation, manuscript editing, revised the manuscript for important intellectual content. NN contributed to data acquisition, participated in tumour sample analysis and manuscript editing. GM participated in tumour sample analysis and revised the manuscript for important intellectual content; ŽK contributed to data acquisition, the interpretation of the results and manuscript editing. NPŠ produced the idea, designed the study, contributed to analysis and interpretation of the results, wrote the manuscript and revised it for important intellectual content, and approved the final version of the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interest.
Kafka A, Bačić M, Tomas D, et al. Different behaviour of DVL1, DVL2, DVL3 in astrocytoma malignancy grades and their association to TCF1 and LEF1 upregulation. J Cell Mol Med. 2019;23:641–655. 10.1111/jcmm.13969
Funding information
This work was supported by grant agreement no. 6625 from Croatian Science Foundation; co‐financed by the European Union through the European Regional Development Fund, Operational Programme Competitiveness and Cohesion, grant agreement no. KK.01.1.1.01.0007, CoRE ‐ Neuro.
REFERENCES
- 1. Ostrom QT, Gittleman H, Stetson L, et al. Epidemiology of gliomas. Cancer Treat Res. 2015;163:1‐14. [DOI] [PubMed] [Google Scholar]
- 2. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803‐820. [DOI] [PubMed] [Google Scholar]
- 3. Perry A, Wesseling P. Histologic classification of gliomas. Handb Clin Neurol. 2016;134:71‐95. [DOI] [PubMed] [Google Scholar]
- 4. The Cancer Genome Atlas (TCGA) Research Network . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nasser MM, Mehdipour P. Exploration of involved key genes and signaling diversity in brain tumors. Cell Mol Neurobiol. 2017;38:393‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang K, Zhang J, Han L, et al. Wnt/beta‐catenin signaling in glioma. J Neuroimmune Pharmacol. 2012;7:740‐749. [DOI] [PubMed] [Google Scholar]
- 7. Pećina‐Šlaus N, Kafka A, Tomas D, et al. Wnt signaling transcription factors TCF‐1 and LEF‐1 are upregulated in malignant astrocytic brain tumors. Histol Histopathol. 2014;29:1557‐1564. [DOI] [PubMed] [Google Scholar]
- 8. Kahlert UD, Suwala AK, Koch K, et al. Pharmacologic Wnt inhibition reduces proliferation, survival, and clonogenicity of glioblastoma cells. J Neuropathol Exp Neurol. 2015;74:889‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pećina‐Šlaus N, Kafka A, Varošanec AM, et al. Expression patterns of Wnt signaling component sFRP3 in astrocytoma and glioblastoma. Mol Med Rep. 2016;13:4245‐4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCord M, Mukouyama YS, Gilbert MR, Jackson S. Targeting WNT signaling for multifaceted glioblastoma therapy. Front Cell Neurosci. 2017;11:318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nusse R, Clevers H. Wnt/β‐catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985‐999. [DOI] [PubMed] [Google Scholar]
- 12. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim W, Kim M, Jho EH. Wnt/β‐catenin signalling: from plasma membrane to nucleus. Biochem J. 2013;450:9‐21. [DOI] [PubMed] [Google Scholar]
- 14. Lee Y‐N, Gao Y, Wang H‐y. Differential mediation of the wnt canonical pathway by mammalian Dishevelleds‐1, ‐2, and 3. Cell Signal. 2008;20:443‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kafka A, Basic‐Kinda S, Pecina‐Slaus N. The cellular story of Dishevelleds. Croat Med J. 2014;55:459‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nagahata T, Shimada T, Harada A, et al. Amplification, up‐regulation and over‐expression of DVL‐1, the human counterpart of the Drosophila disheveled gene, in primary breast cancers. Cancer Sci. 2003;94:515‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uematsu K, He B, You L, et al. Activation of the Wnt pathway in non small cell lung cancer: evidence of disheveled overexpression. Oncogene. 2003;22:7218. [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y, Yang ZQ, Wang Y, et al. Dishevelled‐1 and Dishevelled‐3 affect cell invasion mainly through canonical and noncanonical Wnt pathway, respectively, and associate with poor prognosis in nonsmall cell lung cancer. Mol Carcinog. 2010;49:760‐770. [DOI] [PubMed] [Google Scholar]
- 19. Li X‐Y, Liu S‐L, Cha N, et al. Transcription expression and clinical significance of Dishevelled‐3 mRNA and δ‐catenin mRNA in pleural effusions from patients with lung cancer. Clin Dev Immunol. 2012;2012:904946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang C, Li C, Chen X, et al. Overexpression of dishevelled 2 is involved in tumor metastasis and is associated with poor prognosis in hepatocellular carcinoma. Clin Transl Oncol. 2017;19:1507‐1517. [DOI] [PubMed] [Google Scholar]
- 21. GeneLoc Genome Locator. https://genecards.weizmann.ac.il/geneloc/index.shtml. Accessed September 29, 2015.
- 22. Zadeh MD, Amini R, Firoozray M, Derakhshandeh‐Peykar P. Frequent homozygous deletion of p16/CDKN2A gene in malignant gliomas of Iranian patients. Pak J Biol Sci. 2007;10:4246‐4250. [DOI] [PubMed] [Google Scholar]
- 23. Ono Y, Ueki K, Joseph JT, Louis DN. Homozygous deletions of the CDKN2/p16 gene in dural hemangiopericytomas. Acta Neuropathol. 1996;91:221‐225. [DOI] [PubMed] [Google Scholar]
- 24. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401‐404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98‐W102. 10.1093/nar/gkx247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sareddy GR, Panigrahi M, Challa S, et al. Activation of Wnt/beta‐catenin/Tcf signaling pathway in human astrocytomas. Neurochem Int. 2009;55:307‐317. [DOI] [PubMed] [Google Scholar]
- 28. Pulvirenti T, Van Der Heijden M, Droms LA, et al. Dishevelled 2 signaling promotes self‐renewal and tumorigenicity in human gliomas. Cancer Res. 2011;71:7280‐7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li J, Guo G, Li J, et al. The expression and significance of dishevelled in human glioma. J Surg Res. 2014;192:509‐514. [DOI] [PubMed] [Google Scholar]
- 30. Breivik J, Gaudernack G. Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Semin Cancer Biol. 1999;9:245‐254. [DOI] [PubMed] [Google Scholar]
- 31. Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci U S A. 1996;93:14800‐14803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martinez R, Schackert HK, Appelt H, et al. Low‐level microsatellite instability phenotype in sporadic glioblastoma multiforme. J Cancer Res Clin Oncol. 2005;131:87‐93. [DOI] [PubMed] [Google Scholar]
- 33. Eckert A, Kloor M, Giersch A, et al. Microsatellite instability in pediatric and adult high‐grade gliomas. Brain Pathol. 2007;17:146‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vladimirova V, Denkhaus D, Soerensen N, et al. Low level of microsatellite instability in paediatric malignant astrocytomas. Neuropathol Appl Neurobiol. 2008;34:547‐554. [DOI] [PubMed] [Google Scholar]
- 35. Rodríguez‐Hernández I, Garcia JL, Santos‐Briz A, et al. Integrated analysis of mismatch repair system in malignant astrocytomas. PLoS ONE. 2013;8:e76401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karunasena E, McIver LJ, Rood BR, et al. Somatic intronic microsatellite loci differentiate glioblastoma from lower‐grade gliomas. Oncotarget. 2014;5:6003‐6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li YC, Korol AB, Fahima T, Nevo E. Microsatellites within genes: structure, function, and evolution. Mol Biol Evol. 2004;21:991‐1007. [DOI] [PubMed] [Google Scholar]
- 38. Hause RJ, Pritchard CC, Shendure J, Salipante SJ. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342‐1350. [DOI] [PubMed] [Google Scholar]
- 39. Lee SH, Kim JH, Rhee CH, et al. Loss of heterozygosity on chromosome 10, 13q(Rb), 17p, and p53 gene mutations in human brain gliomas. J Korean Med Sci. 1995;10:442‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. White GRM, Stack M, Santibanez‐Koref M, et al. High levels of loss at the 17p telomere suggest the close proximity of a tumor suppressor. Br J Cancer. 1996;74:863‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Konishi H, Takahashi T, Kozaki K, et al. Detailed deletion mapping suggests the involvement of a tumor suppressor gene at 17p13.3, distal to p53, in the pathogenesis of lung cancers. Oncogene. 1998;17:2095‐2100. [DOI] [PubMed] [Google Scholar]
- 42. Orellana C, Hemandez‐Marti M, Martinez F, et al. Pediatric brain tumors: loss of heterozygosity at 17p and TP53 gene mutations. Cancer Genet Cytogenet. 1998;102:93‐99. [DOI] [PubMed] [Google Scholar]
- 43. Wiper DW, Zanotti KM, Kennedy AW, et al. Analysis of allelic imbalance in chromosome 17p13 in stage I and stage II epithelial ovarian cancers. Gynecol Oncol. 1998;71:77‐82. [DOI] [PubMed] [Google Scholar]
- 44. Hu J, Jiang C, Ng HK, et al. Chromosome 3 may harbour multiple tumor suppressor genes associated with primary glioblastoma multiforme. Chin J Cancer Res. 2002;14:183‐186. [PubMed] [Google Scholar]
- 45. Hui AB, Lo KW, Yin XL, et al. Detection of multiple gene amplifications in glioblastoma multiforme using array‐based comparative genomic hybridization. Lab Invest. 2001;81:717‐723. [DOI] [PubMed] [Google Scholar]
- 46. Dams E, Van de Kelft EJ, Martin JJ, et al. Instability of microsatellites in human gliomas. Cancer Res. 1995;55:1547‐1549. [PubMed] [Google Scholar]
- 47. Zhu J, Guo SZ, Beggs AH, et al. Microsatellite instability analysis of primary human brain tumors. Oncogene. 1996;12:1417‐1423. [PubMed] [Google Scholar]
- 48. Izumoto S, Arita N, Ohnishi T, et al. Microsatellite instability and mutated type II transforming growth factor‐b receptor gene in gliomas. Cancer Lett. 1997;112:251‐256. [DOI] [PubMed] [Google Scholar]
- 49. Leung SY, Chan TL, Chung LP, et al. Microsatellite instability and mutation of DNA mismatch repair genes in gliomas. Am J Pathol. 1998;153:1181‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li XY, Liu SL, Cha N, et al. Transcription expression and clinical significance of Dishevelled‐3 mRNA and δ‐catenin mRNA in pleural effusions from patients with lung cancer. Clin Dev Immunol. 2012;2012:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Girard L, Zöchbauer‐Müller S, Virmani AK, et al. Genome‐wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non‐small cell lung cancer, and loci clustering. Cancer Res. 2000;60:4894‐4906. [PubMed] [Google Scholar]
- 52. Kurashina K, Yamashita Y, Ueno T, et al. Chromosome copy number analysis in screening for prognosis‐related genomic regions in colorectal carcinoma. Cancer Sci. 2008;99:1835‐1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prasad CP, Gupta SD, Rath G, Ralhan R. Wnt signaling pathway in invasive ductal carcinoma of the breast: relationship between beta‐catenin, dishevelled and cyclin D1 expression. Oncology. 2007;73:112‐117. [DOI] [PubMed] [Google Scholar]
- 54. Mizutani K, Miyamoto S, Nagahata T, et al. Upregulation and overexpression of DVL1, the human counterpart of the Drosophila dishevelled gene, in prostate cancer. Tumori. 2005;91:546‐551. [DOI] [PubMed] [Google Scholar]
- 55. Pulvirenti T, Van Der Heijden M, Droms LA, et al. Dishevelled 2 signaling promotes self‐renewal and tumorigenicity in human gliomas. Cancer Res. 2011;71:280‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Metcalfe C, Ibrahim AE, Graeb M, et al. Dvl2 promotes intestinal length and neoplasia in the ApcMin mouse model for colorectal cancer. Cancer Res. 2010;70:6629‐6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gan X, Wang J, Ying X, et al. Nuclear Dvl, c‐Jun, β‐catenin, and TCF form a complex leading to stabilization of β‐catenin‐TCF interaction. J Cell Biol. 2008;180:1087‐1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wei Q, Zhao Y, Yang Z‐Q, et al. Dishevelled family proteins are expressed in non‐small cell lung cancer and function differentially on tumor progression. Lung Cancer. 2008;62:181‐192. [DOI] [PubMed] [Google Scholar]
- 59. Kafka A, Tomas D, Beroš V, et al. Brain metastases from lung cancer show increased expression of DVL1, DVL3 and beta‐catenin and down‐regulation of E‐cadherin. Int J Mol Sci. 2014;15:10635‐10651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sharma M, Castro‐Piedras I, Simmons GE, Pruitt K. Dishevelled: a masterful conductor of complex Wnt signals. Cell Signal. 2018;47:52‐64. 10.1016/j.cellsig.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nguyen A, Rosner A, Milovanović T, et al. WNT pathway component LEF1 mediates tumor cell invasion and is expressed in human and murine breast cancers lacking ErbB2 (her‐2/neu) overexpression. Int J Oncol. 2005;27:949‐956. [PubMed] [Google Scholar]
- 62. Ravindranath A, O'Connel A, Johnston PG, El‐Tanani MK. The role of LEF/TCF factors in neoplastic transformation. Current Mol Med. 2008;8:38‐50. [DOI] [PubMed] [Google Scholar]
- 63. Najdi R, Holcombe RF, Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. J Carcinog. 2011;10:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim K, Lu Z, Hay ED. Direct evidence for a role of beta‐catenin/LEF‐1 signaling pathway in induction of EMT. Cell Biol Int. 2002;26:463‐476. [DOI] [PubMed] [Google Scholar]
- 65. Sareddy GR, Geeviman K, Panigrahi M, et al. Increased beta‐catenin/Tcf signaling in pilocytic astrocytomas: a comparative study to distinguish pilocytic astrocytomas from low‐grade diffuse astrocytomas. Neurochem Res. 2012;37:96‐104. [DOI] [PubMed] [Google Scholar]
- 66. Nikuseva Martić T, Pećina‐Slaus N, Kusec V, et al. Changes of AXIN‐1 and beta‐catenin in neuroepithelial brain tumors. Pathol Oncol Res. 2010;16(1):75‐79. 10.1007/s12253-009-9190-9 [DOI] [PubMed] [Google Scholar]