

ABSTRACT
Morphogenesis of the inner ear epithelium requires coordinated deployment of several signaling pathways, and disruptions cause abnormalities of hearing and/or balance. The FGFR2b ligands FGF3 and FGF10 are expressed throughout otic development and are required individually for normal morphogenesis, but their prior and redundant roles in otic placode induction complicates investigation of subsequent combinatorial functions in morphogenesis. To interrogate these roles and identify new effectors of FGF3 and FGF10 signaling at the earliest stages of otic morphogenesis, we used conditional gene ablation after otic placode induction, and temporal inhibition of signaling with a secreted, dominant-negative FGFR2b ectodomain. We show that both ligands are required continuously after otocyst formation for maintenance of otic neuroblasts and for patterning and proliferation of the epithelium, leading to normal morphogenesis of both the cochlear and vestibular domains. Furthermore, the first genome-wide identification of proximal targets of FGFR2b signaling in the early otocyst reveals novel candidate genes for inner ear development and function.
KEY WORDS: Otocyst, Conditional mutant, Ligand trap, RNA-Seq
Summary: The FGFR2 ligands FGF3 and FGF10 are required continuously for inner ear morphogenesis, and FGFR2b signaling targets are identified as novel candidates for involvement in inner ear development and function.
INTRODUCTION
The membranous labyrinth of the mammalian inner ear is one of the most complex examples of organ morphogenesis. An unremarkable patch of cranial ectoderm is transformed into a structurally intricate sensory apparatus with two functionally distinct compartments: the ventral cochlea and the dorsal vestibular system, responsible for the perception of sound and acceleration, respectively. Within these compartments an exquisitely patterned array of sensory, non-sensory and supporting cell types are poised to transduce auditory and vestibular stimuli through sensory ganglia to the brain. Proper morphogenesis of the labyrinth is essential for normal auditory and vestibular function as indicated by imaging studies of hearing loss patients (Kimura et al., 2018; Sennaroğlu and Bajin, 2017). In light of the advent of cochlear implantation to treat hearing loss in cases of inner ear malformation (Isaiah et al., 2017), elucidating the signals governing otic morphogenesis and appreciation of the spectrum of labyrinthine morphogenetic defects are necessary to advance treatment.
Amniote inner ear development initiates during neurulation when hindbrain proximal ectoderm is induced to thicken, forming the otic placode, the source of both the otic epithelium and the neurons of its sensory ganglia. Next, the placode invaginates, forming a cup that deepens and delaminates neuroblasts, before detaching from the overlying ectoderm to form a spherical vesicle, the otocyst, which at embryonic day (E) 9.5 in mouse is already patterned along the three anatomical axes. Otocyst morphogenesis initiates with dorsomedial budding to form the endolymphatic duct and sac (EDS) anlagen. Vestibular structures initiate by sequential dorsal and lateral evaginations of the epithelium to form vertical and horizontal pouches, which are further sculpted by epithelial fusion and resorption, generating three orthogonal semicircular canals. The utricle and saccule form from anterior/central bulges, and the cochlear duct (CD) emerges as a ventral outgrowth. In mice, it undergoes progressive ventral extension and coiling, reaching 1.75 turns by E15.5, when gross morphogenesis is largely complete (Morsli et al., 1998; Sajan et al., 2007). Cell-type differentiation and functional maturation, however, continue until well after birth (reviewed by Alsina and Whitfield, 2017; Basch et al., 2016; Whitfield, 2015; Wu and Kelley, 2012).
Signals regulating otic placode induction and early otocyst patterning emanate from surrounding tissues and are understood in some detail (Ladher, 2017), but by otocyst stages intrinsic signals are also produced and their roles in driving region-specific morphogenesis are less well understood. Extrinsic signals regulating dorsal morphogenesis include hindbrain WNTs and BMPs. SHH secreted from the ventral hindbrain and notochord initiates ventral morphogenesis. Crosstalk between these signals involves regulation of key region-specific transcription factors (Ohta and Schoenwolf, 2018). FGF signaling is also crucial for otic development, and functions at multiple stages. A cascade of FGFs from endoderm, mesoderm and hindbrain is required for otic placode induction (Alvarez et al., 2003; Ladher et al., 2005; Wright and Mansour, 2003a; Zelarayan et al., 2007). In particular, Fgf3 and Fgf10, encoding ligands that signal through FGF receptor isoforms FGFR1b and FGFR2b (Zhang et al., 2006), are required redundantly for otic placode induction, such that germline double null mutants (F3KO;F10KO) have no inner ear (Alvarez et al., 2003; Wright and Mansour, 2003a). Applications of FGFs and FGFR inhibitors to chick embryos revealed profound influences of FGFs on otic morphogenesis (Chang et al., 2004) and studies of individual mouse mutants revealed roles for Fgf3 and Fgf10 in morphogenesis. Mice lacking Fgf3 (F3KO) fail with variable penetrance and expressivity to form an EDS, and consequently have variable morphogenesis and dysfunction of both the cochlea and vestibule (Hatch et al., 2007; Mansour et al., 1993). Mice lacking Fgf10 (F10KO) have no posterior semicircular canals (PSCCs), and show milder deformations of the anterior and lateral canals. Fgf10 heterozygotes also exhibit PSCC reductions or agenesis (Pauley et al., 2003; Urness et al., 2015). The F10KO CD is also affected, being shorter and narrower than that of heterozygous or wild-type mice owing to loss of non-contiguous non-sensory domains (Urness et al., 2015). Fgfr2b null mutants form otocysts (Pirvola et al., 2000); however, they develop with severe cochlear and vestibular dysmorphology, suggesting that Fgf3 and Fgf10 could have additional and combinatorial roles during morphogenesis.
Here, we define the expression of Fgf3 and Fgf10 in the developing mouse otic epithelium and ganglion, and interrogate their functions after otic placode induction. We employ two complementary genetic strategies: otic placode lineage-restricted gene ablation and timed induction of a soluble dominant-negative FGFR2b ectodomain that acts rapidly as an extracellular ligand trap to block signaling. Together, our data show that Fgf3 and Fgf10 are not required in the placode lineage for otocyst formation, but are required subsequently for otocyst patterning, neuroblast maintenance, epithelial proliferation and both vestibular and cochlear morphogenesis. Furthermore, the first differential RNA-Seq analyses of otocysts revealed FGFR2b signaling targets that define novel candidates for involvement in otic morphogenesis and function.
RESULTS
Fgf3 and Fgf10 are expressed dynamically during otocyst and ganglion formation, and cochlear morphogenesis
To determine Fgf3 and Fgf10 expression during otocyst formation and cochlear morphogenesis, we used RNA in situ hybridization (ISH). Before E9.0, both genes were exclusively periotic (data not shown). Consistent with previous studies (Schimmang, 2007; Wright and Mansour, 2003b), Fgf3 and Fgf10 transcripts were non-overlapping at the otic cup stage, with Fgf3 expressed in hindbrain adjacent to the cup, and Fgf10 expressed in the cup itself, exclusive of the dorsal and lateral-most regions (Fig. 1A,B). Once the otocyst closed, Fgf3 was diminished in the hindbrain and was first seen in the ventrolateral otocyst and in the forming otic ganglion, whereas Fgf10 was expressed in the ventral and medial otocyst (Fig. 1C,D). By E10.25-E11.25, Fgf3 was confined to the ventrolateral otocyst. At this stage, Fgf10 overlapped with and was more extensively expressed in the ventral otocyst than was Fgf3, and also began to be expressed strongly in the ganglion (Fig. 1E-H).
Fig. 1.

Fgf3 and Fgf10 are expressed throughout otocyst formation and cochlear morphogenesis. (A-P) ISH of Fgf3 and Fgf10 probes to wild-type otic cup (A,B), otic vesicle (C-H) and cochlear duct (I-P). Probes are indicated to the left and developmental ages at the top right of each paired column. (A-J) Transverse orientation, directional arrows in A apply to all. (K-P) Sagittal orientation with cochlear apex at the top. Insets show basal-most cochlear turn. cd, cochlear duct; D, dorsal; hb, hindbrain; Ko, Kölliker's organ; L, lateral; oC, organ of Corti; oc, otic cup; og, otic ganglion; ov, otic vesicle; sg, spiral ganglion. Scale bars: 100 µm.
We confirmed overlap of Fgf3 and Fgf10 in the developing vestibular sensory tissues, with Fgf10 expression much stronger than Fgf3 (data not shown; see Pauley et al., 2003; Pirvola et al., 2000). Focusing on the developing CD, from E12.5-E16.5, we found Fgf3 in a progressively limited portion of the CD that appeared by E16.5 to flank the developing sensory organ of Corti. Fgf10 continued expression in a broader domain than Fgf3, resolving to Kölliker's organ by E16.5, and was maintained at high levels in the cochlear ganglion (Fig. 1I-P). These observations suggested that Fgf3 and Fgf10 could have combinatorial roles in morphogenesis and ganglion development.
Epithelial Fgf3 and Fgf10 are not required for otocyst formation, but both are required for vestibular and cochlear morphogenesis
Because F3KO;F10KO embryos lack otocysts, we disrupted these genes individually and combinatorially after otic placode induction using Tg(Pax2-Cre), which is active in the otic placode lineage starting at E8.5 (Fig. 2A; Ohyama and Groves, 2004) and recombines in both the epithelium and ganglion (Fig. 2B,C). We evaluated gross otic morphogenesis by paintfilling at E15.5 and CD development by histology at E18.5. In contrast to the variable F3KO otic phenotypes (Hatch et al., 2007; Mansour et al., 1993), disruption of Fgf3 in the Pax2-Cre lineage (F3cKO) had no effect on otic morphogenesis or on CD histology (Fig. 2D-E′). Indeed, F3cKO animals survived in the expected numbers and had normal auditory thresholds and motor behavior (data not shown). In contrast, F10cKO ears showed both vestibular and cochlear abnormalities, including reduction or loss of the PSCC and variable CD shortening and narrowing, the latter reflecting loss of Reissner's membrane (Fig. 2F-G′). These abnormalities were similar to those of F10KO ears (Urness et al., 2015) and, indeed, immunostaining of E18.5 F10cKO CDs was similar to F10KO CDs (data not shown). Notably, Fgf10−/c Cre-negative ears had mild PSCC shortening, but CD defects appeared only in F10cKO ears (Fig. 2F,F′).
Fig. 2.

Fgf3 and Fgf10 are not required in the Pax2-Cre lineage for otocyst formation, but both are required subsequently for vestibular and cochlear morphogenesis. (A-C) Pax2-Cre otic lineage (blue) at indicated stages. (H-K′) E9.5 left otocysts showing normal development. (D-F′,L-O′) E15.5 paintfilled ears, right lateral views of most common phenotype (enumerated in Table S1). (E-G′,P-S′) E18.5 Hematoxylin and Eosin-stained sagittal cochlear sections. Boxed regions are enlarged in E′,G′,P′-S′. Fgf genotypes are shown above each column and Cre status indicated to the right. Yellow text indicates apparent length reductions, red text indicates expected positions of missing structures. aa, anterior ampula; ascc, anterior semicircular canal; cd, cochlear duct; ed, endolymphatic duct; eds, endolymphatic duct/sac; la, lateral ampula; lscc, lateral semicircular canal; nt, neural tube; nse, non-sensory epithelium; oC, organ of Corti; og, otic ganglion; op, otic placode; ov, otic vesicle; pa, posterior ampula; pscc, posterior semicircular canal; rm, Reissner's membrane; sac, saccule; se, sensory-like epithelium; sg, spiral ganglion; ve, vestibular epithelium; vg, vestibular ganglion. Scale bar in H applies to H-K′; in D to D-F′,L′-O′; in E, to E,G; in E′ to E′,G′; in P to P-S; and in P′ to P′-S′.
Next, we evaluated morphogenesis after varying Fgf3 and Fgf10 dosage. At E9.5, all embryos had otocysts starting to develop an endolymphatic duct, showing that otic placode induction occurred normally, even in the absence of both Fgf genes (Fig. 2H-K′). However, E15.5 ears showed genotype-dependent morphologic defects (Fig. 2L-O′). Even Cre-negative ears had PSCC defects when heterozygous for Fgf10 (Fig. 2N,O). Not surprisingly, all Cre-positive ears had reduced or absent PSCCs, as Fgf10 is at least heterozygous in those cases (Fig. 2L′,M′,N′,O′). Ears were classified according to the scheme used for Fgf10 mutants (Urness et al., 2015) and formed a phenotypic spectrum of increasing severity (Table S1). Most F3cHet;F10cHet ears showed only mild PSCC reductions (Fig. 2L′), whereas most F3cKO;F10cHet ears lacked a PSCC (Fig. 2M′). However, most F3cHet/F10cKO ears had an unfused vestibular pouch, reductions in the saccule and utricle, and more extensive shortening and narrowing of the CD (Fig. 2N′) than in F10cKO ears (Fig. 2F′). Only the EDS appeared normal. The distribution of F3cHet;F10cKO phenotypes was significantly more severe than that of F10cKO phenotypes (P<10−4; Table S1). However, limited cochlear marker gene analyses of E18.5 F3cHet;F10cKO ears did not reveal any exacerbation of changes seen previously in F10KO ears (data not shown; see Urness et al., 2015) and the apparent narrowing of the CD was not significant (Fig. S1). Strikingly, conditional disruption of both Fgf3 and Fgf10 blocked both vestibular and cochlear development, leaving only a small spherical vesicle (Fig. 2O′). F3cHet;F10cHet and F3cKO;F10cHet CD histology (Fig. 2P-Q′) was indistinguishable from that of F3cKO (Fig. 2E,E′), whereas F3cHet;F10cKO CDs were very narrow and lacked Reissner's membrane (Fig. 2R,R′), similar to those of F10cKO (Fig. 2G,G′) and F10KO CDs (Urness et al., 2015). The F3cKO;F10cKO ‘ear’ had an epithelium comprising a thin, non-sensory region and a thickened vestibular-like sensory region. Most notably, these mutants showed no evidence of cochlear or vestibular neurons (Fig. 2S,S′). These data show that both Fgf3 and Fgf10 are required in the Pax2-Cre lineage, not only for vestibular morphogenesis, but also for cochlear morphogenesis and otic gangliogenesis, with the effect of Fgf3 being revealed only in the absence of Fgf10.
Fgf3 and Fgf10 are not required in the Pax2-Cre lineage for early otocyst proliferation, but are required for otocyst patterning and maintenance of otic neuroblasts
As F3cKO;F10cKO embryos ultimately develop very small otic vesicles, and this was first apparent at E10.5-E11.5, we quantified mitotic cells in E10.5 otocyst sections by calculating the number of phosphohistoneH3 (pHH3)-positive cells per otic epithelial area. The difference between control and F3cKO;F10cKO vesicles, however, was not significant (Fig. S2A-C); cleaved caspase 3, a measure of dying cells, was similarly unaffected (Fig. S2C′). This may be because hindbrain Fgf3 (Fig. 1A,C), which is unaffected by Pax2-Cre and is required to form a normally sized otocyst (Fig. S2D-G′), is sufficient to promote otocyst proliferation and survival through E10.5.
To assess otocyst patterning in conditional mutants, we conducted whole-mount ISH of E9.5-E11.5 samples using probes for regionally expressed genes that are known targets of FGF3 and/or FGF10 signaling and/or are required for morphogenesis. To manage the number of samples analyzed, we omitted single conditional mutants and used only a single Cre-negative genotype (Fgf3−/c;Fgf10−/c). At E9.5, most genes tested, as exemplified by Sox9, were unaffected even in F3cKO;F10cKO ears (Fig. 3A). Other genes unaffected by loss of both Fgf3 and Fgf10 alleles at this stage included Dusp6, Spry1, Foxg1, Has2, Gbx2, Hmx3, Sox2 and Pax2 (data not shown), all of which are lost in F3KO;F10KO ears by otic placode stages (Alvarez et al., 2003; Urness et al., 2010; Wright and Mansour, 2003a). However, the common FGF signaling target, Etv5, which has distinct ventromedial and dorsolateral domains in E9.5 control otocysts, showed differential localization in conditional mutants with only a single Fgf3 or Fgf10 allele remaining, and expression was entirely absent from F3cKO;F10cKO ears (Fig. 3B), demonstrating that epithelial FGF3/FGF10 signaling was disrupted within 24 h of CRE activation. The only other gene affected at E9.5 was Tbx1, which had dorsolateral and posteroventral otocyst domains in all genotypes except F3cKO;F10cKO, which lost the dorsolateral domain (Fig. 3C). By E10.5, the dorsolateral Tbx1 domain was lost from both F3cHet;F10cKO and F3cKO;F10cKO otocysts (Fig. 3D).
Fig. 3.

Fgf3 and Fgf10 are required in the Pax2-Cre lineage for otocyst patterning and maintenance of otic neuroblasts. (A-M) Transverse sections through the ov (A,B,E-M), or whole mounts (C,D), of embryos subjected to ISH. Probes and developmental stages are indicated aside each row. Genotypes are shown atop each column. Scale bars (100 µm) apply to each probe/stage group. Arrowheads in H indicate Bmp4 expression. Directional arrows in A and C apply to all sections and whole-mounts, respectively. D, dorsal; M, medial; nt, neural tube; og, otic ganglion; ov, otic vesicle; P, posterior.
At E10.5, expression of several other genes was still unaffected, as exemplified by Gbx2 (Fig. 3E), which is required for vestibular morphogenesis (Lin et al., 2005) and is downregulated at this stage in F3KO mutants (Hatch et al., 2007). Other genes unaffected at E10.5 included Hmx3, Spry2, Gli1, Id1 and Lfng (data not shown). In contrast, Sox2, Foxg1 and Bmp4, which are primarily lateral at E10.5, were extinguished in F3cKO;F10cKO ears (Fig. 3F-H). Other genes lost at E10.5 included Dusp6, Etv5, Etv4 and Spry2 (data not shown), all of which are transcriptional targets of FGF signaling. Curiously, we found that Pax2, which at E10.5 is normally expressed medially, was unchanged in all otocysts except F3cKO/F10cKO, where it was expanded (Fig. 3I). By E11.5 the only tested genes still unaffected in F3cKO;F10cKO otocysts were Gli1, Hmx3, Lfng and Id1 (data not shown), so these are unlikely to be targets of FGF3/FGF10 signaling in the otocyst.
To assess the otic ganglion, we assayed Neurog1 and its target, Neurod1, at both E9.5 and E10.5. At E9.5, all genotypes exhibited similar expression in a ventrolateral epithelial domain and in delaminating neuroblasts (Fig. 3J,L). In contrast, at E10.5 Neurog1 was strongly reduced and Neurod1 was virtually eliminated from the otic epithelium, and otic ganglion development was suppressed specifically in F3cKO/F10cKO otocysts (Fig. 3K,M). These data show that epithelial/ganglion expression of Fgf3 and Fgf10 are required for aspects of gene expression driving otic morphogenesis, particularly in lateral regions, and that they are also required for otic ganglion formation.
Doxycycline induction of a secreted FGFR2b ectodomain phenocopies F3KO;F10KO mutants
FGF3 and FGF10 bind to and signal through ‘b’-type FGF receptors (FGFR2b>FGFR1b; Zhang et al., 2006). To enable simultaneous and inducible inhibition of their signaling activity at any stage, we employed two alleles that together enable doxycycline (DOX)-inducible expression of a secreted, dominant-negative form of FGFR2b (dnFGFR2b), which serves as a ligand trap. Rosa26rtTA drives ubiquitous expression of the reverse tetracycline transactivator (Belteki et al., 2005) and Tg(tetO-dnFgfr2b) encodes a tetO-regulated and secreted FGFR2b ectodomain (Hokuto et al., 2003). This system is validated for temporally controlled inhibition of FGFR2b/1b-dependent mammary gland, tooth, limb and lung development (Al Alam et al., 2015; Danopoulos et al., 2013; Parsa et al., 2010; Parsa et al., 2008).
To validate this system for inner ear studies, we fed DOX-containing chow (DOX-chow) to pregnant females from E5.5-E10.5 and observed gross embryonic phenotypes. Rosa26rtTA/+ (control) embryos appeared normal, but Rosa26rtTA/+;Tg(tetO-dnFgfr2b)/+ (experimental) embryos had a short curly tail, lacked limb buds and had tiny otic vesicles (Fig. 4A,B), phenocopying F3KO;F10KO mutants. Experimental embryos exposed to DOX from E5.5 or E6.5 to E11.5 showed only an otic remnant (Fig. S3) and did not exhibit mid-hindbrain phenotypes characteristic of inhibition of ligands such as FGF8 and FGF17 that signal through ‘c’-type FGFRs (Chi et al., 2003; Sato and Joyner, 2009; Xu et al., 2000).
Fig. 4.

Induction of dnFGFR2b phenocopies F3KO;F10KO mutants and acts rapidly, revealing continuous requirements for FGFR2b ligands in otic morphogenesis. (A,B) E10.5 control and experimental embryos exposed to DOX from E5.5 to E10.5. Affected structures are outlined in black; dashed lines indicate regions behind the embryo. Scale bar in A applies to B. (C-D′) E9.5 control and experimental embryos exposed to DOX for 4 h and hybridized with Etv5. Dashed lines in C,D indicate transverse section planes for C′,D′. Scale bars in panels C,C′ apply to D,D′. (E-J) E15.5 paintfilled inner ears from embryos exposed to DOX for the interval indicated at the bottom of each panel. Number of ears with the phenotype shown is indicated. Additional phenotypes listed below. Scale bar in E applies to F-J. cd, cochlear duct; eds, endolymphatic duct/sac; flb, forelimb bud; hlb, hindlimb bud; ov, otic vesicle; sac, saccule; scc, semicircular canal; ut, utricle; vcp, vertical canal pouch.
Secreted dnFGFR2b acts rapidly to inhibit signaling by FGFR2b ligands
To determine the timing of signaling inhibition, we initiated dnFGFR2b expression by injecting DOX at different stages, providing DOX-chow for various intervals, and assaying for Etv5 expression by ISH. After only 4 h of DOX starting at E9.5, Tg(tetO-dnFgfr2b)/+ embryos showed robust Etv5 expression in numerous sites of FGF signaling, including throughout most of the otic vesicle (Fig. 4C,C′). In contrast, experimental embryos retained expression of Etv5 at many sites, but lacked otic Etv5 (Fig. 4D,D′). In addition, 6 h of DOX starting at E8.25 nearly ablated Etv5 throughout experimental embryos, including in the otic cup (Fig. S4A-B′) and 4 h of DOX starting at E10.25 significantly downregulated Etv5 in the dorsolateral quadrant of the otic vesicle (Fig. S4C-D′). Thus, inhibition of FGFR2b ligands has a rapid onset in otic tissue, consistent with studies of dnFGFR2b induction in the limb (Danopoulos et al., 2013).
FGFR2b ligands are required continuously for otic morphogenesis
Next, we asked when FGFR2b ligands are required for otocyst morphogenesis. We started dnFGFR2b expression on different days of development and sustained the induction through E15.5, when inner ears were paintfilled. We compared Rosa26rtTA/+ control to experimental samples. E8.5-E15.5 DOX exposure completely inhibited inner ear development (n=6/6; data not shown). E9.5-E15.5 DOX exposure had no effect on controls (Fig. 4E), but experimental ears showed three distinct phenotypes: six had a small spheroid chamber similar to that of F3cKO;F10cKO samples (Fig. 2O′), ten had a structure resembling an EDS (Fig. 4F) and two resembled the majority of the E10.5-E15.5 group (Fig. 4G). DOX from E10.5-E15.5 also caused three phenotypic variants: five had a spheroid chamber, 13 had a vertical canal pouch, but no SSCs, a small saccule and utricle, and a dramatically shortened and narrowed CD (Fig. 4G), resembling the most strongly affected F10KO mutants (Urness et al., 2015), and two resembled the E11.5-E15.5 treatment group (Fig. 4H). DOX from E11.5-E15.5 caused consistent phenotypes: all ears had narrowed SSCs, reductions of the utricle and saccule, and a narrow and short CD (Fig. 4H). DOX from E12.5-E15.5 also caused consistent phenotypes: all ears had narrow SSCs, somewhat reduced utricles and saccules, and a narrow CD that nevertheless appeared normally elongated and coiled (Fig. 4I). Even experimental ears exposed to DOX from only E13.5-E15.5 had consistently narrow SCCs, but the rest of the inner ear appeared grossly normal (Fig. 4J). These data show that FGFR2b ligands are required continuously during otic morphogenesis.
Transient activation of dnFGFR2b reveals critical periods for FGFR2b ligands in otic morphogenesis
To determine intervals for FGFR2b ligand requirements in particular events of otic morphogenesis, we treated pregnant dams with different DOX pulses and examined E15.5 inner ears by paintfilling. As expected, all control ears, regardless of DOX exposure had normal morphology (Fig. 5A,E,G,K,M,Q). Experimental ears, however, showed exposure time-dependent abnormalities. A 4-h DOX exposure starting at 09.00 on E8.5 (termed E8.25) caused mild PSCC reductions in three ears, two of which also featured a truncated EDS and thickened CD (Fig. 5B), but had no effect on the remaining ears (not shown). 6-h or 24-h exposures had increasingly severe consequences. Most of the 6-h group and some of the 24-h group lacked an EDS (Fig. 5C′,D). The majority of 24-h exposures blocked most development of the otocyst, leaving a small vesicle (Fig. 5D′) or no ear tissue (not shown). By delaying DOX administration to 21.00 on E8.5 (called E8.75) all experimental samples with any ear tissue (n=17/32) showed at least an EDS and most of these (n=12/32) had a central (vestibular) segment and a linear CD (Fig. 5F,F′). A 2-h DOX pulse starting at 09.00 on E9.5 (called E9.25) had no effect on morphogenesis (data not shown), but 4- or 6-h exposures consistently caused only PSCC defects (Fig. 5H-I′). The 24-h exposures allowed EDS-like outgrowth, but consistently blocked most vestibular and cochlear outgrowth (Fig. 5J,J′). 12-h DOX exposures starting at 21.00 on E9.5 permitted EDS outgrowth and formation of at least a vertical canal pouch, but no SCC formation. In addition, the CD was short and narrow (n=14/16) or not present (n=2/16; Fig. 5L,L′). 4- or 6-h DOX exposures starting at 09.00 on E10.5 (termed E10.25) consistently blocked normal PSCC formation and the 6-h exposure sometimes affected the anterior semicircular canal (ASCC), but had virtually no effect on development of the utricle, saccule or CD (Fig. 5N-O′) until the exposure reached 24 h (Fig. 5P,P′). By starting DOX at 21.00 on E10.5 (termed E10.75), the most severe defects were avoided; nevertheless, the SCCs appeared thin, the utricle and saccule were reduced and the CD was short (Fig. 5R). In summary, we found that the earlier DOX was started and the longer it was present, the more severe were the morphogenesis defects. Furthermore, some of the DOX pulses gave such consistent outcomes that it seemed possible to identify acute epithelial transcriptional targets of FGFR2b ligands mediating particular morphogenetic events.
Fig. 5.

Transient activation of dnFGFR2b reveals critical periods for FGFR2b ligands in otic morphogenesis. (A-R) Paintfilled ears from control and experimental embryos with genotypes indicated above. DOX was provided to pregnant dams at E8.25 (A-D′), E8.75 (E-F′), E9.25 (G-J′), E9.75 (K-L′), E10.25 (M-P′) or E10.75 (Q,R) for the hours indicated above each panel. The number of ears from each treatment showing the same phenotype is indicated at the lower left. Alternate phenotypes are enumerated below. Scale bar in A applies to all panels. ac, anterior canal; cd, cochlear duct; eds, endolymphatic duct/sac; lc, lateral canal; pc, posterior canal; ut, utricle.
RNA-Seq reveals transcriptional targets of FGFR2b ligands during early otic morphogenesis
To identify transcriptional targets of FGFR2b ligands during early otocyst morphogenesis, when they are required for both vestibular and cochlear outgrowth, we chose three overlapping DOX exposures (Fig. 6A) that gave similar morphogenesis outcomes: E9.75+12 h (Seq1, Fig. 5L,L′), E10.25+24 h (Seq2, Fig. 5P,P′) and E9.25+24 h (Seq3, Fig. 5J,J′). Embryonic otocysts were microdissected, cleaned of mesenchyme (Fig. 6B), and pooled into control and experimental groups from each female. RNA was isolated, processed for RNA-Seq and analyzed for differential expression under both unpaired (genotype only) and paired (genotype and litter) statistical models.
Fig. 6.

Differential RNA-Seq reveals expected and novel targets of FGFR2b ligands during early otic morphogenesis and their requirement in otic epithelial proliferation. (A) Schematic of otocyst morphogenesis correlated with the three DOX exposures used for RNA-Seq. Red represents otic tissue, blue represents neural tissue. (B) Microdissected E10.25 otocyst. (C) RNA-Seq1 volcano plot showing significantly downregulated (green) and upregulated (red) genes. Significance is plotted on the y-axis and fold-change on the x-axis. Gene labels highlighted in yellow indicate fold-change >1.5, common FGF target genes, and genes pursued for ISH validation. (D,E) Transverse sections of E10.25 control and dnFgfr2b RNA-Seq1 otocysts hybridized with Etv5. (F,G) Transverse sections of E10.25 control and dnFgfr2b RNA-Seq3 otocysts immunostained for pHH3 (green) and E-cadherin (red). (H) Quantification of pHH3-positive cells per otic epithelial area (shown with mean and 95% c.i.) in RNA-Seq3 control and dnFgfr2 otocysts. Scale bars in D,F apply to E,G. og, otic ganglion; ov, otic vesicle.
Differentially expressed genes with an adjusted (adj) P<0.05 in each paired dataset were visualized with volcano plots (Fig. 6C; Fig. S5). In all three datasets, the maximum fold-changes were relatively modest, perhaps reflecting the short periods of inhibition, but for many genes the differences were highly significant. Fgfr2 and Ighg1 were among the most differentially expressed Seq1 genes (5.3-fold and 633-fold induced, respectively, but Ighg1 was omitted from the plot for legibility; Fig. 6C). Inspection of Fgfr2 reads showed upregulation was due to Fgfr2b splice isoform expression specifically in transgene-containing samples. Ighg1 reads were also transgene-encoded, thus validating the efficacy of the inductions. Excluding Fgfr2 and Ighg1, there were 968 genes >1.5-fold upregulated and 631 genes >1.5-fold downregulated (adjP<0.05) in experimental otocyst RNA. Significantly downregulated genes included well-known transcriptional targets of FGF signaling, Etv1, Etv4, Etv5, Dusp6, Spry2 and Spry1 (Fig. 6C; listed in Table S4, sheet 1). Similar results were obtained for RNA-Seq2 and RNA-Seq3 (Fig. S4; listed in Table S4, sheet 1). To validate an FGF target gene significantly downregulated in all three datasets, we detected Etv5 by ISH of otocyst sections. Seq1 control otocysts showed lateral and ventromedial Etv5 expression, whereas experimental embryos did not express otocyst Etv5 (Fig. 6D,E). Similar results were obtained with Seq3 samples (Fig. S6).
Fgfr1 sequence reads in each dataset showed no changes in level between control and experimental samples, which were similar to control levels of Fgfr2. Interestingly, Fgfr1c, considered mesenchymal (Pirvola et al., 2004), was the predominant splice isoform, but Fgfr1b was also detected. Fgfr3 sequences were present at levels at least 20-fold below those of Fgfr1 or Fgfr2 in control samples and were unchanged by dnFGFR2 induction (Table S4, sheet 1). The only FGFR2b ligand genes expressed at significant levels in control or experimental otocysts were Fgf3 and Fgf10 (Table S4, sheet 1), consistent with ISH surveys (Wright et al., 2003; data not shown). Interestingly, Fgf3 was slightly, but significantly, upregulated in all three datasets. However, as dnFGFR2b inhibition acts at the level of protein, this is unlikely to impact the phenotypes.
FGFR2b ligands promote otocyst cell proliferation
To explore functional relationships between significantly differentially expressed genes in each dataset (either up or downregulated; adjP<0.05), we used Ingenuity Pathway Analysis. In each case, the top 5 affected pathways included cell cycle and DNA damage/repair pathways [Table S4, sheet 2; −log(P-value)=8-13]. In most cases, these genes were downregulated in our datasets. In addition, we used GOrilla software to identify gene ontology terms for processes enriched in the downregulated Seq1 dataset (adjP<0.05), and the results were similar (top 5 shown in Table S4, sheet 2; FDR q-values 1.55×10−21-7.84×10−13). To assess proliferation directly, we quantified pHH3-positive cells per otic epithelial area in Seq3 otocyst sections and found that the mean labeling of experimental samples was reduced significantly to 57% of the control mean (Fig. 6F-H), showing that one role for FGFR2b ligands during E9.25-E10.25 is to control otic epithelial proliferation.
Signaling by FGFR2b ligands in the early otocyst represses genes that function later in otic epithelial development or hearing
Among the significantly upregulated genes in dnFgfr2b samples from Seq1, we noticed a gene required for hearing (Gjb6; Fig. 6C). Other such genes included Cldn14, Tmprss3, Pcdh15 and Gjb2 (Table S4, sheet 1). To determine whether additional such genes, or those responsible for mouse hearing loss and/or cochlear development, were enriched in either the up- or downregulated genes, we conducted a gene set enrichment analysis (Subramanian et al., 2005) on all 16,232 genes detected in the Seq1 paired analysis using two partially overlapping gene lists: 95 human hereditary hearing loss genes identified by Nishio et al. (2015; rank listed in Table S4, sheet 3) and 258 mouse genes involved in inner ear development or function collated by Ohlemiller et al. (2016; rank listed in Table S4, sheet 4). Both gene sets were highly enriched in the upregulated Seq1 dataset (normalized enrichment score=2.09 for the human genes and 2.06 for the mouse genes; both nominal P-values <1×10−3), but not in the downregulated set. Similar results were obtained with the Seq2 dataset (data not shown). Together, these analyses suggest that one role of FGFR2b ligands at this early stage is to prevent premature expression of epithelial genes that have later roles in development or function of the inner ear.
Validation of new genes regulated by FGFR2b ligands during the early stages of otocyst morphogenesis
To validate new FGFR2b target genes, we focused first on downregulated genes and assayed selected genes by ISH based on overlap in multiple RNA-Seq datasets (Table S4, sheet 5), relatively high degree of differential expression, and normalized read count above that of Fgf3 (Table S4, sheet 1), which is difficult to detect by ISH, and, finally, novelty with respect to inner ear development and/or FGF/MAPK signaling. Seven genes validated as downregulated in RNA-Seq1 otocysts are illustrated in Fig. 7. Spred3 was expressed in E10.25 control otocysts in a ventromedial domain, Six2 was detected in the ventral-most region as well as laterally, and Prdm1 (also known as Blimp1) was expressed similarly to Six2 (Fig. 7A-C). All three were strongly downregulated in the corresponding experimental otocysts (Fig. 7A′-C′). Crlf1 was also expressed similarly to Six2 in controls, but ventral expression was absent and lateral expression was downregulated in dnFGFR2b otocysts (Fig. 7D,D′). Tspan15 was expressed in a broad ventrolateral domain in controls, whereas Pou3f3 and Gchfr were expressed in the ventral-most region of controls (Fig. 7E-G) and each was absent from dnFGFR2b otocysts (Fig. 7E′-G′). We observed similar downregulation of Spred3, Prdm1, Crlf1, Tspan15 and Gchfr expression in dnFGFR2b otocysts subjected to the Seq2 and Seq3 DOX exposures (Figs S7 and S8). Six2 and Pou3f3 downregulation was confirmed by ISH in Seq3 otocysts (Fig. S8), but not tested in Seq2 otocysts, as these genes were not significantly affected in the Seq2 dataset.
Fig. 7.
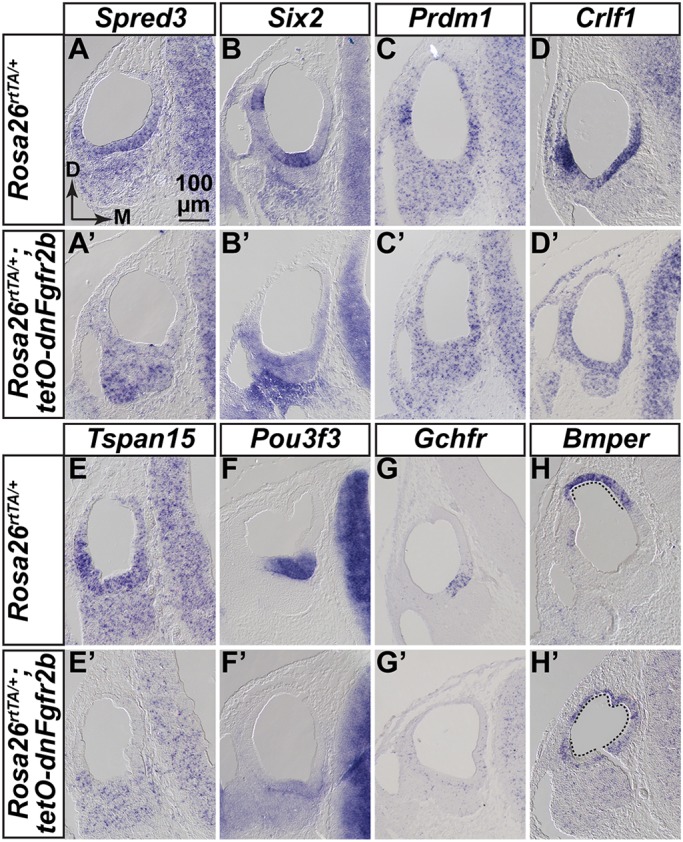
Validation of new genes regulated by FGFR2b ligands during early otocyst morphogenesis. (A-H′) ISH of transverse sections of E10.25 RNA-Seq1 control and dnFgfr2b otocysts (n=3 each). Probes are indicated above each column and genotypes are indicated to the left of each row. Scale and orientation for all panels is indicated in A. D, dorsal; M, medial.
We also tested several genes common to the upregulated lists, but most were widely expressed in controls and any changes in expression levels were not revealed by ISH (data not shown). However, Bmper transcripts were confined to the dorsomedial region of control otocysts, but the expression domain expanded to encompass most of the otic epithelium in experimental otocysts (Fig. 7H,H′). Similar results obtained with Seq2 and Seq3 otocysts (Figs S7 and S8). Therefore, these datasets are a rich source of novel FGFR2b signaling targets in the early otocyst.
DISCUSSION
Fgf3 and Fgf10 are expressed continuously throughout otocyst development
Fgf3 and Fgf10 are likely the only relevant FGFR2b (or FGFR1b) ligand-encoding genes for early otic morphogenesis as, based on RNA-Seq data, the others are either not expressed (Fgf22) or are detected at negligible levels (Fgf1 and Fgf7). We found that Fgf10 has an earlier and broader distribution than Fgf3 in the otic epithelium, and both transcripts are present in otic neuroblasts, but Fgf3 is seen only transiently, whereas once Fgf10 starts to be expressed, it is present continuously at high levels in the cochlear ganglion. Although Fgf10 is expressed in mesenchyme underlying preplacodal ectoderm, neither gene appears in periotic mesenchyme during otic cup formation or later (Schimmang, 2007; Urness et al., 2015; Wright and Mansour, 2003b). ISH data for Fgfr2b and Fgfr1b, which encode the receptors for FGF3 and FGF10, are limited because of the small size of probes that distinguish them from ‘c’ isoforms, but extant data are consistent with the idea that they are also primarily epithelial (Orr-Urtreger et al., 1993; Pirvola et al., 2000; Wright et al., 2003; Wright and Mansour, 2003a) and, indeed, the ‘b’ isoforms are evident in our RNA-Seq datasets. Therefore, taken together, the ligand and receptor expression data are consistent with our findings of continuous and combinatorial roles for Fgf3 and Fgf10 in otic development. They also raise the interesting possibility that signaling involves epithelial and/or ganglion ligands activating epithelial, rather than mesenchymal or ganglionic, receptors, except perhaps as neuroblasts begin delamination from the epithelium. This contrasts with the other epithelial ligands, FGF9 and FGF20, which signal canonically to mesenchymal ‘c’-type receptors, inducing signals that subsequently regulate epithelial proliferation (Huh et al., 2015).
Fgf3 and Fgf10 function continuously to promote vestibular and cochlear morphogenesis and maintenance of otic neuroblasts
Conditional mutant analyses showed that both Fgf3 and Fgf10 are required after otic placode induction, but deletion of Fgf3 alone from the Pax2-Cre lineage was inconsequential. This contrasts with the variably penetrant otic dysmorphologies of F3KO mutants, the most severe of which initiate with alterations of dorsal otocyst patterning, loss of the EDS, and subsequent cystic development of the epithelium, ultimately resulting in hearing loss and circling behavior (Hatch et al., 2007; Mansour et al., 1993). The normal phenotype of F3cKO ears suggests a crucial role for Fgf3 expression in the hindbrain. Indeed, we found that F10KO embryos in which only hindbrain sources of Fgf3 were deleted (using Sox1Cre) had very small otocysts. In contrast, F10cKO ears had abnormalities very similar to those of F10KO ears. This demonstrates that the unique functions of Fgf10 in otic morphogenesis arise from its expression in the placodal lineage rather than earlier in the mesenchyme. Analysis of conditional mutants that separate epithelial from ganglionic sites of Fgf10 expression will be needed to dissect further the spatial requirements for Fgf10 function.
Although Pax2-Cre is active in the placode at E8.5, we found no obvious effects at E9.5 on otocyst morphology in F3cKO;F10cKO embryos, and only two tested genes, Etv5 and Tbx1, were lost or altered in these otocysts. The first major losses in expression of multiple genes required for morphogenesis occurred at E10.5. This shows that both Fgf3 and Fgf10 are required in the placode lineage for normal otocyst morphogenesis and suggests that overlapping expression of Fgf3 and Fgf10 starting at E9.5 is crucial for both cochlear and vestibular outgrowth and morphogenesis. However, the otic phenotypes of embryos with three conditionally mutant alleles point to functional differences between Fgf3 and Fgf10. Both the cochlear and vestibular morphology of F3cKO;F10cHet ears was less severely affected than in F3cHet;F10cKO ears. This may reflect the larger domain and higher level of epithelial Fgf10 than of Fgf3, and may be presaged by the differential effects on Etv5 expression in the two types of E9.5 otocysts. The loss of dorsolateral Etv5 when Fgf10 is the only remaining allele, and of ventromedial Etv5 when Fgf3 is the only remaining allele, suggest that Fgf3 is particularly important dorsally and Fgf10 ventrally, at least initially. The loss of dorsolateral Tbx1 in the two most severely affected genotypes likely reflects effects of FGF3/FGF10 signaling on development of the vertical canal pouch, the derivatives of which (PSCC and ASCC) are strongly affected in these and in Tbx1 mutants (Freyer et al., 2013; Macchiarulo and Morrow, 2017). Whether this is a direct or indirect effect on Tbx1 expression is not yet clear, but it is interesting to note that Tbx1 is slightly, but significantly, downregulated in the Seq2 and Seq3 datasets (Table S4, sheet 1).
The presence of an EDS in embryos with both combinations of three conditionally mutant alleles and normal Gbx2 expression in these mutants and in F3cKO;F10cKO mutants contrasts with findings from F3KO mutants (Hatch et al., 2007), which usually lack an EDS and lose Gbx2 expression by E10.5. This is consistent with the idea that hindbrain, rather than epithelial, Fgf3 induces the EDS. It is possible that the further shortening of F3cHet;F10cKO CDs results from reduced FGF3-stimulated proliferation rather than alterations in molecular patterning. This is supported by preliminary analyses of E18.5 ears that did not reveal any exacerbation of changes to CD marker genes analyzed previously in the F10KO mutant (data not shown; see Urness et al., 2015). However, the timing of proliferative effects in F3cKO;F10cKO mutants must be later than E10.5, when differences in pHH3 labeling between control and F3cKO;F10cKO otocysts were not significant.
We suggested previously that Fgf3 plays a role in otic ganglion development, as the F3KO ganglion, like that of Fgfr2b null mutants (Pirvola et al., 2000), appeared smaller than normal (Mansour et al., 1993). In contrast, F10KO early otic ganglia and later cochlear ganglia appear normal (Urness et al., 2015) despite defects of vestibular innervation consequent to midgestation loss of vestibular sensory epithelia (Pauley et al., 2003). In contrast to zebrafish (Vemaraju et al., 2012), our present data from F3cKO;F10cKO mutants suggest that Fgf3 and Fgf10 are required together for maintenance of Neurog1 and Neurod1 expression, and development of an otic ganglion, rather than specification of otic neuroblasts. Our data do not address whether this requirement involves ligand expression in the epithelium or ganglion or both. However, by restricting dnFGFR2b expression to the placodal lineage by using Pax2-Cre with the unrecombined Rosa26lslrtTA allele, it may be possible to avoid disrupting otic induction and determine whether FGFR2b ligands play any role in mouse otic neuroblast specification. In addition, this paradigm of tissue-restricted and timed induction of dnFGFR2b could also be used to identify candidate genes responsible for neuroblast maintenance.
Temporally controlled inhibition of FGFR2b signaling during otocyst morphogenesis reveals requirements at multiple stages
Ubiquitous and sustained dnFGFR2b expression starting on different days of development revealed that FGFR2b ligands are required continuously for otic development at least until E13.5. Pulses of dnFGFR2b caused highly specific and penetrant otic malformations, supporting the idea that, unlike irreversible CRE-mediated deletion of coding exons, the signaling inhibition effected by dnFGFR2b is reversible.
Together, the sustained and pulsed inhibition paradigms suggest a distinct progression of roles for FGFR2b ligands: first in inducing the placode, then in stimulating EDS, and subsequently vestibular pouch and CD outgrowth, and finally in sculpting the SCCs, outgrowth of the utricle and saccule, and specification of CD non-sensory tissue. By comparison with genetic mutants, some phenotypes suggest the timing of roles for FGFR2b ligands. For example, the 6-24 h period starting at E8.25 proved important for EDS formation, potentially reflecting FGF3 inhibition (Mansour et al., 1993; Hatch et al., 2007) and the 6 h period starting at E9.25 was important for PSCC formation, potentially reflecting the earliest effects of FGF10 inhibition (Pauley et al., 2003; Urness et al., 2015). Finally, inhibition from E9.75+12 h and E10.25+24 h caused phenotypes remarkably similar to those of FGF3cHet;FGF10cKO ears, suggesting a critical interval requiring both FGF3 and FGF10 for SCC maturation and CD elongation. Some vestibular phenotypes are particularly interesting as they reveal potential, but previously unsuspected, functions for FGFR2b ligands. Sustained dnFGFR2b induction starting at E10.5, or 12-24 h pulses starting at E9.75 or E10.25, blocked fusion of vestibular pouches and reduced the utricle and saccule, suggesting requirements for FGFR2b ligands in fusion plate formation and growth of the utricle and saccule. Sustained induction starting E11.5-E13.5 or 12-h pulses starting E9.75-E10.75 caused very thin SCCs, suggesting roles for FGFR2b ligands in canal pouch outgrowth and/or in limiting resorption of fusion plates. Thus, it will be interesting to explore regulatory relationships between FGFR2b signaling and genes already known to regulate vestibular morphogenesis (Alsina and Whitfield, 2017), as well as to induce dnFGFR2b in particular temporal/spatial windows and pursue unbiased identification of effector genes involved in the development of structures of interest.
FGFR2b ligands promote otic epithelial proliferation and prevent premature expression of genes required for hearing
Our RNA-Seq datasets revealed significant downregulation of genes involved in the cell cycle and DNA repair, and, indeed, immunostaining of Seq3 samples showed that mitotic cell numbers were significantly reduced in E10.25 dnFGFR2b-containing otocysts. This result differed from that obtained with E10.5 F3cKO/F10cKO otocysts, which did not show a mitotic defect. Given that hindbrain Fgf3 is unaffected in Pax2-Cre;F3cKO/F10cKO mutants, and is extinguished by E10.5, it is likely that otocyst proliferation defects in these mutants would be detected at later stages of morphogenesis.
The RNA-Seq datasets also revealed significantly upregulated genes. We found that in Seq1 and Seq2, these genes are highly enriched for human hereditary hearing loss genes and mouse genes that are expressed and/or function later in the inner ear. These include Pax2, which was expanded in E10.5 F3cKO;F10cKO otocysts. This suggests that at early stages, FGFR2b signaling normally represses genes important for later development and function of the cochlea, or, alternatively, that the proliferative block imposed by dnFGFR2b expression promotes early differentiation of the epithelium. The latter possibility, however, does not apply to the earliest genes required for sensory cell differentiation (Atoh1, Pou4f3 and Gfi1), which were detected at very low levels and were unaffected in any of the RNA-Seq datasets (Table S4, sheet 1). We suggest that the upregulated gene sets are worth mining for new candidates for hearing loss genes, of which many remain to be identified (Bowl and Brown, 2018).
Although we show that FGFR2b ligands are required to activate Bmp4 and repress a BMP regulatory gene (Bmper), we found no evidence for FGFR2b ligand regulation of key downstream components of the BMP or SHH pathways, suggesting that although FGFR2b ligands may regulate individual components of these pathways, at least at the stages investigated, they are not exclusively upstream of these key programs directing dorsal and ventral otic morphogenesis, respectively.
Novel targets of FGFR2b signaling in early otocyst development
We validated by ISH seven novel genes downregulated and one gene upregulated by FGFR2b ligands in the RNA-seq datasets. Some genes may be regulated directly by the intracellular signaling pathway activated by FGFR2b, as one analysis point was only 12 h after induction. As we could not study more than a few differentially expressed genes, it is difficult to speculate about their combinatorial functions in inner ear development. Nevertheless, it is interesting to note that downregulated genes in our most robust dataset (Seq1) are enriched for transcription factor-coding genes (Table S4, sheet 2), including those validated here, Six2, Prdm1 and Pou3f3. The first two have otocyst expression patterns similar to Spred3, Crlf1 and Tspan15, whereas Pou3f3 expression appears to overlap with Gchfr. This suggests that further mining of the differential expression data and generation of additional targets by employing different windows of FGFR2b inhibition, combined with promoter analysis and genome-wide studies of otocyst chromatin modification could reveal important new gene regulatory networks acting to shape the epithelium.
The only upregulated gene validated by ISH was Bmper, which encodes BMP-binding endothelial regulator, an ortholog of Drosophila crossveinless 2 (cv-2) (Coffinier et al., 2002). Cv-2 and BMPER appear to modulate BMP signaling similarly by enhancing signaling when ligands are low and limiting signaling when ligands are high (Conley et al., 2000; Dyer et al., 2014; Kelley et al., 2009; Serpe et al., 2008). Thus, Bmper null mutants have some phenotypes suggestive of a classic BMP signaling antagonist (Moser et al., 2003) and others suggestive of a BMP signaling agonist (Ikeya et al., 2006). Multiple BMPs and their receptors are expressed in and required for otocyst morphogenesis (Chang et al., 2008; Hwang et al., 2010; Ohyama et al., 2010) and misexpression of BMP ventral to the otic placode blocks outgrowth of the chick cochlea (Ohta et al., 2016). Thus, it will be interesting to determine whether the otic phenotype of a Bmper null mutant reflects a loss or gain of BMP signaling, and whether this differs in different regions. Our results also showed that at early morphogenesis stages, Fgf3 and Fgf10 are required for Bmp4 expression, which is itself required for both vestibular and cochlear development (Chang et al., 2008). Determining whether FGFR2b ligand-dependent upregulation of Bmper functions in this context to further antagonize BMP signaling, or, alternatively, to mitigate Bmp4 reduction by increasing signaling by other BMP ligands will require additional studies of otic Bmp expression and manipulation of Bmper levels in combination with dnFGFR2b induction at different stages.
The identification of several FGFR2b target genes not implicated previously in ear development or hearing loss syndromes provides a tantalizing glimpse into a new set of potential otocyst morphogenetic factors. Given the novelty of these targets, it is tempting to speculate that previously unappreciated regulatory pathways may be at play during otic morphogenesis, as has been postulated for otic placode induction (Anwar et al., 2017). Functional studies will be required to address the roles of each of these new genes.
MATERIALS AND METHODS
Mouse models and genotyping
Mice were maintained and euthanized in accordance with protocols approved by the University of Utah Institutional Animal Care and Use Committee. All Fgf mutant alleles were kept on a mixed genetic background comprised of C57Bl/6 and various 129 substrains. CD-1 outbred mice (Charles River Laboratory) were used to generate embryos for studies of normal expression patterns and for generating embryos for induction of dnFGFR2b. Noon of the day a mating plug was observed was considered E0.5. Embryos were used without regard to sex.
Generation and PCR genotyping of the Fgf3 and Fgf10 null alleles (Fgf3−, formally designated Fgf3tm1.1Sms=MGI:3767558, and Fgf10−, formally designated Fgf10tm1.1Sms=MGI:3526181) and Fgf3 and Fgf10 and conditional alleles [Fgf3c (Fgf3tm1.2Sms=MGI:4456396) and Fgf10c (Fgf10tm1.2Sms=MGI:4456398)] were described previously (Hatch et al., 2007; Urness et al., 2010). Tg(Pax2-Cre) mice [Tg(Pax2-cre)1Akg=MGI:3046196] were obtained from Dr Andrew Groves (Ohyama and Groves, 2004). Tg(Pax2-Cre) was detected by PCR using primers specific to the transgene (5′-GGGGATCCCGACTACAAGG-3′; 5′-TAGTGAGTCGTATTAATTTCGATAAGC-3′). The Sox1Cre allele (Takashima et al., 2007) was transferred from Dr Mario Capecchi with permission from Dr Shin-Ichi Nishikawa (RIKEN, Japan) and genotyped using generic Cre primers. Rosa26lslLacZ reporter mice [Gt(ROSA)26Sortm1Sor=MGI:1861932] (Soriano, 1999) were maintained as homogyzotes.
Single conditional mutants were generated by crossing Fgf3c/c females to Fgf3−/+;Tg(Pax2-Cre)/+ males or Fgf10c/c females to Fgf10−/+;Tg(Pax2-Cre)/+ males. Combinations of Fgf3 and Fgf10 conditional mutants were obtained by crossing Fgf3c/c;Fgf10c/c females to Fgf3−/+;Fgf10−/+;Tg(Pax2-Cre)/+ or for hindbrain deletion of Fgf3 in the Fgf10 null background, Fgf3c/c;Fgf10−/+ females to Fgf3−/+;Fgf10−/+; Sox1Cre/+ males. CRE activity was confirmed by mating Cre-bearing males to Rosa26LacZR/LacZR females, harvesting embryos at the indicated stages and staining with X-gal as described (Yang and Mansour, 1999).
The germline-recombined Rosa26rtTA allele [derived from Gt(ROSA)26Sortm1(rtTA,EGFP)Nagy; MGI:3583817] (Belteki et al., 2005; Parsa et al., 2008) and the Tg(tetO-dnFgfr2b) allele [Tg(tetO-Fgfr2b/Igh1.3Jaw; MGI 5582625] (Hokuto et al., 2003) were transferred from Dr Saverio Bellusci with permission from Dr Jeffery Whitsett (Cincinnati Children's Medical Center, OH, USA). Genotyping primers to detect Tg(tetO-dnFgfr2b) were 5′-CAGGCCAACCAGTCTGCCTGGC-3′ and 5′-CGTCTGAGCTGTGTGCACCTCC-3′. ROSA26rtTA genotyping primers were ROSA5 (5′-GAGTTCTCTGCTGCCTCCTG-3′) and ROSA3 (5′-CGAGGCGGATCACAAGCAATA-3′), which generate a wild-type band of 322 bp and ROSA5 and RTTA3 (5′-AAGACCGCGAAGAGTTTGTC-3′), which generate a 215 bp rtTA-specific product. Rosa26rtTA/+;Tg(tetO-dnFgfr2b)/+ (experimental) embryos were obtained initially by crossing Rosa26rtTA/+ to Tg(tetO-dnFgfr2b)/+. For most studies described here, we crossed wild-type CD-1 females to Rosa26rtTA/rtTA;Tg(tetO-dnFgfr2b)/+ males, generating 50% each of control and experimental genotypes.
RNA in situ hybridization
Embryos were harvested and fixed in 4% paraformaldehyde and stored in methanol at −20°C. RNA ISH to whole-mount embryos or paraffin-embedded sections were performed as described (Urness et al., 2008, 2010). Probes for Sox9, Fgf3, Fgf10, Bmp4, Etv5, Gbx2, Sox2, Foxg1, Pax2, Neurod1, Neurog1 and Crlf1 were generated by transcription of cDNA-containing plasmids. Template plasmids and acknowledgements are shown in Table S2. All other RNA probes were generated by transcription of a PCR-amplified, gene-specific 3′ UTR fragment containing a T7 promoter. The primer sequences are shown in Table S3. Each panel represents the results from three independent hybridizations. Whole embryos were photographed using a stereomicroscope (Zeiss Discovery.V12) fitted with a digital camera (QImaging Micropublisher 5.0). Hybridized tissue sections were photographed under DIC illumination (Zeiss Axioskop) using a digital camera (Zeiss Axiovision or Lumenera Infinity3).
Immunostaining of frozen tissue sections for quantification of mitotic cells in the otocyst
Embryos were fixed in 4% paraformaldehyde solution and cryosectioned in the transverse plane for immunostaining as described (Urness et al., 2015). Rabbit anti-phosphohistone H3 (Millipore, 06-570) was applied at a dilution of 1:400 and mouse monoclonal anti-E-cadherin (cadherin 1) (BD Biosciences, 610181) was diluted 1:60. Rabbit anti-cleaved caspase 3 (Cell Signaling, 9661) was used at a dilution of 1:100. Secondary antibodies were from Invitrogen and diluted 1:400 into phosphate-buffered saline/0.1% Triton X-100/5% normal serum [Alexa Fluor 488 goat anti-rabbit (A11034) and Alexa Fluor 594 goat anti-mouse (A11032)]. DAPI was included in the mounting medium (Vectashield, Vector Labs). Fluorescent signals were observed under epi-illumination on a Zeiss Axioskop and captured using an Infinity3 camera (Lumenera) driven by InfinityAnalyze software. Channels were overlaid using Photoshop CS5. All pHH3-positive cells in the otocysts (defined by E-cadherin staining) were counted from 6 µm (Pax2-Cre cross) or 8 µm (dnFgfr2b cross) sections extending from anterior to posterior by an individual blinded to genotype. n=8 control (either Fgf3−/c;Fgf10−/c or Fgf3c/+;Fgf10c/+;Pax2-Cre/+) and n=6 experimental (Fgf3−/c;Fgf10−/c;Pax2-Cre/+) samples were counted for the Fgf3/Fgf10/Pax2-Cre conditional cross; n=6 control (Rosa26rtTA/+) and n=6 experimental (Rosa26rtTA/+;tetO-dnFgfR2b/+) samples for the dnFGFR2b cross. pHH3-positive cells per ear were normalized to the cross-sectional area counted. Statistical significance was determined using an unpaired Student's t-test (Prism software 7.0) with Welch's correction.
Paintfilling of embryonic inner ears
Filling of embryonic inner ears with latex paint and photography was described previously (Urness et al., 2015). The number of ears with the illustrated phenotype/total ears of the same genotype is shown on each panel.
Induction of dnFGFR2b expression
Initial inductions of dnFGFR2b designed to phenocopy Fgf3/Fgf10 double mutants were achieved by feeding pregnant females DOX-chow (200 mg/kg, Custom Animal Diets, LLC) ad libitum for the indicated time periods (E5.5-E10.5, E5.5-E11.5 or E6.5-E11.5). All subsequent inductions to generate samples for paintfilling, RNA-Seq, ISH or immunostaining were initiated by a single intraperitoneal injection of the pregnant dam with 0.1 ml/10 g body weight of 0.15 mg/ml (1.5 mg/kg body weight) doxycycline hyclate (Sigma-Aldrich) prepared in PBS followed by provision of DOX-chow ad libitum for the indicated time periods. We avoided using female Rosa26rtTA parents, as these seemed to require larger and variable amounts of DOX to see phenotypes than when rtTA was contributed by the male parent, presumably because the widespread, ubiquitous expression of rtTA in females served to sequester DOX. We did not measure the time needed to reactivate signaling after DOX withdrawal, but based on studies of the limb (Danopoulos et al., 2013) we expect that signaling resumes after 12-24 h.
Otic vesicle preparation and RNA isolation
Embryos from timed matings of CD-1 females and Rosa26rtTA/rtTA;Tg(tetO-(s)dnFgfr2b)/+ males, with DOX exposures as specified, were dissected and the yolk sacs saved for genotyping. The otic vesicles, including surrounding mesenchyme, were crudely dissected from the head. Isolation of the vesicles free of mesenchyme was accomplished in a similar manner to methods previously described (Urness et al., 2010) with the following modifications. Otocysts with adherent mesenchyme were incubated in 50 µl ice-cold PT solution [25 mg/ml pancreatin (Sigma), 5 mg/ml trypsin (Sigma) and 5 mg/ml polyvinylpyrrolidone MW360 (Sigma) in Tyrode's solution] for ∼7 min (E10.25) or ∼8 min (E11.25) to promote separation of the mesenchyme. Otocysts were aspirated to Hepes-DMEM-10% fetal bovine serum, so that the digested mesenchyme could be gently teased from the underlying epithelium using fine forceps or tungsten needles, and by ‘rolling’ the vesicle over the bottom of the dish to detach the mesenchyme as it adhered to the plastic. The two otocysts from each embryo were aspirated into 100 µl RNALater (Ambion) and stored at −20°C prior to genotyping. For each of four pregnant females per DOX induction regime, all otocysts of the same genotype were combined into paired control (Rosa26rtTA/+) and experimental [Rosa26rtTA/+;Tg(tetO-(s)dnFgfr2b)/+] pools (n=6-12 otocysts/pool).
Total RNA from each control and experimental otocyst pool was prepared using a Micro RNAeasy kit (Qiagen, 74004) and analyzed for quantity and quality on a BioAnalyzer RNA TapeStation. All 24 samples (2 genotypes×4 females×3 DOX exposures) exceeded a RIN quality control number of 8.
RNA-Seq and bioinformatics
RNA library preparation, sequencing and analyses were conducted by the University of Utah/Huntsman Cancer Institute High-Throughput Genomics and Bioinformatic Analysis Shared Resource. Each RNA library was prepared using a TruSeq Stranded mRNA Sample Prep kit (Illumina) with oligo(dT) selection. 50-cycle single-read sequencing of each library was conducted on an Illumina Hi-Seq 2500. Sequencing reads were aligned to mm10+splice junctions (Ensembl build 74) using Novoalign (v2.08.03). Spliced alignments were converted back to genomic space, sorted and indexed using USeq (v8.8.8) SamTranscriptomeParser. Normalized coverage tracks (coverage per million mapped reads) were generated using USeq Sam2USeq and USeq2UCSCExe. Read counts for each gene were generated using USeq DefinedRegionDifferentialSeq (Nix et al., 2008) and differential expression analysis was performed using DESeq2 (Love et al., 2014) with pairing of samples from the same litters. As Tg(tetO-dnFgfr2b) was generated from a construct in which sequences encoding the FGFR2b extracellular domain were fused to sequences encoding the hinge and Fc regions of mouse IGHG1 (Hokuto et al., 2003), elevated levels of both transgene-encoded sequences are diagnostic of successful induction in experimental tissues.
To inspect Fgfr splicing, we merged each set of control and dnFgfr2b alignments to separate .bam files, uploaded them to IGV 2.4.10 (Robinson et al., 2011; Thorvaldsdottir et al., 2013) and generated Sashimi plots. To identify significantly regulated pathways (P<0.05, Fisher's Exact Test), all differentially expressed genes were loaded into Ingenuity Pathway Analysis (Qiagen, https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). For gene set enrichment analysis (GSEA), two custom gene sets based on human hearing loss genes from Nishio et al. (2015) and mouse inner ear genes from Ohlemiller et al. (2016) were loaded into the Broad Institute GSEA website (Subramanian et al., 2005) and compared with ranked lists of otocyst genes sorted by fold-change from DESeq2.
Supplementary Material
Acknowledgements
We thank Katia Hatch for auditory testing of F3cKO mice, Leslie Slota for unpublished work on marker gene expression in F3cHet;F10cKO cochleae, Saverio Bellusci and Denise Al-Alam for transferring Rosa26rtTA and Tg(tetO-dnFgfr2b) mice, Mario Capecchi for transferring Sox1Cre mice, Tim Mosbrugger and Chris Stubben for bioinformatics support, Shannon Odelberg for help with statistics and Gary Schoenwolf for experimental and editorial advice.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: L.D.U., S.L.M.; Formal analysis: L.D.U., H.D., S.L.M.; Investigation: L.D.U., X.W., H.D., N.S., C.A.N., E.G.-M., R.L., S.L.M.; Writing - original draft: L.D.U., S.L.M.; Writing - review & editing: L.D.U., H.D., S.L.M.; Visualization: L.D.U., X.W., H.D., N.S., C.A.N., E.G.-M., R.L., S.L.M.; Supervision: S.L.M.; Project administration: S.L.M.; Funding acquisition: S.L.M.
Funding
This work was funded by grants from the National Institutes of Health (R01 DC011819 and R01 DC004185 to S.L.M.) and a National Science Foundation-funded SDB CHOOSE Development! Fellowship (IOS-1239422 to E.G.-M.). Deposited in PMC for release after 12 months.
Data availability
The three RNA-seq datasets are deposited in Gene Expression Omnibus under accession number GSE116404.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.170142.supplemental
References
- Al Alam D., El Agha E., Sakurai R., Kheirollahi V., Moiseenko A., Danopoulos S., Shrestha A., Schmoldt C., Quantius J., Herold S. et al. (2015). Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development 142, 4139-4150. 10.1242/dev.109173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsina B. and Whitfield T. T. (2017). Sculpting the labyrinth: morphogenesis of the developing inner ear. Semin. Cell Dev. Biol. 65, 47-59. 10.1016/j.semcdb.2016.09.015 [DOI] [PubMed] [Google Scholar]
- Alvarez Y., Alonso M. T., Vendrell V., Zelarayan L. C., Chamero P., Theil T., Bösl M. R., Kato S., Maconochie M., Riethmacher D. et al. (2003). Requirements for FGF3 and FGF10 during inner ear formation. Development 130, 6329-6338. 10.1242/dev.00881 [DOI] [PubMed] [Google Scholar]
- Anwar M., Tambalo M., Ranganathan R., Grocott T. and Streit A. (2017). A gene network regulated by FGF signalling during ear development. Sci. Rep. 7, 6162 10.1038/s41598-017-05472-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basch M. L., Brown R. M. II, Jen H.-I. and Groves A. K. (2016). Where hearing starts: the development of the mammalian cochlea. J. Anat. 228, 233-254. 10.1111/joa.12314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belteki G., Haigh J., Kabacs N., Haigh K., Sison K., Costantini F., Whitsett J., Quaggin S. E. and Nagy A. (2005). Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 33, e51 10.1093/nar/gni051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowl M. R. and Brown S. D. M. (2018). Genetic landscape of auditory dysfunction. Hum. Mol. Genet. 27, R130-R135. 10.1093/hmg/ddy158 [DOI] [PubMed] [Google Scholar]
- Chang W., Brigande J. V., Fekete D. M. and Wu D. K. (2004). The development of semicircular canals in the inner ear: role of FGFs in sensory cristae. Development 131, 4201-4211. 10.1242/dev.01292 [DOI] [PubMed] [Google Scholar]
- Chang W., Lin Z., Kulessa H., Hebert J., Hogan B. L. M. and Wu D. K. (2008). Bmp4 is essential for the formation of the vestibular apparatus that detects angular head movements. PLoS Genet. 4, e1000050 10.1371/journal.pgen.1000050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi C. L., Martinez S., Wurst W. and Martin G. R. (2003). The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development 130, 2633-2644. 10.1242/dev.00487 [DOI] [PubMed] [Google Scholar]
- Coffinier C., Ketpura N., Tran U., Geissert D. and De Robertis E. M. (2002). Mouse Crossveinless-2 is the vertebrate homolog of a Drosophila extracellular regulator of BMP signaling. Mech. Dev. 119 Suppl. 1, S179-S184. 10.1016/S0925-4773(03)00113-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley C. A., Silburn R., Singer M. A., Ralston A., Rohwer-Nutter D., Olson D. J., Gelbart W. and Blair S. S. (2000). Crossveinless 2 contains cysteine-rich domains and is required for high levels of BMP-like activity during the formation of the cross veins in Drosophila. Development 127, 3947-3959. [DOI] [PubMed] [Google Scholar]
- Danopoulos S., Parsa S., Al Alam D., Tabatabai R., Baptista S., Tiozzo C., Carraro G., Wheeler M., Barreto G., Braun T. et al. (2013). Transient Inhibition of FGFR2b-ligands signaling leads to irreversible loss of cellular beta-catenin organization and signaling in AER during mouse limb development. PLoS ONE 8, e76248 10.1371/journal.pone.0076248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer L., Wu Y., Moser M. and Patterson C. (2014). BMPER-induced BMP signaling promotes coronary artery remodeling. Dev. Biol. 386, 385-394. 10.1016/j.ydbio.2013.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyer L., Nowotschin S., Pirity M. K., Baldini A. and Morrow B. E. (2013). Conditional and constitutive expression of a Tbx1-GFP fusion protein in mice. BMC Dev. Biol. 13, 33 10.1186/1471-213X-13-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch E. P., Noyes C. A., Wang X., Wright T. J. and Mansour S. L. (2007). Fgf3 is required for dorsal patterning and morphogenesis of the inner ear epithelium. Development 134, 3615-3625. 10.1242/dev.006627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokuto I., Perl A.-K. T. and Whitsett J. A. (2003). Prenatal, but not postnatal, inhibition of fibroblast growth factor receptor signaling causes emphysema. J. Biol. Chem. 278, 415-421. 10.1074/jbc.M208328200 [DOI] [PubMed] [Google Scholar]
- Huh S.-H., Warchol M. E. and Ornitz D. M. (2015). Cochlear progenitor number is controlled through mesenchymal FGF receptor signaling. eLife 4, e05921 10.7554/eLife.05921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang C. H., Guo D., Harris M. A., Howard O., Mishina Y., Gan L., Harris S. E. and Wu D. K. (2010). Role of bone morphogenetic proteins on cochlear hair cell formation: analyses of Noggin and Bmp2 mutant mice. Dev. Dyn. 239, 505-513. 10.1002/dvdy.22200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeya M., Kawada M., Kiyonari H., Sasai N., Nakao K., Furuta Y. and Sasai Y. (2006). Essential pro-Bmp roles of crossveinless 2 in mouse organogenesis. Development 133, 4463-4473. 10.1242/dev.02647 [DOI] [PubMed] [Google Scholar]
- Isaiah A., Lee D., Lenes-Voit F., Sweeney M., Kutz W., Isaacson B., Roland P. and Lee K. H. (2017). Clinical outcomes following cochlear implantation in children with inner ear anomalies. Int. J. Pediatr. Otorhinolaryngol. 93, 1-6. 10.1016/j.ijporl.2016.12.001 [DOI] [PubMed] [Google Scholar]
- Kelley R., Ren R., Pi X., Wu Y., Moreno I., Willis M., Moser M., Ross M., Podkowa M., Attisano L. et al. (2009). A concentration-dependent endocytic trap and sink mechanism converts Bmper from an activator to an inhibitor of Bmp signaling. J. Cell Biol. 184, 597-609. 10.1083/jcb.200808064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y., Masuda T. and Kaga K. (2018). Vestibular function and gross motor development in 195 children with congenital hearing loss-assessment of inner ear malformations. Otol. Neurotol. 39, 196-205. 10.1097/MAO.0000000000001685 [DOI] [PubMed] [Google Scholar]
- Ladher R. K. (2017). Changing shape and shaping change: inducing the inner ear. Semin. Cell Dev. Biol. 65, 39-46. 10.1016/j.semcdb.2016.10.006 [DOI] [PubMed] [Google Scholar]
- Ladher R. K., Wright T. J., Moon A. M., Mansour S. L. and Schoenwolf G. C. (2005). FGF8 initiates inner ear induction in chick and mouse. Genes Dev. 19, 603-613. 10.1101/gad.1273605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z., Cantos R., Patente M. and Wu D. K. (2005). Gbx2 is required for the morphogenesis of the mouse inner ear: a downstream candidate of hindbrain signaling. Development 132, 2309-2318. 10.1242/dev.01804 [DOI] [PubMed] [Google Scholar]
- Love M. I., Huber W. and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchiarulo S. and Morrow B. E. (2017). Tbx1 and Jag1 act in concert to modulate the fate of neurosensory cells of the mouse otic vesicle. Biol. Open 6, 1472-1482. 10.1242/bio.027359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour S. L., Goddard J. M. and Capecchi M. R. (1993). Mice homozygous for a targeted disruption of the proto-oncogene int-2 have developmental defects in the tail and inner ear. Development 117, 13-28. [DOI] [PubMed] [Google Scholar]
- Morsli H., Choo D., Ryan A., Johnson R. and Wu D. K. (1998). Development of the mouse inner ear and origin of its sensory organs. J. Neurosci. 18, 3327-3335. 10.1523/JNEUROSCI.18-09-03327.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser M., Binder O., Wu Y., Aitsebaomo J., Ren R., Bode C., Bautch V. L., Conlon F. L. and Patterson C. (2003). BMPER, a novel endothelial cell precursor-derived protein, antagonizes bone morphogenetic protein signaling and endothelial cell differentiation. Mol. Cell. Biol. 23, 5664-5679. 10.1128/MCB.23.16.5664-5679.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio S. Y., Hattori M., Moteki H., Tsukada K., Miyagawa M., Naito T., Yoshimura H., Iwasa Y., Mori K., Shima Y. et al. (2015). Gene expression profiles of the cochlea and vestibular endorgans: localization and function of genes causing deafness. Ann. Otol. Rhinol. Laryngol. 124 Suppl. 1, 6s-48s. 10.1177/0003489415575549 [DOI] [PubMed] [Google Scholar]
- Nix D. A., Courdy S. J. and Boucher K. M. (2008). Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics 9, 523 10.1186/1471-2105-9-523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller K. K., Jones S. M. and Johnson K. R. (2016). Application of Mouse Models to Research in Hearing and Balance. J. Assoc. Res. Otolaryngol. 17, 493-523. 10.1007/s10162-016-0589-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S. and Schoenwolf G. C. (2018). Hearing crosstalk: the molecular conversation orchestrating inner ear dorsoventral patterning. Wiley Interdiscip. Rev. Dev. Biol. 7, e302 10.1002/wdev.302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S., Wang B., Mansour S. L. and Schoenwolf G. C. (2016). BMP regulates regional gene expression in the dorsal otocyst through canonical and non-canonical intracellular pathways. Development 143, 2228-2237. 10.1242/dev.137133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama T. and Groves A. K. (2004). Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis 38, 195-199. 10.1002/gene.20017 [DOI] [PubMed] [Google Scholar]
- Ohyama T., Basch M. L., Mishina Y., Lyons K. M., Segil N. and Groves A. K. (2010). BMP signaling is necessary for patterning the sensory and nonsensory regions of the developing mammalian cochlea. J. Neurosci. 30, 15044-15051. 10.1523/JNEUROSCI.3547-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A., Bedford M. T., Burakova T., Arman E., Zimmer Y., Yayon A., Givol D. and Lonai P. (1993). Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev. Biol. 158, 475-486. 10.1006/dbio.1993.1205 [DOI] [PubMed] [Google Scholar]
- Parsa S., Ramasamy S. K., De Langhe S., Gupte V. V., Haigh J. J., Medina D. and Bellusci S. (2008). Terminal end bud maintenance in mammary gland is dependent upon FGFR2b signaling. Dev. Biol. 317, 121-131. 10.1016/j.ydbio.2008.02.014 [DOI] [PubMed] [Google Scholar]
- Parsa S., Kuremoto K., Seidel K., Tabatabai R., Mackenzie B., Yamaza T., Akiyama K., Branch J., Koh C. J., Al Alam D. et al. (2010). Signaling by FGFR2b controls the regenerative capacity of adult mouse incisors. Development 137, 3743-3752. 10.1242/dev.051672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauley S., Wright T. J., Pirvola U., Ornitz D., Beisel K. and Fritzsch B. (2003). Expression and function of FGF10 in mammalian inner ear development. Dev. Dyn. 227, 203-215. 10.1002/dvdy.10297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U., Spencer-Dene B., Xing-Qun L., Kettunen P., Thesleff I., Fritzsch B., Dickson C. and Ylikoski J. (2000). FGF/FGFR-2(IIIb) signaling is essential for inner ear morphogenesis. J. Neurosci. 20, 6125-6134. 10.1523/JNEUROSCI.20-16-06125.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U., Zhang X., Mantela J., Ornitz D. M. and Ylikoski J. (2004). Fgf9 signaling regulates inner ear morphogenesis through epithelial-mesenchymal interactions. Dev. Biol. 273, 350-360. 10.1016/j.ydbio.2004.06.010 [DOI] [PubMed] [Google Scholar]
- Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G. and Mesirov J. P. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24-26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajan S. A., Warchol M. E. and Lovett M. (2007). Toward a systems biology of mouse inner ear organogenesis: gene expression pathways, patterns and network analysis. Genetics 177, 631-653. 10.1534/genetics.107.078584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T. and Joyner A. L. (2009). The duration of Fgf8 isthmic organizer expression is key to patterning different tectal-isthmo-cerebellum structures. Development 136, 3617-3626. 10.1242/dev.041210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmang T. (2007). Expression and functions of FGF ligands during early otic development. Int. J. Dev. Biol. 51, 473-481. 10.1387/ijdb.072334ts [DOI] [PubMed] [Google Scholar]
- Sennaroğlu L. and Bajin M. D. (2017). Classification and current management of inner ear malformations. Balkan. Med. J. 34, 397-411. 10.4274/balkanmedj.2017.0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe M., Umulis D., Ralston A., Chen J., Olson D. J., Avanesov A., Othmer H., O'Connor M. B. and Blair S. S. (2008). The BMP-binding protein Crossveinless 2 is a short-range, concentration-dependent, biphasic modulator of BMP signaling in Drosophila. Dev. Cell 14, 940-953. 10.1016/j.devcel.2008.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S. et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545-15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y., Era T., Nakao K., Kondo S., Kasuga M., Smith A. G. and Nishikawa S.-I. (2007). Neuroepithelial cells supply an initial transient wave of MSC differentiation. Cell 129, 1377-1388. 10.1016/j.cell.2007.04.028 [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir H., Robinson J. T. and Mesirov J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178-192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urness L. D., Li C., Wang X. and Mansour S. L. (2008). Expression of ERK signaling inhibitors Dusp6, Dusp7, and Dusp9 during mouse ear development. Dev. Dyn. 237, 163-169. 10.1002/dvdy.21380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urness L. D., Paxton C. N., Wang X., Schoenwolf G. C. and Mansour S. L. (2010). FGF signaling regulates otic placode induction and refinement by controlling both ectodermal target genes and hindbrain Wnt8a. Dev. Biol. 340, 595-604. 10.1016/j.ydbio.2010.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urness L. D., Wang X., Shibata S., Ohyama T. and Mansour S. L. (2015). Fgf10 is required for specification of non-sensory regions of the cochlear epithelium. Dev. Biol. 400, 59-71. 10.1016/j.ydbio.2015.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemaraju S., Kantarci H., Padanad M. S. and Riley B. B. (2012). A spatial and temporal gradient of Fgf differentially regulates distinct stages of neural development in the zebrafish inner ear. PLoS Genet. 8, e1003068 10.1371/journal.pgen.1003068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield T. T. (2015). Development of the inner ear. Curr. Opin. Genet. Dev. 32, 112-118. 10.1016/j.gde.2015.02.006 [DOI] [PubMed] [Google Scholar]
- Wright T. J. and Mansour S. L. (2003a). Fgf3 and Fgf10 are required for mouse otic placode induction. Development 130, 3379-3390. 10.1242/dev.00555 [DOI] [PubMed] [Google Scholar]
- Wright T. J. and Mansour S. L. (2003b). FGF signaling in ear development and innervation. Curr. Top. Dev. Biol. 57, 225-259. 10.1016/S0070-2153(03)57008-9 [DOI] [PubMed] [Google Scholar]
- Wright T. J., Hatch E. P., Karabagli H., Karabagli P., Schoenwolf G. C. and Mansour S. L. (2003). Expression of mouse fibroblast growth factor and fibroblast growth factor receptor genes during early inner ear development. Dev. Dyn. 228, 267-272. 10.1002/dvdy.10362 [DOI] [PubMed] [Google Scholar]
- Wu D. K. and Kelley M. W. (2012). Molecular mechanisms of inner ear development. Cold Spring Harb. Perspect. Biol. 4, a008409 10.1101/cshperspect.a008409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Liu Z. and Ornitz D. M. (2000). Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development 127, 1833-1843. [DOI] [PubMed] [Google Scholar]
- Yang W. and Mansour S. L. (1999). Expression and genetic analysis of prtb, a gene that encodes a highly conserved proline-rich protein expressed in the brain. Dev. Dyn. 215, 108-116. [DOI] [PubMed] [Google Scholar]
- Zelarayan L. C., Vendrell V., Alvarez Y., Domínguez-Frutos E., Theil T., Alonso M. T., Maconochie M. and Schimmang T. (2007). Differential requirements for FGF3, FGF8 and FGF10 during inner ear development. Dev. Biol. 308, 379-391. 10.1016/j.ydbio.2007.05.033 [DOI] [PubMed] [Google Scholar]
- Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M. and Ornitz D. M. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 281, 15694-15700. 10.1074/jbc.M601252200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.