

Abstract
Adoptive transfer of T cell receptor–engineered (TCR-engineered) T cells is a promising approach in cancer therapy but needs improvement for more effective treatment of solid tumors. While most clinical approaches have focused on CD8+ T cells, the importance of CD4+ T cells in mediating tumor regression has become apparent. Regarding shared (self) tumor antigens, it is unclear whether the human CD4+ T cell repertoire has been shaped by tolerance mechanisms and lacks highly functional TCRs suitable for therapy. Here, TCRs against the tumor-associated antigen NY-ESO-1 were isolated either from human CD4+ T cells or from mice that express a diverse human TCR repertoire with HLA-DRA/DRB1*0401 restriction and are NY-ESO-1 negative. NY-ESO-1–reactive TCRs from the mice showed superior recognition of tumor cells and higher functional activity compared with TCRs from humans. We identified a candidate TCR, TCR-3598_2, which was expressed in CD4+ T cells and caused tumor regression in combination with NY-ESO-1–redirected CD8+ T cells in a mouse model of adoptive T cell therapy. These data suggest that MHC II–restricted TCRs against NY-ESO-1 from a nontolerant nonhuman host are of optimal affinity and that the combined use of MHC I– and II–restricted TCRs against NY-ESO-1 can make adoptive T cell therapy more effective.
Keywords: Immunology, Oncology
Keywords: Cancer immunotherapy, T cell development, T-cell receptor
Introduction
The adoptive transfer of T cell receptor–engineered (TCR-engineered) tumor-reactive T cells is a promising approach to treat solid tumors. One crucial factor for successful therapy is the affinity of the therapeutic TCR to its cognate peptide-MHC molecule on the tumor. However, isolating highly functional TCRs against shared (self) tumor antigens can be difficult because tolerance mechanisms during T cell development may lead to their deletion in the host if the antigen is expressed in the thymus (1, 2). To overcome this limitation, we generated a mouse model in which T cells rearrange a diverse human TCR repertoire but because the mice lack human tumor antigens, they are not subject to the mechanisms of tolerance to such antigens (3, 4). Isolated HLA-A*02:01–restricted (HLA-A2–restricted) TCRs from this mouse model against tumor antigens MAGE-A1 and New York esophageal squamous cell carcinoma 1 (NY-ESO-1) showed higher functional activity compared with TCRs against the same epitopes from the human repertoire (3). Hence, we defined an optimal-affinity TCR operationally as a TCR that recognizes MHC as self and the peptide as foreign, the same way protective pathogen-specific TCRs recognize target cells.
NY-ESO-1 is a tumor-associated antigen that is expressed in a variety of cancers including melanoma but has not been detected in normal tissues except for testis, placenta, and thymus (2, 5). The expression of NY-ESO-1 in medullary thymic epithelial cells may lead to deletion of high-avidity T cells and could be one of the reasons why immunization strategies had little clinical success (6, 7). Likewise, it can be hypothesized that occasionally observed spontaneous anti–NY-ESO-1 IgG antibodies and CD4+ T cell responses in cancer patients are of low avidity and therefore do not lead to clinical benefit (8–10). NY-ESO-1 has been targeted by adoptive T cell therapy (ATT) employing an affinity-matured HLA-A2–restricted TCR (11, 12). These trials showed objective clinical responses but were not as successful as ATT for hematological malignancies targeting CD19 (13). One underlying reason may be that solid tumors require CD4+ T cell help via MHC class II (MHC II) to initiate a full immune response able to achieve major clinical effects. NY-ESO-1 may serve as a suitable target for CD4+ and CD8+ T cells in combination bearing optimal-affinity TCRs.
Although clinical studies have focused on CD8+ T cells, the importance of CD4+ T cells for antitumor immunity has long been known. CD4+ T cells not only exert helper function to support CD8+ T cell responses, they also show tumoricidal activity by their own means irrespective of MHC II expression on the cancer cells (14–17). Combining the potential of CD4+ and CD8+ T cells for ATT can induce bystander killing of antigen-negative tumor cells (18). A combined CD4+ and CD8+ T cell therapy may lead to more effective immune responses against the target, thereby avoiding selection of antigen-negative variants, as has been seen in a previous trial targeting NY-ESO-1 (11).
In this study, we raised MHC II–restricted TCRs against NY-ESO-1 in human TCRαβ gene loci/HLA-DRA-IE/HLA-DRB1*0401-IE–transgenic (ABabDR4) mice (4, 19, 20). The advantage of this model is that CD4+ T cells in ABabDR4 mice bear a diverse human HLA-DRA/DRB1*0401–restricted (HLA-DR4–restricted) TCR repertoire but are not influenced by NY-ESO-1–specific tolerance, as mice do not express sequences homologous to the HLA-DR4–restricted NY-ESO-1 epitope used in this study. TCRs against NY-ESO-1 isolated from ABabDR4 mice were compared with TCRs isolated from human CD4+ T cell in vitro cultures and showed greater peptide sensitivity and superior melanoma recognition. These data suggest that MHC II–restricted TCRs against NY-ESO-1 from a nontolerant nonhuman host are of optimal affinity and are necessary to accomplish tumor regression in conjunction with an NY-ESO-1–specific MHC I–restricted TCR.
Results
NY-ESO-1–reactive TCRs isolated from a nontolerant host.
The aim was to isolate NY-ESO-1–reactive TCRs with MHC II restriction from a T cell repertoire unaffected by any tolerance mechanisms implicated by NY-ESO-1 expression in the host. For this purpose, we employed ABabDR4 mice, which were generated to rearrange human TCRs with HLA-DR4 restriction and do not express the human tumor antigen NY-ESO-1. ABabDR4 mice were shown to express a diverse CD4+ T cell repertoire and are thus a useful model for raising human TCRs in a nonhuman and therefore nontolerant host (19).
To elicit T cell responses against NY-ESO-1, we immunized ABabDR4 mice with the peptide NY-ESO-1116, which has been described as immunogenic in an HLA-DR4 restriction setting and has no sequence homology in mice (10). A distinct CD4+ T cell response to NY-ESO-1116 but not to an irrelevant peptide appeared upon peptide restimulation of blood from immunized ABabDR4 mice (Figure 1A). To confirm natural processing of the NY-ESO-1116 epitope, we also immunized ABabDR4 mice with NY-ESO-1 full-length DNA. Similarly, CD4+ T cells responded specifically to NY-ESO-1116 (Figure 1A).
Figure 1. NY-ESO-1–reactive TCRs generated from ABabDR4 mice.

(A) PBLs from an ABabDR4 mouse immunized with NY-ESO-1116 peptide or NY-ESO-1 DNA were pulsed with NY-ESO-1116 or irrelevant peptide and were stained intracellularly after overnight incubation. The results shown are representative of >10 (Peptide imm.) and 3 (DNA imm.) mice. (B) For sorting by flow cytometry, NY-ESO-1–reactive CD4+ T cells were labeled by the IFN-γ capture method following NY-ESO-1116 restimulation (TCR-3598/3598_2) or by DR4/NY-ESO-1116 tetramer staining following a 1-week culture period in the presence of 10–8 M NY-ESO-1116 (TCR-3600 and -5712) or αCD3/CD28 beads (TCR-5713). (C) Splenocytes from an ABabDR4 mouse immunized with NY-ESO-1 DNA were pulsed with NY-ESO-1116 or irrelevant peptide and stained intracellularly after 6 hours of incubation. (D) Human CD4+ T cells were transduced with NY-ESO-1–reactive TCRs isolated from CD4+ T cells shown in B and stained with DR4/NY-ESO-1116 tetramer and for mouse TCRβ constant region (mTCRβ). α and β sequences of NY-ESO-1–reactive TCRs are listed in Table 1. Plotted cells were gated on lymphocytes, live cells, and CD3+ cells (A and B), and CD4+ cells (C and D). Results are representative of 2 independent experiments (C and D). Values represent percentages. See also Supplemental Figure 1.
To isolate NY-ESO-1–reactive TCRs, we used splenocytes from NY-ESO-1116 or NY-ESO-1 DNA–immunized ABabDR4 mice. First, NY-ESO-1–reactive CD4+ T cells were labeled by the IFN-γ capture method and isolated by flow cytometry (Figure 1B). Two predominant TCRα and 3 predominant TCRβ chains were identified, which were matched by combinatorial expression and DR4/NY-ESO-1116 tetramer staining and revealed TCR-3598 and TCR-3598_2 (Table 1 and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI120391DS1). Three subsequent TCR isolations were conducted from DR4/NY-ESO-1116 tetramer–stained and sorted cells after 1-week in vitro expansion and yielded one predominant TCRα and -β chain combination each — TCR-3600, -5712, and -5713 (Figure 1B and Table 1). As ABabDR4 mice were boosted several times, narrowing the T cell response to a few clones that may be of optimal affinity is an expected outcome (21). Although the TCRs were isolated from cells labeled by different methods (IFN-γ capture method or DR4/NY-ESO-1116 tetramer), the final prerequisite for all TCRs to enter further analysis was positive DR4/NY-ESO-1116 tetramer staining.
Table 1. V/J gene segments and CDR3 regions of α and β chains of ABabDR4-derived TCRs.
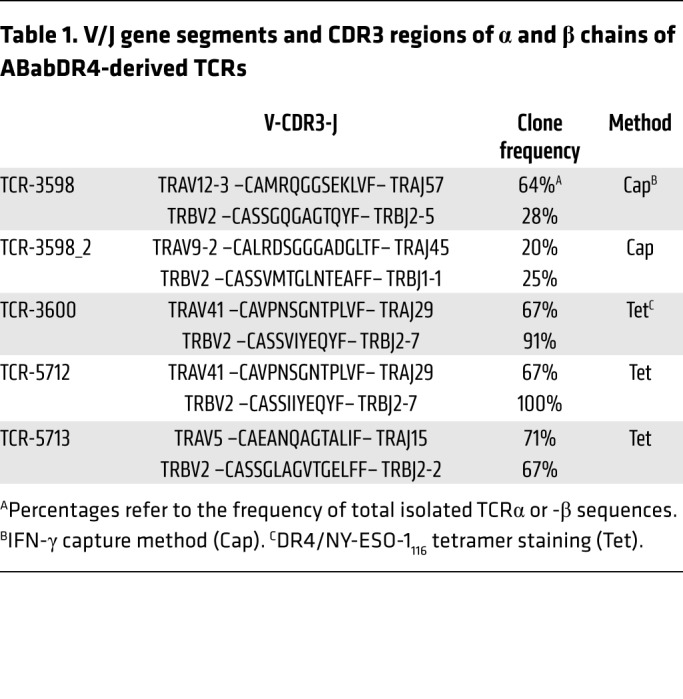
Of note, all identified TCRβ chains used the TCRβ variable 2 (TRBV2) gene segment. To confirm that the immune response to NY-ESO-1116 is dominated by the TRBV2 gene segment, we stained peptide-restimulated splenocytes from an immunized ABabDR4 mouse for TRBV2 and IFN-γ. Exclusively TRBV2+ CD4+ T cells responded to NY-ESO-1116 (Figure 1C).
Human peripheral blood leukocytes (PBLs) were retrovirally transduced with the TCRs containing mouse constant regions (22) and stained with DR4/NY-ESO-1116 tetramer (Figure 1D). In the TCR-3598_2–transduced sample, not all mouse TCRβ+ (mTCRβ+) CD4+ T cells bound the DR4/NY-ESO-1116 tetramer. This phenomenon was also observed in transduced Jurkat 76/CD4 cells that did not express endogenous TCRs (Supplemental Figure 1) and was therefore not primarily due to formation of mixed TCR dimers composed of endogenous and transduced TCR chains, but was at least partly due to inefficient tetramer binding. Taken together, 5 different NY-ESO-1–reactive TCRs were identified from 4 immunized ABabDR4 mice.
NY-ESO-1–reactive TCRs isolated from human CD4+ T cells.
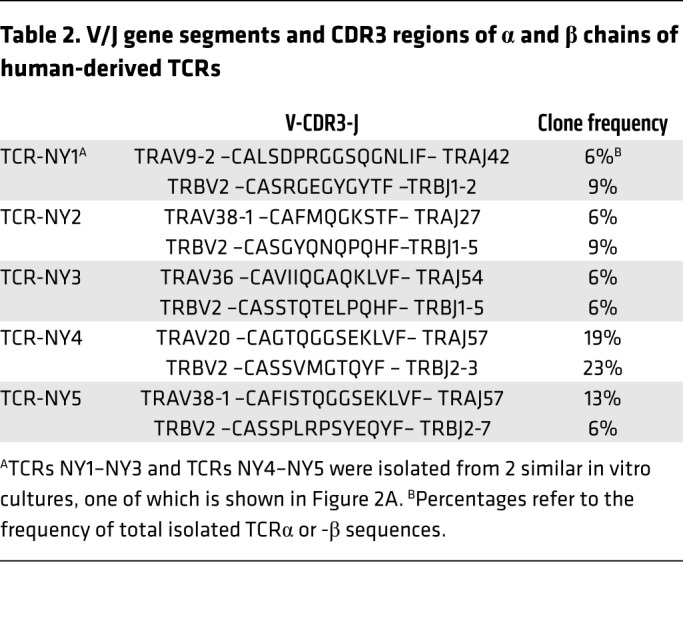
To compare the NY-ESO-1–reactive TCRs derived from ABabDR4 mice with TCRs from humans, in whom NY-ESO-1 is a self-protein potentially leading to a skewed CD4+ T cell repertoire, we isolated TCRs from human CD4+ T cells. Based on the dominant TRBV2 gene segment usage of the NY-ESO-1–reactive CD4+ T cells in ABabDR4 mice (Table 1), we enriched human PBLs from an HLA-DR4+ donor for CD4+ T cells expressing TRBV2 and cultured them in the presence of NY-ESO-1116. CD4+ T cells depleted of TRBV2+ cells were cultured in the same manner as control. After 2 weeks, more than 2% of the CD4+ T cells stained with DR4/NY-ESO-1116 tetramer, while only few cells stained in the TRBV2-depleted fraction (Figure 2A). From 2 similar in vitro cultures, DR4/NY-ESO-1116 tetramer+ CD4+ T cells were sorted by flow cytometry, and TCR sequences were isolated. In total, 6 functional TCRs were identified by combinatorial expression of single TCRα and -β chains and subsequent DR4/NY-ESO-1116 tetramer staining (Supplemental Figure 2). Five TCRs, named NY1–NY5, were chosen for further analyses (Table 2). CD4+ T cells transduced with those TCRs were stained with DR4/NY-ESO-1116 tetramer (Figure 2B).
Figure 2. NY-ESO-1–reactive TCRs generated from CD4+ T cells from human donors.

(A) PBLs from an HLA-DR4+ donor were enriched by magnetic cell sorting for CD4+ T cells expressing TRBV2 and cultured in the presence of 2 μM NY-ESO-1116 and irradiated CD4– cells as feeders. As a control the CD4+ fraction depleted of TRBV2+ cells was cultured in the same manner. After 2 weeks, NY-ESO-1–reactive CD4+ T cells were stained with DR4/NY-ESO-1116 tetramer and sorted by flow cytometry for isolation of the TCR chains. One of 2 similar in vitro cultures is shown. Cells were gated on lymphocytes, live cells, CD3+ cells, and CD4+ and CD8– cells. (B) Human CD4+ T cells were transduced with the NY-ESO-1–reactive TCRs, which were identified by combinatorial expression (Supplemental Figure 2) and stained with DR4/NY-ESO-1116 tetramer and for mTCRβ. The results are representative of 2 independent experiments. α and β sequences of NY-ESO-1–reactive TCRs are listed in Table 2. The untransduced sample is the same as that shown in Figure 1D, since experiments in Figure 1D and 2B were performed in parallel. Plotted cells were gated on lymphocytes, live cells, and CD3+ and CD4+ cells.
Table 2. V/J gene segments and CDR3 regions of α and β chains of human-derived TCRs.
ABabDR4-derived TCRs recognized NY-ESO-1 more efficiently than human-derived TCRs.
First, we tested the NY-ESO-1–reactive TCRs for function by coculturing TCR-transduced CD4+ T cells with NY-ESO-1116–loaded or NY-ESO-1–transduced cell lines. NY-ESO-1 protein expression by the transduced or naturally expressing melanoma cell lines was confirmed by Western blot analysis, and IFN-γ–pretreated melanoma cell lines showed HLA-DR expression comparable to that in the untreated lymphoblastoid B cell line (LCL) BSM (Figure 3, A and B). All TCRs recognized the HLA-DR4+ melanoma cell line FM3 loaded with NY-ESO-1116 (Figure 3C). However, the NY-ESO-1–transduced lines FM3 and BSM (HLA-DR4+) were recognized by all ABabDR4-derived TCRs but not, or to a lesser extent, by the human-derived TCRs. The recognition of the NY-ESO-1–transduced lines was blocked by αHLA-DR antibody, confirming HLA-DR4–mediated recognition (Figure 3C). Taken together, TCRs from both settings were able to recognize loaded NY-ESO-1116, but ABabDR4-derived TCRs were better at recognizing processed NY-ESO-1 in transduced cell lines.
Figure 3. ABabDR4-derived TCRs recognized NY-ESO-1 more efficiently than human-derived TCRs.

(A) Protein lysates from cell lines used for coculture experiments in C and D were assessed for the presence of NY-ESO-1 protein. β-Actin was stained as protein loading control. (B) Melanoma cell lines pretreated with IFN-γ and the LCL BSM were stained for HLA-DR (dark gray) or isotype control (light gray) and were measured by flow cytometry. (C and D) TCR-transduced CD4+ T cells were cocultured with the LCL BSM (HLA-DR4+) and the melanoma cell lines FM3 (NY-ESO-1–, HLA-DR4+), FM6 (NY-ESO-1+, HLA-DR4–), FM82, and FM56 (NY-ESO-1+, HLA-DR4+). Cell lines FM3-NY and BSM-NY were transduced to express NY-ESO-1; BSM was transduced with mCherry (BSM-mCh) as a control. NY-ESO-1116 (NY116), PMA and ionomycin (P/I), and blocking antibody αHLA-DR or αHLA-ABC were added where indicated. After overnight incubation, IFN-γ or IL-2 was measured in the supernatant. Mean values of intra-assay duplicates with SD are shown. The results are representative of 3 independent experiments performed with PBLs from different donors (B, C, and D).
Next, we analyzed the NY-ESO-1–reactive TCRs for recognition of naturally NY-ESO-1–expressing melanoma cell lines. Four of 5 ABabDR4-derived TCRs (3598_2, 3600, 5712, and 5713) recognized the NY-ESO-1– and HLA-DR4–expressing melanoma cell lines FM56 and FM82, while 2 of the human-derived TCRs (NY2 and NY3) recognized, albeit weakly, the melanoma cell line FM56 (Figure 3D). Again, αHLA-DR antibody blocked recognition. As expected, the NY-ESO-1–negative cell line FM3 and the HLA-DR4–negative cell line FM6 were not recognized, and addition of PMA/ionomycin elicited IFN-γ secretion in all samples (Figure 3D).
To investigate the peptide sensitivity of the NY-ESO-1–reactive TCRs, we tested TCR-transduced CD4+ T cells in a peptide titration assay with NY-ESO-1119–133 (NY-ESO-1119), comprising the epitope recognized by all TCRs. All ABabDR4-derived and 2 human-derived TCRs showed recognition up to 10–10 M NY-ESO-1119 with EC50 values between 1.5 × 10–9 and 2.1 × 10–9, while 3 human-derived TCRs were less sensitive and showed recognition up to 10–9 M NY-ESO-1119 with EC50 values between 9.8 × 10–9 and 4.1 × 10–8 (Figure 4A). ABabDR4-derived TCRs elicited higher maximal IFN-γ concentrations than the human-derived TCRs. Further, we analyzed TCR-transduced CD4+ T cells for DR4/NY-ESO-1116 tetramer MFI, taken as a rough measure of TCR affinity (Figure 4B). While MFI for mTCRβ constant region, representing TCR expression level, was similar in both TCR groups, ABabDR4-derived TCRs displayed higher DR4/NY-ESO-1116 tetramer MFI. This was also the case for TCR-transduced Jurkat 76/CD4+ T cells, in which mixed TCR dimers do not play a role, because endogenous TCR expression is lacking (Figure 4B). Taken together, the results indicate that ABabDR4-derived TCRs more efficiently recognized NY-ESO-1–positive cell lines and showed on average higher functional activity compared with the human-derived TCRs.
Figure 4. ABabDR4-derived TCRs showed higher functional activity compared with human-derived TCRs.

(A) TCR-transduced CD4+ T cells were cocultured with K562/DR4 cells loaded with decreasing concentrations of NY-ESO-1119 (NY119). After overnight incubation, IFN-γ was measured in the supernatant. The data were fitted in 3-parameter dose-response curves. (B) TCR-transduced CD4+ T cells or Jurkat 76/CD4 (Jurkat) cells were stained with DR4/NY-ESO-1116 tetramer and for mTCRβ as shown for CD4+ T cells in Figure 1D and Figure 2B and analyzed for MFI. t test was performed for statistical analysis. *P < 0.05. The results are representative of 3 (A) and 2 (B) independent experiments.
ABabDR4-derived TCR-3598_2 showed no alloreactivity or cross-reactivity.
To analyze the NY-ESO-1–reactive TCRs for potential MHC alloreactivity, we cocultured TCR-transduced CD4+ T cells with a panel of 14 LCLs expressing different MHC I and II molecules (Supplemental Table 1). Four ABabDR4-derived TCRs (TCR-3598, -3600, -5712, and -5713) and 1 human-derived TCR (TCR-NY4) reacted to 2 or more LCLs (Figure 5). Which MHC molecule was recognized by the cross-reactive TCRs was not further analyzed. The ABabDR4-derived TCR-3598_2 did not react to any LCL. Further analysis with peptide-loaded K562 cells expressing different HLA-DR molecules showed that recognition by TCR-3598_2 was restricted to HLA-DR4 (Supplemental Figure 3).
Figure 5. ABabDR4-derived TCR-3598_2 showed no alloreactivity.

TCR-transduced CD4+ T cells were cocultured with a panel of LCLs expressing different MHC class I and II molecules (Supplemental Table 1). As a positive control, PMA and ionomycin were added to the T cells. After overnight incubation, IL-2 was measured in the supernatant. Background, defined as the highest cytokine secretion observed for untransduced T cells, is indicated by the dotted line. Mean values of intra-assay duplicates with SD are shown. In the grid below, cross-reactions to LCLs are marked by an X. The results are representative of 3 independent experiments performed with PBLs from different donors. See also Supplemental Figure 3.
For further analysis of potential cross-reactivity, we identified the recognition motifs of the NY-ESO-1–reactive TCRs. NY-ESO-1119 was sequentially mutated to alanine at each position, and recognition by the TCRs was tested (Figure 6A). An amino acid was rated as a recognition site when the response to the respective alanine exchanged peptide was less than one-third as compared with the unchanged NY-ESO-1119. In total, 9 different recognition motifs were identified from the 10 TCRs. While all TCRs required the K in the sixth position, all but TCR-5713 required the E in the seventh position, and all but TCR-NY5 required the F in the eighth position. Of note, there was no clear-cut difference in recognition by the ABabDR4-derived compared with the human-derived TCRs (Figure 6A).
Figure 6. ABabDR4-derived TCR-3598_2 showed no cross-reactivity.

(A) TCR-transduced CD4+ T cells were cocultured with K562/DR4 cells that were loaded with NY-ESO-1119 containing single alanine exchanges at 10–7 M. After overnight incubation, IFN-γ concentration (conc.) was measured in the supernatant. An amino acid was identified as a recognition site when the response to the respective alanine exchanged peptide was less than one-third as compared with the unchanged NY-ESO-1119 as indicated by the dotted line. (B) TCR-3598_2–transduced CD4+ T cells were incubated with K562/DR4 cells loaded with 50 different peptides (Supplemental Table 2) containing the recognition motif -L-K-E-F. Peptides were included if they had a predicted affinity to HLA-DR4 of less than 500 nM and are present in the human but not the mouse proteome. After overnight incubation, IFN-γ was measured in the supernatant. (C) TCR-3598_2-transduced CD4+ T cells were incubated with HLA-DR4+ BSM transduced to express XRRA1 (XR) or NY-ESO-1 (NY). NY-ESO-1116 and PMA and ionomycin were added where indicated. Shown are IL-2 levels in the supernatant after overnight incubation. Mean values of 3 independent experiments with SD (A) or mean values of intra-assay duplicates with SD (B and C) are shown. The results are representative of 2 independent experiments performed with PBLs from different donors (B and C).
As in the ABabDR4 mouse TCRs are negatively selected on mouse and not human self-peptides, peptides that are not present in the mouse are potentially cross-reactive. TCR-3598_2, which did not show alloreactivity in the LCL coculture, was further tested for peptides containing its recognition motif (X-X-X-X-L-K-E-F-X-X-X-X-X-X-X; Supplemental Table 2). From 50 tested peptides, one peptide, X-ray radiation resistance-associated protein 1 (XRRA1)729–743 was recognized at 10–6 M but not 10–7 M by TCR-3598_2 (Figure 6B). To exclude that this cross-reaction is relevant in a physiological setting where XRRA1 must be processed and loaded onto MHC II, we cocultured TCR-3598_2–transduced CD4+ T cells with full-length XRRA1-transduced HLA-DR4+ BSM cells. XRRA1-transduced BSM cells were recognized only when loaded externally with NY-ESO-1116, while NY-ESO-1–transduced BSM cells were recognized as expected (Figure 6C). Thus, no relevant cross-reactivity of TCR-3598_2 was detected.
NY-ESO-1–specific MHC I– and II–restricted TCRs synergize in tumor regression.
To confirm the functionality of TCR-3598_2 in an in vivo setting, we set up a model of ATT in which tumor-bearing mice were treated with NY-ESO-1–specific CD4+ and CD8+ T cells in combination. One month after DR4xRag–/– mice received a subcutaneous injection of fibrosarcoma cells (Tet-TagLuc-NY-ESO-1-HHD clone 1), tumors were palpable, and the mice were treated with TCR-3598_2–transduced CD4+ T cells and CD8+ T cells transduced with a TCR recognizing NY-ESO-1157–165 on HLA-A2, named TCR-ESO (3). Thus, in this model CD8+ T cells recognize only antigen-presenting HHD (HLA-A2-H-2Db chimeric) on cancer cells but not antigen cross-presented by tumor stromal cells, while CD4+ T cells recognize only antigen cross-presented by stroma cells, since HHD is only present on the cancer cells and HLA-DR4 is only present on the host cells. As controls, NY-ESO-1–specific CD4+ or CD8+ T cells alone or CD4+ and CD8+ T cells expressing an irrelevant TCR were given. TCR transduction rates were at least 23%, and the phenotype of CD4+ T cells was central memory (CD62L+, CD44+), while the phenotype of CD8+ T cells was partly central memory and effector/effector memory (CD62L–, CD44+; Supplemental Figure 4). In group 1, which received NY-ESO-1–specific CD4+ T cells alone, tumor growth slowed down, while in group 2, which received NY-ESO-1–specific CD8+ T cells alone, 4 of 10 tumors regressed (Figure 7A). In control group 4, which received CD4+ T cells transduced with an irrelevant TCR together with NY-ESO-1–specific CD8+ T cells, 5 of 8 tumors regressed, while all tumors grew out in groups 5 and 6, which received a combination of irrelevant CD4+ and CD8+ T cells or no T cells, respectively. Only in group 3, which received a combination of NY-ESO-1–specific CD4+ and CD8+ T cells, did all tumors (10 of 10) regress (Figure 7A). Although eventually antigen-negative variants grew out in most mice of this group, survival was significantly longer (Supplemental Figure 5).
Figure 7. TCR-3598_2–transduced CD4+ T cells in combination with TCR-ESO–transduced CD8+ T cells caused tumor regression.

(A) Tumor-bearing mice were treated with TCR-3598_2–transduced CD4+ T cells and/or TCR-ESO–transduced CD8+ T cells on day 30, when the tumors were palpable. TCR-1367–transduced CD4+ and/or CD8+ T cells were injected as controls (CD4/CD8-irrelevant) where indicated. Shown are tumor sizes on the indicated days after tumor cell injection. Results from 2 independent experiments were combined. (B) Adoptively transferred CD8+ and CD4+ T cells were detected in the blood 9 days after treatment. Group numbers refer to A. Each dot represents data derived from one individual mouse. One-way ANOVA followed by Bonferroni’s post hoc test was performed for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.005. (C) CD11b+ stromal cells isolated from tumor material were recognized by TCR-3598_2–transduced CD4+ T cells. As positive controls, CD11b+ stromal cells were loaded with NY-ESO-1116 or anti-CD3/CD28 activator beads (act. beads) were added to the T cells. Intra-assay duplicates with mean values are shown. The results are representative of 3 independent experiments. See also Supplemental Figures 4–8.
Analysis of the T cells in peripheral blood of the treated mice revealed a higher number of CD8+ T cells in group 3, which received NY-ESO-1–specific CD4+ and CD8+ T cells in combination, compared with all other groups. Also CD4+ T cell numbers were higher in group 3 compared with groups 4 and 5, which received irrelevant CD4+ T cells (Figure 7B). Moreover, more CD8+ and CD4+ T cells were found within the tumors of mice in group 3, which received NY-ESO-1–specific CD4+ and CD8+ T cells in combination, compared with groups 1 and 2, which received only NY-ESO-1–specific CD4+ or CD8+ T cells, and compared with the control group, which received irrelevant CD4+ and CD8+ T cells (Supplemental Figure 6). To investigate the persistence of the adoptively transferred cells, we challenged mice from group 3 with injection of Tet-TagLuc-NY-ESO-1-HHD clone 1 cells into the flank opposite the regressed tumor (Supplemental Figure 7). All mice (4 of 4) rejected the tumor cells, while control mice, which were not treated previously, developed tumors.
Finally, we cocultured TCR-transduced CD4+ T cells with CD11b+ stromal cells purified from a control-treated tumor to analyze cross-presentation of NY-ESO-1 from tumor cells (Figure 7C and Supplemental Figure 8). The two ABabDR4-derived and 2 human-derived TCRs that performed best in the peptide sensitivity assay (Figure 4A) were selected for analysis. TCR-3598_2–transduced CD4+ T cells, used in the tumor model, recognized the CD11b+ stromal cells (Figure 7C), as well as CD4+ T cells transduced with the ABabDR4-derived TCR-5713 and one of both tested human-derived TCRs (TCR-NY3) (Supplemental Figure 8). Peptide-loaded CD11b+ cells elicited cytokine secretion by all TCR-transduced CD4+ T cells, and αCD3/CD28-activating beads elicited responses in the untransduced CD4+ T cells as well. Taken together, the results demonstrate the in vivo functionality of TCR-3598_2 in a mouse model of ATT.
Discussion
In this study, we observed better performance of NY-ESO-1–reactive MHC II–restricted TCRs from a nontolerant host compared with TCRs from a human donor regarding recognition of melanoma cell lines and functional activity. These results extend a finding in a similar mouse model transgenic for the human TCR gene loci and HLA-A2 (ABabDII mice), in which we have shown that a TCR raised against the HLA-A2 epitope NY-ESO-1157–165 is of higher functional activity than 1G4, a TCR obtained from a melanoma patient (3).
It is important to note that CD4+ T cell tolerance as well as immunity have been observed for different self-antigens in mouse models. While the CD4+ T cell repertoire against carcinoembryonic antigen (CEA) was shown to be tolerant, this was not the case for the CD4+ T cell repertoire against p53 (1, 23). Abundance of the antigen and availability for MHC II presentation might explain why the uniformly expressed but tightly regulated p53 does not lead to tolerance formation, while the transgenic protein CEA, which was also shown to be expressed in the thymus, does (1, 23). Using Cre recombinase as an artificial tissue-restricted self-antigen, it was shown that CD4+ T cell tolerance is not deletional and can be broken upon immune challenge (24). In this study, however, no Cre expression was detected in the thymus. As NY-ESO-1 expression has been detected in the thymus (2), a likely outcome is tolerance formation, which is supported by our data herein.
CD4+ T cell responses against NY-ESO-1116 in ABabDR4 mice were dominated by TRBV2 usage. As TRBV2 is not overrepresented in the ABabDR4 TCRβ repertoire (19), it seems to play a predominant role in NY-ESO-1116 recognition. Overrepresentation of TRBV2 by NY-ESO-1116–reactive TCRs was taken advantage of for in vitro expansion of human NY-ESO-1116–reactive CD4+ T cells, where clearly only the TRBV2-enriched cultures expanded DR4/NY-ESO-1116 tetramer+ cells. Prompt expansion of NY-ESO-1–reactive CD4+ T cells in culture was also observed in other reports (25, 26). Lower functional activity of the NY-ESO-1116–reactive TCRs isolated from the human PBLs herein suggests that CD4+ T cell tolerance to NY-ESO-1 is present in humans and that remaining CD4+ T cells are of lower avidity.
In other reports, MHC II–restricted TCRs were isolated from human Tregs, thereby avoiding the repertoire hole that may have been imposed by tolerance (27). In the human T cell primings herein, Tregs were not depleted. However, expansion of Tregs is unlikely under the given culture conditions, including low IL-2. We cannot exclude that high-affinity TCRs against NY-ESO-1 can be isolated from human Tregs, as CD4+ T cells recognizing self-antigens can develop into Tregs instead of being negatively selected (28). However, we hypothesize that ABabDR4 mice as a TCR source bear the advantage of natural selection for the fittest clonotypes in the memory response (21), thus generating optimal-affinity TCRs.
ABabDR4-derived TCRs recognized HLA-DR4/NY-ESO-1+ melanoma cell lines. Although recognition of MHC II–presented endogenous antigens does not comply with the classical antigen presentation pathway, in which exogenous antigens are endocytosed and loaded onto MHC II, NY-ESO-1+ melanoma cell lines have been recognized by CD4+ T cells in several cases (29–31). To what extent direct recognition of cancer cells by CD4+ T cells plays a role in tumor rejection is unclear, as MHC II–negative tumor cells can be controlled by CD4+ T cells in mouse models (15, 17, 32). However, unlike for MHC I–presented epitopes, it is very difficult to predict which precise epitopes are presented on MHC II molecules. We think the most (and probably only) important readout for the therapeutic efficacy of an MHC II–restricted TCR is recognition of an endogenously processed and presented epitope from naturally expressed NY-ESO-1 in human cancer cells. In this regard, the ABabDR4-derived TCRs were superior to the human-derived TCRs, suggesting that they may have better therapeutic efficacy.
Several NY-ESO-1 TCRs isolated from mouse or human in the present study showed alloreactivity toward one or more LCLs. Since the TCRs originate from individuals that bear only a limited number of MHC molecules and it has been shown that up to 10% of T cells from a naive polyclonal repertoire are alloreactive, this finding is not unexpected but underscores the need for alloreactivity testing (33). The presence of more MHC molecules in the thymus is thought to lead to a higher number of T cells affected by negative selection and thereby fewer alloreactive T cells (34). Thus, the reason that there were more alloreactive ABabDR4-derived TCRs in comparison to human-derived TCRs in this study may be that the ABabDR4 mouse expresses only one MHC II molecule.
ABabDR4-derived TCR-3598_2–transduced CD4+ T cells in combination with NY-ESO-1–specific CD8+ T cells caused regression of palpable tumors in a mouse model of ATT. This combined treatment, in which CD4+ T cells recognized NY-ESO-1 on host cells and CD8+ T cells recognized it on cancer cells, was more effective than treatment with CD4+ or CD8+ T cells alone, thus showing their synergistic effect in ATT of cancer. As CD4+ T cells alone had only a limited effect in vivo, we think that their role was primarily in helping CD8+ T cells. This is in line with our finding of significantly more CD8+ T cells in the blood and in the tumor of mice treated with CD4+ and CD8+ T cells in combination. We hypothesize that mechanistically CD4+ T cells find the tumor by cross-presented antigen on stromal cells and provide help to CD8+ T cells via cytokines, e.g., IFN-γ and IL-2, either directly or indirectly through induction of an inflammatory milieu. Whether recognition of stromal CD11b+ cells also caused their destruction is unclear, but the effect was not strong enough to achieve tumor regression by CD4+ T cells alone. Synergy of CD4+ and CD8+ T cell lines with specificity for NY-ESO-1 has been shown to delay tumor growth in a xenograft mouse model (30). This model, however, does not allow for recognition of cross-presented antigen, which was shown to be crucial for tumor rejection through bystander killing of antigen-negative cancer cells (18).
Transfer of NY-ESO-1–specific CD4+ T cells alone delayed tumor growth but did not lead to tumor rejection. In vivo models in which CD4+ T cells alone can eradicate transplanted MHC II–negative tumors either involve very early treatment on day 0 or 1 following tumor injection or treatment in conjunction with chemotherapy (15, 32, 35). Thus, failure to reject tumors by CD4+ T cells alone was not surprising, as in the model used herein, tumors established for 1 month and were palpable before the mice were treated.
Transfer of NY-ESO-1–specific CD8+ T cells alone or with irrelevant CD4+ T cells caused tumor regression only in some of the mice. Although CD8+ T cells have been shown to reject even large established tumors, this was the case when the target was a strong model antigen or when the target was overexpressed as trimer minigene (36, 37). The Tet-TagLuc cells used in this study express the target NY-ESO-1 as a full-length protein, which is more reminiscent of the physiologic situation. Moreover, in this model CD8+ T cells cannot recognize cross-presented antigen on stromal cells because of the absence of HLA-A2 in the host mice. Insufficient rejection by CD8+ T cells alone was therefore not unexpected.
Although NY-ESO-1–specific CD4+ and CD8+ T cells in combination caused tumor regression initially, antigen-negative variants grew out in most mice. Thus, recognition of cross-presented antigen by CD4+ T cells in addition to CD8+ T cells allowed tumor regression in the first place but did not eradicate antigen-loss variants, unlike in other models (18). Among the possible reasons, insufficient antigen expression or lack of cross-presentation to CD8+ T cells in our model may play a role. We hypothesize that irradiation or chemotherapy concomitant with ATT may increase the extent of cross-presentation to CD4+ T cells, as shown in other models (38), thereby compensating for insufficient antigen expression or lack of cross-presentation to CD8+ T cells and preventing relapse of antigen-negative variants.
The NY-ESO-1–specific TCR-3598_2 derived from the ABabDR4 mouse model showed a favorable safety profile, with no detectable alloreactivity or cross-reactivity but high functional activity toward NY-ESO-1. TCR-3598_2 is a promising candidate for clinical application to be used in combination with NY-ESO-1–specific CD8+ T cells to treat solid tumors.
Methods
Supplemental Methods are available online with this article.
Cell lines.
The human melanoma cell lines FM-82, FM-56 (NY-ESO-1+, HLA-DR4+), FM-3 (NY-ESO-1–, HLA-DR4+), and FM-6 (NY-ESO-1+, HLA-DR4–) were provided by the European Searchable Tumor Cell Bank and Database (ESTDAB). Jurkat 76/CD4 cells are TCR deficient and were generated by introducing human CD4 into the Jurkat 76 clone (39). The murine cell line T.54ζ17 is TCR deficient and expresses human CD4 and murine ζ chain (58/CD4 cells; ref. 40). K562 cells expressing different HLA-DR molecules were generated by transducing K562 cells with HLA-DRA and HLA-DRB1*0401 (K562/DR4 cells), -DRB1*0101, -DRB1*0701, -DRB1*1101, or -DRB3*0101 (41). All cell lines were cultured in RPMI supplemented with 10% FCS (PAN Biotech) and 1× antibiotic-antimycotic. The retroviral packaging cell lines 293GP-GLV and Plat-E (producing amphotropic and ecotropic retroviral vectors, respectively) were cultured in DMEM supplemented with 10% FCS (42, 43). The panel of Epstein-Barr virus–transformed LCLs were cultured in RPMI supplemented with 10% FCS, 1× antibiotic-antimycotic, 1 mM sodium pyruvate, and 1× nonessential amino acids. All cell culture reagents were purchased from Life Technologies unless otherwise indicated.
Mice.
ABabDR4 mice express a diverse TCR repertoire and a mouse/human chimeric MHC II molecule, HLA-DRA-IE/HLA-DRB1*0401-IE, while neither mouse TCRs nor mouse MHC II molecules are expressed (19). The TCR-transgenic mouse lines OTII and P14 were purchased from the Jackson Laboratory and bred to Rag-deficient mice to generate OTII × Rag+/– and P14 × Rag–/– strains. C57BL/6J (B6) mice were purchased from the Jackson Laboratory. HLA-DR4 mice were purchased from Taconic and bred to Rag-deficient mice to generate DR4 × Rag–/– mice. Male and female mice were taken at age 6–16 weeks for experiments. All mouse lines were bred in-house under specific pathogen–free conditions.
Immunization of ABabDR4 mice.
For peptide immunization, 100 μg of the NY-ESO-1 peptide 116–135 (lpvpgvllkeftvsgnilti, NY-ESO-1116, GenScript) and 50 μg CpG (CpG 1826, MOLBIOL) were prepared in a 1:1 emulsion of incomplete Freund’s adjuvant and PBS and injected subcutaneously. Immunizations were repeated with at least 4-week intervals. For DNA immunizations, the Helios Gene Gun system from Bio-Rad was used. pcDNA3.1 vectors containing NY-ESO-1 cDNA or GM-CSF cDNA were precipitated on gold microcarriers of 1-μm diameter and loaded into GoldCoat tubing using a Tubing Prep Station (Bio-Rad) for cartridge preparation. Using helium pressure, DNA was delivered into the skin at the abdomen of the mouse. One week after immunizations CD4+ T cell responses were analyzed by culturing peripheral blood samples with 1 μg/ml NY-ESO-1116 peptide, Padre as negative control (44), or 4 × 105 Dynabeads T activator CD3/28 (Invitrogen) as positive control. Brefeldin A (BD GolgiPlug) was added to the cultures, and intracellular IFN-γ staining was performed as a readout after overnight incubation (see Flow cytometry).
TCR isolation.
For T cell isolation, cells from spleen and draining lymph nodes were prepared from mice 10–14 days after the last immunization. After 8 hours incubation in the presence of NY-ESO-1116 at 1 μg/ml, responding cells were labeled by Mouse IFN-γ Secretion Assay (Milteny Biotec) and sorted by flow cytometry. For in vitro expansion of NY-ESO-1–reactive CD4+ T cells from immunized mice, spleen and lymph node cells were cultured for 7–9 days in the presence of 10–8 M NY-ESO-1116 or Dynabeads T activator CD3/CD28 at a ratio of 1:1 and 10 U/ml IL-2, followed by tetramer staining and cell sorting by flow cytometry. To isolate NY-ESO-1–reactive TCRs from human T cells, PBLs from an HLA-DR4+ donor were enriched for CD4 and TRBV2 expression by subsequent magnetic separation (Milteny Biotec) yielding 68% TRBV2+ CD4+ T cells. 1.9 × 106 TRBV2-enriched CD4+ T cells were cultured with 2.5 × 106 irradiated (30 Gy) autologous CD4– cells in the presence of 2 μM NY-ESO-1116 and 10 U/ml IL-2. After 14 days, NY-ESO-1–reactive CD4+ T cells were stained by tetramer and sorted by flow cytometry. From all T cell sorts (yielding between 500 and 10,000 cells), total RNA was extracted (RNeasy Micro Kit, QIAGEN) and 5′RACE PCR was performed using a SMARTer RACE cDNA Amplification Kit (Clontech) with the following reverse primers: 5′-CGGCCACTTTCAGGAGGAGGATTCGGAAC-3′ (for TCRα) and 5′-CCGTAGAACTGGACTTGACAGCGGAAGTGG-3′ (for TCRβ). The RACE PCR products were cloned into TOPO vectors (Zero Blunt TOPO PCR Cloning Kit, Invitrogen) to transform competent E. coli. Six to 32 clones per TCR chain were analyzed, and the most frequent TCRαβ chain pairs were analyzed.
Retroviral transduction.
Virus supernatant for retroviral transduction was produced with the packaging lines 293GP-GLV (amphotropic) or Plat-E (ecotropic) by transfecting the retroviral vector MP71-PRE (45) containing a TCR expression cassette or the single TCR chains using Lipofectamine 2000 reagent (Thermo Fisher Scientific). TCR expression cassettes contained codon-optimized TCRβ and -α chains linked by the porcine teschovirus-1–derived self-cleaving peptide P2A and murinized in their constant regions (22, 46). Single TCR chains were cloned from TOPO vectors and equipped with murinized constant regions by overlap extension PCR. Viral supernatant was harvested after 48 and 72 hours following transfection and used directly for transducing target cells. Freshly isolated human PBLs were activated on anti-CD3 (OKT3)/anti-CD28–coated (CD28.2; BD Pharmingen) plates and cultured in hTCM (300 U/ml IL-2). The first transduction was performed 48 hours after T cell activation by spinoculating the T cells with virus supernatant for 90 minutes at 800 g. The second transduction was performed the following day by spinoculating the T cells 30 minutes at 800 g on virus-preloaded retronectin–coated (Takara) plates. After expansion of the T cells for 1 week, they were cultured in hTCM supplemented with low IL-2 (30 U/ml) for 3 days, before being used in in vitro experiments. Jurkat 76/CD4 cells and 58/CD4 cells were transduced twice on 2 subsequent days by spinoculation with virus supernatant.
Coculture experiments.
All coculture experiments with human cells were performed by incubating 1 × 104 transduced TCR+ CD4+ T cells with 5 × 104 target cells 16–18 hours. IFN-γ or IL-2 levels were measured in the supernatant by ELISA (BD OptEIA; BD Biosciences). As a positive control, 50 ng/ml PMA and 5 μg/ml ionomycin were added. NY-ESO-1 peptides (NY-ESO-1116–135 or NY-ESO-1119–133) were added at 10–6 M or at indicated concentrations. Alanine-exchanged NY-ESO-1119 peptides (GenScript, >95% purity) were added at 10–7 M. Peptides containing the TCR-3598_2 recognition motif -L-K-E-F- (JPT Peptide Technologies, unpurified) were added to the coculture at 10–6 or 10–7 M as indicated. HLA-DR and HLA-ABC blocking antibodies (L243, W6/32; BioLegend) were added to the target cells at 20 μg/ml at least 1 hour prior to addition of the T cells.
Flow cytometry.
The following antibodies were used for staining at 1:100 dilution and purchased from BioLegend unless otherwise indicated: anti-mCD4–FITC (RM4-5), anti-mCD4–BV421 (RM4-5), anti-mCD8–PECy7 (53-6.7, BD), anti-mCD8–BV421 (clone 53-6.7), anti-mCD3–PE (145-2C11, 1:200), anti–mIL-2–APC (JES6-5H4), anti–mIFN-γ–BV421 (XMG1.2), anti–mTCRβ chain–APC (H57-597), anti-CD44–APC (IM7), anti-mCD62L–PE (MEL-14, BD), anti-hCD4–PE/Cy7 (OKT4), anti-hCD3–APC (SK7), anti-hTRBV2–FITC (IMMU 546, 1:20, Beckman Coulter), anti-hTRBV12–FITC (56C5.2, 1:20, Beckman Coulter), anti-hTRBV28–FITC (CH92, 1:20, Beckman Coulter), anti-hTRBV28–PE (Jovi-3, Ancell), anti–HLA-DR–APC (L243).
Immune responses of immunized ABabDR4 mice were analyzed by intracellular IFN-γ staining of peptide-restimulated blood samples. After Fc blocking (anti-mCD16/32, clone 93, BioLegend), cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (1:1000 in PBS, Life Technologies). Samples were fixed (BD Cytofix/Cytoperm) and stained with antibodies for 30 minutes at 4°C.
For tetramer staining, cells were incubated in hTCM containing 10 μg/ml DR4/NY-ESO-1116 tetramer or DR4/CLIP87–101 control tetramer for 1 hour at 37°C. Subsequently, antibodies were directly added, and the cells were incubated 30 minutes on ice. PE-labeled DR4/NY-ESO-1116 and DR4/CLIP87–101 tetramers were obtained through the NIH Tetramer Core Facility at Emory University.
Adoptively transferred T cells were analyzed in peripheral blood and tumor tissue. Antibodies were added into the blood sample, followed by lysing of erythrocytes or into tumor cell suspension. The entire samples were analyzed by flow cytometry, and the numbers of T cells were calculated according to the volume of peripheral blood sample or according to the weight of the tumor piece used for staining.
FACSAria II was used for sorting of cells, and FacsCanto II or LSRFortessa was used to analyze cells by flow cytometry (all BD).
Mouse model of ATT of cancer.
A fibroblast line originating from a TREloxPstoploxPTagLuc transgenic mouse (Tet-TagLuc cells; ref. 47) was retrovirally transduced with HLA-A2 harboring the murine H-2Db α3 domain and fused to human β2m (HHD; ref. 48) and full-length NY-ESO-1 with mCherry reporter expression from IRES to create Tet-TagLuc-NY-ESO-1-HHD cells. A cell clone (Tet-TagLuc-NY-ESO-1-HHD clone 1) was used for subcutaneous tumor challenge of DR4 × Rag2–/– recipient mice. After 30 days, tumors were palpable, and the mice were treated with 1 × 106 CD4+ T cells from OTII × Rag+/– mice transduced with TCR-3598_2 and/or 1 × 106 CD8+ T cells from P14 × Rag1–/– mice transduced with TCR-ESO (3). As controls CD4+ and CD8+ T cells were transduced with TCR-1367 recognizing MAGE-A1 on HLA-A2 (3). Tumor sizes were measured 2 or 3 times per week.
For analysis of tumor-infiltrating lymphocytes and isolation of CD11b+ stromal cells, cell suspensions from tumor material were made. Tumor pieces were incubated at 37°C in medium containing 1 mg/ml collagenase II (Gibco), 1 mg/ml dispase II, and 10 μg/ml DNase I (Roche). After 1 hour, trypsin-EDTA (Gibco) was added for a further incubation of 30 minutes. The filtered cell suspensions were analyzed for tumor-infiltrating lymphocytes by antibody staining or stained for CD11b-PE (M1/70, BioLegend) and subjected to magnetic cell separation using anti-PE microbeads (Milteny) for isolation of CD11b+ cells.
Coculture of TCR-transduced CD4+ T cells and CD11b+ stromal cells was performed with 1 × 105 cells each. 2 × 105 Dynabeads T activator CD3/28 (Invitrogen) or NY-ESO-1116 peptide were added as a positive control. IFN-γ and IL-2 levels were measured in the supernatant by ELISA (BD OptEIA) after 16 hours of incubation.
Statistics.
Log-rank (Mantel-Cox) test, 2-tailed t test, and 1-way ANOVA followed by Bonferroni’s post hoc test were performed using GraphPad Prism 7. P values of 0.05 or less were considered significant.
Study approval.
All animal experiments were approved by the responsible state office (Landesamt für Gesundheit und Soziales).Informed consent was obtained from all human blood donors after project approval by the local ethics committee of the Charité.
Author contributions
LP planned and performed experiments, analyzed data, and wrote the manuscript. XC generated TCR-ESO used in the tumor model and revised the manuscript. FKML generated K562 cells expressing HLA-DR molecules and revised the manuscript. TB supervised the project, analyzed data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank C. Westen, M. Manzke, A. Gärtner, and I. Becker for technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft through SFB-TR36 and by the Berlin Institute of Health through a collaborative research grant.
Version 1. 12/10/2018
Electronic publication
Version 2. 01/02/2019
Print issue publication
Footnotes
Conflict of interest: LP, XC, and TB are named inventors on a patent application (PCT/EP2016/055242) covering the NY-ESO-1–specific T cell receptors described in this study held by the Max Delbrück Center for Molecular Medicine.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(1):324–335.https://doi.org/10.1172/JCI120391.
See the related Commentary at High-affinity T cell receptors for adoptive cell transfer.
Contributor Information
Lucia Poncette, Email: lucia.poncette@mdc-berlin.de.
Xiaojing Chen, Email: xiaojing.chen@mdc-berlin.de.
Thomas Blankenstein, Email: tblanke@mdc-berlin.de.
References
- 1.Bos R, et al. Expression of a natural tumor antigen by thymic epithelial cells impairs the tumor-protective CD4+ T-cell repertoire. Cancer Res. 2005;65(14):6443–6449. doi: 10.1158/0008-5472.CAN-05-0666. [DOI] [PubMed] [Google Scholar]
- 2.Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J Exp Med. 2004;199(2):155–166. doi: 10.1084/jem.20031677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Obenaus M, et al. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat Biotechnol. 2015;33(4):402–407. doi: 10.1038/nbt.3147. [DOI] [PubMed] [Google Scholar]
- 4.Li LP, et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med. 2010;16(9):1029–1034. doi: 10.1038/nm.2197. [DOI] [PubMed] [Google Scholar]
- 5.Chen YT, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A. 1997;94(5):1914–1918. doi: 10.1073/pnas.94.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fourcade J, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res. 2014;74(4):1045–1055. doi: 10.1158/0008-5472.CAN-13-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeoka T, et al. NY-ESO-1 Protein cancer vaccine with poly-ICLC OK-432: rapid strong induction of NY-ESO-1-specific immune responses by poly-ICLC. J Immunother. 2017;40(4):140–147. doi: 10.1097/CJI.0000000000000162. [DOI] [PubMed] [Google Scholar]
- 8.Gnjatic S, et al. Survey of naturally occurring CD4+ T cell responses against NY-ESO-1 in cancer patients: correlation with antibody responses. Proc Natl Acad Sci U S A. 2003;100(15):8862–8867. doi: 10.1073/pnas.1133324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stockert E, et al. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med. 1998;187(8):1349–1354. doi: 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng G, Touloukian CE, Wang X, Restifo NP, Rosenberg SA, Wang RF. Identification of CD4+ T cell epitopes from NY-ESO-1 presented by HLA-DR molecules. J Immunol. 2000;165(2):1153–1159. doi: 10.4049/jimmunol.165.2.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapoport AP, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914–921. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robbins PF, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21(5):1019–1027. doi: 10.1158/1078-0432.CCR-14-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maude SL, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. 2010;70(21):8368–8377. doi: 10.1158/0008-5472.CAN-10-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenberg PD, Cheever MA, Fefer A. Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2- lymphocytes. J Exp Med. 1981;154(3):952–963. doi: 10.1084/jem.154.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quezada SA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207(3):637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin Z, Blankenstein T. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12(6):677–686. doi: 10.1016/S1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 18.Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med. 2010;207(11):2469–2477. doi: 10.1084/jem.20092450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Poncette L, Blankenstein T. Human TCR-MHC coevolution after divergence from mice includes increased nontemplate-encoded CDR3 diversity. J Exp Med. 2017;214(11):3417–3433. doi: 10.1084/jem.20161784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito K, et al. HLA-DR4-IE chimeric class II transgenic, murine class II-deficient mice are susceptible to experimental allergic encephalomyelitis. J Exp Med. 1996;183(6):2635–2644. doi: 10.1084/jem.183.6.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10(4):485–492. doi: 10.1016/S1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 22.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66(17):8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lauwen MM, et al. Self-tolerance does not restrict the CD4+ T-helper response against the p53 tumor antigen. Cancer Res. 2008;68(3):893–900. doi: 10.1158/0008-5472.CAN-07-3166. [DOI] [PubMed] [Google Scholar]
- 24.Legoux FP, et al. CD4+ T cell tolerance to tissue-restricted self antigens is mediated by antigen-specific regulatory T cells rather than deletion. Immunity. 2015;43(5):896–908. doi: 10.1016/j.immuni.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kayser S, et al. Rapid generation of NY-ESO-1-specific CD4+ THELPER1 cells for adoptive T-cell therapy. Oncoimmunology. 2015;4(5):e1002723. doi: 10.1080/2162402X.2014.1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poli C, Raffin C, Dojcinovic D, Luescher I, Ayyoub M, Valmori D. MHC class II/ESO tetramer-based generation of in vitro primed anti-tumor T-helper lines for adoptive cell therapy of cancer. Haematologica. 2013;98(2):316–322. doi: 10.3324/haematol.2012.071712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao X, et al. Isolation and characterization of an HLA-DPB1*04: 01-restricted MAGE-A3 T-cell receptor for cancer immunotherapy. J Immunother. 2016;39(5):191–201. doi: 10.1097/CJI.0000000000000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol. 2012;12(3):157–167. doi: 10.1038/nri3155. [DOI] [PubMed] [Google Scholar]
- 29.Fonteneau JF, Brilot F, Münz C, Gannagé M. The tumor antigen NY-ESO-1 mediates direct recognition of melanoma cells by CD4+ T cells after intercellular antigen transfer. J Immunol. 2016;196(1):64–71. doi: 10.4049/jimmunol.1402664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuzaki J, et al. Direct tumor recognition by a human CD4(+) T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci Rep. 2015;5:14896. doi: 10.1038/srep14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Zheng Z, Khong HT, Rosenberg SA, Morgan RA. Transduction of an HLA-DP4-restricted NY-ESO-1-specific TCR into primary human CD4+ lymphocytes. J Immunother. 2006;29(4):398–406. doi: 10.1097/01.cji.0000203082.20365.7f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mumberg D, et al. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc Natl Acad Sci U S A. 1999;96(15):8633–8638. doi: 10.1073/pnas.96.15.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. 2001;166(2):973–981. doi: 10.4049/jimmunol.166.2.973. [DOI] [PubMed] [Google Scholar]
- 34.Ni PP, Wang Y, Allen PM. Both positive and negative effects on immune responses by expression of a second class II MHC molecule. Mol Immunol. 2014;62(1):199–208. doi: 10.1016/j.molimm.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Diez A, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood. 2007;109(12):5346–5354. doi: 10.1182/blood-2006-10-051318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leisegang M, Kammertoens T, Uckert W, Blankenstein T. Targeting human melanoma neoantigens by T cell receptor gene therapy. J Clin Invest. 2016;126(3):854–858. doi: 10.1172/JCI83465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schreiber K, et al. Spleen cells from young but not old immunized mice eradicate large established cancers. Clin Cancer Res. 2012;18(9):2526–2533. doi: 10.1158/1078-0432.CCR-12-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang B, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204(1):49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heemskerk MH, et al. Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specific T-cell receptor complexes expressing a conserved alpha joining region. Blood. 2003;102(10):3530–3540. doi: 10.1182/blood-2003-05-1524. [DOI] [PubMed] [Google Scholar]
- 40.Kieback E, et al. Thymus-derived regulatory T cells are positively selected on natural self-antigen through cognate interactions of high functional avidity. Immunity. 2016;44(5):1114–1126. doi: 10.1016/j.immuni.2016.04.018. [DOI] [PubMed] [Google Scholar]
- 41.Lozzio BB, Lozzio CB. Properties of the K562 cell line derived from a patient with chronic myeloid leukemia. Int J Cancer. 1977;19(1):136. doi: 10.1002/ijc.2910190119. [DOI] [PubMed] [Google Scholar]
- 42.Ghani K, et al. Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum Gene Ther. 2009;20(9):966–974. doi: 10.1089/hum.2009.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7(12):1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 44.Alexander J, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1(9):751–761. doi: 10.1016/S1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 45.Engels B, et al. Retroviral vectors for high-level transgene expression in T lymphocytes. Hum Gene Ther. 2003;14(12):1155–1168. doi: 10.1089/104303403322167993. [DOI] [PubMed] [Google Scholar]
- 46.Leisegang M, et al. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J Mol Med. 2008;86(5):573–583. doi: 10.1007/s00109-008-0317-3. [DOI] [PubMed] [Google Scholar]
- 47.Anders K, et al. Oncogene-targeting T cells reject large tumors while oncogene inactivation selects escape variants in mouse models of cancer. Cancer Cell. 2011;20(6):755–767. doi: 10.1016/j.ccr.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA, Pérarnau B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. J Exp Med. 1997;185(12):2043–2051. doi: 10.1084/jem.185.12.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.