

Abstract
Levothyroxine (LT4) is a form of thyroid hormone used to treat hypothyroidism. In the brain, T4 is converted to the active form T3 by type 2 deiodinase (D2). Thus, it is intriguing that carriers of the Thr92Ala polymorphism in the D2 gene (DIO2) exhibit clinical improvement when liothyronine (LT3) is added to LT4 therapy. Here, we report that D2 is a cargo protein in ER Golgi intermediary compartment (ERGIC) vesicles, recycling between ER and Golgi. The Thr92-to-Ala substitution (Ala92-D2) caused ER stress and activated the unfolded protein response (UPR). Ala92-D2 accumulated in the trans-Golgi and generated less T3, which was restored by eliminating ER stress with the chemical chaperone 4-phenyl butyric acid (4-PBA). An Ala92-Dio2 polymorphism–carrying mouse exhibited UPR and hypothyroidism in distinct brain areas. The mouse refrained from physical activity, slept more, and required additional time to memorize objects. Enhancing T3 signaling in the brain with LT3 improved cognition, whereas restoring proteostasis with 4-PBA eliminated the Ala92-Dio2 phenotype. In contrast, primary hypothyroidism intensified the Ala92-Dio2 phenotype, with only partial response to LT4 therapy. Disruption of cellular proteostasis and reduced Ala92-D2 activity may explain the failure of LT4 therapy in carriers of Thr92Ala-DIO2.
Keywords: Endocrinology, Metabolism
Keywords: Behavior, Cell stress, Thyroid disease
Introduction
Hypothyroidism results from autoimmune destruction or surgical removal of the thyroid gland. Hence, symptoms are due to insufficient levels of thyroid hormones, which affects tens of millions worldwide (1). T4 is the main hormone secreted by the normal thyroid gland, but it needs to be converted to T3 in order to gain full biological activity (2, 3). In healthy individuals, the thyroid gland contributes only a small fraction of daily T3 production (<20%); the bulk of T3 production happens outside the thyroid parenchyma via conversion from T4 by 2 deiodinases, D1 and D2. Thus, therapy consisting of daily tablets of levothyroxine (LT4) to treat hypothyroidism is commonsensical, having evolved to be the standard of care for this disease (1, 4).
About 15% of LT4-treated hypothyroid patients remain symptomatic, with impaired cognition and reduced physical activity despite appropriate treatment (5). The origin of these symptoms is not clear, with some speculating that LT4 therapy is not sufficient for all patients; thus, symptoms may be due to residual hypothyroidism. In fact, LT4-treated hypothyroid patients lack thyroidal T3 secretion; all T3 is produced in tissues other than the thyroid gland, mostly via D2. While it has been assumed that deiodinases restore T3 homeostasis in LT4-treated patients (6), preclinical and clinical studies indicate that this pathway alone is not sufficient to fully restore daily T3 production during LT4 therapy (7–10).
Thyroidectomized rats on LT4 exhibit hypothyroidism in the liver, skeletal muscle, and brain, despite normal serum thyroid-stimulating hormone (TSH) levels (8). Individuals on LT4 with normal serum TSH exhibit higher BMI and tend to experience greater use of beta blockers, statins, or antidepressant medication (9) and lower energy expenditure (11); they also exhibit difficulty in weight management, fatigue, or low energy levels and problems with mood and memory (12). The brain, in particular, depends on T3 produced via the D2 pathway, which is located in glial cells. Indeed, most T3 bound to nuclear thyroid hormone receptors (TR) in the brain is produced locally via the D2 pathway (13). This occurs via a paracrine-signaling mechanism in which glial cell–derived T3 activates neuronal gene expression (14). Thus, adequate D2 functionality is critical in LT4 treatment for hypothyroid patients, producing most circulating T3 and also directly affecting intracellular T3 levels.
D2 is a type I ER-resident protein, which enables D2-generated T3 to access and bind TRs (3). D2 is atypical among the deiodinases in that it displays a relatively short half-life as a result of ubiquitination and proteasomal degradation. This is caused by an exclusive 18-residue loop that mediates binding to 2 ubiquitin ligases (15–17). This loop also harbors a Thr92-to-Ala substitution (Ala92-D2) caused by a SNP present in 12%–36% of the population (18). Not surprisingly, the single amino acid substitution slows down the rate of D2 turnover; it is also associated with ectopic presence of D2 in the Golgi apparatus (19). Clinically, carriers of the Thr92Ala-DIO2 polymorphism were found more likely to have hypertension, insulin resistance, type 2 diabetes, bipolar disorder, mental retardation, low IQ, susceptibility to lung injury, osteoarthritis, Alzheimer’s disease (20), and increased bone turnover, but perhaps unsurprisingly, these associations have not been reproduced in all population studies (6, 21). It is intriguing, however, that carriers of the Thr92Ala polymorphism in the D2 gene (DIO2) exhibited symptomatic improvement when liothyronine (LT3) was added to LT4 therapy (22, 23), suggesting that Thr92Ala-DIO2 carriers do not produce sufficient amounts of T3 via D2. Indeed, 2 studies suggest that Thr92Ala-D2 is less catalytically active (24, 25), potentially explaining why only a small percentage of the LT4-treated hypothyroid patients exhibit residual symptoms.
Here, we studied the Thr92Ala polymorphism in cell and animal models, having found that D2 is a cargo protein in ER Golgi intermediary compartment (ERGIC) vesicles, recycling between ER and Golgi. Ala92-D2 causes ER stress and activates the unfolded protein response (UPR) in different brain areas. Our findings suggest that disruption of cellular proteostasis and reduced Ala92-D2 catalytic activity explain the failure of LT4 therapy in carriers of the Thr92Ala-DIO2 polymorphism; the data support a need for further studies with chemical chaperones and combination therapy with LT3 in humans.
Results
To understand how Thr92Ala-DIO2 affects D2, we studied HEK-293 cells stably expressing either form of D2 double tagged with His and yellow fluorescent protein (YFP) (19). Thr92-D2HY distributed predominantly to the ER with low-level colocalization with the cis-Golgi marker GM130 (Figure 1, A–C) and the trans-Golgi marker p230 (Figure 1, D–F). Whereas Ala92-D2HY also exhibited low-level colocalization with the cis-Golgi marker (Figure 1, G–I), it was clearly present in the trans-Golgi (Figure 1, J–L).
Figure 1. D2 recycles between Golgi apparatus and ER.

(A–F) Immunofluorescence of Thr92-D2HY stably expressing cells using the indicated antibodies. On the far right is the Pearson’s plot for each immunofluorescence image. The right top number is the Pearson’s coefficient for that specific cell. Pearson’s coefficient was calculated as follows: Thr92-DIO2 × α-GM130 (0.32 ± 0.06); Ala92-DIO2 × α-GM130 (0.36 ± 0.07). (G–L) Same as A–F, except that cells stably express Ala92-D2HY. Arrows point to Golgi AlaD2 staining. Pearson’s coefficient: Thr92-DIO2 × α-p230 (0.33 ± 0.05); Ala92-DIO2 × α-p230 (0.65 ± 0.12; P < 0.01 versus Thr92-DIO2 × α-p230). (M and N) Thr92-D2HY (Thr) or Ala92-D2HY (Ala) pulldown, followed by Western blot analysis with the indicated antibodies. (O) Same as M and N, except that cells transiently express Δ18-D2HY. (P–R) Same as A–C except that cells transiently express Δ18-D2HY. (S) Pearson’s coefficient between the indicated D2 proteins and cis-Golgi marker GM130; Δ18-D2 is Δ18-D2HY (P–R). D2T is Thr92-D2HY (A–C). ΔC-D2T is Δ10C-Thr92-D2HY (T–V). ΔC-D2A is Δ10C-Ala92-D2HY (W–Y). (T–Y) Same as A–C, except that cells transiently express Δ10C-Thr92-D2HY or Δ10C-Ala92-D2HY. Original magnification, APO ×60/1.40 oil objective. Values are shown in box-and-whiskers plot indicating median and quartiles. n = 21/group. Statistical analysis used was Mann-Whitney U test in comparison with D2T. ***P ≤ 0.0001.
A network of ERGIC vesicles exists that shuttles proteins back and forth between ER and cis-Golgi, namely the COPII and COPI vesicles (26, 27). COPII vesicles recognize, concentrate, and export ER proteins to the Golgi (28) based on the presence of export signals, none of which can be found in D2. Other proteins are concentrated in COPII vesicles via specific transport adaptors (29). To find out whether D2 traffics through this network, we analyzed pulldowns of both Thr92-D2HY and Ala92-D2HY. While we failed to identify COPII proteins (data not shown), these pulldowns contained the transport adaptor ERGIC53 (30) along with p97 and UBXD-1. The ATP-driven chaperone p97 is involved in quality control and uses adaptors to process ubiquitinated proteins for recycling or degradation (31). For example, p97 and the adaptor Atx-3 bind to and direct D2 to the proteasomes (32). Here, we saw that both Thr92-D2HY and Ala92-D2HY pulldowns contained the p97 adaptor UBXD-1, known to bind ERGIC53 (33) (Figure 1, M and N). Thus, both forms of D2 can be directed to the ERGIC via interaction with the p97/UBXD-1/ERGIC53 complex. The 18-residue loop in D2 (15) is also critical for interaction with p97/UBXD-1: its truncation (Δ18-D2HY) prevents ERGIC53 pulldown (Figure 1O) and reduces by 30% colocalization with GM130 (Figure 1, P–S).
Scanning the D2 sequence led us to 2 carboxyl target dibasic peptide ER-retrieval motifs, i.e., SKRUKKTR, where underlines indicate dibasic peptides (34, 35). Indeed, truncation of this sequence (Δ10C-ThrD2HY or Δ10C-AlaD2HY) increased colocalization with GM130 by approximately 50% (Figure 1, S–Y). Together, these studies indicate that a bidirectional traffic of D2 between ER and Golgi exists, with both Thr92-D2HY and Ala92-D2HY recycling between ER and Golgi. These observations, however, do not explain why or how only Ala92-D2HY accumulates in the trans-Golgi network.
ER stress pushes Ala92-D2HY to the trans-Golgi.
Ala92-D2HY exhibits a longer half-life (19), an indication that the Ala92-D2 slows D2 targeting to the proteasome and could lead to its detrimental accumulation in the ER. Indeed, we detected UPR in Ala92-D2HY cells (Figure 2, A–N). In these cells, there is mostly activation of the IRE1α and ATF6 pathways, which increase protein-folding ability via chaperones and degradation of misfolded proteins. There was an approximately 2-fold increase in IRE1α phosphorylation (Figure 2, A and B) and an approximately 3-fold increase in spliced XBP1 (sXBP1) mRNA levels (Figure 2C) in cells expressing Ala92-D2HY. While levels of uncleaved ATF6 protein remained stable (Figure 2D), there was an approximately 2-fold increase in cleaved ATF6 protein (Figure 2, D and E) and the mRNA levels of its downstream targets CHOP (~3-fold; Figure 2F) as well as the ER chaperone/folding protein BIP (~4-fold; Figure 2, G–I). PERK phosphorylation (Figure 2, J and K), which attenuates protein synthesis, as well as its downstream targets, such as EIF2α phosphorylation (Figure 2, L and M) and ATF4 mRNA levels, was not affected (Figure 2N).
Figure 2. Expression of Ala92-D2 causes ER stress, triggers UPR response.

UPR markers in Thr92-D2HY–expressing (Thr) or Ala92-D2HY–expressing (Ala) cells. (A) Western blot of pIRE1α. (B) Quantification of pIRE1α shown in A. (C) sXBP1 mRNA levels. RQ, relative quantification. (D) Western blot of uncleaved (uc) and cleaved (c) ATF6. (E) Quantification of cATF6 shown in D. (F) CHOP mRNA levels. (G) Western blot of BIP. (H) Quantification of BIP shown in G. (I) BIP mRNA levels. (J) Western blot of pPERK. (K) Quantification of pPERK shown in J. (L) Western blot of pEIF2α. (M) Quantification of pEIF2α shown in L. (N) ATF4 mRNA levels. (O) Western blot of ERGIC53. (P) Quantification of ERGIC53 shown in O. (Q) ERGIC53 mRNA levels. (R–W) Immunofluorescence of Thr92-D2HY or Ala92-D2HY stably expressing cells using the indicated antibodies. Original magnification, APO ×60/1.40 oil objective. Values are shown in a box-and-whiskers plot indicating median and quartiles. n = 4–5/group. Statistical analysis used was the Mann-Whitney U test or Kruskall-Wallis test followed by the Dunn’s multiple comparison test. *P ≤ 0.05; **P ≤ 0.01.
In some settings, the Golgi quality control contributes to UPR by capturing misfolded proteins that emanate from the ER, diverting them for lysosomal degradation (36). In fact, it is known that cleaved ATF6 induces ERGIC53 expression (37) and its redistribution so that it is closer to the cis-Golgi (38), both of which are features present in cells expressing Ala92-D2HY. Here, we found a 2.2-fold increase in ERGIC53 protein levels (Figure 2, O and P) and a 50% increase in ERGIC53 mRNA levels (Figure 2Q). The images confirm an increase in ERGIC53 protein (Figure 2, R and S), revealing a 35% higher colocalization with GM130 (Figure 2, T–W) and indicating redistribution to cis-Golgi. Furthermore, colocalization between Ala92-D2HY and GM130 was approximately 30% lower in ERGIC53–/– cells (Figure 3, A–H), confirming that ERGIC53 is involved in the distribution of Ala92-D2HY to the Golgi.
Figure 3. ERGIC53 and SCAP play a role in Ala92-D2 trafficking.

(A) Western blot of control HEK-293 and HEK-293-ERGIC–/– cells using the indicated antibodies. (B–D) Immunofluorescence of control HEK-293 cells transiently expressing Ala92-D2HY using the indicated antibodies; on the far right is the Pearson’s plot for each immunofluorescence image. The right top number is the Pearson’s coefficient for that specific cell. (E–G) Immunofluorescence of HEK-293-ERGIC–/– cells transiently expressing Ala92-D2HY; arrows point to Golgi AlaD2 staining. (H) Pearson’s coefficient between the indicated D2 proteins and cis-Golgi marker GM130. (I) FRET in HEK293 cells transiently expressing either D2T-EYFP (Thr) or D2A-EYFP (Ala) and SCAP constructs containing the YFP and CFP fluorophores at the indicated positions; cells were treated with HPβCD to cause cholesterol deprivation, as indicated. (J) Same as I, except that SCAP-ΔWD40 was used. (K–P) Immunofluorescence of Thr92-D2HY- or Ala92-D2HY-expressing cells, which were incubated overnight with 500 μM 4-PBA. (Q) Pearson’s coefficient between the indicated D2 proteins and trans-Golgi marker p230 as shown in K–P. (R–V, X) same as K–P, except the cells were incubated overnight with 10 μg/ml cholesterol. (W) Same as in Q, except that data are from R–V, X. Original magnification, APO ×60/1.40 oil objective. Values are shown in a box-and-whiskers plot indicating median and quartiles. n = 9–69/group. Statistical analysis used was the Mann-Whitney U test or the Kruskall-Wallis test, followed by Dunn’s multiple comparison test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
The 18-residue loop in D2 mediates binding to the WD40 domain in WSB1, a D2 ubiquitin ligase (15, 16, 39). Typically, WD40 domains exhibit low levels of sequence conservation, making them promiscuous binding partners (40). Therefore, we tested to determine whether Ala92-D2HY interacts with other ER WD40-containing proteins that could provide an additional exit route to the Golgi. The SREBP cleavage-activating protein (SCAP) is a WD40-containing ER cholesterol sensor that transports SREBP1 from the ER, via ERGIC (41), to the Golgi (42), a pathway that is activated by ER stress (43, 44). Fluorescence resonance energy transfer (FRET) studies in live cells indicated that Ala92-D2HY, but not Thr92-D2HY, interacted with SCAPECFP (ECFP in the carboxyl end) in cells depleted of cholesterol (Figure 3I) through a mechanism that required the WD40 domain in SCAP (Figure 3J).
To find out how much the presence of Ala92-D2HY in the trans-Golgi depends on ER stress and/or SCAP activation, we explored whether overnight incubation of Ala92-D2HY cells with (a) the chemical chaperone 4-phenyl butyric acid (4-PBA) or (b) cholesterol excess affected colocalization of Ala92-D2HY and p230. Indeed, exposure to 4-PBA normalized UPR markers (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI123176DS1) and brought colocalization of Ala92-D2HY and p230 to the levels observed in Thr92-D2HY cells (Figure 3, K–Q). At the same time, saturation with cholesterol reduced p230/Ala92-D2HY colocalization by approximately 20% (Figure 3, R–X). These data strengthen the argument that distribution of Ala92-D2HY to the trans-Golgi is caused by ER stress and could involve the SCAP pathway.
Brain UPR in a mouse carrier of the Thr92Ala-Dio2 polymorphism.
Five of the 6 critical amino acids in D2’s 18-residue loop are conserved (17) in humans (92-TEGGDN-97) and mice (92-PESGNN-97): 93-E, 95-G, and 97-N are the same, and 96-D/N is a conserved substitution. Because P occupies position 92 in murine D2, Thr92-Dio2 and Ala92-Dio2 mice were created using a CRISPR/Cas9 system on a B6 background. Ala92-Dio2 mice bred and grew normally (Supplemental Figure 1A), exhibiting normal circulating levels of TSH, T4, and T3 (Supplemental Figure 1B) and D2 kinetics with a Km(T4) of approximately 1.9 nM (Supplemental Figure 1C). No differences were observed in body weight and food intake (Supplemental Figure 1, A and D). Systemic euthyroidism was further documented by absence of hypothyroid-like features in the Ala92-Dio2 skeleton (Supplemental Figure 1, E–V).
Discrete brain areas were studied using an unbiased whole-transcript microarray approach (Ala92-Dio2 homozygotes versus Thr92-Dio2 homozygotes; Supplemental Tables 2–11). Midline 1 (Mid1), which encodes a microtubule-associated protein with E3 ubiquitin ligase activity involved in the Opitz syndrome (45), was the only common gene differentially expressed in all 5 brain areas: 2.4- to 3.7-fold lower mRNA levels in the Ala92-Dio2 mouse brain (P < 0.001). Gene set enrichment analysis (GSEA) (P < 1%; Supplemental Tables 12–21) identified several gene sets related to ERGIC, Golgi, or the ubiquitin proteasome system in the amygdala (Supplemental Table 12), cerebellum (Supplemental Table 14), prefrontal cortex (Supplemental Table 18), and striatum (Supplemental Table 20), including the top 2 gene sets in the prefrontal cortex, which were ERGIC related (Supplemental Table 18). The more sensitive real-time quantitative PCR (RT-qPCR) indicated widespread elevation of Bip mRNA levels (Table 1). Other UPR-related genes were also activated in the cortex, particularly in the Atf6 pathway (Table 1), and increased levels of cleaved ATF6 in sonicates of Ala92-Dio2 cortex (Figure 4, A and B); sXbp1 mRNA levels were also elevated by 40% in the cortex of Ala92-Dio2 mice (Table 1). The amygdala was particularly affected, with 51% elevation in Bip, 54% in Ergic53 and Pdi, and 59% in sXbp1 mRNA levels (Table 1).
Table 1. ER stress–related gene mRNA in brain areas of Thr92-Dio2 and Ala92-Dio2 mice.

Figure 4. ER stress in Ala92-Dio2 cerebral cortex and slower rate of T4 to T3 conversion in Ala92-D2 expressing cells.

(A) Western blot of cerebral cortex sonicates of Thr92-Dio2 (Thr) and Ala92-Dio2 (Ala) mice utilizing the indicated antibodies; each lane represents an independent mouse sample. (B) Quantification of uncleaved and cleaved ATF6. (C) In vivo deiodination in intact Thr92-D2HY–expressing (Thr) or Ala92-D2HY–expressing (Ala) cells. (D) Immunoprecipitation followed by Western blot of T and A cells utilizing α-YFP and α-cyclophilin B. (E) In vitro deiodination in T and A cell sonicates. (F) Same as C, except that cells were treated for 24 hours with 500 μM 4-PBA or 10 μg/ml cholesterol. Values are shown in a box-and-whiskers plot indicating median and quartiles or mean ± SEM. n = 3–10/group. Statistical analysis used was the Mann-Whitney U test or the Kruskall-Wallis test, followed by the Dunn’s multiple comparison test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Slower rate of T4-to-T3 conversion in intact cells expressing Ala92-D2HY.
The in vitro kinetics (Km[T4]) of Ala92-D2HY and Thr92-D2HY are indistinguishable in cell sonicates (25). However, here, we saw that in intact cells, the ability of D2 to convert T4 to T3 was diminished by the Thr92Ala substitution (Figure 4C). Ala92-D2HY cells incubated with 10 pM (physiological) or 50 pM free 125I-T4 produced approximately 30% less T3 over 24 hours when compared with Thr92-D2HY cells. At the same time, Ala92-D2HY protein levels (Figure 4D) and activity (Figure 4E) in cell sonicates were indistinguishable from those of Thr92-D2HY sonicates. These contrasting results could reflect limited availability or efficacy of the in vivo cofactor; when assayed in vitro, cells were broken and cofactor was abundant in the form of DTT. To determine whether Golgi distribution affects Ala92-D2HY activity, we measured T3 production in intact Ala92-D2HY cells that had been treated overnight with 4-PBA or cholesterol (as in Figure 3, K and W). Treatment with 4-PBA increased the T3 production rate in Ala92-D2HY cells to the level observed in Thr92-D2HY cells (Figure 4F). The effect of cholesterol saturation was less clear, as T3 production in cholesterol-treated Ala92-D2HY cells remained below control levels (Figure 4F).
There is localized hypothyroidism in the Ala92-Dio2 brain.
To study T3-TR signaling in the different brain areas, 2 custom-made gene sets containing 33 T3-responsive genes were prepared for GSEA: (a) 19 positively regulated genes (Supplemental Table 22) and (b) 14 negatively regulated genes (Supplemental Table 23). There is evidence of reduced T3-TR signaling in the Ala92-Dio2 striatum (Supplemental Figure 2): positively T3-regulated genes were enriched in the Thr92-Dio2 brain, whereas negatively T3-regulated genes were enriched in the Ala92-Dio2 brain. These striatal differences were confirmed by measuring a positively and a negatively T3-regulated gene, i.e., Ier5 and Odf4 via RT-qPCR (Supplemental Table 24). In the Ala92-Dio2 amygdala and prefrontal cortex, only the positively T3-regulated genes were affected (Supplemental Figure 2). In the hippocampus and cerebellum, differences between Thr92-Dio2 and Ala92-Dio2 animals were observed at the FDR q value level (0.15–0.24), but not at the much stricter P value level (Supplemental Figure 2).
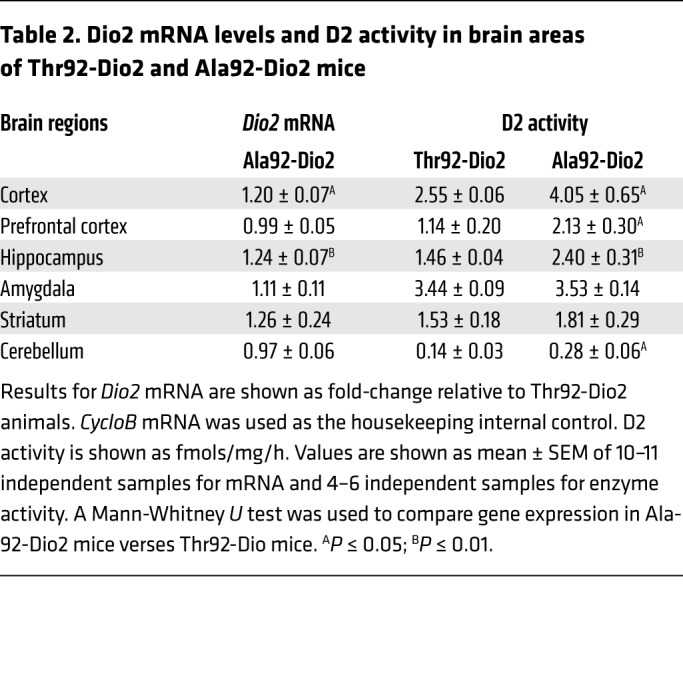
A component of the brain adaptation to hypothyroidism is a regional-specific increase in D2 activity via transcriptional and posttranscriptional mechanisms that mitigates the effects of hypothyroidism on the brain (46). Indeed, the Ala92-Dio2 mouse exhibits a 1.6- to 2.0-fold increase in Dio2 mRNA and D2 activity in prefrontal cortex, hippocampus, and cerebellum, whereas in the striatum and amygdala — the 2 areas that exhibit decreased T3-TR signaling — such a response was not observed (Table 2).
Table 2. Dio2 mRNA levels and D2 activity in brain areas of Thr92-Dio2 and Ala92-Dio2 mice.
Ala92-Dio2 mice are sensitive to an anxiogenic environment.
The open-field (OF) test was performed under low-intensity red light to minimize anxiety (Supplemental Figure 3, A–F). Ambulatory activity was not different (Supplemental Figure 3, B and C), but exploratory activity was higher in Ala92-Dio2 mice (Supplemental Figure 3, D and E). Moving the animals to the elevated plus maze (EPM), a typical anxiogenic platform, resulted in Ala92-Dio2 mice exhibiting higher ambulatory/exploratory activities (Supplemental Figure 3, G–L) and more risk-assessment behavior (Supplemental Figure 3M) when compared with Thr92-Dio2 mice. The pattern of higher mobility in Ala92-Dio2 mice was maintained during the highly anxiogenic tail-suspension (TS) studies (Supplemental Figure 3, N and O).
Once settled, Ala92-Dio2 mice refrain from physical activity and exhibit sleepiness and impaired memory.
The hypersensitivity to the environment prompted us to study Ala92-Dio2 mice in their home cages, while they were housed in a comprehensive lab animal monitoring system (CLAMS) and monitored 24 hours a day. Once settled in an environment of low anxiety, Ala92-Dio2 mice traveled approximately 30% shorter distances, exhibited an approximately 9% slower nightly oxygen consumption (VO2) rate, and slept approximately 4.2 times longer when compared with Thr92-Dio2 mice (Tables 3 and 4). This was not due to an impaired motor function, given that in separate experiments, mice ran on a treadmill for approximately 21 minutes and reached speeds of approximately 28 m/min, regardless of genotype. These measurements were repeated while animals had voluntary access to a spinning wheel for 3 days. Ala92-Dio2 mice used the wheel about half as much as Thr92-Dio2 mice (Tables 3 and 5), with a reduced nightly VO2 rate during the same time period (Tables 3 and 5). Despite the exercise stimulus, Ala92-Dio2 mice continued to travel approximately 15% less and slept 3.6 times longer compared with Thr92-Dio2 mice (Tables 3 and 5).
Table 3. Metabolic parameters in Thr92-Dio2 and Ala92-Dio2 mice.

Table 4. Summary of observed differences in metabolic parameters between Ala92-Dio2 and Thr92-Dio2 mice undergoing the indicated treatments.
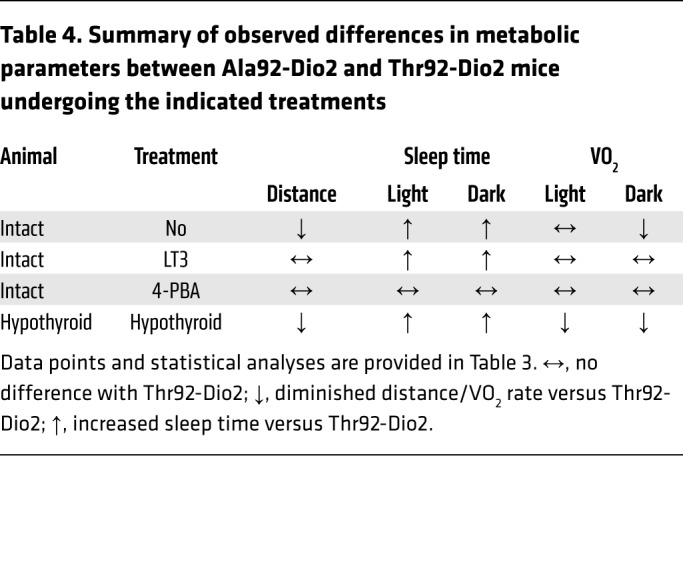
Table 5. Summary of observed differences in metabolic parameters with free access to spinning wheels between Ala92-Dio2 and Thr92-Dio2 mice undergoing the indicated treatments.

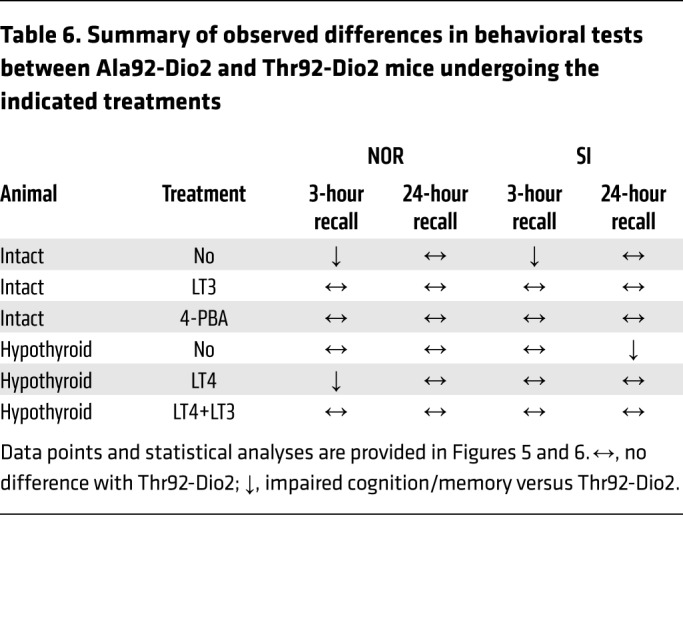
Cognition was tested through the novel object recognition (NOR) and the valence-based social interaction (SI) tests, which rely on exploratory behavior and assess memory and preference for novelty. In the NOR and SI tests, animals of both genotypes explored the objects/subjects similarly during the familiarization period (Supplemental Figures 4 and 5), with Thr92-Dio2 mice passing the 3-hour recall by focusing preferentially on the new object (Figure 5A). In contrast, the Ala92-Dio2 mice failed the 3-hour recall test, dedicating similar amounts of time to old and new objects (Figure 5A). Later, both Ala92-Dio2 and Thr92-Dio2 mice did well on the second recall 24 hours later (Figure 5B). The SI test uses conspecific animals, and thus, memory formation is strengthened by emotionally salient experiences. While Thr92-Dio2 mice passed the 3-hour and 24-hour recalls (Figure 6, A and B), Ala92-Dio2 mice barely passed the 3-hour recall (statistically borderline; Figure 6A), but did well on the 24-hour recall (Figure 6B).
Figure 5. Impaired new object recognition in Ala92-Dio2 mouse.

NOR memory test displayed as preference index (%) of Thr92-Dio2 (Thr) and Ala92-Dio2 (Ala) mice. (A and B) Intact animals. N, new object; O, old object. (C and D) Intact+LT3 animals. (E and F) Intact+4-PBA animals. (G and H) Hypothyroid animals. (I and J) Hypothyroid+LT4 animals. (K–L) Hypothyroid+LT4+LT3 animals. (A, C, E, G, I, and K) 3-hour recall. (B, D, F, H, J, and L) 24-hour recall. Values are shown in a box-and-whiskers plot indicating median and quartiles. Statistical analysis used was Mann-Whitney U test. n = 5–11/group. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Figure 6. Impaired new social recognition in Ala92-Dio2 mouse.

SI memory test displayed as preference index (%) of Thr92-Dio2 (Thr) and Ala92-Dio2 (Ala) mice. (A and B) Intact animals. (C and D) Intact+LT3 animals. (E and F) Intact+4-PBA animals. (G and H) Hypothyroid animals. (I and J) Hypothyroid+LT4 animals. (K–L) Hypothyroid+LT4+LT3 animals. (A, C, E, G, I, and K) 3-hour recall. (B, D, F, H, J, and L) 24-hour recall. Values are shown in a box-and-whiskers plot indicating median and quartiles. Statistical analysis used was Mann-Whitney U test. n = 5–11/group. **P ≤ 0.01; ***P ≤ 0.001.
Short-term treatment with LT3 partially rescues the Ala92-Dio2 mouse phenotype.
In an attempt to increase T3-TR signaling and rescue the Ala92-Dio2 mouse phenotype, Thr92-Dio2 and Ala92-Dio2 mice were treated with LT3 (1 μg/d for 10 days); as expected, serum TSH became suppressed (Table 3). LT3 dissipated differences in distance traveled and VO2 rate, but Ala92-Dio2 mice continued to sleep approximately 2 times longer when compared with Thr92-Dio2 mice (Tables 3 and 4). During the 3-day exercise period, Ala92-Dio2 mice traveled around the cage as much as Thr92-Dio2 mice, while exhibiting a similar VO2 rate, but still slept approximately 2.1 times longer (Tables 3 and 4) and used the spinning wheel about half as much as the Thr92-Dio2 mice (Tables 3 and 5). LT3 improved performance of Ala92-Dio2 mice on the NOR (Figure 5, C and D) and SI tests (Figure 6, C and D), although LT3-treated Ala92-Dio2 mice continued to perform more poorly than Thr92-Dio2 mice in the 3-hour recall (Figure 5C). As before, no differences between genotypes were observed at the 24-hour recall (Figure 5D). Performance of both groups of LT3-treated animals was indistinguishable during the 3-hour and 24-hour recalls in the SI test (Figure 6, C and D).
Treatment with 4-PBA rescues the Ala92-Dio2 mouse phenotype.
In order to evaluate how much of the Ala92-Dio2 mouse phenotype is due to ER stress and UPR activation, intact Thr92-Dio2 and Ala92-Dio2 mice were given 4-PBA (0.25 g/kg BW/d for 20 days) to restore proteostasis; 4-PBA normalized UPR markers in different brain areas (Supplemental Table 25). 4-PBA–treated Ala92-Dio2 animals traveled distances that were similar to those of Thr92-Dio2 mice and slept similarly (Tables 3 and 4). VO2 rate was also not different between groups (Tables 3 and 4). Similarly, during the 3-day exercise period, Ala92-Dio2 mice traveled in the home cages as much as Thr92-Dio2 mice (Tables 3 and 5). A dramatic effect of 4-PBA treatment was to dissipate differences in utilization of the spinning wheel and VO2 rate between Ala92-Dio2 and Thr92-Dio2 mice (Tables 3 and 5). Treatment with 4-PBA also normalized performance of Ala92-Dio2 mice on the NOR (Figure 5, E and F) and SI tests (Figure 6, E and F).
Differences between Thr92-Dio2 and Ala92-Dio2 mice remain during hypothyroidism.
After 5 to 6 weeks on methimazole (MMI), all animals developed overt hypothyroidism, with high serum TSH levels (Table 3). Hypothyroid Ala92-Dio2 mice traveled approximately 20% less as compared with hypothyroid Thr92-Dio2 mice and maintained a 7.5%–10% slower VO2 rate (Tables 3 and 4). Sleeping duration increased by approximately 460% in hypothyroid Thr92-Dio2 mice and by approximately 25% in Ala92-Dio2 mice, eliminating the differences between genotypes (Tables 3 and 4). Exercise in the spinning wheels stimulated motor activity in all animals, but hypothyroid Ala92-Dio2 mice continued to travel shorter distances in the cage (~50% less) when compared with Thr92-Dio2 mice (Tables 3 and 5). Notably, only in hypothyroid Thr92-Dio2 mice did exercise shorten the sleeping duration by half (Tables 3 and 5). Likewise, Ala92-Dio2 mice used the spinning wheel about half as much as Thr92-Dio2 mice (Tables 3 and 5) and exhibited slightly slower nightly VO2 rate (Tables 3 and 5). Cognition was markedly affected by hypothyroidism, regardless of genotype. All hypothyroid animals failed the 3-hour and 24-hour recall on the NOR test (Figure 5, G and H). While all animals failed the 3-hour recall on the SI test, only Thr92-Dio2 mice managed to pass the 24-hour recall (Figure 6, G and H).
Chronic treatment with LT4 minimally improves memory in hypothyroid Ala92-Dio2 mice; full response with LT4+LT3.
All hypothyroid animals were implanted with subcutaneous pellets to deliver stable amounts of LT4 or LT4+LT3 for an additional 6 weeks, which respectively normalized (Thr92-Dio2: 3.5 ± 0.45 versus Ala92-Dio2: 2.02 ± 0.69; n = 5–6) or suppressed serum TSH (Thr92-Dio2: 0.04 ± 0.002 versus Ala92-Dio2: 0.03 ± 0.001; n = 6). Treatment with LT4 eliminated the differences between Thr92-Dio2 and Ala92-Dio2 mice previously observed in home-cage assessment, including movement, sleeping time, and VO2 rate (Supplemental Table 26). However, LT4 alone failed to fully restore performance in the memory tests. Thr92-Dio2 mice responded to LT4 by passing the NOR and SI 3-hour recall tests, but failed the 24-hour recall on both tests (Figure 5, I and J, and Figure 6, I and J). In contrast, Ala92-Dio2 mice did not respond to LT4 and failed the NOR test 3-hour and 24-hour recalls; only a partial improvement in the SI test 3-hour recall was observed in Ala92-Dio2 mice (Figure 5, I and J, and Figure 6, I and J). Notably, treatment with LT4+LT3 normalized memory scores in all animals, regardless of genotype (Figures 5, K and L, and Figure 6, K and L).
Discussion
Previous studies in which added N- or O-glycosylation sequences failed to direct Golgi-specific D2 glycosylation led to the conclusion that D2 is retained in the ER (17). Here, we specifically studied the Golgi apparatus, having found that Thr92-D2HY indeed resides in the ER, but also recycles between ER and cis-Golgi via ERGIC, keeping a minimal presence in the cis-Golgi (Figure 1, A–F). In contrast, Ala92-D2HY causes ER stress and can be easily visualized in the trans-Golgi (Figure 1, G–L). This occurs as a result of protective mechanisms that restored cellular proteostasis and increased expression of ERGIC53 and activation of the SCAP pathway.
A slower enzyme reactivation could explain the ensuing ER stress in Ala92-D2HY cells. Conformational changes in D2 during catalysis include formation of an intramolecular selenenylsulfide that requires cofactor-mediated resolution before the enzyme is ready for a new round of deiodination (47, 48). That Ala92-D2HY–expressing cells produce less T3 (Figure 4C) suggests that reactivation of Ala92-D2 is slower, given that Ala92-D2 exhibits similar Km(T4) (Supplemental Figure 1C), and in Ala92-D2HY cells, D2 protein (Figure 4D) and in vitro activity (Figure 4E) are preserved. Furthermore, an extended life of the oxidized selenenylsulfide linkage also explains the finding of the altered redox state in Ala92-D2HY cells (19). Therefore, Ala92-D2 is likely trapped in an altered molecular configuration (the selenenylsulfide) with slower catalytic activity, which in turn triggers ER stress and activates UPR to restore proteostasis. This is supported by the observation that the enhanced folding capacity in Ala92-D2HY cells provided by 4-PBA ends ER stress/UPR (Supplemental Table 1), reduces Ala92-D2HY in the trans-Golgi (Figure 3, K–Q), accelerates D2-mediated T3 production (Figure 4E), and dissipates most of the Ala92-Dio2 phenotype in vivo (Tables 3–6 and Figure 5).
Table 6. Summary of observed differences in behavioral tests between Ala92-Dio2 and Thr92-Dio2 mice undergoing the indicated treatments.
The buildup of Ala92-D2HY in the trans-Golgi likely results from binding to the p97/Ubdx1 protein complex that delivers Ala92-D2HY to ERGIC53 (33). Normally, Thr92-D2HY is retrotranslocated to the cytoplasm via interaction with the p97/ataxin-3 complex, deubiquitinated, and delivered to the proteasomes (32). However, the extended Ala92-D2HY half-life indicates that this process is impaired (19). Under ER stress, the 2- to 3-fold higher levels of ERGIC53 (Figure 2, O–Q) enhance the formation of the UBXD1/ERGIC53 complex (33) that binds to Ala92-D2HY/p97 (Figure 1, M and N), directing Ala92-D2HY to the Golgi instead of the proteasomes. The critical role played by ERGIC53 in this process is illustrated by findings in ERGIC53–/– cells, which exhibit less colocalization of Ala92-D2HY with the cis-Golgi marker GM130 (Figure 3, A–H). SCAP (Figure 3, I and J) might constitute an additional pathway, which is also activated by ER stress (43, 44). However, the fact that cholesterol saturation reduced Ala92-D2HY colocalization only slightly with the trans-Golgi marker p230 (Figure 3, R–X) suggests that this is a secondary mechanism.
There is ample evidence that ER stress/UPR (49) and disruption of T3-TR signaling (50) can interfere with neuronal function and lead to impaired cognition. For example, mice with astrocyte-specific inactivation of Dio2 are systemically euthyroid, but exhibit anxiety-depressive–like behavior due to generalized hypothyroidism in the brain (51). In addition, a transgenic mouse bearing a mutant TRβ1 is systemically euthyroid, but displays behavioral abnormalities, such as inattention, hyperactivity, and impulsivity, a phenotype that is dynamic and sensitive to environmental changes (52); this is reminiscent of our findings in the Thr92Ala-Dio2 mouse. That the Ala92-Dio2 brain exhibits hypothyroid areas was confirmed by the observation that short-term treatment with LT3 improved cognition (Table 6 and Figures 5 and 6) and physical activity (Tables 3, 4, and 5) as compared with that in controls. However, treatment with LT3 caused systemic thyrotoxicosis, as evidenced by suppressed serum TSH (Table 3), indicating that the clinical improvement with LT3 administration may carry side effects. The present studies, however, did not allow complete discrimination of how much of the Thr92Ala-Dio2 phenotype is due to localized brain ER stress/UPR, hypothyroidism, or both. In fact, in addition to restoring proteostasis, 4-PBA also restores Ala92-D2HY–mediated T3 production and may normalize T3-TR signaling in vivo (Figure 4F). The fact that treatment with LT3 markedly improved cognition (Figure 5, C and D, and Figure 6, C and D), whereas 4-PBA restored both motivation (Tables 3 and 4) and cognition (Figure 5, E and F, and Figure 6, E and F), indicates that the Thr92Ala-Dio2 phenotype is multifactorial and includes more than one area of the brain, with both ER stress and decreased T3-TR signaling playing an important role.
Despite the obvious phenotype of the Ala92-Dio2 mice, carriers of the Thr92Ala-DIO2 polymorphism are reportedly asymptomatic when surveyed through quality of life or thyroid-specific questionnaires (53). A phenotype has only been reported in connection with diagnosis and treatment for hypothyroidism (22, 23). In contrast with the Ala92-Dio2 mice, carriers of the Thr92Ala-DIO2 polymorphism have the potential to correct defects through lifelong learning and training, a phenomenon seen in animals (54) and individuals with neurodevelopmental disorders (55). However, it is conceivable that, once carriers of the Thr92Ala-DIO2 polymorphism become hypothyroid and are treated with LT4, these adaptive mechanisms are exhausted, bringing out a phenotype that is similar to the Ala92-Dio2 mice. For example, the approximately 10% lower serum T3 levels observed in adequately LT4-treated hypothyroid patients (7, 9, 56) could be the key element that tips the balance toward behavioral and cognitive dysfunction.
Hypothyroid Thr92-Dio2 mice developed the expected behavior and cognitive and metabolic phenotype that brought them closer to the Ala92-Dio2 phenotype (Tables 3, 4, and 5; Figure 5, G–H, and Figure 6, G–H). Notably, their responsiveness to chronic treatment with LT4 was variable. While the metabolic parameters responded similarly to LT4 regardless of the genotype, cognitive parameters were only partially improved (Figure 5, I and J, and Figure 6, I and J). Considering that hypothyroidism therapy is typically lifelong, further improvements with LT4 could have been observed with even longer observation times. Remarkably, treatment with LT4+LT3 normalized all memory scores in all animals (Figure 5, K and L, and Figure 6, K and L) at the expense of suppressing serum TSH.
In depth studies of carriers of the Thr92Ala-DIO2 polymorphism suggest subtle changes in thyroid hormone homeostasis, compatible with a slower D2 velocity (57). In addition, depending on race, carriers might be at a higher risk for brain degenerative disease, even as they keep a normal thyroid gland function (20). We found that African Americans (AAs) with Thr92Ala-DIO2 had approximately 1.3 times higher odds of developing Alzheimer’s disease or cognitive impairment not demented, whereas no association was seen in European Americans (EAs); microarray studies confirmed transcriptional patterns of Alzheimer’s disease in the temporal pole of AA carriers of the Thr92Ala-DIO2 polymorphism (20). AAs are known to have a 2- to 3-fold higher prevalence of cognitive impairment and Alzheimer’s disease (58), which could explain their greater susceptibility to the Thr92Ala-DIO2 polymorphism. In addition, the Thr92Ala-DIO2 polymorphism has been associated with increased BMI, insulin resistance (59), and greater risk for type 2 diabetes (18, 60). This suggests that Ala92-DIO2–induced ER stress/localized hypothyroidism could play a broader metabolic role, particularly in tissues that express DIO2, such as human skeletal muscle and brown adipose tissue. Indeed, ER stress is known for reducing insulin receptor signaling, linking obesity, insulin signaling, and diabetes (61).
We are hopeful that understanding these mechanisms will accelerate development of new therapeutic approaches for the millions of patients with hypothyroidism and provide justification for clinical studies to assess the utility of customization of thyroid hormone replacement therapy based on the patients’ Thr92Ala-DIO2 status. Utilization of 4-PBA in symptomatic hypothyroid patients is an interesting possibility that should be further investigated. In the meantime, given that some of the Ala92-Dio2 phenotype is reversed by treatment with LT3, albeit at the expense of serum TSH suppression, it would seem logical to explore further the sensible use of LT3 when treating hypothyroid carriers of the Thr92Ala-DIO2 polymorphism. Of course, the challenge is to deliver T3 in ways that provide stable plasma levels while maintaining serum TSH within normal range and avoiding the risks of systemic thyrotoxicosis.
Methods
Cell lines and treatments.
HEK-293 cells stably expressing Thr92-D2HY and Ala92-D2HY were described earlier (19). An HEK-293 cell line in which the ERGIC53 gene was inactivated (HEK-ERGIC53–/–) and the appropriate HEK-293 control cell line were a gift from Jay Yang (University of Wisconsin-Madison, Madison, Wisconsin, USA) (62). Unless specified otherwise, all cells were cultured in high-glucose DMEM (Life Technologies) supplemented with 10% FBS (Atlanta Biologicals) and studied at 70%–80% confluency. Whenever cells were incubated with defined amounts of T4 or 125IT-4, FBS in the medium was replaced with 0.1% BSA. As indicated, some cells were treated overnight with 100–500 μM PBA or 10 μg/ml cholesterol. Conversion of T4 to T3 was assessed by incubating cells plated on 6-well dishes (1 million cells/well) with 10 pM or 50 pM I125-T4 (300,000 cpm/well) and measuring 125I, which reflects T4 to T3 conversion, in media at 16 and 40 hours, (63).
FRET studies.
Hydroxypropyl-β-cyclodextrin (HPβCD; Sigma-Aldrich) and cholesterol (Sigma-Aldrich) were dissolved in cell media at, respectively, 1.5% and 625 μM (64). FRET was performed as described (65), using cells that were plated and transfected (X-tremeGENE HP, Roche) in 35 mm glass-bottom dishes (MatTek Co.). FRET measurement was performed on the second day after transfection using acceptor-photobleaching on a Nikon A1R laser-scanning confocal system in spectral detector mode equipped with a Tokai Hit stage top incubator and Supertech temperature controller. The following parameters were applied: 457 nm argon-laser for ECFP excitation and 464–500 nm range for detection; 514 nm argon lasers for EYFP excitation and 516–540 nm range for detection. Cells were treated with HPβCD-containing media 2 hours before imaging. Only cells that exhibited at least 80% bleach efficiency (decrease in EYFP intensity) were analyzed. Calculation of FRET efficiency was based on the increase of the cyan fluorescent protein (CFP) donor signal after photobleaching the YFP acceptor using the following equation: FRET = (ECFP(postbleach) – ECFP(prebleach))/ECFP(postbleach). Data normalization was performed by expressing FRET activity as a percentage of that of the ECFP-EYFP (C-Y) tandem construct (66). ECFP and EYFP (C and Y) monomers were used to determine background. The construct names reflect the relative position of fluorescent proteins within the fusion (e.g., D2T-EYFP indicates C-terminal YFP fusion to D2 carrying threonine in amino acid position 92). D2T-EYFP and D2A-EYFP were previously described (19, 39). SCAP-ECFP was generated by PCR amplification of human SCAP coding sequences (CDS) using the following primers: forward, 5′-ATTGCTCGAGCCACCATGACCCTGACTGAAAGGC-3′; reverse, 5′-CGGGATCCCGGTCCAGCTTCTCCAGCACAG-3′; fragment was inserted between XhoI-BamHI sites of pECPF-N1 (Clontech). ECFP-SCAP was generated using the following primers: forward, 5′-ATTGCTCGAGCTATGACCCTGACTGAAAGGC-3′; reverse, 5′-CGGGATCCCGTCAGTCCAGCTTCTCCAGC-3′; fragment was inserted between XhoI-BamHI sites of pECPF-C1 (Clontech). WD40 domain deletion was generated by exchanging the SCAP CDS in the previous constructs to WD40 domain–deleted fragments generated by PCR and using the following primers: SCAPΔWD-ECFP, forward, 5′-CGGGATCCCGCAGCTCCCCGCGCCTCCGCCG-3′; ECFP-SCAPΔWD, forward 5′-CGGGATCCCGGCTCAGCTCCCCGCGCCTCCGCCG-3′. D2T and D2A were generated by PCR amplification of human D2 from D2T-EYFP and D2A-EYFP, respectively, using the following primers: forward, 5′-GGAATTCATTATGGGCATCCTCAGCGTAGACTTGCTGATCA-3′; reverse, 5′-ATAAGAATGCGGCCGCTTAACCAGCTAATCTAGTTTTCTTACATCTCTTGCT-3′. The amplified fragments were then inserted into pCI-Neo between the EcoRI and NotI sites.
Immunofluorescence studies.
Cells were plated on poly-d-lysine–coated chamber slides; fixation, imaging, and colocalization studies were described earlier (67). After fixation in 4% paraformaldehyde (Electron Microscopy Science) for 20 minutes, cells were permeabilized with 0.3% Triton X-100 for 10 minutes and blocked with Fish Skin Gelatin (Biotium) for 1 hour. Primary antibodies were added at concentrations ranging from 0.5 to 5 μg/ml at 4°C overnight for 72 hours when necessary (p230 antibody). Primary antibody dilutions were as follows: 1 μg/ml αYFP (Rockland Immunochemicals, catalog 600-401-215); α-Golgi antibodies: 0.5 μg/ml α-GM130 (Cell Signaling, catalog 12480S) or 2 μg/ml p230 (BD Biosciences, catalog 611280). Secondary antibodies (Life Technologies) were incubated for 1 hour in the dark; cells were made into a slide using SlowFade Gold Mounting Medium (Life Technologies). Images were acquired using a Nikon Eclipse Ti microscope with a C1 confocal system using an APO ×60/1.40 oil objective, and 7 or more z-sections were captured by EZ-C1 software. The images were then analyzed by NIS-Element AR (Nikon Instruments) or ImageJ software (NIH).
Immunoprecipitation studies.
Cells were harvested, suspended in PBS with cOmplete Mini EDTA-free Protease Inhibitor (Roche) at 4°C, and lysed with 1% Triton X-100 at 4°C for 30 minutes. After lysis, the pH of lysate was adjusted to 7.5. The crosslinker dithiobis(succinimidyl propionate) (Thermo Fisher Scientific, Pierce) in DMSO was added to a final concentration of 5 mM, and cell lysates were incubated on ice for 3 hours. After crosslinking, Tris buffer (pH 7.4) was added to the final concentration of 100 mM and incubated on ice for 30 minutes for quenching. Cell homogenates were centrifuged for 10 minutes at 800 g, and supernatant fractions, which contained cytoplasm and membrane proteins, were collected. The supernatant was used for D2 immunoprecipitation. Rabbit αYFP (1 μg) (Rockland Immunochemicals, catalog 600-401-215) or control rabbit IgG (Sigma-Aldrich) was added with 30 μl of protein A agarose and incubated at 4°C overnight. After washing with 1 ml of 1% Triton X-100 three times, the beads were treated with SDS-sample buffer for Western blot. D2HY immunoprecipitation and Western blot were performed as described (32).
Western blot studies.
Western blot analyses utilized 30 μg of total protein or immunoprecipitate pellets. Proteins were resolved on a 4%–12% SDS-PAGE gel and transferred to a PVDF membrane (Immobilon-FL, Millipore), incubated with indicated antibodies overnight at 4°C and subsequently quantitated with the LiCOR Odyssey instrument with Odyssey Image Studio software using 2 different infrared channels. The following antibodies were used at indicated dilutions: 1:1000, p-PERK (Cell Signaling, catalog 3179S); 1:500, p-EIF2α (Thermo Fisher Scientific, catalog MAS-15133); 1:500, ATF6 (Santa Cruz Biotechnology Inc., catalog 166659); 1:1000, p-IRE1α (Cell Signaling, catalog 3294S); 1:1000, ERGIC53 (Sigma-Aldrich, catalog AV46614); 1:1000, p97 (Cell Signaling, catalog 2648S); 1:500, Ubxd1 (Abcam, catalog SAB2107374); 1:1000 Bip (Abcam, catalog Ab21685); 1:2500 Dio2 (Abcam, catalog Ab87527); when appropriate, a 1:1000 dilution of α-actin (Santa Cruz Biotechnology Inc., catalog SC-8432) and 1:2500 α–cyclophilin B (Abcam, catalog Ab16045) were used as housekeeping internal controls.
Animals.
Residue 92 in the murine Dio2 gene is a proline; thus, Applied Stemcell Inc. (Milpitas) was contracted for creation of Thr92-Dio2 and Ala92-Dio2 knockin mice, including generation of guide RNA and utilization of the CRISPR/Cas9 system on a C57BL/6 background (68, 69). Two Thr92-Dio2 and 3 Ala92-Dio2 founders were obtained; the genotype PCR products (~532 bp) of all F1 and F2 mice were sequenced, and only those that differed from PP C57BL/6 by the desired SNP at position 92 were bred further. All other animals were euthanized. Founders were mosaic males and were crossed with C57BL/6 WT females (Pro92-D2). For genotyping, DNA was extracted with the AccuStart II Mouse Genotyping Kit (Quanta Biosciences) and processed through PCR with forward (TGACCATCCTTTATATTGCCTGAC) and reverse (CGGTGCTTCTTAACCTCAAAAGA) primers. The PCR product was subjected to EcoNI restriction enzyme digestion. PCR and digestion products were resolved by electrophoresis: the Pro92 DNA was digested by EcoNI (~250 bp), whereas Thr92 and Ala92 were not (~532 bp). Heterozygotes (Thr92/Pro92 or Ala92/Pro92) displayed 2 distinct bands. Identity of PCR products was confirmed by sequencing. Heterozygote F1s (Thr92/Pro92 and Ala92/Pro92) were crossbred with littermate F1s of identical genotype. Mice were bred and maintained as homozygous Thr92-Dio2 and Ala92-Dio2 colonies. F3 or greater progeny were used for experiments. Unless otherwise specified, male, 8- to 10-week-old animals were used throughout experiments. We are aware that hypothyroidism is more prevalent in females. However, here we focused on males because mood and cognition tests are affected by estrous cycles (70–72). All animals were kept at room temperature (22°C) on a 12-hour dark/12-hour light cycle and maintained on a chow diet or low iodine diet, as indicated.
Studies in CLAMS.
Twenty-four–hour food intake was measured at the indicated times using the Oxymax feed scale device (Columbus Instruments). VO2 rate and locomotor activity were continuously monitored using the Oxymax system 5.35, CLAMS (Columbus Instruments). Locomotor activity was determined by X (front to back), Y (right to left), Z (rearing), and distance (ft). Animals were placed in the CLAMS with free access to food and water and allowed to acclimatize in individual metabolic cages for 48 hours. Subsequently, metabolic profiles were generated in successive 26-minute cycles over 48 hours with locked wheels and 36 hours with unlocked wheels (voluntary exercise). Sleep detection information was collected using the CLAMS-HC program. The application specifies a set of criteria based on animal activity and time that establishes a threshold below which the onset of a sleep event is triggered. Activity is sensed by the traditional IR beam method. Beam interruptions are scored as counts. The sleep detection parameters used were as follows: 4 epoch(s) × 60 seconds per epoch = 240 seconds, with an activity threshold = 0 counts. LT3 was given at 1 μg/d for 10 days. 4-PBA was given at 0.25 g/kg body weight/d for 20 days. Hypothyroidism was induced by adding MMI (0.05%) to the drinking water for 6 weeks. Some animals received a subcutaneous implant of an LT4 pellet (0.05 mg/pellet) or LT4+LT3 (0.025 mg/pellet T4 and 0.01 mg/pellet T3) that lasted 60 days.
Cognitive and behavioral testing.
All animals were studied through standard mood and behavior tests, OF (73), EPM (73), and TS (73). NOR assesses recognition memory by evaluating preference for objects based on their previous encounter with 2 identical objects or a new object. Results are expressed as a preference index (ratio of the amount of time spent exploring any one of the 2 objects over the total time spent exploring both objects × 100 [%]) (74). SI is performed similarly to NOR, except that animals are used instead of objects (75).
Thyroid function tests, tissue samples, and D2 activity assay.
Animals were euthanized by asphyxiation with CO2 followed by cervical dislocation. Blood was collected and serum levels of TSH, T4, and T3 measured using a MILLIPLEX Rat Thyroid Hormone Panel Kit (Millipore), and the assay was read on a BioPlex (Bio-Rad). Whole brain was obtained and dissected for further analyses; pituitary gland and brown adipose tissue were also obtained. Bone samples, as indicated, were harvested from 16-week-old male mice, and phenotyping studies were performed as described (76, 77). D2 activity was assessed in cerebral cortex sonicates from male Thr92-Dio2 and Ala92-Dio2 mice in the presence of 10 mM dithiotreitol, 0.25 M sucrose, and 1 nM 125I-(5′) T4 (PerkinElmer); 1 mM propylthiouracil was added to inhibit potential D1 activity and 10 nM T3 to saturate D3 (63).
Microarray studies.
RNA of 5 different brain regions of Thr92-Dio2 and Ala92-Dio2 mice was extracted with the RNeasy Mini Kit (QIAGEN) and processed for microarray at the Joslin Diabetes Center Genomics Core Laboratory (Boston, Massachusetts, USA). Gene expression was determined using the Clariom S Mouse Array (Affymetrix). Data were preprocessed using the Affymetrix Expression Console. Differential expression analysis was performed with the Affymetrix Transcriptome Analysis Console to identify genes with more than 2-fold or less than 2-fold change and a nominal P value of less than 0.05. Gene ontology analysis was used to determine differences in gene set enrichment (GSEA, Broad Institute, Cambridge, Massachusetts, USA) with an FDR of less than 25%, a NES of more than 1.5, and a nominal P value of less than 0.01. All original microarray data were deposited in the NCBI’s Gene Expression Omnibus database (GEO GSE119351).
RT-qPCR studies.
Total RNA was extracted from cell lines or brain areas (hippocampus, cerebellum, amygdala, prefrontal cortex, and cerebral cortex) using the RNeasy Lipid Tissue Mini Kit (QIAGEN) and quantified with a NanoDrop. From 0.5 to 1.0 μg was used for cDNA synthesis using the Transcriptor First-Strand cDNA Synthesis Kit (Roche). RT-qPCR conditions were as described (25). Primers were designed to span exon-exon and/or intron-spanning sequences or were previously validated (https://pga.mgh.harvard.edu/primerbank/). Sequences are listed elsewhere (Supplemental Table 27). Genes of interest were analyzed (StepOne; Applied Biosystems) using PerfeCTa SYBR Green FastMix, ROX (Quantabio). The melting curve was used to verify the specificity of the amplicon. Standard curves consisted of 4 to 5 points of serially diluted mixture of experimental and control group cDNA. Cyclophilin A (CycloA), cyclophilin B (CycloB), 18S rRNA, or β-actin was used as a housekeeping control. The coefficient of correlation was more than 0.99 for all curves, and the amplification efficiency ranged between 80% and 110%. Results were expressed as the ratio of test mRNA to housekeeping mRNA.
Statistics.
All data were analyzed by Prism software (GraphPad). Unless otherwise indicated, data are presented as mean ± SEM (tables) or through box-and-whiskers plots (figures). Mann-Whitney U test was used when the experiment contained 2 independent groups. In other cases, the Kruskall-Wallis test was used followed by the Dunn’s multiple comparison test. P < 0.05 was considered significant.
Study approval.
The IACUC at Rush University Medical Center approved all experiments (16-077 and 15-033).
Author contributions
SJ conducted in vitro experiments with cells, prepared all Western blots, prepared figures, and analyzed data. TLF and BMLCB conducted in vivo experiments,analyzed data, and prepared figures. GWF performed microarray analysis and PCRs. JPWDC and DLI performed the Km Dio2 experiments and dissected bone samples. EAM assisted with the development of polymorphic animals and dissection of bone samples. APB performed brain area PCRs. RRC performed exercise tests. PE and DN performed FRET experiments. MMB and MOR assisted in the analysis of behavioral phenotypes. VDL, NSM, KFC, and NCB performed bone experiments. JHDB and GRW performed bone experiments and revised the manuscript. CF and BG performed plasmid construction and FRET experiments and revised the manuscript. ACB planned and directed all studies and wrote the manuscript. BG contributed to the design of the Thr92-Dio2 and Ala92-Dio2 mice.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grants DK58538 and DK65055; EU H2020 THYRAGE no. 666869; the Hungarian Brain Research Program (2.0–2017-1.2.1-NKP-2017-00002); and a Wellcome Trust Joint Investigator Award (110141/Z/15/Z); Pró-Reitoria de Extensão (PROEX) 0653/2018; and the São Paulo Research Foundation (FAPESP) 2017/18277-0.
Version 1. 10/23/2018
In-Press Preview
Version 2. 12/03/2018
Electronic publication
Version 3. 01/02/2019
Print issue publication
Funding Statement
Support from NIDDK DK58538 and DK65055
EU H2020 “Thyrage” No. 666869, the Hungarian Brain Research Program 2.0 - 2017-1.2.1-NKP-2017-00002
The Wellcome Trust Joint Investigator Award 110141/Z/15/Z.
Footnotes
Conflict of interest: ACB is a consultant for Sentier LLC and Synthonics Inc.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(1):230–245.https://doi.org/10.1172/JCI123176.
See the related Commentary at Cognitive function in hypothyroidism: what is that deiodinase again?.
Contributor Information
Sungro Jo, Email: Sungro_Jo@Rush.edu.
Tatiana L. Fonseca, Email: tatianafonseca@uchicago.edu.
Gustavo W. Fernandes, Email: gustavo_fernandes@rush.edu.
Elizabeth A. McAninch, Email: Elizabeth_A_McAninch@rush.edu.
Anaysa P. Bolin, Email: anaysa.bolin@yahoo.com.br.
Rodrigo R. Da Conceição, Email: rodriguescontato1@hotmail.com.
Daniele L. Ignacio, Email: daniignacio@oi.com.br.
Péter Egri, Email: egri.peter.koki@gmail.com.
Dorottya Németh, Email: nemeth.dorottya@koki.mta.hu.
Csaba Fekete, Email: fekete.csaba@koki.hu.
Maria Martha Bernardi, Email: marthabernardi@gmail.com.
Victoria D. Leitch, Email: v.leitch@imperial.ac.uk.
Naila S. Mannan, Email: naila.mannan12@imperial.ac.uk.
Katharine F. Curry, Email: k.curry@imperial.ac.uk.
Natalie C. Butterfield, Email: natalie.butterfield@imperial.ac.uk.
J.H. Duncan Bassett, Email: d.bassett@imperial.ac.uk.
Graham R. Williams, Email: graham.williams@imperial.ac.uk.
Balázs Gereben, Email: gereben.balazs@koki.hu.
Miriam O. Ribeiro, Email: miriam.ribeiro@mackenzie.br.
Antonio C. Bianco, Email: abianco1@uchicago.edu.
References
- 1.Chaker L, Bianco AC, Jonklaas J, Peeters RP. Hypothyroidism. Lancet. 2017;390(10101):1550–1562. doi: 10.1016/S0140-6736(17)30703-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122(9):3035–3043. doi: 10.1172/JCI60047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gereben B, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29(7):898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jonklaas J, et al. Guidelines for the treatment of hypothyroidism: prepared by the american thyroid association task force on thyroid hormone replacement. Thyroid. 2014;24(12):1670–1751. doi: 10.1089/thy.2014.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saravanan P, Visser TJ, Dayan CM. Psychological well-being correlates with free thyroxine but not free 3,5,3’-triiodothyronine levels in patients on thyroid hormone replacement. J Clin Endocrinol Metab. 2006;91(9):3389–3393. doi: 10.1210/jc.2006-0414. [DOI] [PubMed] [Google Scholar]
- 6.Gereben B, McAninch EA, Ribeiro MO, Bianco AC. Scope and limitations of iodothyronine deiodinases in hypothyroidism. Nat Rev Endocrinol. 2015;11(11):642–652. doi: 10.1038/nrendo.2015.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gullo D, Latina A, Frasca F, Le Moli R, Pellegriti G, Vigneri R. Levothyroxine monotherapy cannot guarantee euthyroidism in all athyreotic patients. PLoS One. 2011;6(8):e22552. doi: 10.1371/journal.pone.0022552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Werneck de Castro JP, et al. Differences in hypothalamic type 2 deiodinase ubiquitination explain localized sensitivity to thyroxine. J Clin Invest. 2015;125(2):769–781. doi: 10.1172/JCI77588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson SJ, McAninch EA, Bianco AC. Is a normal TSH synonymous with “euthyroidism” in levothyroxine monotherapy? J Clin Endocrinol Metab. 2016;101(12):4964–4973. doi: 10.1210/jc.2016-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escobar-Morreale HF, Obregón MJ, Escobar del Rey F, Morreale de Escobar G. Replacement therapy for hypothyroidism with thyroxine alone does not ensure euthyroidism in all tissues, as studied in thyroidectomized rats. J Clin Invest. 1995;96(6):2828–2838. doi: 10.1172/JCI118353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samuels MH, Kolobova I, Smeraglio A, Peters D, Purnell JQ, Schuff KG. Effects of levothyroxine replacement or suppressive therapy on energy expenditure and body composition. Thyroid. 2016;26(3):347–355. doi: 10.1089/thy.2015.0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson SJ, et al. An online survey of hypothyroid patients demonstrates prominent dissatisfaction. Thyroid. 2018;28(6):707–721. doi: 10.1089/thy.2017.0681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crantz FR, Silva JE, Larsen PR. An analysis of the sources and quantity of 3,5,3’-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology. 1982;110(2):367–375. doi: 10.1210/endo-110-2-367. [DOI] [PubMed] [Google Scholar]
- 14.Freitas BC, et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest. 2010;120(6):2206–2217. doi: 10.1172/JCI41977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dentice M, et al. The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol. 2005;7(7):698–705. doi: 10.1038/ncb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zavacki AM, et al. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol Cell Biol. 2009;29(19):5339–5347. doi: 10.1128/MCB.01498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeöld A, et al. Metabolic instability of type 2 deiodinase is transferable to stable proteins independently of subcellular localization. J Biol Chem. 2006;281(42):31538–31543. doi: 10.1074/jbc.M604728200. [DOI] [PubMed] [Google Scholar]
- 18.Dora JM, Machado WE, Rheinheimer J, Crispim D, Maia AL. Association of the type 2 deiodinase Thr92Ala polymorphism with type 2 diabetes: case-control study and meta-analysis. Eur J Endocrinol. 2010;163(3):427–434. doi: 10.1530/EJE-10-0419. [DOI] [PubMed] [Google Scholar]
- 19.McAninch EA, et al. Prevalent polymorphism in thyroid hormone-activating enzyme leaves a genetic fingerprint that underlies associated clinical syndromes. J Clin Endocrinol Metab. 2015;100(3):920–933. doi: 10.1210/jc.2014-4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McAninch EA, et al. A common DIO2 polymorphism and Alzheimer disease dementia in African and European Americans. J Clin Endocrinol Metab. 2018;103(5):1818–1826. doi: 10.1210/jc.2017-01196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McAninch EA, Bianco AC. New insights into the variable effectiveness of levothyroxine monotherapy for hypothyroidism. Lancet Diabetes Endocrinol. 2015;3(10):756–758. doi: 10.1016/S2213-8587(15)00325-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panicker V, et al. Common variation in the DIO2 gene predicts baseline psychological well-being and response to combination thyroxine plus triiodothyronine therapy in hypothyroid patients. J Clin Endocrinol Metab. 2009;94(5):1623–1629. doi: 10.1210/jc.2008-1301. [DOI] [PubMed] [Google Scholar]
- 23.Carlé A, Faber J, Steffensen R, Laurberg P, Nygaard B. Hypothyroid patients encoding combined MCT10 and DIO2 gene polymorphisms may prefer L-T3 + L-T4 combination treatment — data using a blind, randomized, clinical study. Eur Thyroid J. 2017;6(3):143–151. doi: 10.1159/000469709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castagna MG, et al. DIO2 Thr92Ala reduces deiodinase-2 activity and serum-T3 levels in thyroid-deficient patients. J Clin Endocrinol Metab. 2017;102(5):1623–1630. doi: 10.1210/jc.2016-2587. [DOI] [PubMed] [Google Scholar]
- 25.Canani LH, et al. The type 2 deiodinase A/G (Thr92Ala) polymorphism is associated with decreased enzyme velocity and increased insulin resistance in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2005;90(6):3472–3478. doi: 10.1210/jc.2004-1977. [DOI] [PubMed] [Google Scholar]
- 26.Lorente-Rodríguez A, Barlowe C. Entry and exit mechanisms at the cis-face of the Golgi complex. Cold Spring Harb Perspect Biol. 2011;3(7):a005207. doi: 10.1101/cshperspect.a005207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119(Pt 11):2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- 28.Barlowe C, et al. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77(6):895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 29.Baines AC, Zhang B. Receptor-mediated protein transport in the early secretory pathway. Trends Biochem Sci. 2007;32(8):381–388. doi: 10.1016/j.tibs.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Hauri HP, Kappeler F, Andersson H, Appenzeller C. ERGIC-53 and traffic in the secretory pathway. J Cell Sci. 2000;113(Pt 4):587–596. doi: 10.1242/jcs.113.4.587. [DOI] [PubMed] [Google Scholar]
- 31.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14(2):117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 32.Arrojo E Drigo R, Egri P, Jo S, Gereben B, Bianco AC. The type II deiodinase is retrotranslocated to the cytoplasm and proteasomes via p97/Atx3 complex. Mol Endocrinol. 2013;27(12):2105–2115. doi: 10.1210/me.2013-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haines DS, et al. Protein interaction profiling of the p97 adaptor UBXD1 points to a role for the complex in modulating ERGIC-53 trafficking. Mol Cell Proteomics. 2012;11(6):M111.016444. doi: 10.1074/mcp.M111.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaynor EC, te Heesen S, Graham TR, Aebi M, Emr SD. Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J Cell Biol. 1994;127(3):653–665. doi: 10.1083/jcb.127.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schutze MP, Peterson PA, Jackson MR. An N-terminal double-arginine motif maintains type II membrane proteins in the endoplasmic reticulum. EMBO J. 1994;13(7):1696–1705. doi: 10.1002/j.1460-2075.1994.tb06434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu JW, et al. Unfolded protein response regulates yeast small GTPase Arl1p activation at late Golgi via phosphorylation of Arf GEF Syt1p. Proc Natl Acad Sci U S A. 2016;113(12):E1683–E1690. doi: 10.1073/pnas.1518260113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renna M, Caporaso MG, Bonatti S, Kaufman RJ, Remondelli P. Regulation of ERGIC-53 gene transcription in response to endoplasmic reticulum stress. J Biol Chem. 2007;282(31):22499–22512. doi: 10.1074/jbc.M703778200. [DOI] [PubMed] [Google Scholar]
- 38.Qin SY, Kawasaki N, Hu D, Tozawa H, Matsumoto N, Yamamoto K. Subcellular localization of ERGIC-53 under endoplasmic reticulum stress condition. Glycobiology. 2012;22(12):1709–1720. doi: 10.1093/glycob/cws114. [DOI] [PubMed] [Google Scholar]
- 39.Sagar GD, et al. Ubiquitination-induced conformational change within the deiodinase dimer is a switch regulating enzyme activity. Mol Cell Biol. 2007;27(13):4774–4783. doi: 10.1128/MCB.00283-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mishra AK, Muthamilarasan M, Khan Y, Parida SK, Prasad M. Genome-wide investigation and expression analyses of WD40 protein family in the model plant foxtail millet (Setaria italica L.) PLoS ONE. 2014;9(1):e86852. doi: 10.1371/journal.pone.0086852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Espenshade PJ, Li WP, Yabe D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc Natl Acad Sci U S A. 2002;99(18):11694–11699. doi: 10.1073/pnas.182412799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of Scap. Annu Rev Biochem. 2018;87:783–807. doi: 10.1146/annurev-biochem-062917-011852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kammoun HL, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119(5):1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Werstuck GH, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest. 2001;107(10):1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winter J, Basilicata MF, Stemmler MP, Krauss S. The MID1 protein is a central player during development and in disease. Front Biosci (Landmark Ed) 2016;21:664–682. doi: 10.2741/4413. [DOI] [PubMed] [Google Scholar]
- 46.Peeters R, et al. Regional physiological adaptation of the central nervous system deiodinases to iodine deficiency. Am J Physiol Endocrinol Metab. 2001;281(1):E54–E61. doi: 10.1152/ajpendo.2001.281.1.E54. [DOI] [PubMed] [Google Scholar]
- 47.Schweizer U, Schlicker C, Braun D, Köhrle J, Steegborn C. Crystal structure of mammalian selenocysteine-dependent iodothyronine deiodinase suggests a peroxiredoxin-like catalytic mechanism. Proc Natl Acad Sci U S A. 2014;111(29):10526–10531. doi: 10.1073/pnas.1323873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Callebaut I, et al. The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J Biol Chem. 2003;278(38):36887–36896. doi: 10.1074/jbc.M305725200. [DOI] [PubMed] [Google Scholar]
- 49.Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13(8):477–491. doi: 10.1038/nrneurol.2017.99. [DOI] [PubMed] [Google Scholar]
- 50.Williams GR. Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol. 2008;20(6):784–794. doi: 10.1111/j.1365-2826.2008.01733.x. [DOI] [PubMed] [Google Scholar]
- 51.Bocco BM, et al. Type 2 deiodinase disruption in astrocytes results in anxiety-depressive-like behavior in male mice. Endocrinology. 2016;157(9):3682–3695. doi: 10.1210/en.2016-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siesser WB, Zhao J, Miller LR, Cheng SY, McDonald MP. Transgenic mice expressing a human mutant beta1 thyroid receptor are hyperactive, impulsive, and inattentive. Genes Brain Behav. 2006;5(3):282–297. doi: 10.1111/j.1601-183X.2005.00161.x. [DOI] [PubMed] [Google Scholar]
- 53.Wouters HJ, et al. No effect of the Thr92Ala polymorphism of deiodinase-2 on thyroid hormone parameters, health-related quality of life, and cognitive functioning in a large population-based cohort study. Thyroid. 2017;27(2):147–155. doi: 10.1089/thy.2016.0199. [DOI] [PubMed] [Google Scholar]
- 54.Lee H, Dvorak D, Kao HY, Duffy ÁM, Scharfman HE, Fenton AA. Early cognitive experience prevents adult deficits in a neurodevelopmental schizophrenia model. Neuron. 2012;75(4):714–724. doi: 10.1016/j.neuron.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ullman MT, Pullman MY. A compensatory role for declarative memory in neurodevelopmental disorders. Neurosci Biobehav Rev. 2015;51:205–222. doi: 10.1016/j.neubiorev.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abdalla SM, Bianco AC. Defending plasma T3 is a biological priority. Clin Endocrinol (Oxf) 2014;81(5):633–641. doi: 10.1111/cen.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butler PW, et al. The Thr92Ala 5’ type 2 deiodinase gene polymorphism is associated with a delayed triiodothyronine secretion in response to the thyrotropin-releasing hormone-stimulation test: a pharmacogenomic study. Thyroid. 2010;20(12):1407–1412. doi: 10.1089/thy.2010.0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Potter GG, et al. Cognitive performance and informant reports in the diagnosis of cognitive impairment and dementia in African Americans and whites. Alzheimers Dement. 2009;5(6):445–453. doi: 10.1016/j.jalz.2009.04.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mentuccia D, et al. Association between a novel variant of the human type 2 deiodinase gene Thr92Ala and insulin resistance: evidence of interaction with the Trp64Arg variant of the beta-3-adrenergic receptor. Diabetes. 2002;51(3):880–883. doi: 10.2337/diabetes.51.3.880. [DOI] [PubMed] [Google Scholar]
- 60.Nair S, Muller YL, Ortega E, Kobes S, Bogardus C, Baier LJ. Association analyses of variants in the DIO2 gene with early-onset type 2 diabetes mellitus in Pima Indians. Thyroid. 2012;22(1):80–87. doi: 10.1089/thy.2010.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ozcan U, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 62.Duellman T, Burnett J, Shin A, Yang J. LMAN1 (ERGIC-53) is a potential carrier protein for matrix metalloproteinase-9 glycoprotein secretion. Biochem Biophys Res Commun. 2015;464(3):685–691. doi: 10.1016/j.bbrc.2015.06.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bianco AC, et al. American Thyroid Association guide to investigating thyroid hormone economy and action in rodent and cell models. Thyroid. 2014;24(1):88–168. doi: 10.1089/thy.2013.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christian AE, et al. Comparison of the capacity of beta-cyclodextrin derivatives and cyclophanes to shuttle cholesterol between cells and serum lipoproteins. J Lipid Res. 1999;40(8):1475–1482. [PubMed] [Google Scholar]
- 65.Egri P, Gereben B. Minimal requirements for ubiquitination-mediated regulation of thyroid hormone activation. J Mol Endocrinol. 2014;53(2):217–226. doi: 10.1530/JME-14-0156. [DOI] [PubMed] [Google Scholar]
- 66.Cicchetti G, Biernacki M, Farquharson J, Allen PG. A ratiometric expressible FRET sensor for phosphoinositides displays a signal change in highly dynamic membrane structures in fibroblasts. Biochemistry. 2004;43(7):1939–1949. doi: 10.1021/bi035480w. [DOI] [PubMed] [Google Scholar]
- 67.Jo S, et al. Neuronal hypoxia induces Hsp40-mediated nuclear import of type 3 deiodinase as an adaptive mechanism to reduce cellular metabolism. J Neurosci. 2012;32(25):8491–8500. doi: 10.1523/JNEUROSCI.6514-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aida T, et al. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 2015;16:87. doi: 10.1186/s13059-015-0653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Singh P, Schimenti JC, Bolcun-Filas E. A mouse geneticist’s practical guide to CRISPR applications. Genetics. 2015;199(1):1–15. doi: 10.1534/genetics.114.169771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McEwen BS, Alves SE. Estrogen actions in the central nervous system. Endocr Rev. 1999;20(3):279–307. doi: 10.1210/edrv.20.3.0365. [DOI] [PubMed] [Google Scholar]
- 71.Qiu LR, et al. Hippocampal volumes differ across the mouse estrous cycle, can change within 24 hours, and associate with cognitive strategies. Neuroimage. 2013;83:593–598. doi: 10.1016/j.neuroimage.2013.06.074. [DOI] [PubMed] [Google Scholar]
- 72.Marques AA, Bevilaqua MC, da Fonseca AM, Nardi AE, Thuret S, Dias GP. Gender differences in the neurobiology of anxiety: focus on adult hippocampal neurogenesis. Neural Plast. 2016;2016:5026713. doi: 10.1155/2016/5026713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hodgson RA, et al. Characterization of a novel vasopressin V1b receptor antagonist, V1B-30N, in animal models of anxiety-like and depression-like behavior. Eur J Pharmacol. 2014;730:157–163. doi: 10.1016/j.ejphar.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 74.Antunes M, Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cogn Process. 2012;13(2):93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J, Woodgett JR. Assessment of social interaction behaviors. J Vis Exp. 2011;(48):2473. doi: 10.3791/2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bassett JH, et al. Rapid-throughput skeletal phenotyping of 100 knockout mice identifies 9 new genes that determine bone strength. PLoS Genet. 2012;8(8):e1002858. doi: 10.1371/journal.pgen.1002858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bassett JH, et al. Optimal bone strength and mineralization requires the type 2 iodothyronine deiodinase in osteoblasts. Proc Natl Acad Sci U S A. 2010;107(16):7604–7609. doi: 10.1073/pnas.0911346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.