

Abstract
Mutations in CDCA7 and HELLS that respectively encode a CXXC-type zinc finger protein and an SNF2 family chromatin remodeler cause immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome types 3 and 4. Here, we demonstrate that the classical nonhomologous end joining (C-NHEJ) proteins Ku80 and Ku70, as well as HELLS, coimmunoprecipitated with CDCA7. The coimmunoprecipitation of the repair proteins was sensitive to nuclease treatment and an ICF3 mutation in CDCA7 that impairs its chromatin binding. The functional importance of these interactions was strongly suggested by the compromised C-NHEJ activity and significant delay in Ku80 accumulation at DNA damage sites in CDCA7- and HELLS-deficient HEK293 cells. Consistent with the repair defect, these cells displayed increased apoptosis, abnormal chromosome segregation, aneuploidy, centrosome amplification, and significant accumulation of γH2AX signals. Although less prominent, cells with mutations in the other ICF genes DNMT3B and ZBTB24 (responsible for ICF types 1 and 2, respectively) showed similar defects. Importantly, lymphoblastoid cells from ICF patients shared the same changes detected in the mutant HEK293 cells to varying degrees. Although the C-NHEJ defect alone did not cause CG hypomethylation, CDCA7 and HELLS are involved in maintaining CG methylation at centromeric and pericentromeric repeats. The defect in C-NHEJ may account for some common features of ICF cells, including centromeric instability, abnormal chromosome segregation, and apoptosis.
Keywords: Cell Biology, Genetics
Keywords: Genetic instability, Molecular pathology, Monogenic diseases
Introduction
Immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome is a rare autosomal recessive disorder characterized by reduced immunoglobulin levels in the serum and recurrent infection (1). ICF patients possess naive B cells but lack mature B cells in the peripheral blood, the former of which may bear immune receptors with long complementarity-determining region 3’s (CDR3s) composed of full-length diversity (DH) gene segments (2). Centromeric instability manifests as stretched heterochromatin, chromosome breaks, and multiradial configurations involving the centromeric regions of chromosomes 1, 9, and 16 in activated lymphocytes (3). The cytological defects are accompanied by CG hypomethylation in pericentromeric satellite-2 and -3 repeats of these chromosomes. Approximately half of ICF patients have mutations in the DNA methyltransferase 3B (DNMT3B) gene (ICF syndrome type 1: ICF1, OMIM #242860) (4–6). In ICF1 cells, CG hypomethylation is observed in pericentromeric repeats and subtelomeric regions, the latter of which are often abnormally short and vulnerable to DNA damage (7–11). In contrast, the remaining ICF patients show hypomethylation in pericentromeric repeats and centromeric α-satellite repeats (12), but not in subtelomeric regions (13), and are classified as type 2 (ICF2, OMIM #614069), type 3 (ICF3, OMIM #616910), type 4 (ICF4, OMIM #616911), and type X (ICFX). ICF2, ICF3, and ICF4 patients possess mutations in the zinc finger and BTB domain containing 24 (ZBTB24), cell division cycle associated 7 (CDCA7), and helicase lymphoid specific (HELLS) genes, respectively (14–17). The causative gene for ICFX remains unknown. A recent study also identified other regions that distinguish ICF1 from ICF2, 3, and 4 patients by CG methylation status (18).
Among the ICF2–4 genes, ZBTB24 encodes a protein with a BTB domain, an AT hook, and eight C2H2-type zinc finger motifs. CDCA7 encodes a protein with four CXXC-type zinc finger motifs, while HELLS (also known as LSH, PASG, or SMARCA6) encodes a protein with an SNF2 family ATPase domain. It has been reported that ZBTB24 acts as a transcriptional activator of CDCA7 (19). Furthermore, Xenopus Cdca7e, an egg-specific paralog of Cdca7, recruits Hells directly to chromatin and supports its nucleosome remodeling activity (20). These findings suggest that the 3 proteins work in the same biological pathway. Among these, HELLS, together with its homolog (DDM1) in Arabidopsis, is involved in DNA methylation, perhaps through its nucleosome remodeling activity that opens histone H1–containing heterochromatin (20–26). HELLS also promotes the efficient repair of DNA double-strand breaks (DSBs) (27). Perhaps because of this, murine embryonic fibroblasts with a Hells mutation display excessive numbers of centrosomes and abnormal mitosis (28). Mouse mutants homozygous for a Hells deletion die soon after birth (29), and their hematopoietic cells poorly contribute to T and B cells in recipient mice (30). In contrast, the involvement of CDCA7 in ICF pathology and regulation of DNA methylation is poorly understood.
Here, we report that, in human embryonic kidney (HEK) 293T cells, CDCA7 interacts with HELLS, the classical nonhomologous end joining (C-NHEJ) proteins Ku80 (XRCC5 or Ku86) and Ku70 (XRCC6), and phosphorylated H2AX (γH2AX, a DSB marker) in an ICF mutation–sensitive manner. Various cytological and molecular changes that likely resulted from the defect in DNA repair were observed in CDCA7 and HELLS mutant HEK293 cells and also in DNMT3B and ZBTB24 mutant cells. Furthermore, CDCA7 or HELLS deficiency caused a C-NHEJ defect and delay in Ku80 accumulation at DNA damage sites. Our results suggest that the defect in C-NHEJ accounts for some of the common features of ICF cells, including instability of satellite repeats, abnormal chromosome configuration, reduced proliferation rate, and apoptosis.
Results
HELLS and C-NHEJ proteins coimmunoprecipitate with CDCA7.
To understand the molecular function of CDCA7, we attempted to identify proteins that potentially interact with CDCA7 in HEK293T cells. To this end, we prepared expression vectors for FLAG-tagged WT CDCA7 and mutant (R274C) protein (FLAG-CDCA7_WT and _R274C, respectively). This ICF3 mutation is located in the zinc finger domain (ref. 15 and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI99751DS1), and a corresponding amino acid substitution in Xenopus Cdca7e attenuates its DNA and chromatin binding (20). Endogenous proteins that coimmunoprecipitated with FLAG-CDCA7_WT and/or _R274C proteins were identified by mass spectrometry. Table 1 provides a list of proteins that coimmunoprecipitated with FLAG-CDCA7 regardless of the mutation (peptide number ≥ 5, R274C/WT ≥ 0.6, n = 17). An extended list (peptide number ≥ 2, n = 36) is available in Supplemental Table 1. The list included HELLS and 14-3-3 proteins, which are the known interactors of CDCA7 (20, 31). These data support the validity of our experiment. Other notable proteins in the list were linker histones H1.4 and H1.3, as the homolog of HELLS in Arabidopsis (DDM1) is known to open H1-containing heterochromatin for DNA methylation (25).
Table 1. Proteins coimmunoprecipitate with FLAG-CDCA7 (mutation-insensitive) (peptide ≥ 5, R274C/WT ≥ 0.6).
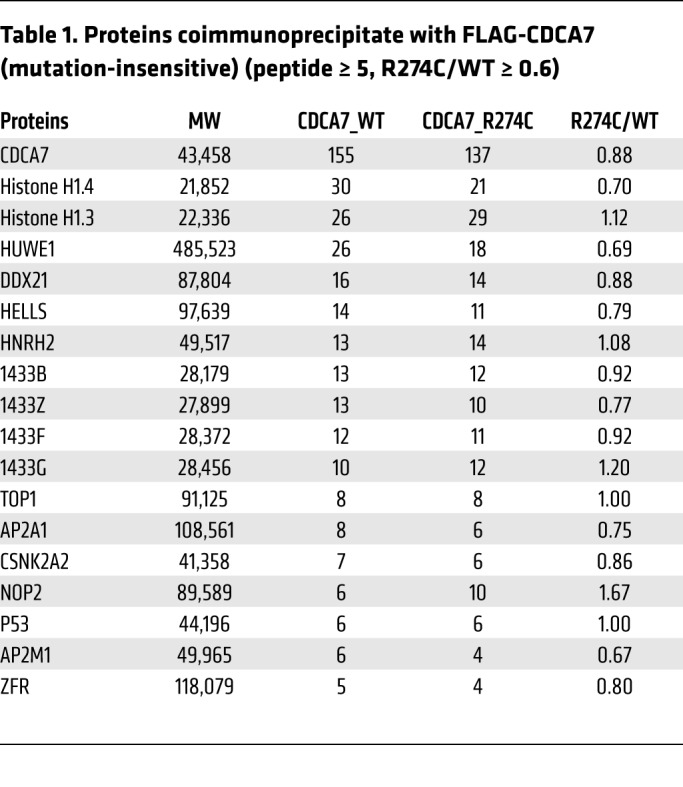
Table 2 shows proteins that coimmunoprecipitated with FLAG-CDCA7 in an ICF3 mutation–sensitive manner (peptide number ≥ 5, R274C/WT < 0.6, n = 11). An extended list (peptide number ≥ 2, n = 50) is shown in Supplemental Table 2. Consistent with the reported mutation-sensitive interaction of Xenopus Cdca7e with nucleosomes (20), the list included core histones (H3.1, H4, and H2B1C). Intriguingly, the list also included Ku80 and PRKDC (the catalytic subunit of DNA-dependent protein kinase [DNA-PK]), which are involved in C-NHEJ, V(D)J recombination, and immunoglobulin class switch recombination (32–35). Although the peptide number was below our cutoff level, Ku70, which forms a heterodimer with Ku80, and H2AX, of which the phosphorylated form (γH2AX) is a DSB marker, also coimmunoprecipitated with FLAG-CDCA7 in a mutation-sensitive manner. In addition, chromatin remodelers involved in DSB repair were included in the list. They included SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 (SMARCA5); SPT16 homolog, facilitates chromatin remodeling subunit (SUPT16H); and structure specific recognition protein 1 (SSRP1). The latter two form the facilitates chromatin transcription (FACT) complex (36, 37). These results provided a hint that CDCA7 might have a role in DNA repair, especially DSB repair.
Table 2. Proteins coimmunoprecipitate with FLAG-CDCA7 (mutation-sensitive) (peptide ≥ 5, R274C/WT < 0.6).
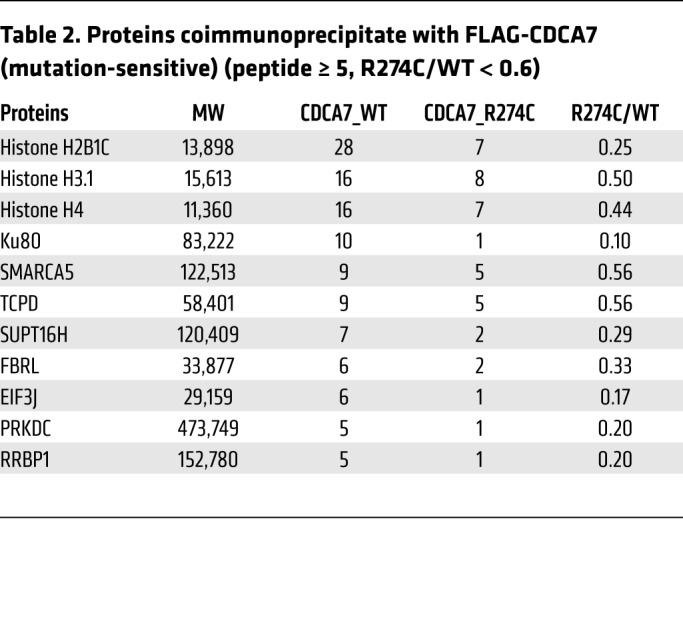
We also determined proteins that potentially interact with FLAG-tagged WT and/or mutant (Q699R) HELLS proteins (FLAG-HELLS_WT and _Q699R). The mutation corresponded to the only amino acid substitution identified in ICF4 patients and is located in the helicase C-terminal domain (Supplemental Figure 1 and ref. 15). Only histone H2A1A coimmunoprecipitated with FLAG-HELLS (peptide number ≥ 5), and this interaction was mutation-sensitive (Supplemental Tables 3 and 4).
The coimmunoprecipitation of the above proteins was confirmed by Western blotting in the presence and absence of benzonase nuclease, which cleaves nucleosome-free DNA and RNA. Endogenous HELLS coimmunoprecipitated with both FLAG-CDCA7_WT and _R274C, regardless of the treatment (Figure 1). Likewise, CDCA7 coimmunoprecipitated with both FLAG-HELLS_WT and _Q699R, albeit with a lower efficiency, perhaps reflecting stoichiometry (Supplemental Figure 2A). In contrast, the coimmunoprecipitation of Ku80, Ku70, SMARCA5, SUPT16H, histone H3, and H2AX with FLAG-CDCA7 was sensitive to the R274C substitution, consistent with the mass spectrometry data (Figure 1 and Supplemental Table 2). Notably, Western blotting using a modification-specific antibody revealed the mutation-sensitive coimmunoprecipitation of γH2AX. Importantly, the coimmunoprecipitation of Ku proteins with FLAG-CDCA7 was sensitive to benzonase, but not RNase A, suggesting that the interaction involves DNA (Figure 1 and Supplemental Figure 2B). In summary, the findings indicated that CDCA7 interacts with HELLS, and that this interaction is resistant to R274C substitution and nuclease treatment. However, the interaction of CDCA7 with SMARCA5, SUPT16H, histone H3, and γH2AX is clearly mediated by the zinc finger domain and may or may not involve DNA. In contrast, the interaction of CDCA7 with Ku proteins involves both the zinc finger domain and DNA.
Figure 1. HELLS and C-NHEJ proteins Ku80 and Ku70 coimmunoprecipitate with CDCA7.

Coimmunoprecipitation of indicated proteins with FLAG-CDCA7 confirmed by Western blotting in the presence (+) or absence (–) of benzonase. PCNA was a negative control. To detect Ku80, mouse monoclonal 5C5 antibody was used.
To exclude the possibility that the observed interactions were due to CDCA7 overexpression, we prepared two HEK293 stable clones expressing FLAG-CDCA7_WT and _R274C at near-endogenous levels and confirmed the coimmunoprecipitation of some of the proteins (Supplemental Figure 2C). Furthermore, we excluded the possibility of artificial interaction of abundant Ku proteins with CDCA7 via DNA fragments generated during the protein extraction procedure. We combined lysates prepared separately from FLAG-CDCA7_WT– and GFP-Ku80–expressing cells, performed immunoprecipitation with anti-FLAG antibody, and confirmed negligible coimmunoprecipitation (Supplemental Figure 2D). Finally, we examined whether the presence of DSBs had any effect on the interaction between CDCA7 and HELLS. The amount of HELLS that coimmunoprecipitated with FLAG-CDCA7_WT apparently increased upon bleomycin treatment (Supplemental Figure 2E). However, the level of HELLS protein, but not its mRNA, increased upon bleomycin treatment (Supplemental Figure 2E), likely accounting for the increased interaction. Interestingly, this upregulation was abrogated when CDCA7 was depleted (Supplemental Figure 2F).
Mutations in ICF genes cause defects in cell proliferation and chromosome segregation.
We generated HEK293 cells carrying a mutation in each of the 4 causative genes by genome editing. We chose HEK293 cells because they express all ICF causative genes at relatively high levels. We obtained 2 null mutant clones for each ICF gene by targeting exons mutated in patients (15, 16) (Supplemental Table 5 and Supplemental Figure 1). Since HEK293 cells are hypotriploid (chromosome numbers 62–70) (38), some mutant cells had 3 mutated alleles. We also tried to derive Ku80 mutant cells and obtained only 1 hypomorphic clone (mut24) that possessed 2 alleles, each carrying a frameshift followed by a premature stop codon, with the third allele carrying amino acid substitutions at consecutive positions (K183E, G184R) (Supplemental Table 5 and Supplemental Figure 1). The predicted truncated CDCA7, HELLS, and Ku80 proteins were not detected by Western blotting and therefore seemed unstable (Figure 2A and Supplemental Figure 3). In many mutant lines, the mRNA level of the targeted gene decreased to below 60% of the WT level (Figure 2B), possibly as a result of nonsense-mediated mRNA decay (39). Consistent with the recent report that ZBTB24 is a transcriptional activator of CDCA7 (19), the levels of endogenous CDCA7 mRNA and protein decreased in ZBTB24 mutant cells (Figure 2, A and B, and Supplemental Figure 3), and were restored by the expression of WT ZBTB24 (Supplemental Figure 4, A and B).
Figure 2. Establishment of cells carrying mutations in ICF genes and Ku80.

(A) Representative blots from biological triplicate showing expression of DNMT3B, ZBTB24, CDCA7, HELLS, and Ku80 proteins in WT and mutant HEK293 cells. ACTB was an internal control. Null mutant cells were designated knockout (KO, plus clone number), and a Ku80 hypomorphic mutant was designated mut24. The overall structure of each protein and regions recognized by the antibody (immunogen) are shown in Supplemental Figure 1. Full Western blot images show that no truncated proteins are present (Supplemental Figure 3). To detect Ku80, rabbit monoclonal EPR3467 antibody was used. (B) Levels of mRNAs in the mutant cells were measured by real-time qPCR in biological triplicate and technical triplicate (n = 9). The mRNA abundance (ΔCt) of each gene was calculated in comparison with that of ACTB and is shown as percent relative expression (2–ΔCt × 100). Data are mean ± SEM. *P < 0.0011 and **P < 0.0002 (Student’s 2-tailed t test) were considered statistically significant at the 5% and 1% levels, respectively, after Bonferroni correction. The exact P value, which was significant (P < 0.01) before the correction, is shown for reference.
During the establishment of the mutant cells, we recognized their poor proliferation. The doubling times of ZBTB24, CDCA7, HELLS, and Ku80 mutant cells were 1.3- to 1.4-fold longer than that of WT cells (Figure 3A). DNMT3B mutant cells were less affected. Restoration of WT proteins in DNMT3B and CDCA7 mutant cells, but not in HELLS or ZBTB24 mutant cells, recovered the proliferation rate (Supplemental Figure 4, A and C). This suggests that irreversible changes might have occurred in HELLS and ZBTB24 mutant cells (see later). In all mutant cells, the sub-G1 population (less than 3n), corresponding to the apoptotic fraction, and the aneuploid population with excessive DNA content (greater than 6n) increased significantly (Figure 3B). The existence of apoptotic cells and aneuploid cells was confirmed by a TUNEL assay and metaphase spreads (Figure 3C and Supplemental Figure 5A). The latter examination also revealed that the chromosomes were significantly stretched and fragile (Supplemental Figure 5, A–F). Possible chromosome fusions were also detected (Supplemental Figure 5A).
Figure 3. ICF mutant cells display proliferation defects, aneuploidy, and apoptosis.

(A) Proliferation curves of WT and mutant cells. Since 2 mutant clones per each gene showed similar results, a curve obtained from only 1 clone is shown. Fifty thousand cells were seeded, cultured, and counted at indicated time points (mean ± SEM, n = 4 for each clone). The average doubling time is shown on the right. (B) Cell cycle analysis based on DNA content measured by flow cytometry. Experiments were performed in biological triplicate and technical triplicate (n = 9). Because HEK293 cells are hypotriploid (38), those in G1 phase are just below 3n, and those in G2/M phase are near 6n. *P < 0.0011 and **P < 0.0002 (Mann-Whitney U test) were considered statistically significant at the 5% and 1% levels, respectively, after Bonferroni correction. The exact P values, which were significant (P < 0.01) before the correction, are shown for reference. (C) Representative images from biological duplicate showing apoptotic cells. DNA fragmentation was analyzed by TUNEL assay (green), and nuclei were counterstained with DAPI. Scale bars: 20 μm.
In CDCA7, HELLS, and Ku80 mutant cells, the size of the nuclei and number of centrosomes were significantly increased, with frequent appearance of micronuclei and giant aggregated centrosomes (>5 μm) (Figure 4, A and B). The frequency of anaphase bridges was also increased (Figure 4C), suggesting that CDCA7, HELLS, and Ku80 are required for proper chromosome segregation. These results are consistent with the previously reported abnormal centrosome number in HELLS mutant cells and increased chromosomal aberrations observed in Ku80 mutant cells (28, 40, 41). Although less frequent and prominent, DNMT3B and ZBTB24 mutant cells also showed similar aberrations (Figure 4, A–C).
Figure 4. ICF mutant cells display enlarged nuclei, centrosome amplification, and abnormal chromosome segregation.

(A) Representative images from biological triplicate showing immunostaining of centrosomes with anti-pericentrin antibody (green) in WT and mutant cells. Nuclei of CDCA7, HELLS, and Ku80 mutant cells were significantly enlarged (DAPI staining, blue). Arrows and arrowheads indicate giant aggregated centrosomes and metaphase chromosome spreads with an aberrant number of centrosomes, respectively. Scale bars: 20 μm. The right panel shows the distribution of areas (μm2) of individual nuclei measured using ZEN 2012 software. At least 30 nuclei (the exact number is shown in parentheses) were measured for each clone. Each box indicates 25th to 75th percentile, and a bar in the box indicates the median. X indicates the mean. *P < 0.0033 and **P < 0.0007 (Mann-Whitney U test) were considered statistically significant at the 5% and 1% levels, respectively, after Bonferroni correction. The exact P value, which was significant (P < 0.01) before the correction, is shown for reference. (B) Enlarged images of representative metaphase chromosome spreads and centrosomes from A. The right panel shows the percentage composition of metaphase cells with indicated centrosome numbers. At least 40 metaphase cells (the exact number is shown in parentheses) were analyzed for each clone. Scale bars: 5 μm. (C) Representative images from biological triplicate showing anaphase chromosomes stained with DAPI. At least 50 anaphase-nuclei (the exact number is shown in parentheses) were observed for each clone (mean ± SEM). The right panel shows the percentage anaphase images with bridges. *P < 0.0033 (Student’s 2-tailed t test) was considered statistically significant at the 5% level after Bonferroni correction. The exact P values, which were significant (P < 0.05) before the correction, are shown for reference. Scale bars: 5 μm.
Mutations in ICF genes cause accumulation of γH2AX.
The phenotype of the mutant cells described above was consistent with the presence of a DNA repair defect. Strikingly, in the absence of DNA-damaging reagent, all mutant cells, including Ku80 mutant cells, accumulated γH2AX signals (Figure 5, A and B), a marker of DSB, particularly in the centromeric, pericentromeric, and telomeric regions (Supplemental Figure 6, A and B). This was observed in cells not undergoing apoptosis (see Figure 5 legend and ref. 42). If the telomeric regions did accumulate DSBs in Ku80 mutant cells, as suggested by the strong γH2AX signals, this might explain the reported role of this protein in telomere maintenance (43). Importantly, the γH2AX accumulation was reversed upon restoration of WT proteins in all mutant cells (Figure 5, A and B). The predominant γH2AX signals in satellite repeats suggest the importance of the ICF proteins in maintaining the integrity of such regions.
Figure 5. ICF mutant cells and ICF lymphoblastoid cells accumulate γH2AX.

(A) Representative images from biological triplicate (more than 5 images were taken for each time) showing accumulation of γH2AX signals (green) in mutant cells with or without restoration of WT proteins. The restored mutant cells were designated rescue (Resc, plus clone number). Nuclei were stained with DAPI (blue). The exposure time was fixed for all images. Scale bars: 50 μm. (B) Percentage of cells with at least 10 γH2AX foci (mean ± SEM). The total cell number examined (n) is shown in parentheses. Apoptotic cells identified by DNA fragmentation were removed from the count. **P < 0.0017 (Mann-Whitney U test) was considered statistically significant at the 1% level after Bonferroni correction. (C) Family trees of ICF patients from whom the lymphoblastoid cells were derived (15, 17, 65). (D) Representative images from 5 images for each clone except for ICF3 (pC), of which only 2 images were available, showing accumulation of γH2AX signals (green) in lymphoblastoid cells from 2 healthy volunteers (HEV0190 and L1) and 5 ICF patients. Scale bars: 20 μm. (E) Percentage of cells with at least 10 γH2AX foci (mean ± SEM). The total cell number examined (n) is shown in parentheses. Statistical analysis was based on a comparison with control cells. *P < 0.0033 (Mann-Whitney U test) was considered statistically significant at the 5% level after Bonferroni correction. NA, not applicable.
We next examined whether γH2AX accumulation is observed in lymphoblastoid cells from ICF patients. The patients included an ICF1 patient with heterozygous DNMT3B mutations (Q42X/R832Q) (P1), ICF2 siblings homozygous for a ZBTB24 mutation (H132Q fsX19/H132Q fsX19) (pD and pV), an ICF3 patient homozygous for a CDCA7 mutation (G294V/G294V) (pC), and an ICF4 patient homozygous for a HELLS mutation (L801del/L801del) (pU) (ref. 15, 17, 44, 45, and Figure 5C). All these lymphoblastoid cells, especially those from the ICF3 patient, proliferated poorly and frequently died (Supplemental Figure 7, A–D). Like mutant HEK293 cells, lymphoblastoid cells from ICF2, ICF3, and ICF4 patients showed γH2AX accumulation (Figure 5, D and E). Although less significant, lymphoblastoid cells from the ICF1 patient also accumulated γH2AX signals.
CDCA7 and HELLS contribute to C-NHEJ.
The results so far suggest that ICF proteins may have a role in DSB repair. Considering the coimmunoprecipitation of Ku80, Ku70, and PRKDC with CDCA7 (Table 2 and Figure 1), C-NHEJ, the major DSB repair pathway, is likely affected. Thus, we examined the involvement of CDCA7 and HELLS in C-NHEJ. We established 3 independent HEK293 clones with stable integration of a C-NHEJ reporter possessing two I-SceI homing endonuclease recognition sites (pIRES-TK-GFP-dA3-1) (ref. 46 and Figure 6A, left). As expected, I-SceI expression decreased the uncut DNA and increased the joined DNA in all clones (Figure 6A, right). In this setup, successful C-NHEJ would excise the herpes simplex virus–thymidine kinase gene, leading to expression of enhanced green fluorescent protein (EGFP). We treated the cells with siRNAs against CDCA7 or HELLS (Figure 6B) and evaluated the C-NHEJ activity by counting EGFP-positive cells. In all clones, CDCA7 and HELLS knockdown respectively compromised C-NHEJ (Figure 6, C and D), indicating that CDCA7 and HELLS are positively involved in C-NHEJ at 3 independent genomic sites. Although the dominant repair pathway can vary during the cell cycle (47, 48), the cell cycle profiles were grossly normal in all knockdown cells except HELLS–double knockdown cells, which showed a slightly increased early S population (Figure 6E).
Figure 6. CDCA7 and HELLS are involved in C-NHEJ.

(A) Schematic representation of the C-NHEJ assay (46) (left), and detection of uncut and joined DNA after 2 days of I-SceI expression in three HEK293 clones carrying a C-NHEJ reporter (pIRES-TK-EGFP-dA3-1) (right). PCR was performed using genomic DNA from cells with (+) or without (–) I-SceI expression. CMV, cytomegalovirus promoter/enhancer; TK, herpes simplex virus–thymidine kinase; IRES, internal ribosome entry site; pA or polyA, polyadenylation signal. (B) Representative blots from biological duplicate showing confirmation of CDCA7 and HELLS knockdown. The stable clones were treated with indicated siRNAs, cultured for 2 days, and subjected to Western blotting. (C) Representative data from biological triplicate and technical triplicate (n = 9) showing flow cytometric analysis of EGFP-positive cells. Two days after siRNA treatment, cells were transfected with pCBASceI, cultured for an additional 2 days, and subjected to the analysis. Percentage of EGFP-positive cells is indicated. SSC, side scatter. (D) Relative frequency of EGFP-positive cells in the knockdown cells shown in C (mean ± SEM, n = 9). The percentage of EGFP-positive cells in I-SceI– cells (background) was subtracted from that in I-SceI+ cells or I-SceI expressed cells. **P < 0.0017 (Mann-Whitney U test) was considered statistically significant at the 1% level after Bonferroni correction. (E) Cell cycle profiles of the knockdown cells. The stable clones were treated with a siRNA(s), cultured for 4 days, and subjected to flow cytometric analysis.
We then treated ICF mutant cells with an inhibitor of C-NHEJ (a DNA-PK inhibitor, NU7026) to examine the extent of the additional effect on cell viability. The reduction in viability in comparison with untreated cells was not appreciably different in WT, ICF mutant, and Ku80 mutant cells (Supplemental Figure 8A), suggesting a compensation by the other DSB repair mechanisms including homologous recombination (HR). We also treated the cells with a poly(ADP-ribose) polymerase (PARP) 1/2 inhibitor (olaparib), which induces apoptosis in HR-deficient cells at a high frequency (49). The ICF mutant cells were insensitive to the inhibitor, as were WT and Ku80 mutant cells (Supplemental Figure 8B), suggesting that HR is preserved. In addition to the 2 inhibitors, we also treated the mutant cells with mitomycin C (MMC) and temozolomide (TMZ). MMC causes DNA interstrand cross-links, which can be repaired by the Fanconi anemia pathway, HR, nucleotide excision repair, and/or translesion synthesis (50). The ICF mutant cells were similarly sensitive to this compound compared with WT and Ku80 mutant cells (Supplemental Figure 8C). TMZ generates alkyl adducts to DNA, which are removed by O6-methylguanine methyltransferase or base excision repair (51). The TMZ sensitivity of the ICF mutant cells was also similar to that of WT and Ku80 mutant cells (Supplemental Figure 8D). Taken together, the data indicate that C-NHEJ is affected in ICF mutant cells, but other DNA repair pathways, including HR, are largely intact.
CDCA7 and HELLS facilitate accumulation of Ku80 at DSBs.
Because chromatin remodeling is required for DSB repair (36, 37, 52–56), we hypothesized that the CDCA7/HELLS chromatin remodeling complex (15) might be involved in the recruitment of Ku80 to DSB sites. The recruitment of GFP-Ku80 was examined in CDCA7 and HELLS mutant cells by live-cell imaging after laser microirradiation (56). While we observed prompt and strong accumulation of GFP-Ku80 at DNA damage sites (presumed DSBs) within 10 seconds in WT cells, the accumulation was significantly delayed and attenuated in CDCA7 and HELLS mutant cells (Figure 7, A–D). This suggests that CDCA7 and HELLS contribute to C-NHEJ by facilitating the accumulation of Ku80 at DSBs.
Figure 7. CDCA7 and HELLS facilitate accumulation of Ku80 at DNA damage sites.

(A) GFP-Ku80 accumulation in WT, CDCA7 mutant, and HELLS mutant cells after laser microirradiation. Representative images from biological duplicate are shown. ROI, region of interest. Scale bars: 5 μm. (B) Schematic representation of the kinetics analysis. Signal intensities (mean ROIs) of damage sites (region-1) and background (region-2, next to region-1) were obtained using ZEN 2012 SP5 software. The time required for the signal intensity of region-1 to become 1.2-fold that of region-2 is set as the time (seconds) required for GFP-Ku80 accumulation. (C) Percentage of cells that required the indicated times for GFP-Ku80 accumulation. More than 60 cells (the exact number is in parentheses) were analyzed. (D) Maximum fold change of signal intensity. The same data examined in C were reanalyzed. Each box indicates 25th to 75th percentile, and a bar in the box indicates the median. X indicates the mean. **P < 0.0017 (Mann-Whitney U test) was considered statistically significant at the 1% level after Bonferroni correction.
Defect in C-NHEJ alone does not induce CG hypomethylation of satellite repeats.
To explore whether CG hypomethylation of satellite repeats in ICF cells involves defective C-NHEJ, we examined the CG methylation levels of centromeric α-satellite and pericentromeric satellite-2 repeats by bisulfite sequencing (Supplemental Figure 9A) in peripheral blood leukocytes from an ICF2 patient homozygous for a ZBTB24 mutation (R320X/R230X) (P7), an ICF4 patient homozygous for a HELLS mutation (Ser762Argfs*4/Ser762Argfs*4) (2.2), and the parents of the 2 patients (refs. 15, 16, and Supplemental Figure 9B). The CG methylation levels of both repeats significantly decreased in the cells from the patients, and the decrease was most significant in satellite-2 repeats of the ICF4 patient (Supplemental Figure 9C).
Despite the global CG hypomethylation seen in most cultured cells (57), HEK293 cells maintained relatively high levels of CG methylation in these repeats, especially in α-satellite repeats (Figure 8, A and B). Like ICF leukocytes, ZBTB24, CDCA7, and HELLS mutant cells showed CG hypomethylation in both repeats after 4 months of culture, corresponding to approximately 96–116 cell divisions (Figure 8, A and B). The reduction was progressive (compare data at 2 and 4 months) and was more drastic in satellite-2 than in α-satellite repeats. In contrast, the CG methylation levels of these repeats were not affected in Ku80 mutant cells, suggesting that a C-NHEJ defect alone is not sufficient to induce CG hypomethylation (Figure 8, A and B). The CG methylation level of α-satellite repeats was recovered by the expression of the corresponding WT proteins in ZBTB24, CDCA7, and HELLS mutant cells (Figure 8A). In contrast, the CG methylation level of satellite-2 repeats was not recovered in any mutant cells (Figure 8B). This suggests the difficulty in recovering a normal epigenetic state of the pericentromeric repeats. These findings suggest that the molecular basis of CG methylation regulation is different between α-satellite and satellite-2 repeats.
Figure 8. The C-NHEJ defect alone does not cause CG hypomethylation at satellite repeats.

(A and B) CG methylation (percent) at centromeric α-satellite (A) and pericentromeric satellite-2 repeats (B) in mutant cells. The methylation levels were measured by bisulfite sequencing (PCR products were cloned into TA vector and at least 10 clones were sequenced for each condition) at 2 and ≥4 months after the introduction of the mutation. Because the doubling time of the mutant cells was 25–30 hours (Figure 3A), 2 and ≥4 months roughly corresponded to 48–58 and ≥96–116 cell divisions, respectively. Restoration of the methylation levels in the “rescue” clones was evaluated at 6 weeks after the introduction of WT genes. Data are mean ± SEM. Statistical analysis was performed separately for the data obtained at 2 and ≥4 months and the data obtained after 6 weeks of WT restoration. For the former data sets, **P < 0.0007 (Mann-Whitney U test) was considered statistically significant at the 1% level after Bonferroni correction. For the latter data set, **P < 0.0004 (Mann-Whitney U test) was considered statistically significant at the 1% level after Bonferroni correction. The exact P values, which were significant (P < 0.05) before the correction, are shown for reference. See also Supplemental Figure 9.
Discussion
It was recently reported that CDCA7 knockdown impairs chromatin recruitment of HELLS in HeLa cells and that Xenopus Cdca7e stimulates the chromatin remodeling activity of Hells through direct interaction (20). In the present study, in addition to HELLS, core histones including γH2AX, the C-NHEJ proteins Ku80 and Ku70, and other chromatin-related proteins coimmunoprecipitated with CDCA7 in HEK293T cells. HEK293 cells carrying mutations in ICF genes displayed poor proliferation, abnormal chromosome segregation, increased apoptotic and aneuploid cells, and accumulation of γH2AX, all of which are attributable to impaired DSB repair (58–60). The cellular defects were shared by lymphoblastoid cells derived from ICF patients to varying degrees. Importantly, reporter assays showed that CDCA7 depletion as well as HELLS depletion compromised the C-NHEJ activity of the cells. Furthermore, live-cell imaging after laser microirradiation revealed that accumulation of Ku80 at DSBs was significantly delayed and attenuated in CDCA7 and HELLS mutant cells, suggesting that these proteins promote C-NHEJ through facilitation of Ku80 recruitment.
The above findings indicate a general C-NHEJ defect in ICF mutant cells. The various repeat-associated defects observed in ICF mutant cells may reflect their exceptional susceptibility to DNA breaks and/or specific dependency on C-NHEJ for maintaining the integrity of the repeats. In the absence of Ku80, alternative repair pathways such as the HR pathway substitute for the defective C-NHEJ (61). Our experiments using small-molecule inhibitors suggest that HR is functional in ICF mutant cells. Since HR involves strand exchange between homologous sequences, predominant DSB repair by this mechanism could cause structural instability of satellite repeats, which is a hallmark of ICF cells (1, 3, 10, 11, 62). DNA replication stress may be one cause of DSBs in satellite repeats, as they are well-known hot spots (62, 63). Since Ku80 mutant cells as well as ICF mutant cells show anaphase chromosome bridges, the defect in C-NHEJ appears to cause chromosome missegregation likely due to HR-mediated DNA rearrangements at these repeats. We speculate that this chromosome segregation error further induces apoptosis and chromosome breaks.
A recent study showed that CDCA7 directly binds DNA and that this binding is disrupted by an ICF3 mutation within the zinc finger domain (R274C) (18). We found that the same mutation affects the coimmunoprecipitation of core histones including γH2AX and C-NHEJ proteins. Thus, CDCA7 likely interacts via the zinc finger domain with nucleosomal DNA, which is resistant to benzonase. The coimmunoprecipitation of the C-NHEJ proteins was sensitive to benzonase (but not to RNase A), suggesting their direct association with DSB sites and indirect association with CDCA7 via nucleosome-free DNA. As described above, CDCA7 recruits HELLS to chromatin and stimulates the chromatin remodeling activity of HELLS through direct interaction (20). Therefore, it is tempting to speculate that the CDCA7/HELLS complex facilitates Ku80 recruitment to DSB sites through its chromatin remodeling activity, although how this occurs remains an open question.
The defect in the C-NHEJ pathway in Ku80 mutant cells was not sufficient to induce centromeric or pericentromeric CG hypomethylation, suggesting that the hypomethylation phenotype of ICF patients is not caused by the C-NHEJ defect. We observed that centromeric and pericentromeric repeats lose and gain CG methylation with different kinetics upon depletion and restoration of ICF proteins in HEK293 cells, respectively. Together with the fact that DNMT3B mutations (ICF1) affect only pericentromeric repeats (7–9, 12), this suggests that different mechanisms establish and/or maintain CG methylation in centromeric and pericentromeric repeats. The CDCA7/HELLS complex is perhaps a common component required for both repeats, and DNMT3B is a downstream effector specific to pericentromeric repeats. The nucleosome remodeling activity of the CDCA7/HELLS complex might also be required for recovering CG methylation in repaired DNA segments as part of the postrepair process (20–26).
Finally, it is known that B cells from ICF patients undergo apoptosis after in vitro activation (2) and that both T and B cells are significantly reduced in the peripheral blood of Hells mutant mice (30). Consistent with these reports, our mutant HEK293 cells and ICF lymphoblastoid cells showed poor proliferation and increased apoptosis. However, since C-NHEJ proteins are also involved in V(D)J and class switch recombination (33–35), and since impaired IgE class switch induction has been reported in a non-ICF1 patient (2), it would be interesting to investigate whether ICF3 and ICF4 immune cells have a defect in such cellular processes.
In conclusion, our results suggest that the defect in C-NHEJ may account for some of the common features of ICF cells, including instability of satellite repeats, abnormal chromosome configuration, poor proliferation, and apoptosis. Further studies are required to understand how the C-NHEJ defect is related to the CG hypomethylation and immunological defects of ICF syndrome.
Methods
Cells.
A lymphoblastoid cell line from a healthy female (HEV0190) was purchased from the RIKEN Cell Bank (64), and a line from an ICF1 patient (P1) was obtained from a cell bank maintained in Saitama Children’s Medical Center (44). Details of the other lymphoblastoid cell lines from a healthy male (L1) and 4 ICF patients (pD, pV, pC, and pU) are described elsewhere (15, 17, 65). The lymphoblastoid cells were maintained in RPMI 1640 supplemented with 20% FBS and penicillin/streptomycin at 37°C in a 5% CO2 incubator. HEK293 and HEK293T cells were obtained from the American Type Culture Collection and maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin at 37°C in a 5% CO2 incubator. HEK293T cells were used for transient expression of FLAG-CDCA7_WT, FLAG-CDCA7_R274C, FLAG-HELLS_WT, and FLAG-HELLS_Q699R. HEK293 cells were used to generate stable clones by transfection of plasmids and DNMT3B, ZBTB24, CDCA7, HELLS, and Ku80 mutant cells using the CRISPR/Cas9 system.
Stable HEK293 clones with an approximately endogenous level of FLAG-CDCA7_WT and FLAG-CDCA7_R274C were established by transfection of the expression plasmid vector of each protein (p3xFLAG-CMV-10). After selection using Geneticin (600 μg/ml; Santa Cruz Biotechnology) for 2 weeks, several clones were established. One clone for each tagged protein was selected based on the expression level (Supplemental Figure 2C). Stable HEK293 clones for the C-NHEJ assay were established by transfection with pIRES-TK-dA3-1 plasmid. After selection using puromycin (2 μg/ml; Gibco) for a month, 3 clones were established (Figure 6A).
For derivation of DNMT3B, ZBTB24, CDCA7, HELLS, and Ku80 mutant cells, a single guide RNA per each gene was designed using the CRISPR Design Tool (http://crispr.mit.edu/) (66). The sequences were: DNMT3B exon 6, 5′-GAGACGGCGGGCAACAGCAT-3′; ZBTB24 exon 4, 5′-GTGATCAATGCGGAAAATAT-3′; CDCA7 exon 7, 5′-TCAGAAGACTATTGATACCA-3′; HELLS exon 19, 5′-GAACCCCCAGTCGGATCTTC-3′; and Ku80 exon 6, 5′-GATGGCCCCTTTCGCTTAGG-3′. The guide sequences were cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid vectors (67) (Addgene, catalog 42230). HEK293 cells were cotransfected with the pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid vector and a plasmid containing the puromycin resistance gene using FuGene HD transfection reagent (Promega). Transfected cells were selected in the presence of puromycin, and each clone was established from a single cell. Mutations were determined by Sanger sequencing using an Applied Biosystems 3730 or 3130xl genetic analyzer (Life Technologies). hSpCas9 integration into the genome was excluded by a lack of PCR products from the Cas9 sequence. Primers are shown in Supplemental Table 6.
Mutant cells with restoration of WT proteins, which were designated as “rescue” (Resc), were established by transfection of DNMT3B KO31, ZBTB24 KO26, CDCA7 KO4, and HELLS KO11 cells with FLAG-DNMT3B_WT, FLAG-ZBTB24_WT, FLAG-CDCA7_WT, and FLAG-HELLS_WT expression plasmid vectors (p3xFLAG-CMV-10), respectively. After selection using Geneticin (600 μg/ml) for 2 weeks, 2 clones per each mutant were established, except for DNMT3B KO31, from which only 1 clone was obtained. Restoration of WT proteins was confirmed by Western blotting (Supplemental Figure 4A) and quantitative PCR (qPCR).
Plasmids and antibodies.
CDCA7 and HELLS cDNAs were amplified by PCR using a high-fidelity polymerase, KOD Plus (TOYOBO), and cloned into p3xFLAG-CMV-10 (Sigma-Aldrich) expression plasmids. GFP-Ku80 expression plasmid and pIRES-TK-dA3-1 reporter plasmid were gifts from Niels Mailand (University of Copenhagen, Copenhagen, Denmark), Ayako Ui (Tokyo University of Technology, Tokyo, Japan), and Takashi Kohno (National Cancer Center Research Institute, Tokyo, Japan). pCBASceI (68, 69) was purchased from Addgene (catalog 26477). The antibodies used in this study are summarized in Supplemental Table 7. We used 2 anti-Ku80 antibodies. One (Abcam, catalog ab79391) was for detection of both WT and mutant Ku80 (K183E/G184R), and the other (Abcam, catalog ab119935) was for detection of WT Ku80.
Cell viability assay using small-molecule compounds.
WT or mutant HEK293 cells (1 × 104) were seeded into 96-well plates and cultured in 50 μl DMEM supplemented with 10% FBS and penicillin/streptomycin overnight. Next day, 50 μl of the medium containing 2 times the final concentration of NU7026 (Wako, catalog 144-09651), olaparib (LKT Laboratories, catalog O4492), MMC (Nacalai Tesque, catalog 20898-21), or TMZ (Wako, catalog 206-19991) was gently added into each well and cultured for 48 hours. Then, 10 μl of Cell Count Reagent SF (Nacalai Tesque, catalog 07553) was added to each well and incubated for 1 hour, and the absorbances at 450 and 600 nm (for calibration) were measured using a Multiskan GO microplate spectrophotometer (Thermo Fisher Scientific). Normalization of cell viability was done using the values obtained from untreated cells for each genotype. The assay was performed in biological triplicate and technical triplicate, and P values were determined by Welch’s t test.
Immunoprecipitation.
HEK293T cells were harvested after 48 hours of transfection and lysed in 0.1% NP-40 lysis buffer (150 mM NaCl, 0.1% NP-40, and 50 mM Tris-HCl, pH 8.0). Cells were homogenized by sonication (until DNA fragments were <1.4 kb) and incubated on ice for 30 minutes. The supernatant was collected by centrifugation. Benzonase nuclease (Sigma-Aldrich), which digests all forms of DNA and RNA (except for DNA on mononucleosome) under various conditions (pH 6–10, 0°C–42°C), was added to the supernatant (≥250 U/sample) throughout the following steps (total incubation time >2 hours), if required. Precleaning was performed by addition of Protein A/G PLUS-Agarose (Santa Cruz Biotechnology) and normal mouse IgG (Santa Cruz Biotechnology) to the supernatant. After 1 hour of rotation at 4°C, the precleaned samples were collected and immunoprecipitation was performed using FLAG M2 affinity agarose gel (Sigma-Aldrich) for 1 hour at 4°C. After washing of the agarose with the lysis buffer 5 times, immunoprecipitated proteins were eluted using 3× FLAG peptides (Sigma-Aldrich) and subjected to Western blotting or mass spectrometry analysis. As a control, protein lysate from untransfected HEK293T cells was prepared and the same assay was performed.
Silver staining and mass spectrometry analysis.
Eluted samples were dissolved in SDS sample buffer, fractionated by SDS-PAGE on an 8% gel, and stained with silver. Individual lanes of the stained gel were sliced into 13 pieces, and proteins within the pieces were subjected to in-gel digestion with trypsin. The resulting peptides were subjected to liquid chromatography–tandem mass spectrometry analysis with an Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific). The obtained data sets were analyzed using Mascot Daemon (Matrix Science). As a control, a protein lysate from untransfected HEK293T cells was prepared and the same assay was performed. Ribosome and proteasome components were removed from the analysis.
qPCR.
Primers used for qPCR are summarized in Supplemental Table 6. PCR reactions were performed using a Thermal Cycler Dice Real Time System Single (TaKaRa Bio) according to the manufacturer’s protocol. Amplification conditions were 30 seconds at 95°C and then 40 cycles each consisting of 5 seconds at 95°C and 30 seconds at 60°C. The ACTB mRNA level was used for normalization. The mRNA abundance (ΔCt) of each gene was calculated by comparison with that of ACTB and shown as percentage relative expression level (2–ΔCt × 100).
Immunofluorescence.
Immunofluorescence staining was performed as follows, except for metaphase chromosome spreads. Adherent cells were seeded in chamber slides 1 day before immunofluorescence, and lymphoblastoid cells were embedded in Smear Gell (GenoStaff) on a glass slide. The cells were fixed with 4% paraformaldehyde (PFA) for 30 minutes, permeabilized with 0.5% Triton X for 30 minutes, and blocked with Block Ace (DS Pharma Biomedical) for 1 hour at room temperature. Cells were incubated with the indicated antibodies for 1 hour at room temperature. After washing with PBS, cells were incubated with CF488 donkey anti-mouse IgG (H + L) antibody (Biotium, catalog 20014; 1:1,000) for 1 hour at room temperature. Nuclei were visualized using VECTASHIELD Antifade mounting medium containing DAPI (Vector Laboratories). Fluorescent images were taken using an LSM700 confocal laser scanning microscope (Carl Zeiss). The area of nuclei (μm2) and length of chromosomes (μm) were measured using the ZEN 2012 SP5 (black edition) software program (Carl Zeiss).
γH2AX, Giemsa, and DAPI staining of metaphase chromosome spreads.
γH2AX staining of metaphase spreads was performed as described previously (11) with minor modifications. Briefly, cells were treated with KaryoMAX Colcemid (Thermo Fisher Scientific, catalog 15212012) in culture medium (final concentration, 0.04 μg/ml) for 50 minutes. Cells were then harvested and incubated in hypotonic buffer (0.2% KCl, 0.2% sodium citrate) for 30 minutes at room temperature. For Giemsa (Nacalai Tesque) or DAPI (Vector Laboratories) staining, cells were fixed with Carnoy’s solution, dropped on glass slides, dried on a heat block at 50°C, and stained. For γH2AX staining, unfixed cells were dropped on glass slides, fixed with 4% PFA for 30 minutes at room temperature, rinsed in water, and permeabilized with KCM buffer (120 mM KCl, 20 mM NaCl, 10 mM Tris, pH 7.5, 0.1% Triton X) for 10 minutes. The slides were blocked with 5% BSA (Sigma-Aldrich) for 1 hour at room temperature and incubated with anti-γH2AX mouse monoclonal antibody for 1 hour at 37°C. After washing of the slides with PBS, the cells were incubated with CF488 donkey anti-mouse IgG (H + L) antibody (Biotium, catalog 20014; 1:1,000) for 1 hour at 37°C. Chromosomes were stained with VECTASHIELD containing DAPI. Fluorescent images were taken using an LSM700 confocal laser scanning microscope.
TUNEL assay.
Apoptotic cells were detected using the ApopTag kit (Merck) and fluorescence microscopy according to the manufacturer’s protocol.
Cell cycle analysis.
Cells were grown in a 60-mm dish or 24-well plate, harvested, and treated with Cycle TEST PLUS (BD Biosciences) according to the manufacturer’s protocol. For flow cytometric analysis, FACSCalibur (BD Biosciences) was used. The percentage of nuclei in G0/G1, S, and G2/M phases of the cell cycle, and any sub-G1 population or polyploidy, were determined from 20,000 ungated cells.
C-NHEJ assay.
Three HEK293 dA3-1 clones (nos. 5, 6, and 11) were transfected with a control siRNA or siRNAs against CDCA7 or HELLS mRNA (final concentration 60 nM) using Lipofectamine RNAiMax (Invitrogen) and cultured for 2 days in puromycin-free media. The siRNAs (all from Sigma-Aldrich) included a universal negative control, SIC-001-25; CDCA7, SASI_Hs01_00212366 (si-1) and SASI_Hs01_00212367 (si-2); and HELLS, SASI_Hs01_00026584 (si-1) and SASI_Hs01_00026585 (si-2). The cells were transfected with or without pCBASceI, together with siRNAs, or with pmaxGFP (Lonza) alone, using Lipofectamine 2000 (Invitrogen), cultured for an additional 2 days, fixed with 3% formaldehyde, and subjected to flow cytometric analysis using FACSVerse (BD Biosciences). EGFP-positive cells were determined from 10,000 ungated cells. We used pmaxGFP-transfected cells for the evaluation of the transfection efficiency, which always exceeded 90%. All assays were performed in biological triplicates, with each in technical triplicate. We subtracted the number of EGFP-positive cells transfected with siRNA(s) alone as a background, which were thought to be autofluorescent cells, from EGFP-positive cells transfected with the same siRNA(s) with I-SceI. PCR analysis was done with genomic DNA extracted from cells 2 days after I-SceI expression. The locations and sequences of primers used for the PCR were described in a previous report (ref. 46; Figure 6A, left, and Supplemental Table 6). The PCR amplification condition was 30 seconds at 98°C and then 28 cycles each consisting of 15 seconds at 98°C, 15 seconds at 56°C, and 30 seconds at 72°C.
Laser microirradiation and live-cell imaging.
We transfected WT, CDCA7 KO4, and HELLS KO11 cells with a GFP-Ku80 plasmid and established stable clones after Geneticin (Santa Cruz Biotechnology) selection for 2 weeks (600 μg/ml). Then the cells with stable expression of GFP-Ku80 were seeded in chambered coverglass (Lab-Tek II, Merck), cultured for 16 hours, and sensitized for DNA damage by treatment with 10 μg/ml Hoechst 33342 (PromoKine) for 10 minutes. Laser microirradiation was performed using an LSM700 confocal scanning laser microscope. At least 60 cells were microirradiated with a 405-nm pulse laser (the irradiation iteration fixed at 20 scans) for each genotype, and the kinetics of GFP-Ku80 accumulation was recorded every 0.5 seconds. The signal intensities (mean region of interest) in the damage region (region-1) and background region (region-2; adjacent to the damage region with the same size) were measured using the ZEN 2012 SP5 (black edition) software program (Carl Zeiss). Time required for accumulation of GFP-Ku80 (seconds) was determined by the time point at which the signal intensity in the damage region reached 1.2-fold that of the background. Fold change (region-1/region-2) was calculated every 0.5 seconds after laser microirradiation, and the maximum fold change during the first 30 seconds was determined.
DNA methylation analysis.
Genomic DNA was subjected to bisulfite conversion as previously described (70). PCR was performed using primers shown in Supplemental Table 6 with ExTaq Hot Start DNA Polymerase (TaKaRa Bio) for satellite-2 and with KAPA2G Robust HS RM with dye (KK5706; Nippon Genetics) for α-satellite. PCR products were electrophoresed, purified using a QIAquick Gel Extraction Kit (Qiagen), and cloned into T-vector pMD20 (TaKaRa Bio). More than 12 clones for each genotype or condition were sequenced using an Applied Biosystems 3730 or 3130xl genetic analyzer (Life Technologies). DNA methylation patterns were visualized using a quantification tool for methylation analysis (QUMA) (71). The bisulfite conversion rate was evaluated by the conversion rate at non-CG sites in the same PCR fragment, which always exceeded 99%.
Statistics.
Mann-Whitney U test, Student’s t test (2-tailed), or Welch’s t test were performed for statistical analysis based on sample numbers and data distributions. Bonferroni correction was applied for multiple comparisons. Significant P values at the 5% and 1% levels for each analysis are described in figure legends.
Study approval.
All clinical samples were obtained in an anonymized manner, and written informed consent was received from participants prior to inclusion in the study. The study was approved by the Kyushu University Institutional Review Board for Human Genome/Gene Research (no. 599-01) and the local ethics committee of Necker-Enfants Malades Hospital, Paris, France.
Author contributions
MU designed the research, conducted all experiments, analyzed data, and wrote the manuscript. GV and CF provided lymphoblastoid cell lines. HF and HS interpreted data and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Ayako Ui (Tokyo University of Technology) and Takashi Kohno (National Cancer Center Research Institute) for their gift of pIRES-TK-dA3-1 plasmid and useful advice; Niels Mailand (University of Copenhagen) for his gift of GFP-Ku80 plasmid; Mizuki Ohno, Yoshimichi Nakatsu, and Yoshihiro Baba (Kyushu University) for their useful advice; Christopher Jenness (The Rockefeller University) for sharing his unpublished data; Mizuho Oda and Emiko Koba (Laboratory for Technical Support, Medical Institute of Bioregulation, Kyushu University) for their technical assistance with mass spectrometry; and Kenji Ishikawa (Shinkouseiki. Co., Ltd.) for his technical advice with confocal microscopy. This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI grants JP26253020 (to HS) and JP18K06961 (to MU) and QR Program of Kyushu University (to MU). HF is supported by a grant (R01GM075249) from the NIH.
Version 1. 10/11/2018
In-Press Preview
Version 2. 11/19/2018
Electronic publication
Version 3. 01/02/2019
Print issue publication
Funding Statement
Hironori Funabiki needs to comply with NIHMS’s open access policy by linking this grant to the manuscript, which should be submitted to the NIHMS system.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
License: Copyright 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(1):78–92.https://doi.org/10.1172/JCI99751.
Contributor Information
Motoko Unoki, Email: unokim@bioreg.kyushu-u.ac.jp.
Hironori Funabiki, Email: funabih@rockefeller.edu.
Guillaume Velasco, Email: guillaume.velasco@univ-paris-diderot.fr.
Claire Francastel, Email: claire.francastel@univ-paris-diderot.fr.
Hiroyuki Sasaki, Email: hsasaki@bioreg.kyushu-u.ac.jp.
References
- 1.Ehrlich M, Jackson K, Weemaes C. Immunodeficiency, centromeric region instability, facial anomalies syndrome (ICF) Orphanet J Rare Dis. 2006;1:2. doi: 10.1186/1750-1172-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blanco-Betancourt CE, et al. Defective B-cell-negative selection and terminal differentiation in the ICF syndrome. Blood. 2004;103(7):2683–2690. doi: 10.1182/blood-2003-08-2632. [DOI] [PubMed] [Google Scholar]
- 3.Maraschio P, Zuffardi O, Dalla Fior T, Tiepolo L. Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: the ICF syndrome. J Med Genet. 1988;25(3):173–180. doi: 10.1136/jmg.25.3.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 5.Xu GL, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402(6758):187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 6.Hansen RS, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 1999;96(25):14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smeets DF, et al. ICF syndrome: a new case and review of the literature. Hum Genet. 1994;94(3):240–246. doi: 10.1007/BF00208277. [DOI] [PubMed] [Google Scholar]
- 8.Jeanpierre M, et al. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum Mol Genet. 1993;2(6):731–735. doi: 10.1093/hmg/2.6.731. [DOI] [PubMed] [Google Scholar]
- 9.Miniou P, et al. Abnormal methylation pattern in constitutive and facultative (X inactive chromosome) heterochromatin of ICF patients. Hum Mol Genet. 1994;3(12):2093–2102. doi: 10.1093/hmg/3.12.2093. [DOI] [PubMed] [Google Scholar]
- 10.Yehezkel S, Segev Y, Viegas-Péquignot E, Skorecki K, Selig S. Hypomethylation of subtelomeric regions in ICF syndrome is associated with abnormally short telomeres and enhanced transcription from telomeric regions. Hum Mol Genet. 2008;17(18):2776–2789. doi: 10.1093/hmg/ddn177. [DOI] [PubMed] [Google Scholar]
- 11.Sagie S, et al. Telomeres in ICF syndrome cells are vulnerable to DNA damage due to elevated DNA:RNA hybrids. Nat Commun. 2017;8:14015. doi: 10.1038/ncomms14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang YL, et al. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum Mutat. 2005;25(1):56–63. doi: 10.1002/humu.20113. [DOI] [PubMed] [Google Scholar]
- 13.Toubiana S, et al. Subtelomeric methylation distinguishes between subtypes of Immunodeficiency, Centromeric instability and Facial anomalies syndrome. Hum Mol Genet. 2018;27(20):3568–3581. doi: 10.1093/hmg/ddy265. [DOI] [PubMed] [Google Scholar]
- 14.de Greef JC, et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet. 2011;88(6):796–804. doi: 10.1016/j.ajhg.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thijssen PE, et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat Commun. 2015;6:7870. doi: 10.1038/ncomms8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nitta H, et al. Three novel ZBTB24 mutations identified in Japanese and Cape Verdean type 2 ICF syndrome patients. J Hum Genet. 2013;58(7):455–460. doi: 10.1038/jhg.2013.56. [DOI] [PubMed] [Google Scholar]
- 17.Chouery E, et al. A novel deletion in ZBTB24 in a Lebanese family with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Clin Genet. 2012;82(5):489–493. doi: 10.1111/j.1399-0004.2011.01783.x. [DOI] [PubMed] [Google Scholar]
- 18.Velasco G, et al. Comparative methylome analysis of ICF patients identifies heterochromatin loci that require ZBTB24, CDCA7 and HELLS for their methylated state. Hum Mol Genet. 2018;27(14):2409–2424. doi: 10.1093/hmg/ddy130. [DOI] [PubMed] [Google Scholar]
- 19.Wu H, et al. Converging disease genes in ICF syndrome: ZBTB24 controls expression of CDCA7 in mammals. Hum Mol Genet. 2016;25(18):4041–4051. doi: 10.1093/hmg/ddw243. [DOI] [PubMed] [Google Scholar]
- 20.Jenness C, Giunta S, Müller MM, Kimura H, Muir TW, Funabiki H. HELLS and CDCA7 comprise a bipartite nucleosome remodeling complex defective in ICF syndrome. Proc Natl Acad Sci U S A. 2018;115(5):E876–E885. doi: 10.1073/pnas.1717509115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren J, et al. The ATP binding site of the chromatin remodeling homolog Lsh is required for nucleosome density and de novo DNA methylation at repeat sequences. Nucleic Acids Res. 2015;43(3):1444–1455. doi: 10.1093/nar/gku1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu W, et al. Genome-wide DNA methylation patterns in LSH mutant reveals de-repression of repeat elements and redundant epigenetic silencing pathways. Genome Res. 2014;24(10):1613–1623. doi: 10.1101/gr.172015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu H, et al. Lsh is involved in de novo methylation of DNA. EMBO J. 2006;25(2):335–345. doi: 10.1038/sj.emboj.7600925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myant K, Stancheva I. LSH cooperates with DNA methyltransferases to repress transcription. Mol Cell Biol. 2008;28(1):215–226. doi: 10.1128/MCB.01073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zemach A, et al. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell. 2013;153(1):193–205. doi: 10.1016/j.cell.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyons DB, Zilberman D. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. Elife. 2017;6:e30674. doi: 10.7554/eLife.30674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burrage J, Termanis A, Geissner A, Myant K, Gordon K, Stancheva I. The SNF2 family ATPase LSH promotes phosphorylation of H2AX and efficient repair of DNA double-strand breaks in mammalian cells. J Cell Sci. 2012;125(pt 22):5524–5534. doi: 10.1242/jcs.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan T, et al. Lsh-deficient murine embryonal fibroblasts show reduced proliferation with signs of abnormal mitosis. Cancer Res. 2003;63(15):4677–4683. [PubMed] [Google Scholar]
- 29.Geiman TM, Tessarollo L, Anver MR, Kopp JB, Ward JM, Muegge K. Lsh, a SNF2 family member, is required for normal murine development. Biochim Biophys Acta. 2001;1526(2):211–220. doi: 10.1016/s0304-4165(01)00129-5. [DOI] [PubMed] [Google Scholar]
- 30.Geiman TM, Muegge K. Lsh, an SNF2/helicase family member, is required for proliferation of mature T lymphocytes. Proc Natl Acad Sci U S A. 2000;97(9):4772–4777. doi: 10.1073/pnas.97.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gill RM, Gabor TV, Couzens AL, Scheid MP. The MYC-associated protein CDCA7 is phosphorylated by AKT to regulate MYC-dependent apoptosis and transformation. Mol Cell Biol. 2013;33(3):498–513. doi: 10.1128/MCB.00276-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Gent DC, van der Burg M. Non-homologous end-joining, a sticky affair. Oncogene. 2007;26(56):7731–7740. doi: 10.1038/sj.onc.1210871. [DOI] [PubMed] [Google Scholar]
- 33.Casellas R, et al. Ku80 is required for immunoglobulin isotype switching. EMBO J. 1998;17(8):2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rolink A, Melchers F, Andersson J. The SCID but not the RAG-2 gene product is required for S mu-S epsilon heavy chain class switching. Immunity. 1996;5(4):319–330. doi: 10.1016/S1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- 35.Björkman A, et al. DNA-PKcs is involved in Ig class switch recombination in human B cells. J Immunol. 2015;195(12):5608–5615. doi: 10.4049/jimmunol.1501633. [DOI] [PubMed] [Google Scholar]
- 36.Smeenk G, et al. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J Cell Sci. 2013;126(pt 4):889–903. doi: 10.1242/jcs.109413. [DOI] [PubMed] [Google Scholar]
- 37.Kari V, Shchebet A, Neumann H, Johnsen SA. The H2B ubiquitin ligase RNF40 cooperates with SUPT16H to induce dynamic changes in chromatin structure during DNA double-strand break repair. Cell Cycle. 2011;10(20):3495–3504. doi: 10.4161/cc.10.20.17769. [DOI] [PubMed] [Google Scholar]
- 38.Bylund L, Kytölä S, Lui WO, Larsson C, Weber G. Analysis of the cytogenetic stability of the human embryonal kidney cell line 293 by cytogenetic and STR profiling approaches. Cytogenet Genome Res. 2004;106(1):28–32. doi: 10.1159/000078556. [DOI] [PubMed] [Google Scholar]
- 39.Hwang J, Kim YK. When a ribosome encounters a premature termination codon. BMB Rep. 2013;46(1):9–16. doi: 10.5483/BMBRep.2013.46.1.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Acilan C, Potter DM, Saunders WS. DNA repair pathways involved in anaphase bridge formation. Genes Chromosomes Cancer. 2007;46(6):522–531. doi: 10.1002/gcc.20425. [DOI] [PubMed] [Google Scholar]
- 41.Difilippantonio MJ, et al. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature. 2000;404(6777):510–514. doi: 10.1038/35006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem. 2000;275(13):9390–9395. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 43.Wang Y, Ghosh G, Hendrickson EA. Ku86 represses lethal telomere deletion events in human somatic cells. Proc Natl Acad Sci U S A. 2009;106(30):12430–12435. doi: 10.1073/pnas.0903362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirohzu H, et al. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am J Med Genet. 2002;112(1):31–37. doi: 10.1002/ajmg.10658. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, et al. Sequence-specific microscopic visualization of DNA methylation status at satellite repeats in individual cell nuclei and chromosomes. Nucleic Acids Res. 2013;41(19):e186. doi: 10.1093/nar/gkt766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogiwara H, et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30(18):2135–2146. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- 47.Rothkamm K, Krüger I, Thompson LH, Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23(16):5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7(18):2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Shi Y, Huang D, Guan X. Emerging therapeutic modalities of PARP inhibitors in breast cancer. Cancer Treat Rev. 2018;68:62–68. doi: 10.1016/j.ctrv.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 50.Lee YJ, Park SJ, Ciccone SL, Kim CR, Lee SH. An in vivo analysis of MMC-induced DNA damage and its repair. Carcinogenesis. 2006;27(3):446–453. doi: 10.1093/carcin/bgi254. [DOI] [PubMed] [Google Scholar]
- 51.Marchesi F, Turriziani M, Tortorelli G, Avvisati G, Torino F, De Vecchis L. Triazene compounds: mechanism of action and related DNA repair systems. Pharmacol Res. 2007;56(4):275–287. doi: 10.1016/j.phrs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152(6):1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Q, Goldstein M. Small RNAs recruit chromatin-modifying enzymes MMSET and Tip60 to reconfigure damaged DNA upon double-strand break and facilitate repair. Cancer Res. 2016;76(7):1904–1915. doi: 10.1158/0008-5472.CAN-15-2334. [DOI] [PubMed] [Google Scholar]
- 54.Aydin ÖZ, Vermeulen W, Lans H. ISWI chromatin remodeling complexes in the DNA damage response. Cell Cycle. 2014;13(19):3016–3025. doi: 10.4161/15384101.2014.956551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saito Y, Zhou H, Kobayashi J. Chromatin modification and NBS1: their relationship in DNA double-strand break repair. Genes Genet Syst. 2016;90(4):195–208. doi: 10.1266/ggs.15-00010. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe R, et al. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014;74(9):2465–2475. doi: 10.1158/0008-5472.CAN-13-3608. [DOI] [PubMed] [Google Scholar]
- 57.Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220(4601):1055–1057. doi: 10.1126/science.6844925. [DOI] [PubMed] [Google Scholar]
- 58.Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141(1):81–93. doi: 10.1016/j.cell.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ye C, et al. Radiation-induced cellular senescence results from a slippage of long-term G2 arrested cells into G1 phase. Cell Cycle. 2013;12(9):1424–1432. doi: 10.4161/cc.24528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang XR, Liu YA, Sun F, Li H, Lei SW, Wang JF. p21 is responsible for ionizing radiation-induced bypass of mitosis. Biomed Environ Sci. 2016;29(7):484–493. doi: 10.3967/bes2016.064. [DOI] [PubMed] [Google Scholar]
- 61.Mansour WY, et al. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res. 2008;36(12):4088–4098. doi: 10.1093/nar/gkn347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Forsburg SL, Shen KF. Centromere stability: the replication connection. Genes (Basel) 2017;8(1):E37. doi: 10.3390/genes8010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gelot C, Magdalou I, Lopez BS. Replication stress in mammalian cells and its consequences for mitosis. Genes (Basel) 2015;6(2):267–298. doi: 10.3390/genes6020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iwakawa M, et al. DNA repair capacity measured by high throughput alkaline comet assays in EBV-transformed cell lines and peripheral blood cells from cancer patients and healthy volunteers. Mutat Res. 2005;588(1):1–6. doi: 10.1016/j.mrgentox.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 65.Velasco G, et al. Germline genes hypomethylation and expression define a molecular signature in peripheral blood of ICF patients: implications for diagnosis and etiology. Orphanet J Rare Dis. 2014;9:56. doi: 10.1186/1750-1172-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Richardson C, Moynahan ME, Jasin M. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 1998;12(24):3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13(20):2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomizawa S, et al. Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development. 2011;138(5):811–820. doi: 10.1242/dev.061416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36(Web Server issue):W170–W175. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.