

Abstract
Treatment of hypothyroidism involves the endogenous conversion of thyroxine (T4) to 3,5,3′-triiodothyronine (T3) and may not be optimal in some cases when based on T4 alone. In the current issue of the JCI, Jo et al. present results that explain the reduced enzymatic activity of a common genetic variant of the enzyme responsible for this conversion, type 2 deiodinase (DIO2). The authors further explore the functional consequences of this variant on brain T3 activity, endoplasmic reticulum stress in glial cells, and cognitive function. These findings have important implications for the clinical treatment of hypothyroidism and for susceptibility to other neurological and metabolic diseases.
Role of DIO2 in thyroid hormone therapy
Whether due to autoimmune thyroid disease or secondary to the removal of the thyroid gland, hypothyroidism is a highly prevalent condition that affects 5% to 10% of the adult population (1). Thyroid hormone therapy, which is based on the administration of thyroxine (T4, 3,3′,5,5′-tetraiodothyronine), is used to help hypothyroid patients normalize their metabolism, cardiac function, mood, and cognition. However, to attain its full ability to regulate gene transcription in target cells, T4 must be converted to 3,5,3′-triiodothyronine (T3), the thyroid hormone that binds specific nuclear receptors with high affinity (2). This conversion process is largely accomplished by type 2 deiodinase (DIO2 or D2) (3).
Although this treatment is generally efficient, about 15% of patients are resistant and show persistent neurological symptoms, including deficits in memory and mood, even after T4 dosage is individually adjusted (4). Given the heavy reliance of this treatment on DIO2 function, investigators have explored DIO2 further and have been intrigued by the presence of a SNP in the human DIO2 gene (5). This SNP is present in up to a third of the human population and results in the change of a threonine (Thr) to an alanine (Ala) in amino acid 92 of the DIO2 polypeptide chain. The substitution affects a small and conserved domain of the protein involved in binding ubiquitin ligases (6), directing the enzyme for proteasomal degradation in a process that depends on the relative abundance and binding of its substrate, T4 (7).
Although initial assays on sonicates from transfected cells showed no deficits in the activity of Ala92-DIO2, studies in intact cells indicated reduced deiodination (8, 9), suggesting that the function of Ala-DIO2 is impaired in a physiological context. This notion was further supported by the clinical benefit that hypothyroid patients carrying this DIO2 SNP obtained from a combination therapy of T4 and T3 (10, 11).
Reduced functionality of Ala92-DIO2 explored
Why would Ala92-DIO2 not be fully functional in vivo? What would be the implications for brain thyroid hormone homeostasis and neurological function? In a remarkable bedside-to-bench work published in this issue of the JCI, Sungro Jo and Tatiana Fonseca, together with colleagues from a multiinstitutional team lead by Antonio Bianco, provide some critical answers to those questions and expand on the clinical implications of the Thr92Ala-DIO2 SNP (12).
In addition to confirming that intact transgenic cells expressing Ala-DIO2 exhibit a reduction in T3 production from T4, the authors observed that DIO2 is recycled between the ER and the Golgi using ER Golgi intermediary compartment (ERGIC) vesicles. They further showed (Figure 1) that the presence of this SNP causes ER stress and impairs ERGIC-mediated shuttling of Ala92-DIO2, which is then abnormally accumulated in the central Golgi, where it is enzymatically inactive.
Figure 1. Pathological basis of Ala92-DIO2.
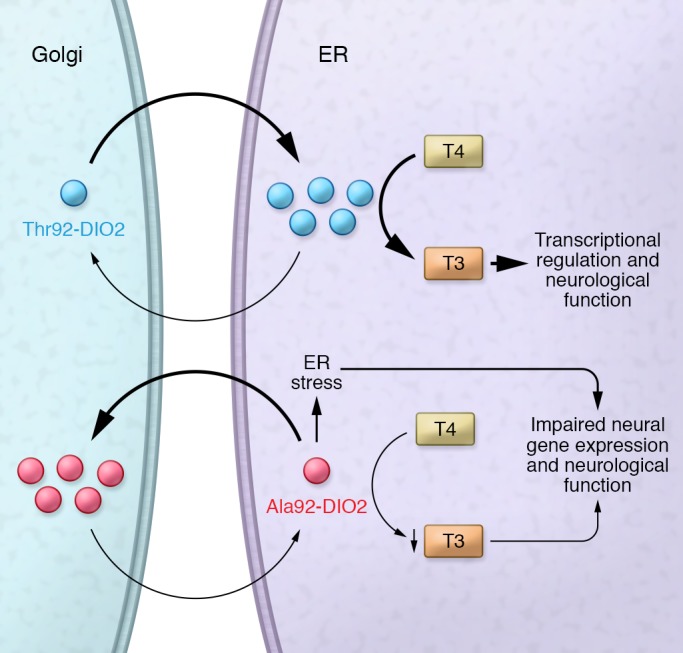
Thr92-DIO2 localizes in the ER where, with the support of appropriate cofactors, it is able to convert T4 into T3 for transcriptional regulation in the nucleus. DIO2 is recycled through the Golgi. Ala92-DIO2 causes ER stress and impairs the recycling of DIO2, which aberrantly accumulates in the Golgi, where it is not active. ER stress and decreased T3 production exert biological effects on glial cells and neurons, affecting neurological function.
If Ala92-DIO2 enzymatic activity is impaired in vivo, what are the functional consequences for the brain, a tissue highly dependent on T3 generated locally by DIO2 (13)? DIO2 is present in glial cells, where it also provides T3 for biological activity in neurons (14). This critical role has been further underscored by the abnormalities in behavior and brain gene expression observed in mice with astrocyte-specific DIO2 deficiency (15). Using “humanized” transgenic mice, Jo et al. showed that the subcellular abnormalities of Ala92-DIO2 were associated with decreased T3 action in the brain. Mice expressing the Ala92-DIO2 variant are euthyroid, based on serum parameters. However, areas of their brains exhibited decreased T3-dependent gene expression and abnormalities in the expression of genes related to ER stress and the ERGIC system. Furthermore, these mice exhibited impaired short-term memory, reduced levels of spontaneous physical activity, and increased sleep, characteristics that resemble those of clinical hypothyroidism.
The gene expression changes in the Ala92-DIO2 brains may seem too modest to cause a neurological phenotype. However, the brain is notorious for its strict and spatial-specific regulation of thyroid hormone levels. In fact, when Ala92-DIO2 mice were treated with T3, their levels of physical activity were normalized, demonstrating that at least some of their neurological deficits were actually caused by local T3 deficiency. Furthermore, the authors examined neurological function after rendering the mice hypothyroid with antithyroid drugs, a model that recapitulates the clinical condition of many hypothyroid patients and eliminates the biological contribution of the T3 secreted by the thyroid gland. When hypothyroid, both Thr92-DIO2 and Ala92-DIO2 mice showed neurological deficits affecting memory, sleep, and locomotor activity. However, after T4 administration, this phenotype was rescued only in Thr92-DIO2 mice and not in Ala92-DIO2 mice. In the latter, neurological function was normalized only when the mice were treated with a combination of T4 and T3. These findings again highlight the deficit in brain T3 associated with Ala92-DIO2, a situation that could become even more critical in the context of hypothyroidism secondary to thyroid disease, as seen in human patients.
Broader implications of the study
There are additional and highly intriguing aspects of this work. As mentioned above, in intact Ala92-DIO2 mice, T3 treatment rescues only one of the neurological phenotypes examined. However, normalizing protein homeostasis with an ER stress inhibitor fully restores the neurological functions. This observation suggests that, at least under serum euthyroid conditions, part of the neurological pathophysiology of Ala92-DIO2 mice is primarily due to of ER stress. In this scenario, ER stress can potentially lead, not only to mislocalization of DIO2 and reduced T3 generation, but also to glial cell dysfunction, potentially influencing neuronal physiology. This possibility is consistent with epidemiological studies showing increased prevalence of bipolar disorder and Alzheimer’s disease in individuals carrying the Ala92-DIO2 polymorphism (16). It would be interesting to determine whether Ala92-DIO2 contributes to age-related cognitive decline and neurodegeneration, especially in the context of the natural changes in thyroid function associated with age.
It is also conceivable that other neurological abnormalities of Ala92- DIO2 mice not identified in this work could partly originate during development, either as a consequence of ER stress alone or combined with reduced local T3 generation. DIO2 expression in most rodent brain regions peaks during neonatal stages, when T3 action and responsiveness are highest in the central nervous system (17). It is then possible that impaired T3 generation in Ala92-DIO2 carriers may influence brain development and explain the increased incidence of mental retardation associated with this DIO2 variant (16).
The extent to which hypothyroid patients with T4-resistant neurological symptoms coincide with those carrying Ala92-DIO2 is not yet clear. Even if this is only partially the case, the presence of Ala92-DIO2 appears as an important risk factor for suboptimal treatment of hypothyroidism, and the present work is a substantial advance in understanding the cellular and molecular basis of this risk.
Clinical implications of Ala92-DIO2 SNP pathology
The high prevalence of hypothyroidism, the relatively high abundance of the Ala92-DIO2 variant in the human population, and the presence of DIO2 in many areas of the central nervous system and other tissues confer broad clinical relevance to the present work and strongly warrant additional studies. Work examining the interacting proteins responsible for the altered subcellular homeostasis of Ala92-DIO2 and defining the thyroid status in which these interactions are most critical to pathophysiology could yield important mechanistic insights. Research defining the pathological contributions of ER stress and T3 deficiency or focusing on specific brain regions and additional neurological functions is also warranted. Finally, given the associations of this SNP with increased metabolic abnormalities, it is also possible that disease susceptibility in Ala92-DIO2 carriers is partly the result of thyroid hormone alterations in nonneural, DIO2-expressing tissues. Our understanding of the pathological basis of the Ala92-DIO2 SNP has just begun, and clinical implications may extend well beyond the treatment of hypothyroidism.
Acknowledgments
This work was supported by grants MH096050 and DK095908 from the NIH.
Version 1. 12/03/2018
Electronic publication
Version 2. 01/02/2019
Print issue publication
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Reference information: J Clin Invest. 2019;129(1):55–57. https://doi.org/10.1172/JCI125203.
See the related article at Type 2 deiodinase polymorphism causes ER stress and hypothyroidism in the brain.
References
- 1.Chaker L, Bianco AC, Jonklaas J, Peeters RP. Hypothyroidism. Lancet. 2017;390(10101):1550–1562. doi: 10.1016/S0140-6736(17)30703-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. 2012;122(9):3035–3043. doi: 10.1172/JCI60047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gereben B, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29(7):898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saravanan P, Visser TJ, Dayan CM. Psychological well-being correlates with free thyroxine but not free 3,5,3′-triiodothyronine levels in patients on thyroid hormone replacement. J Clin Endocrinol Metab. 2006;91(9):3389–3393. doi: 10.1210/jc.2006-0414. [DOI] [PubMed] [Google Scholar]
- 5.Dora JM, Machado WE, Rheinheimer J, Crispim D, Maia AL. Association of the type 2 deiodinase Thr92Ala polymorphism with type 2 diabetes: case-control study and meta-analysis. Eur J Endocrinol. 2010;163(3):427–434. doi: 10.1530/EJE-10-0419. [DOI] [PubMed] [Google Scholar]
- 6.Zavacki AM, et al. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol Cell Biol. 2009;29(19):5339–5347. doi: 10.1128/MCB.01498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagar GD, et al. Ubiquitination-induced conformational change within the deiodinase dimer is a switch regulating enzyme activity. Mol Cell Biol. 2007;27(13):4774–4783. doi: 10.1128/MCB.00283-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canani LH, et al. The type 2 deiodinase A/G (Thr92Ala) polymorphism is associated with decreased enzyme velocity and increased insulin resistance in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2005;90(6):3472–3478. doi: 10.1210/jc.2004-1977. [DOI] [PubMed] [Google Scholar]
- 9.Castagna MG, et al. DIO2 Thr92Ala reduces deiodinase-2 activity and serum-T3 levels in thyroid-deficient patients. J Clin Endocrinol Metab. 2017;102(5):1623–1630. doi: 10.1210/jc.2016-2587. [DOI] [PubMed] [Google Scholar]
- 10.Panicker V, et al. Common variation in the DIO2 gene predicts baseline psychological well-being and response to combination thyroxine plus triiodothyronine therapy in hypothyroid patients. J Clin Endocrinol Metab. 2009;94(5):1623–1629. doi: 10.1210/jc.2008-1301. [DOI] [PubMed] [Google Scholar]
- 11.Carlé A, Faber J, Steffensen R, Laurberg P, Nygaard B. Hypothyroid patients encoding combined MCT10 and DIO2 gene polymorphisms may prefer L-T3 + L-T4 combination treatment — data using a blind, randomized, clinical study. Eur Thyroid J. 2017;6(3):143–151. doi: 10.1159/000469709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jo S, et al. Type 2 deiodinase polymorphism causes ER stress and hypothyroidism in the brain. J Clin Invest. 2019;129(1):230–245. doi: 10.1172/JCI123176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crantz FR, Silva JE, Larsen PR. An analysis of the sources and quantity of 3,5,3′-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology. 1982;110(2):367–375. doi: 10.1210/endo-110-2-367. [DOI] [PubMed] [Google Scholar]
- 14.Freitas BC, et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest. 2010;120(6):2206–2217. doi: 10.1172/JCI41977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bocco BM, et al. Type 2 deiodinase disruption in astrocytes results in anxiety-depressive-like behavior in male mice. Endocrinology. 2016;157(9):3682–3695. doi: 10.1210/en.2016-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McAninch EA, et al. A Common DIO2 Polymorphism and Alzheimer Disease Dementia in African and European Americans. J Clin Endocrinol Metab. 2018;103(5):1818–1826. doi: 10.1210/jc.2017-01196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernal J. Thyroid hormones and brain development. Vitam Horm. 2005;71:95–122. doi: 10.1016/S0083-6729(05)71004-9. [DOI] [PubMed] [Google Scholar]