

Abstract
AU‐rich elements (ARE) exist in the 3′‐untranslated regions of the mRNA transcribed from cell growth‐related genes such as proto‐oncogenes, cyclin‐related genes, and growth factors. HuR binds and stabilizes ARE‐mRNA. HuR is expressed abundantly in cancer cells and related malignant phenotypes. HuR knockdown attenuates the malignant phenotype of oral cancer cells. In this study, we screened 1570 compounds in the approved drug library by differential scanning fluorimetry (DSF) to discover a HuR‐targeted compound. Firstly, 55 compounds were selected by DSF. Then, 8 compounds that showed a shift in the melting temperature value in a concentration‐dependent manner were selected by DSF. Of them, suramin, an anti‐trypanosomal drug, binds to HuR, exhibiting fast‐on and fast‐off kinetic behavior on surface plasmon resonance (SPR). We confirmed that suramin significantly decreased mRNA and protein expression of cyclin A2 and cyclin B1. The cyclin A2 and cyclin B1 mRNAs were destabilized by suramin. Furthermore, the motile and invasive activities of a tongue carcinoma cell line treated with suramin were markedly lower than those of control cells. The above findings suggest that suramin binds to HuR and inhibits its function. We also showed that the anticancer effects of suramin were caused by the inhibition of HuR function, indicating its potential as a novel therapeutic agent in the treatment of oral cancer. Our results suggest that suramin, via its different mechanism, may effectively suppress progressive oral cancer that cannot be controlled using other anticancer agents.
Keywords: AU‐rich elements, drug repositioning, HuR, screening, suramin
1. INTRODUCTION
Regulation of mRNA decay is one of the important steps of gene expression, as with transcription and translation. AU‐rich elements (ARE) are instability elements that are commonly present in the 3′‐untranslated regions (UTR) of mRNA encoding the so‐called early response genes such as proto‐oncogenes, cyclin‐related genes, and growth factors.1, 2, 3 A number of proteins are known to interact with ARE and modulate either the stabilization or destabilization of ARE‐mRNA.1, 2, 4, 5
HuR is an RNA‐binding protein, and it stabilizes not only ARE‐mRNA, but also mRNA not containing ARE.6, 7 HuR has three RNA recognition motifs (RRMs),8 which are highly abundant RNA‐binding domains in eukaryotes.9 The first two RRMs form a cleft that facilitates binding of mRNA8, 10, 11, 12 and they also interact to form a HuR homodimer.13 The hinge region between RRM2 and RRM3 contains the nuclear localization signal.4 RRM3 is important for stabilizing the RNA‐protein complex and mediating protein‐protein interactions,12 such as interactions with the poly‐A tail of target mRNA.10 Moreover, it shows terminal adenosyltransferase activity.14
HuR stabilizes ARE‐mRNAs involved in multiple biological events such as carcinogenesis, differentiation, and inflammation.15, 16 HuR is overexpressed in cancers, such as in oral, colon, ovarian, brain, breast, and pancreatic cancers.17, 18, 19, 20, 21, 22, 23 In particular, the abundant cytoplasmic expression of HuR is associated with the malignant phenotype of several types of carcinomas.19 We previously reported that HuR knockdown attenuated the malignant phenotype by inhibiting the stabilization of ARE‐mRNA. Inhibition of HuR‐ARE interaction can be an attractive strategy in developing new cancer therapeutics.7 Recently, RRMs have been considered for drug design approaches.11, 14 Some compounds have been reported to target RRM1 and RRM2.13, 24, 25 In addition, compounds targeting RRM3 have been reported.14, 24 However, the clinical application of these compounds as anticancer drugs will require a long time. To shorten the time until the clinical application of HuR‐targeted drugs, drug repositioning from an approved drug is effective.26
In the present study, we screened an approved drug library using differential scanning fluorimetry (DSF) to identify a HuR‐targeted compound. We identified suramin as a suitable candidate that bound to HuR and inhibited HuR function in HSC‐3 and SAS cells, a tongue carcinoma cell line. Given its potential to be used clinically, suramin activities were further evaluated in this study.
2. MATERIALS AND METHODS
2.1. Expression and purification of HuR protein
BL21 competent Escherichia coli (C2530H, NEB, Ipswich, MA, USA) cells were transformed with pGEX‐6P‐1 vectors containing sequences coding for full‐length HuR (326 residues) and incubated in Overnight ExpressTM lnstant LB medium (Merck Millipore, Billerica, MA, USA). E. coli was resuspended in Bug Buster Master Mix (Merck Millipore). Insoluble cell debris was removed by centrifugation at 16 000 × g for 20 minutes at 4°C. Recombinant GST‐HuR protein was pulled down by Glutathione Sepharose 4B (GST SpinTrap and GSTrap 4B; GE Healthcare, Uppsala, Sweden) column. HuR protein was separated from GST with PreScission Protease (GE Healthcare) and dissolved in HN buffer (20 mmol/L HEPES and 150 mmol/L NaCl buffer at pH 7.4). When the concentration of HuR protein was low, the protein was concentrated with Amicon Ultra centrifugal filters (Merck Millipore). The protein concentration was determined by TaKaRa Bradford Protein Assay Kit (TaKaRa, Kusatsu, Shiga, Japan). The solubility of GST‐HuR, HuR, and GST proteins was confirmed with gel staining and Western blot (Figure S1).
2.2. Differential scanning fluorimetry (DSF)
Thermal unfolding of HuR constructs was monitored by DSF in the presence of fluorescent SYPRO Orange dye (Invitrogen, Eugene, OR, USA) by using a CFX96 Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA, USA). An approved compound library, Pharmakon (MicroSource Discovery Systems, Gaylordsville, CT, USA), was used. The compounds were transferred to 96‐well microplates [10 mmol/L, dissolved in dimethyl sulfoxide (DMSO)]. Because 30 compounds of the library could not be imported due to ban on drugs import, DMSO was added to the wells instead of these compounds. HuR protein (5 μg) and the dye in HN buffer were used. Quercetin, which has been reported to bind HuR,27 was used as a positive control. DMSO was used as a negative control. The thermal unfolding process was monitored between 20 and 90°C, by increasing the temperature at a rate of 0.5°C. The values of melting temperature (T m) were calculated. The compounds were used at 200 μmol/L in the final 1% of DMSO for HuR protein (5 μg) in the first DSF and 467 μmol/L in the final 0.47% of DMSO for HuR protein (5 μg) in the second DSF.
2.3. Surface plasmon resonance (SPR)
The SPR experiment was performed on a BIACORE T200 instrument (GE Healthcare) equipped with a CM5 sensor chip. The sensor chip was activated via an amine‐coupling reaction with EDC and NHS. Then, HuR protein (36 kD) was applied to a flow cell with 10 mmol/L HEPES buffer at pH 7.0. HuR protein was immobilized at a density of 483 RU (response units). The surface was blocked with 1 mol/L ethanolamine hydrochloride at pH 8.5. To collect kinetic binding data, suramin in 10 mmol/L HEPES and 150 mmol/L NaCl (pH 7.4) was injected into the flow cell at the indicated concentrations at 25°C.
2.4. Cells
HSC‐3 and SAS, human tongue squamous cell carcinoma cell lines, were purchased from RIKEN BRC Cell Bank (Tsukuba, Japan) and JCRB Cell Bank (Osaka, Japan), respectively. The cells were cultured at 37°C under a 5% CO2 atmosphere in Dulbecco's modified Eagle medium (DMEM, Nissui Pharmaceutical, Ueno, Tokyo, Japan) containing 10% fetal bovine serum (FBS) and penicillin/streptomycin/amphotericin B (Sigma, St Louis, MO, USA).
2.5. Western blot analysis
Western blot analysis was performed on cells treated with different concentrations of suramin sodium (suramin) (Wako Pure Chemical Industries, Osaka, Japan) (0, 20, 50, and 100 μmol/L) for 24 hours in DMEM without FBS. The cells were homogenized in RIPA buffer (25 mmol/L Tris‐HCl, pH 7.6, 150 mmol/L NaCl, 1% NP‐40, 1% sodium deoxycholate, and 0.1% SDS) with protease inhibitor cocktail (p8340, Sigma). The protein concentration was determined by the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA). Samples (20 μg) were separated on 10% TGX FastCast™ acrylamide gel (Bio‐Rad) and transferred to a Amasham™ Hybond™ 0.2 μm polyvinylidene fluoride (PVDF) membrane (GE Healthcare). Blocking of the membrane was performed using PBS with 5% skim milk powder (Wako Pure Chemical Industries). The antibodies used in this study were specific to HuR (sc‐5261, 1:2000, Santa Cruz, Santa Cruz, CA, USA), cyclin A (611268, 1:2000), cyclin B1 (554178, 1:2000), Cox2 (610203, 1:2000, BD Biosciences, Cambridge, MA, USA), and β‐actin (A5441, 1:10 000, Sigma). The secondary antibody was horseradish peroxidase‐conjugated IgG (Jackson ImmunoResearch Laboratories, 1:5000‐10 000, West Grove, PA, USA). The protein bands were visualized with the Amersham™ ECL™ start or Prime Western Blotting Detection Reagent (GE Healthcare) and detected by LAS‐4000 mini (GE Healthcare).
2.6. Real‐time quantitative reverse transcription PCR (real‐time qRT‐PCR)
qRT‐PCR was performed on cells treated with different concentrations (0, 20, 50, and 100 μmol/L) of suramin for 24 hours in DMEM without FBS. Total cellular RNA was isolated using TRI reagent (Molecular Research Center, Inc, Montgomery Rd, OH, USA) according to the manufacturer's protocol. Selected total RNA samples (1 μg) were reverse‐transcribed using ReverTra Ace (Toyobo, Osaka, Japan). CFX96 Real‐Time PCR Detection System (Bio‐Rad) and the SSO Advanced™ Universal CYBR Green Supermix (Bio‐Rad) were used for qRT‐PCR. cDNA was amplified using the following primers: for cyclin A2: 5′‐AGCTGCCTTTCATTTAGCACTC‐3′, 5′‐TGCTTTGAGGTAGGTCTGGTG‐3′; for cyclin B1: 5′‐TGTGGATGCAG ‐AAGATGGAG‐3′, 5′‐AACCGATCAATAATGGAGACAG‐3′; for c‐fos: 5′‐CCAACCTGCTGAAGGAGAAG‐3′, 5′‐GCTGCTGATGCTCTTGACAG‐ 3′; for c‐myc: 5′‐CTCCTGGCAAAAGGTCAGAG‐3′, 5′‐TCGGTTGTTGCT‐ GATCTGTC‐3′; for COX‐2:5′‐TGAGCATCTACGGTTTGCTG‐3′, 5′‐TGC‐ TTGTCTGGAACAACTGC‐3′; for cdk1: 5′‐TTCAGGATGTGCTTATGCAG‐ GA‐3′, 5′‐AGTGACAAAACACAATCCCCTGTAG‐3′ and for GAPDH: 5′‐ATCCTGGGCTACACTGAGCA‐3′, 5′‐TGCTGTAGCCAAATTCGTTG‐3′. GAPDH was used for normalization. Relative quantification was performed using the 2−ΔΔ Ct method.
To evaluate the half‐life of total ARE‐mRNA, both control cells and 50 μmol/L suramin‐treated cells were treated with 5 μg/mL actinomycin d‐mannitol (Sigma) for 2 or 4 hours. The RNA extracted after treatment with actinomycin d‐mannitol was subjected to qRT‐PCR.
2.7. MTS assay
The CellTiter 96R Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) was used to detect the viable cells in the proliferation and cytotoxicity assay. HSC‐3 cells (5 × 103) were harvested in 96‐well plates. After 24 hours, the culture medium was discarded and the cells were washed in phosphate‐buffered saline (PBS). Then, DMEM without FBS containing suramin at various concentrations (0, 5, 10, 20, 50, 75, 100, 200, 400, 600, 800, and 1000 μmol/L) was added to the wells. After 24 hours, MTS reagents were added for 4 hours, and the absorbance at optical density (OD) 490 nm was recorded using a microplate reader (Bio‐Rad).
2.8. Wound healing assay
HSC‐3 and SAS cells were wounded with a 200‐μL pipette tip and washed with PBS. The cells were then incubated for 24 hours with 0, 10, 20, 35, 50, and 100 μmol/L of suramin in DMEM without FBS. Migration of the wounded cells was evaluated after 0, 12, 24, and 48 hours with an inverted OLYMPUS CKX41 microscope.
2.9. In vitro invasion assays
Corning Biocoat Matrigel Invasion Chamber (Corning, Two Oak Park, MA, USA) was used for the invasion assays. HSC‐3 and SAS cells were washed in PBS and then suspended in DMEM without FBS. The cells (1.0 × 105) were added to the upper chamber. The lower chamber was filled with 0, 20, 50, and 100 μmol/L of suramin in DMEM without FBS. After incubation for 24 hours, the cells were fixed with 10% formaldehyde neutral buffer solution (Sigma) for 20 minutes at room temperature, before being stained with crystal violet (0.05% in distilled water) (Katayama Chemical, Osaka, Japan) for 10 minutes. The invading cells were counted using an inverted OLYMPUS CKX41 microscope at ×40 magnification.
2.10. Statistics
Statistical analyses of significance were performed using unpaired Student's t test for comparison between two groups (control vs suramin treatment). The results are shown as the mean ± SD; P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Screening of the approved drug library for targeting of HuR by DSF
To identify HuR‐interacting compounds, we screened 1570 compounds in the approved drug library by DSF. The compounds were used at 200 μmol/L for HuR (5 μg) in the first DSF. The T m of 55 compounds was shifted by more than 1.0°C positively or negatively (Figure 1A). The concentration‐dependent thermal shift of T m was confirmed for 55 compounds. The compounds were used at 467 μmol/L for HuR (5 μg) in the second DSF. The shift in the T m of eight compounds was more than that in the first DSF (2.0°C positively or negatively; Figure 1B). Eight compounds of irregular waveform were excluded. Only suramin showed a positive shift; all other compounds (antibiotics, surface acting agent, hormone, etc) showed a negative shift (Figure 1C). Furthermore, to confirm HuR interaction of suramin, DSF was performed at each concentration of suramin. The difference in T m was the highest from 20 μmol/L to 50 μmol/L, and the T m at 100 and 500 μmol/L was almost the same (Figure 1D). These data show that suramin interacted with HuR in a concentration‐dependent manner.
Figure 1.
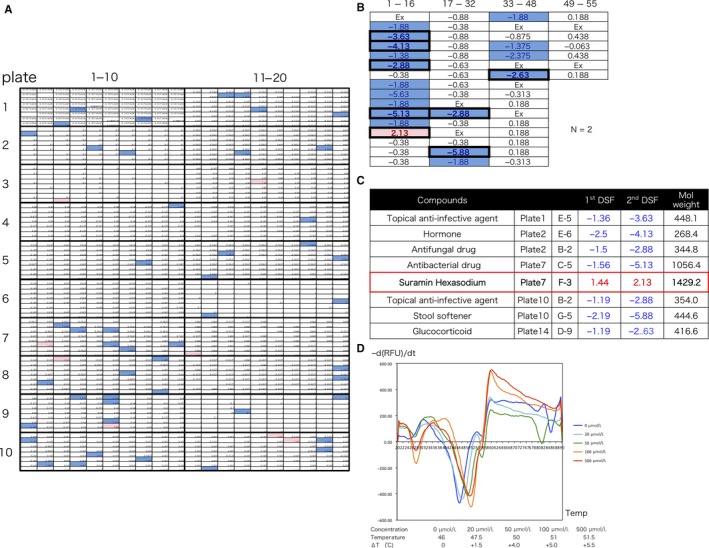
Screening of approved drug library for HuR targets by DSF. A, Screening of 1570 compounds in the approved drug library by DSF. The compounds were used at 200 μmol/L for HuR (5 μg) in the 1st DSF. Red boxes show T m increase by more than 1.0°C. Blue boxes show T m decrease by less than 1.0°C. A total of 80 compounds were placed in one plate. First DSF selected 55 compounds. B, Concentration‐dependent thermal shift was confirmed for 55 compounds. The compounds were used at 467 μmol/L for HuR (5 μg) in the 2nd DSF. The 2nd DSF selected eight compounds. Exclusion (Ex) of irregular waveform. C, Only suramin was shifted positively. All other compounds were shifted negatively. D, DSF for each concentration of suramin
3.2. Suramin binds to HuR on SPR
SPRis label‐free and enables real‐time quantification of ligand‐binding affinities and kinetics.28 We performed SPR to evaluate the ligand‐binding affinities and kinetics between suramin and HuR. The SPR data are shown in Figure 2. The binding interaction between suramin and HuR was concentration‐dependent. The KD value (2.4 × 10−4 mol/L) was obtained from the steady‐state phase data. The sensorgrams showed square wave appearance, which shows that the interactions reached a steady state rapidly upon injection and also showed rapid dissociation from the binding site. Moreover, the sensorgrams exhibited fast‐on and fast‐off kinetic behavior.29 These results show that suramin binds to HuR, but with low affinity.
Figure 2.
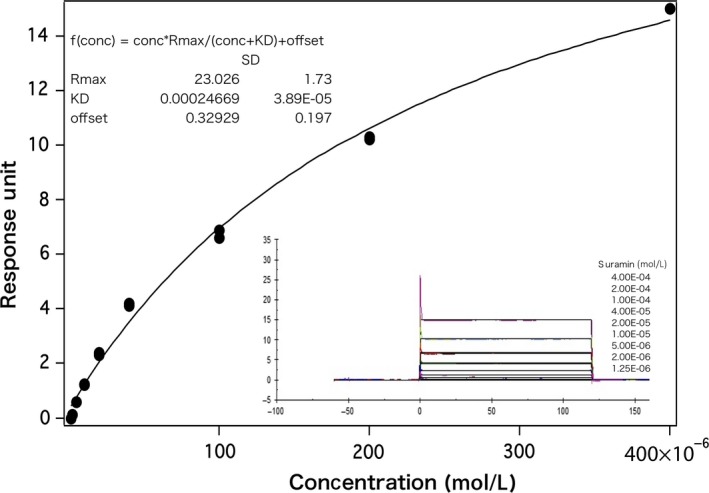
Suramin binds to HuR as shown by SPR. The binding interaction was concentration‐dependent. The KD value (2.4 × 10−4 mol/L) was obtained from the steady‐state phase data of the response versus concentration plot for nine different samples. The sensorgram showed square wave appearance exhibiting fast‐on and fast‐off kinetic behavior
3.3. Suramin inhibits HuR functions
HuR knockdown decreases the expression and stabilization of ARE‐mRNA such asc‐fos, c‐myc, and COX‐2, and mRNAs not containing ARE such as cdk1 and c‐fms.7, 17, 30 The expression and stabilization of these mRNAs decreased when suramin inhibited HuR function. To confirm the expression of ARE‐mRNAs such as c‐fos, c‐myc, COX‐2, cyclin A2, and cyclin B1, and mRNAs not containing ARE such as cdk1, we performed qRT‐PCR on HSC‐3 and SAS cells treated with different concentrations of suramin. The expression of all mRNAs decreased in HSC‐3 cells. A significant decrease was observed in the expression of COX‐2, cyclin A2, and cyclin B1. The expression of cdk1 showed a slight decrease (Figure 3A). Similarly, the expression of all mRNAs except COX‐2 decreased in SAS cells. A significant decrease was observed in the expression of c‐fos, cyclin A2, and cyclin B1. However, the expression of COX‐2 increased in SAS cells (Figure 3B). Suramin did not decrease the binding of all mRNA to HuR. It showed different effect in different cell lines, such as on COX‐2 levels. Furthermore, to confirm the stabilization of ARE‐mRNA, we determined the half‐life (t 1/2) of cyclin A2 and cyclin B1 mRNA in the control‐ and suramin‐treated cells. The expression of cyclin A2 and cyclin B1 mRNA was estimated at 0, 2, and 4 hours after treatment with actinomycin D (an inhibitor of RNA polymerase II) by qRT‐PCR, and the half‐life of these mRNAs was calculated (Figure 3C,D). In suramin‐treated HSC‐3 cells, the half‐life of cyclin A2 and cyclin B1 mRNA was 3.32 and 2.52 hours, respectively, whereas in the control cells, the half‐life was 4.97 and 3.10 hours, respectively (Figure 3C). In suramin‐treated SAS cells, the half‐life of cyclin A2 and cyclin B1 mRNA was 3.78 and 3.29 hours, whereas in the control cells, the half‐life was 4.03 and 3.35 hours, respectively (Figure 3D). To confirm that HuR overexpression inhibits decrease in expression of ARE‐mRNA for treatment of suramin, we performed to transfection in the HSC‐3 cells with plasmid of pcDNA3 or pcDNA3‐HuR and treatment of difference concentration of suramin. Though it is no significant difference, decrease rate of expression of the cyclin A2 and cyclin B1 mRNA in the suramin‐treated cells for the control cells was smaller in the cells transfected pcDNA3‐HuR than in the cells transfected control plasmid (Figure S2). Next, to confirm the location of HuR, we performed immunofluorescence staining. Suramin did not change the localization of HuR (Figure S3). These data indicate that suramin decreases the expression and stabilization of ARE‐mRNA but does not affect nucleo‐cytoplasmic transport, suggesting that suramin inhibits HuR functions, such as mRNA binding.
Figure 3.
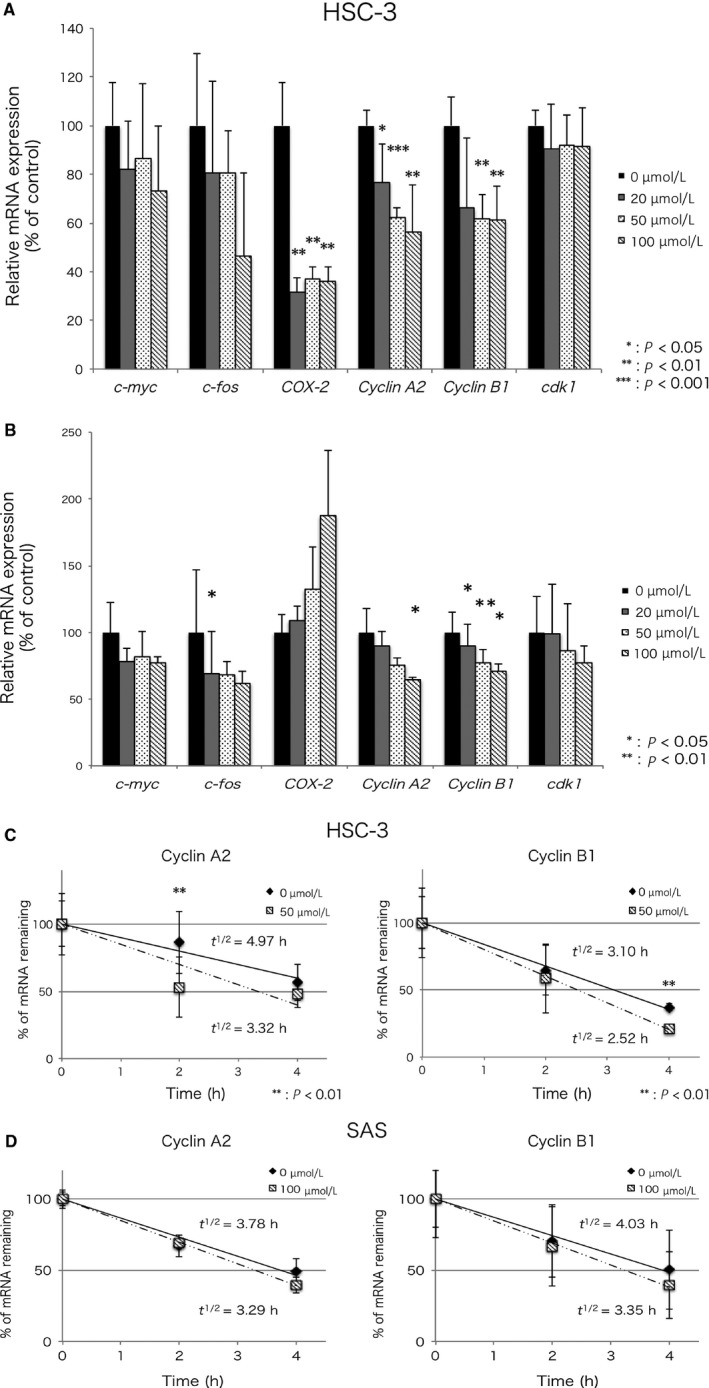
Suramin decreased the expression and stabilization of HuR‐binding mRNA. A,B, The accumulation of c‐fos, c‐myc, COX‐2, cyclin A2, cyclin B1, and cdk1 was estimated by qRT‐PCR. The data represent the mean of four independent experiments in HSC‐3 cells and three independent experiments in SAS cells. C,D, Stabilization of ARE‐mRNA. The amount of each ARE‐mRNA was estimated at the indicated actinomycin D treated time point by qRT‐PCR. The t 1/2 value indicates the half‐life of mRNA. The data represent the mean of three independent experiments. Error bars, SD. Student's t test, *P < 0.05, **P < 0.01, ***P < 0.001
3.4. Suramin decreased the levels of proteins translated from ARE‐mRNA
We performed Western blotting to confirm whether the levels of proteins translated from ARE‐mRNA decreased in suramin‐treated HSC‐3 cells. COX2, cyclin A2, and cyclin B1 protein levels decreased in suramin‐treated cells compared with the control cells (Figure 4). Suramin did not decrease the HuR protein level (Figure 4). These results indicate that suramin decreased COX‐2, cyclin A2, and cyclin B1 protein levels to inhibit HuR functions, such as mRNA stabilization.
Figure 4.
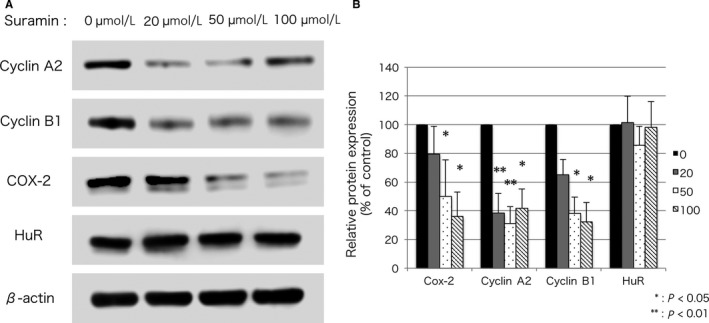
Suramin decreased the levels of proteins translated from ARE‐mRNA. A, HSC‐3 cells were treated with or without suramin, and the expression of COX‐2, cyclin A, cyclin B1, and HuR was estimated by Western blotting. B, Results of densitometry. Error bars, SD. Student's t test, *P < 0.05, **P < 0.01
3.5. Suramin decreases cell viability in a concentration‐dependent manner
MTS assay was performed to confirm the cytotoxicity of suramin against HSC‐3 cells. Although suramin decreased the viability of HSC‐3 cells in a concentration‐dependent manner, it showed low cytotoxicity (IC50 of suramin = 732 µmol/L). The cell viability at the highest treated concentration of suramin (1000 µmol/L) was 42% (Figure 5). These results suggest that suramin exhibited low cytotoxicity and did not induce necrosis strongly.
Figure 5.
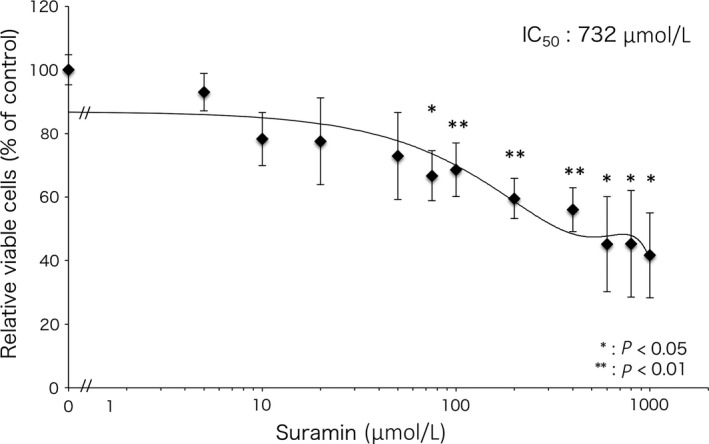
Survival of HSC‐3 cells treated with suramin by MTS assay. The survival rate of HSC‐3 cells treated with suramin was assessed by MTS assay. The concentration dependence of suramin decreased the survival rate of HSC‐3 cells. The IC50 value of suramin is 732 µmol/L. The data represent the mean of three independent experiments. Error bars, SD. Student's t test, *P < 0.05, **P < 0.01
3.6. Suramin attenuated motile and invasive activities of HSC‐3 and SAS cell line
In our previous report, we showed that HuR knockdown attenuates motile and invasive activities.7 We presumed that suramin attenuates the motile and invasive activities of HSC‐3 and SAS cells as well as HuR knockdown HSC‐3 cells. To prove this, wound healing and invasion assays were performed. HSC‐3 and SAS cells treated with each concentration of suramin were scratched and observed after 12, 24, and 48 hours. At a concentration of 35 µmol/L or above, suramin suppressed the motile activity of HSC‐3 cells significantly (P < 0.05 or 0.01; Figure 6A,B). At a concentration of 50 µmol/L or above, suramin suppressed the motile activity of SAS cells significantly (P < 0.05 or 0.01; Figure 6A,B). Next, HSC‐3 and SAS cells were seeded in invasion chambers containing Matrigel‐coated membranes, and the lower chamber was filled with different concentrations of suramin. Treatment of HSC‐3 cells with suramin at concentrations above 50 µmol/L strongly suppressed the invasive activity compared to control cells (P < 0.05; Figure 6C). Suramin (20 µmol/L) tended to suppress the invasive activity of HSC‐3 cells (P = 0.051). Treatment of SAS cells with suramin at 100 µmol/L strongly suppressed the invasive activity as compared to control cells (P < 0.05; Figure 6C). These results suggest that suramin strongly attenuated the motile and invasive activities and thus the malignant phenotype of HSC‐3 and SAS cells.
Figure 6.
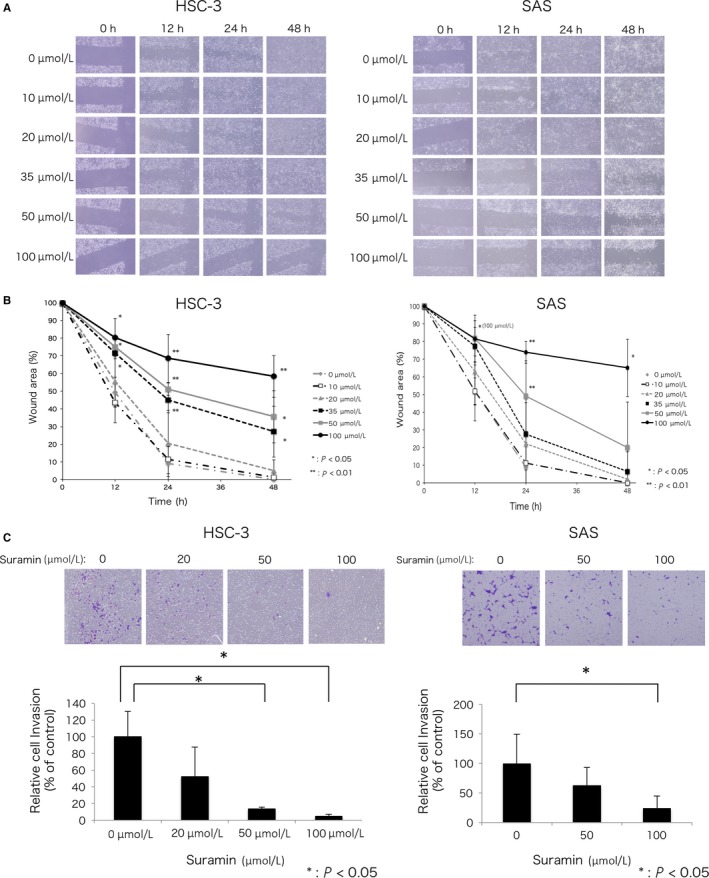
Suramin attenuated motility and invasion. A, Phase images of HSC‐3 and SAS cells treated with different concentrations of suramin at 0, 12, 24, and 48 h after wounding. B, Quantification of cell motility analyzed by wound healing assay. The data indicate the mean of three independent experiments. C, Invasion rate of HSC‐3 and SAS cells treated with different concentrations of suramin by using the Corning Biocoat Matrigel Invasion Chamber. After 24 h, the cells on the lower side of the membrane were fixed, stained, and counted. The data represent the mean of three independent experiments. Error bars, SD. Student's t test, * P < 0.05, **P < 0.01
4. DISCUSSION
In this study, we screened 1570 compounds in the approved drug library by DSF and selected suramin as a potential HuR‐targeted anticancer drug. Suramin exhibited anticancer effects by inhibiting HuR function in HSC‐3 cells. HuR binds and stabilizes the ARE‐mRNA‐related malignant phenotype, such as proliferation, invasion, and metastasis.3 In our previous studies,7, 17 we showed that HuR was expressed abundantly in oral cancer cells, and HuR knockdown attenuated the malignant phenotype of these cells. Therefore, HuR‐targeted compounds that inhibit HuR functions such as ARE‐mRNA stabilization can be used as novel anticancer agents. Recently, certain HuR inhibitors were reported, such as MS‐444,13 quercetin,27 azaphilone‐9,24 and some other small molecule compounds.14, 25 As HuR inhibitors are still in the initial stages of development, it will take a long time to reach the clinical trial stage.
Drug development is time‐consuming and expensive. However, repositioning of approved drugs can function more efficiently and minimize the costs and risks compared to experimental drug development from the start.26 Pharmakon‐1600 is an approved drug library, which combines US drug collection and international drug collection. The US drug collection comprised FDA approved drugs. The international drug collection is used in Europe and Asia, but not approved for use in the United States. Therefore, we chose the approved drug library to screen compounds.
We used DSF for identifying HuR‐interacting compounds in the library. DSF is a high‐throughput and inexpensive screening method to identify low‐molecular‐weight ligands that bind and stabilize purified proteins.31 In the first and second DSF, eight compounds were selected from 1570 compounds (Figure 1A,B). Only suramin shifted the T m positively in a concentration‐dependent manner (Figure 1C,D). In most cases, if a compound binds to a protein, the free energy contribution of ligand‐binding results in an increase in Gibbs free energy of unfolding, and this might cause an increase in T m. The stabilizing effect of compounds on binding is proportional to the concentration and affinity of the ligands.31 The results of DSF suggest that suramin interacts and binds to HuR. We performed SPR to confirm the affinities and kinetics between suramin and HuR. As shown by SPR, suramin binds to HuR, exhibiting fast‐on and fast‐off kinetic behavior. Therefore, we selected suramin as a potential HuR inhibitor.
Suramin is a polysulfonated naphthylurea introduced in the 1920s for the treatment of African trypanosomiasis and onchocerciasis by inhibiting DNA polymerases and reverse transcriptase. At the end of the 1970s, it was found to competitively inhibit retroviral reverse transcriptase.32 Since the late 1980s, the activity of suramin as an anticancer agent in hormone‐refractory prostate cancer (HRPC) patients has been appreciated.33, 34 Suramin exerts antitrypanosomal, anticancer, and antiviral activities.32, 33, 34, 35 It is capable of blocking the binding of growth factors, such as EGF, TGF‐β, PDGF, bFGF, and VEGF, to their surface receptors.33, 36, 37, 38 In addition, suramin decreases the expression of cell proliferation markers such as cyclin A, cyclin D1, and cyclin E, arrests cells in G1, and decreases the number of cells in S‐phase moderately.35 However, the degree to which the anticancer effects of suramin are mediated by the above mechanisms is unclear. We presumed that the anticancer effects of suramin are caused by the inhibition of HuR function and confirmed that suramin decreases the expression and stabilization of HuR‐binding mRNA (Figure 3A). Cdk1 expression was slightly decreased compared to that of other mRNAs in HSC‐3 cells. COX‐2 expression significantly decreased in HSC‐3 cells, but increased in SAS cells. Similarly, suramin treatment showed different effects on ARE‐mRNA in different cell lines.
We found that suramin decreased the protein levels of COX‐2, cyclin A2, and cyclin B1 in HSC‐3 cells (Figure 4). It affected the growth factors and cyclin‐related proteins33, 35, 36, 37, 38 that translated from HuR‐binding ARE‐mRNA.3 HuR knockdown led to G1 arrest39 in cancer cells as well as suramin‐treated cells,35 because HuR regulates cell cycle through stabilization of cell cycle‐related ARE‐mRNA.40, 41 These reports suggested the association between suramin and HuR. Recently, it was reported that suramin improved language and social interaction, and decreased restricted or repetitive behaviors of autism spectrum disorder (ASD).42 However, the mechanism underlying the effects of suramin in ASD is unclear. HuR regulated the translation of mRNA transcribed from autism‐associated genes, Foxp1 and Foxp2.43 Suramin may have improved ASD by inhibiting HuR‐Foxp1 and/or HuR‐Foxp2 binding. Thus, from these findings, it can be suggested that suramin inhibits HuR functions.
Noncytotoxic concentration of suramin was found to be less than 50 µmol/L, whereas it showed high cytotoxicity at concentrations more than 200 µmol/L.44 Effective plasma concentration of suramin was reported to be between 100 and 200 µmol/L.45 Therefore, MTS, wound healing, and invasion assays were performed to find the concentration of suramin that affects the malignant phenotype of oral cancer cells. In MTS assay, suramin did not induce cell death strongly at low‐moderate concentrations (<100 µmol/L). However, low‐moderate concentrations of suramin (50 and 100 µmol/L) attenuated motile and invasive activities markedly in HSC‐3 cells with high metastatic potential. Suramin at 100 µmol/L attenuated these activities in SAS cells. These results showed that low‐moderate concentration of suramin exhibits low cytotoxicity, but affects motile and invasive activities strongly in oral cancer cells.
Large clinical trials of suramin for cancer have been reported previously. A double‐blind placebo‐controlled phase III trial was undertaken to evaluate the efficacy of suramin plus hydrocortisone (HC) in patients with symptomatic HRPC.34 The adrenal suppressive properties of suramin require the concomitant administration of HC. Compared to patients who received placebo plus HC, those who received suramin plus HC showed delayed disease progression, decline in prostate‐specific antigen (PSA), and effective reduction of pain. In the trials, most adverse events were mild or moderate intensity and easily managed medically. The suppression of disease progression and reduction of pain can be explained by the decrease in cyclin‐related and COX‐2 protein levels in HSC‐3 cells by suramin. Although suramin has not been used for the treatment of oral cancer, our results suggest that suramin can suppress disease progression and invasion and may reduce pain by decreasing COX‐2 protein levels depending on cell line. Oral cancer leads to poor quality of life due to intense pain and significantly impairs speech, swallowing, and masticatory functions.46 Oral cancer pain management is difficult. Therefore, suramin, which reduces pain, can be used effectively for oral cancer therapy.
Our study had some limitation. Firstly, higher affinity is ideally preferred for an HuR inhibitor; however, suramin binds to HuR with low affinity (KD value 2.4 × 10−4 mol/L). Secondly, cells were treated with suramin in the absence of serum to exclude the effect of other serum components. Thirdly, suramin has been reported to have anticancer effects in vivo, but we could not report whether suramin could inhibit HuR function in vivo.
In conclusion, in this report, we show that suramin binds to HuR and inhibits HuR functions such as ARE‐mRNA stabilization and translation. In addition, suramin markedly attenuates the malignant phenotype of HSC‐3 and SAS cells. Therefore, suramin, a HuR‐targeted drug, has potential as a new therapeutic agent for oral cancer therapy. Our results suggest that suramin, via its different mechanism, may effectively suppress progressive oral cancer that cannot be controlled using other anticancer agents.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
This research was partially supported by the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from Japan Agency for Medical Research and Development (AMED) and by Hokkaido University, Global Facility Center (GFC), Pharma Science Open Unit (PSOU), funded by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) under “Support Program for Implementation of New Equipment Sharing System.” We would like to thank Editage (www.editage.jp) for English language editing.
Kakuguchi W, Nomura T, Kitamura T, et al. Suramin, screened from an approved drug library, inhibits HuR functions and attenuates malignant phenotype of oral cancer cells. Cancer Med. 2018;7:6269–6280. 10.1002/cam4.1877
Funding information
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI [Grant Number 15K20822].
REFERENCES
- 1. Chen CY, Shyu AB. AU‐rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465‐470. [DOI] [PubMed] [Google Scholar]
- 2. Jacobson A, Peltz SW. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu Rev Biochem. 1996;65:693‐739. [DOI] [PubMed] [Google Scholar]
- 3. Khabar KS. Hallmarks of cancer and AU‐rich elements. Wiley Interdiscip Rev RNA. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58:266‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Antic D, Keene JD. Embryonic lethal abnormal visual RNA‐binding proteins involved in growth, differentiation, and posttranscriptional gene expression. Am J Hum Genet. 1997;61:273‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Campos AR, Grossman D, White K. Mutant alleles at the locus elav in Drosophila melanogaster lead to nervous system defects. A developmental‐genetic analysis. J Neurogenet. 1985;2:197‐218. [DOI] [PubMed] [Google Scholar]
- 7. Kakuguchi W, Kitamura T, Kuroshima T, et al. HuR knockdown changes the oncogenic potential of oral cancer cells. Mol Cancer Res. 2010;8:520‐528. [DOI] [PubMed] [Google Scholar]
- 8. Ma WJ, Cheng S, Campbell C, Wright A, Furneaux H. Cloning and characterization of HuR, a ubiquitously expressed Elav‐like protein. J Biol Chem. 1996;271:8144‐8151. [DOI] [PubMed] [Google Scholar]
- 9. Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA‐binding platform to regulate post‐transcriptional gene expression. FEBS J. 2005;272:2118‐2131. [DOI] [PubMed] [Google Scholar]
- 10. Ma WJ, Chung S, Furneaux H. The Elav‐like proteins bind to AU‐rich elements and to the poly(A) tail of mRNA. Nucleic Acids Res. 1997;25:3564‐3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benoit RM, Meisner NC, Kallen J, et al. The x‐ray crystal structure of the first RNA recognition motif and site‐directed mutagenesis suggest a possible HuR redox sensing mechanism. J Mol Biol. 2010;397:1231‐1244. [DOI] [PubMed] [Google Scholar]
- 12. Grammatikakis I, Abdelmohsen K, Gorospe M. Posttranslational control of HuR function. Wiley Interdiscip Rev RNA. 2017;8(1):e1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meisner NC, Hintersteiner M, Mueller K, et al. Identification and mechanistic characterization of low‐molecular‐weight inhibitors for HuR. Nat Chem Biol. 2007;3:508‐515. [DOI] [PubMed] [Google Scholar]
- 14. Meisner NC, Hintersteiner M, Seifert JM, et al. Terminal adenosyl transferase activity of posttranscriptional regulator HuR revealed by confocal on‐bead screening. J Mol Biol. 2009;386:435‐450. [DOI] [PubMed] [Google Scholar]
- 15. López de Silanes I, Lal A, Gorospe M. HuR: post‐transcriptional paths to malignancy. RNA Biol. 2005;2:11‐13. [DOI] [PubMed] [Google Scholar]
- 16. Hinman MN, Lou H. Diverse molecular functions of Hu proteins. Cell Mol Life Sci. 2008;65:3168‐3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hasegawa H, Kakuguchi W, Kuroshima T, et al. HuR is exported to the cytoplasm in oral cancer cells in a different manner from that of normal cells. Br J Cancer. 2009;100:1943‐1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dixon DA, Tolley ND, King PH, et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase‐2 expression in colon cancer cells. J Clin Invest. 2001;108:1657‐1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. López de Silanes I, Fan J, Yang X, et al. Role of the RNA‐binding protein HuR in colon carcinogenesis. Oncogene. 2003;22:7146‐7154. [DOI] [PubMed] [Google Scholar]
- 20. Yi X, Zhou Y, Zheng W, Chambers SK. HuR expression in the nucleus correlates with high histological grade and poor disease‐free survival in ovarian cancer. Aust N Z J Obstet Gynaecol. 2009;49:93‐98. [DOI] [PubMed] [Google Scholar]
- 21. Heinonen M, Fagerholm R, Aaltonen K, et al. Prognostic role of HuR in hereditary breast cancer. Clin Cancer Res. 2007;13:6959‐6963. [DOI] [PubMed] [Google Scholar]
- 22. Costantino CL, Witkiewicz AK, Kuwano Y, et al. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up‐regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009;69:4567‐4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Filippova N, Yang X, Wang Y, et al. The RNA‐binding protein HuR promotes glioma growth and treatment resistance. Mol Cancer Res. 2011;9:648‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaur K, Wu X, Fields JK, et al. The fungal natural product azaphilone‐9 binds to HuR and inhibits HuR‐RNA interaction in vitro. PLoS ONE. 2017;12:e0175471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu X, Lan L, Wilson DM, et al. Identification and validation of novel small molecule disruptors of HuR‐mRNA interaction. ACS Chem Biol. 2015;10:1476‐1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lotfi Shahreza M, Ghadiri N, Mousavi SR, Varshosaz J, Green JR. A review of network‐based approaches to drug repositioning. Brief Bioinform. 2018;19(5):878‐892. [DOI] [PubMed] [Google Scholar]
- 27. Chae MJ, Sung HY, Kim EH, et al. Chemical inhibitors destabilize HuR binding to the AU‐rich element of TNF‐alpha mRNA. Exp Mol Med. 2009;41:824‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patching SG. Surface plasmon resonance spectroscopy for characterisation of membrane protein‐ligand interactions and its potential for drug discovery. Biochim Biophys Acta. 2014;1838:43‐55. [DOI] [PubMed] [Google Scholar]
- 29. Liu C, Liu Z, Wang J. Uncovering the molecular and physiological processes of anticancer leads binding human serum albumin: A physical insight into drug efficacy. PLoS ONE. 2017;12:e0176208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woo HH, Zhou Y, Yi X, et al. Regulation of non‐AU‐rich element containing c‐fms proto‐oncogene expression by HuR in breast cancer. Oncogene. 2009;28:1176‐1186. [DOI] [PubMed] [Google Scholar]
- 31. Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212‐2221. [DOI] [PubMed] [Google Scholar]
- 32. Zaniboni A. Suramin: the discovery of an old anticancer drug. Med Oncol Tumor Pharmacother. 1990;7:287‐290. [DOI] [PubMed] [Google Scholar]
- 33. Stein CA, LaRocca RV, Thomas R, McAtee N, Myers CE. Suramin: an anticancer drug with a unique mechanism of action. J Clin Oncol. 1989;7:499‐508. [DOI] [PubMed] [Google Scholar]
- 34. Small EJ, Meyer M, Marshall ME, et al. Suramin therapy for patients with symptomatic hormone‐refractory prostate cancer: results of a randomized phase III trial comparing suramin plus hydrocortisone to placebo plus hydrocortisone. J Clin Oncol. 2000;18:1440‐1450. [DOI] [PubMed] [Google Scholar]
- 35. Qiao L, Pizzolo JG, Melamed MR. Effects of suramin on expression of proliferation associated nuclear antigens in DU‐145 prostate carcinoma cells. Biochem Biophys Res Commun. 1994;201:581‐588. [DOI] [PubMed] [Google Scholar]
- 36. Coffey RJ, Leof EB, Shipley GD, Moses HL. Suramin inhibition of growth factor receptor binding and mitogenicity in AKR‐2B cells. J Cell Physiol. 1987;132:143‐148. [DOI] [PubMed] [Google Scholar]
- 37. Little PJ, Rostam MA, Piva TJ, et al. Suramin inhibits PDGF‐stimulated receptor phosphorylation, proteoglycan synthesis and glycosaminoglycan hyperelongation in human vascular smooth muscle cells. J Pharm Pharmacol. 2013;65:1055‐1063. [DOI] [PubMed] [Google Scholar]
- 38. Waltenberger J, Mayr U, Frank H. Hombach V. Suramin is a potent inhibitor of vascular endothelial growth factor. A contribution to the molecular basis of its antiangiogenic action. J Mol Cell Cardiol. 1996;28:1523‐1529. [DOI] [PubMed] [Google Scholar]
- 39. Kullmann M, Göpfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5ʹUTR. Genes Dev. 2002;16:3087‐3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doller A, Schlepckow K, Schwalbe H, Pfeilschifter J, Eberhardt W. Tandem phosphorylation of serines 221 and 318 by protein kinase Cdelta coordinates mRNA binding and nucleocytoplasmic shuttling of HuR. Mol Cell Biol. 2010;30:1397‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000;19:2340‐2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Naviaux RK, Curtis B, Li K, et al. Low‐dose suramin in autism spectrum disorder: a small, phase I/II, randomized clinical trial. Ann Clin Transl Neurol. 2017;4:491‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Popovitchenko T, Thompson K, Viljetic B, et al. The RNA binding protein HuR determines the differential translation of autism‐associated FoxP subfamily members in the developing neocortex. Sci Rep. 2016;6:28998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gan Y, Lu J, Yeung BZ, Cottage CT, Wientjes MG, Au JL. Pharmacodynamics of telomerase inhibition and telomere shortening by noncytotoxic suramin. AAPS J. 2015;17:268‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y, Song S, Yang F, Au JL, Wientjes MG. Nontoxic doses of suramin enhance activity of doxorubicin in prostate tumors. J Pharmacol Exp Ther. 2001;299:426‐433. [PubMed] [Google Scholar]
- 46. Viet CT, Schmidt BL. Biologic mechanisms of oral cancer pain and implications for clinical therapy. J Dent Res. 2012;91:447‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials