

Summary
The invasion of Chlamydia trachomatis, an obligate intracellular bacterium, into epithelial cells is driven by a complex interplay of host and bacterial factors. To comprehensively define the host genes required for pathogen invasion, we undertook a fluorescence-activated cell sorting (FACS)-based CRISPR screen in human cells. A genome-wide loss-of-function library was infected with fluorescent C. trachomatis and then sorted to enrich for invasion-deficient mutants. The screen identified heparan sulfate, a known pathogen receptor, as well as coatomer complex I (COPI). We found that COPI, through a previously unappreciated role, promotes heparan sulfate cell surface presentation, thereby facilitating C. trachomatis attachment. The heparan sulfate defect does not fully account for the resistance of COPI mutants. COPI also promotes the activity of the pathogen's type III secretion system. Together, our findings establish the requirement for COPI in C. trachomatis invasion and the utility of FACS-based CRISPR screening for the elucidation of host factors required for pathogen invasion.
Subject Areas: Molecular Mechanism of Behavior, Medical Microbiology, Methodology in Biological Sciences, Cell Biology, Host-pathogen Interactions, Molecular Microbiology, Genetic Engineering, Genetic Screens
Graphical Abstract
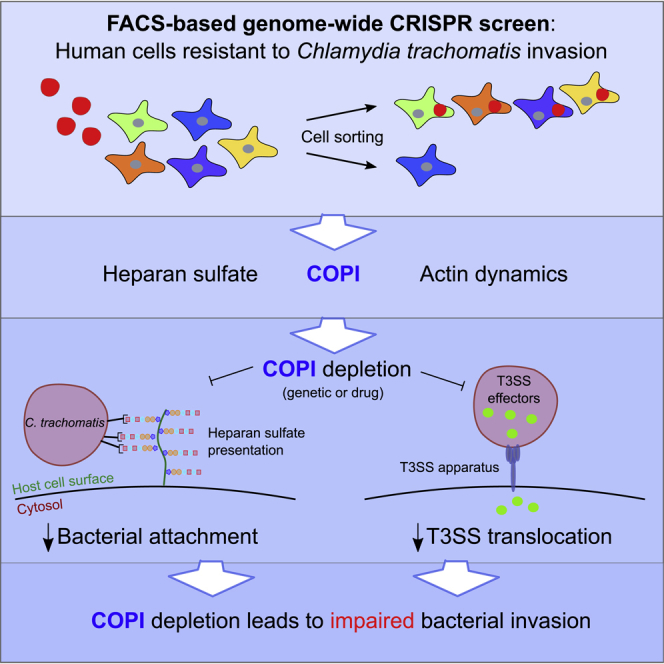
Highlights
-
•
FACS-based CRISPR screen to identify host factors required for C. trachomatis invasion
-
•
Candidate genes comprise heparan sulfate biosynthesis, actin remodeling, and COPI
-
•
COPI regulates heparan sulfate cell surface presentation and C. trachomatis attachment
-
•
COPI is also required for efficient C. trachomatis T3SS translocation
Molecular Mechanism of Behavior; Medical Microbiology; Methodology in Biological Sciences; Cell Biology; Host-pathogen Interactions; Molecular Microbiology; Genetic Engineering; Genetic Screens
Introduction
Chlamydia trachomatis, a major cause of ocular and genital tract infection in humans, is a gram-negative obligate intracellular bacterium. C. trachomatis serovars are distinguished by their tissue-specific tropism and associated pathology. Infection of the ocular conjunctival epithelium with C. trachomatis serovars A–C can lead to trachoma and thus blindness (Hu et al., 2010). Sexually transmitted infection (STI) of the genitourinary epithelium, caused by C. trachomatis serovars D–K, is associated with pelvic inflammatory disease, ectopic pregnancy, and infertility (Haggerty et al., 2010). C. trachomatis serovars L1–3 cause lymphogranuloma venereum, another STI, which is characterized by chronic lymphadenopathy in lymphatic tissues surrounding the genital area. Because C. trachomatis is the leading cause of infectious blindness as well as bacterial STI worldwide (Centers for Disease Control and Prevention Chlamydia, 2016, Mariotti et al., 2009, World Health Organization, 2011), understanding the molecular mechanisms of C. trachomatis pathogenesis has important implications for the development of therapeutics. In particular, identification of host factors necessary for C. trachomatis infection may provide a new avenue for therapeutic intervention.
The developmental cycle of C. trachomatis is biphasic, with the pathogen alternating between the extracellular elementary body (EB) and the intracellular reticulate body (RB) forms. The EB is the infectious form. The invasion of C. trachomatis into epithelial cells is driven by a complex interplay between host and bacterial factors that enable pathogen attachment and internalization. Invasion is initiated by EB attachment to and penetration into host cells as a membrane-bound structure. Attachment of EBs to host cells is mediated by engagement with host cell surface sulfated proteoglycans, particularly heparan sulfate (Su et al., 1996, Elwell et al., 2008, Rosmarin et al., 2012), although C. trachomatis serovar E attachment is not dependent on heparan sulfate (Taraktchoglou et al., 2001). Subsequently, EB uptake into a vesicular compartment is likely initiated through interactions with growth factor receptors (Elwell et al., 2008, Kim et al., 2011) and effector-mediated changes to the host actin cytoskeleton (Carabeo et al., 2002). Within 6–12 hr of invasion, EBs begin to differentiate into RBs and undergo binary fission, leading to the formation of a large parasitophorous vacuole known as the inclusion (Brunham and Rey-Ladino, 2005, Elwell et al., 2016). The nascent chlamydial vesicle does not acquire typical endocytotic vesicular markers, but instead fuses with a subset of sphingomyelin-containing exocytic vesicles (Scidmore et al., 2003).
Essential to the developmental cycle of C. trachomatis is a type III secretion system (T3SS), a multicomponent bacterial apparatus for the injection of proteinaceous “effectors” into the host cytoplasm (Portaliou et al., 2016). T3SS injection not only begins from EBs, which harbor a pre-synthesized pool of effectors (Saka et al., 2011), but also actively continue from RBs across the inclusion membrane (Mueller et al., 2014). Continued T3SS injection by RBs allows C. trachomatis to manipulate host pathways that are critical for its intracellular survival and expansion of the inclusion. Several key host regulators of vesicular membrane dynamics, such as Rab GTPases, are co-opted in this process and accumulate at the inclusion periphery (Damiani et al., 2014, Moore et al., 2011). Identification of bacterial- and host-derived molecules interacting at the inclusion membrane has been furthered by proteomic (Aeberhard et al., 2015, Mirrashidi et al., 2015) and chemical genetic (Kokes et al., 2015) approaches, deepening our understanding of the C. trachomatis host-pathogen interface.
Although there has been recent progress in creating genetic tools for C. trachomatis (Johnson and Fisher, 2013, Kannan et al., 2013, Mueller et al., 2016, Wang et al., 2011), its obligate intracellular lifestyle has made genetic manipulation difficult. Consequently, several studies have focused on identifying host factors contributing to the C. trachomatis invasion process (Derré et al., 2007, Elwell et al., 2008, Elwell et al., 2016). Toward this end, genome-wide, loss-of-function screens in human cells provide a robust forward genetics approach for unbiased identification of host genetic loci required for bacterial pathogenesis. Elwell et al. (2008), utilizing Chlamydia muridarum and an RNA interference (RNAi) screen in Drosophila melanogaster S2 cells, identified genes involved in heparan sulfate biosynthesis, as well as the role of the platelet-derived growth factor receptor pathway. Another RNAi-based screen revealed the contribution of the MEK-ERK pathway to C. trachomatis replication (Gurumurthy et al., 2010). Rosmarin et al. (2012) conducted a haploid-cell-based screen for null mutants resistant to C. trachomatis cytotoxicity, which enriched for mutants deficient in heparan sulfate. Finally, in an RNAi screen in Drosophila cells for Chlamydia caviae infection, Derré et al. (2007) revealed a novel role for the mitochondrial Tom complex in C. caviae replication and also identified candidates genes in the COPI vesicular trafficking pathway, although the latter's role was not investigated further.
COPI is a heptameric protein complex composed of α, β, β′, δ, ɛ, γ1/γ2, and ζ1/ζ2 subunits (Waters et al., 1991); together they form a cage-like lattice structure that is characteristic of all membrane-associated coat systems, including clathrin and COPII (Hughson, 2010). Membrane association of COPI is regulated by ADP ribosylation factors such as Arf1, which must be activated into their GTP-bound state by the nucleotide exchange factor GBF1 (Béthune et al., 2006, Kawamoto et al., 2002, Nickel et al., 2002). The best characterized function of COPI-coated vesicles is in the retrograde transport of cargo from the Golgi apparatus to the endoplasmic reticulum (ER) (Cosson and Letourneur, 1994, Letourneur et al., 1994), although additional roles of COPI in anterograde ER-to-Golgi traffic (Gaynor and Emr, 1997, Pepperkok et al., 1993), endosome function (Whitney et al., 1995), and lipid droplet regulation (Wilfling et al., 2014) have been reported.
The advent of CRISPR/Cas9 editing technology has enabled a new approach for carrying out genome-wide loss-of-function screens (Hartenian and Doench, 2015, Shalem et al., 2014, Wang et al., 2014). Here, to identify host factors primarily required for efficient C. trachomatis invasion, we conducted an fluorescence-activated cell sorting (FACS)-based screen, employing fluorescent C. trachomatis serovar L2. Following infection of a CRISPR/Cas9 loss-of-function pool, host cells with minimal fluorescence were sorted as a means to enrich for invasion-deficient mutants. Besides identifying most of the genes required for biogenesis of heparan sulfate, the screen also yielded several genes encoding subunits of the COPI vesicular trafficking complex. In subsequent validation studies, we found that COPI facilitates C. trachomatis attachment to host cells. In particular, we discovered that COPI plays a previously unappreciated role in promoting surface presentation of heparan sulfate. Furthermore, we found that independent of C. trachomatis attachment, COPI is also required for efficient T3SS effector translocation. In addition to revealing the importance of COPI in facilitating both C. trachomatis attachment and type III system secretion, our findings illustrate the potency of FACS-based CRISPR screens for the identification of host factors that facilitate pathogen invasion.
Results
A FACS-Based CRISPR Screen for Host Factors that Facilitate C. trachomatis Entry
A high-throughput FACS-based screen was developed to identify host proteins required for C. trachomatis invasion (i.e., attachment and entry) into human cells (Figure 1A). Initially, HT29 intestinal epithelial cells, in which we had previously created a genome-wide CRISPR loss-of-function library (Blondel et al., 2016), were infected with C. trachomatis L2 constitutively expressing mCherry. The infection time and multiplicity of infection (MOI) were optimized to limit cytotoxicity while maintaining a high proportion of fluorescent cells. Host cell fluorescence was associated with bacterial internalization and inclusion formation, which was discernible by flow cytometry and fluorescence microscopy (Figure 1B). With an MOI of 50, at 21 hr post infection (hpi), greater than 90% of cells exhibited mCherry fluorescence (mCherry+, based on a gate drawn at the 99th percentile of fluorescence in uninfected control cells; Figure 1B, top panel). Analysis of sorted cells by microscopy confirmed that the fluorescence of most cells was indeed attributable to internalized, not attached, bacteria, and that the leftmost peak was composed of cells lacking detectable intracellular bacteria (Figure 1B i–iv). Infection of a sulfation-deficient SLC35B2-null cell line (Blondel et al., 2016), which was expected to have reduced Chlamydia attachment (Rosmarin et al., 2012), resulted in a greater proportion of cells in the leftmost peak (Figure 1B, bottom panel), consistent with the idea that the cells in this peak correspond to uninvaded cells. Thus infection with fluorescent bacteria coupled with FACS allowed for enrichment of mutagenized cells that were not permissive for C. trachomatis entry.
Figure 1.

Identification of Host Factors Facilitating C. trachomatis Entry
(A) Schematic of FACS-based screening strategy. The Avana CRISPR library of HT29 cells (Blondel et al., 2016) was infected with mCherry-expressing C. trachomatis L2. After 21 hr, a FACS-based positive selection for non-invaded cells was performed. MAGeCK (Li et al., 2014) was used to calculate the statistical significance of enriched guide sgRNAs.
(B) FACS plots showing profiles of wild-type (top) or SLC35B2 mutant (bottom) HT29 cells infected with fluorescent bacteria, compared with uninfected control (gray curve). Cells were designated mCherry+ if they exhibited fluorescence intensity greater than that measured in the 99th percentile of the uninfected control cells (dotted line). The percentage of mCherry+ cells is shown in red. Cells with different fluorescence intensities were sorted (i–iv) and their intracellular fluorescence was assessed using confocal microscopy (representative cells are shown). Scale bar, 5 μm.
(C) Heatmap plot showing p values of top genes (listed in rows) identified in the four screen replicates (across columns). As a negative control, SRY (a Y chromosome-encoded gene) is shown; HT29 cells are derived from a female (Kawai et al., 2002). Genes are color coded according to the categorization in Figure 1D.
(D) Hits clustered according to corresponding biological process. The radii of the circles vary with the number of genes in the category. Genes implicated in heparan sulfate biosynthesis were identified based on known functions of biosynthetic enzymes (see Figure S1) or in a previous haploid cell screen (TMEM165 and COG2;Jae et al., 2013).
We infected an Avana CRISPR loss-of-function pool of HT29 Cas9 cells (Blondel et al., 2016, Doench et al., 2016) with mCherry-expressing C. trachomatis using the optimized invasion protocol. The screen was performed in quadruplicate; two pools mutagenized with the same single guide RNA (sgRNA) library (biological replicates, A and B) were each screened twice (technical replicates). Infected cells were harvested at 21 hpi, and mCherry− cells were collected. At this time point, it is expected that bacteria will have invaded the host cells and that mCherry− cells will be enriched for candidate genes that modulate attachment and/or entry, although steps downstream of invasion, i.e., EB-to-RB conversion and early RB replication, will also have begun. The positive selection was consistently strong across the replicates, with between 3% and 6% of the pool of HT29 cells being selected (Figure S1A). For comparative analyses, the library was also directly harvested without infection. Deep sequencing of the sgRNAs in the sorted cells as well as in the uninfected cells enabled identification of sgRNAs that were strongly enriched in the sorted population. Analysis of sgRNA abundances in the input and output HT29 pools demonstrated a strong enrichment of a subset of sgRNAs, whereas a majority of the sgRNAs became depleted (Figure S1B). We used the MAGeCK algorithm (Li et al., 2014) to evaluate the statistical significance of the enriched guides at the gene level. MAGeCK takes into account the magnitude of enrichment of the four sgRNAs per gene and compares their performance against a null distribution of control guides present in the library. A list of genes, ranked by p value, from each of the four screen replicates was compiled (Table S1). Finally, to integrate the data across these replicates, we further selected the genes whose p value met a stringent threshold (p < 0.001) in at least two of the four replicates. Seventeen genes met this criterion and constituted the list of concordant candidate genes, or “hits” (Figure 1C).
Strikingly, six of the hits corresponded to genes encoding nearly all the required glycosyltransferases within the heparan sulfate biosynthetic pathway (EXT2, EXTL3, XYLT2, B3GALT6, B3GAT3, and EXT1) (Figure S1). B4GALT7, which encodes another glycosyltransferase important for heparan sulfate biosynthesis was enriched, but did not reach the stringent cutoff. As heparan sulfate is a well-characterized cell surface receptor for C. trachomatis serovar L2 attachment (Rosmarin et al., 2012, Yabushita et al., 2002), our screen's comprehensive enrichment for mutants associated with the pathway for heparan sulfate biosynthesis provided confidence in the screen's capacity to identify biologically relevant candidate genes. Although SLC35B2, the Golgi transporter of the sulfate donor PAPS, is required for the biosynthesis of several sulfated glycosaminoglycans (GAGs), the absence of hits in glycosyltransferases involved in polymerizing GAGs other than heparan sulfate (e.g., chondroitin sulfate or dermatan sulfate) suggested that SLC35B2 likely contributes to infection via its role in heparan sulfate biosynthesis. In support of this idea, previous studies have shown that C. trachomatis binding can be blocked by addition of heparan sulfate, but not by addition of chondroitin sulfate (Su et al., 1996). Interestingly, our screen also yielded two candidate genes, TMEM165 and COG2, which were identified in a previous screen for genes implicated in cell surface heparan sulfate presentation in haploid cells (Jae et al., 2013). Because the products of these genes are not thought to act as glycosyltransferases, we hypothesize that these proteins may indirectly influence the cell surface presentation of heparan sulfate. For example, COG-deficient cells have a severe glycosylation defect due to mislocalization of several glycosyltransferases (Pokrovskaya et al., 2011). Altogether, 9 of 17 of our top candidate genes have been implicated in heparan sulfate biosynthesis or presentation (Figure 1D), emphasizing the key role of this particular sulfated GAG in C. trachomatis binding. However, despite heparan sulfate genes being identified as the strongest host candidate factors in our screen, the decrease in invasion of the sgSLC35B2 mutant (which is deficient in heparan sulfate biogenesis) was only ∼20% (Figure 1B), suggesting that there may be no single genetic knockout that confers absolute resistance against C. trachomatis invasion.
Three genes with roles in actin dynamics, ARPC4, ARPC4-TTLL3, and RAC1, were also hits in the screen. C. trachomatis internalization depends on polymerization of actin at sites of entry (Carabeo et al., 2002), and the participation of actin-modulating factors such as Arp2/3 (Hybiske and Stephens, 2007) and Rac1 (Carabeo et al., 2004) is essential. Thus hits in ARPC4 (encoding a subunit of the Arp2/3 complex) and RAC1 are consistent with these previous observations. We additionally identified ARPC4-TTLL3, which encodes a fusion protein resulting from readthrough transcription of ARPC4 with TTLL3, a tubulin glycine ligase. We cannot rule out that the latter gene may independently facilitate C. trachomatis infection, because microtubule dynamics has been implicated in the pathogen's internalization and perinuclear migration (Clausen et al., 1997). Additional candidate genes were in SLC39A9, a Golgi-resident zinc transporter (Matsuura et al., 2009), and UNC50, a factor that has been implicated in the early endosome-to-Golgi trafficking of Shiga toxin (Selyunin et al., 2017); the contributions of these factors to infection were not further explored in this study.
The remaining three top candidate genes—COPA, COPG1, and TMED10—are all associated with the coatomer complex I (COPI), which has not been previously implicated in C. trachomatis infection, and hence became the focus of our investigation. COPA and COPG1, which encode indispensable subunits of the COPI coat, and TMED10, which encodes a subunit of p24 trafficking protein (a type I transmembrane protein thought to recruit COPI through its cytoplasmic tail), were among the highest scoring candidate genes in the screen (Figures 1C and 1D). Two additional COPI subunits—COPB2 and COPE—did not reach our cutoff threshold, but were, nonetheless, significant hits in individual replicates (Table S1). In contrast to COPI, genes encoding components of the two other analogous coat systems, COPII and clathrin, which regulate vesicular traffic in the anterograde ER-to-Golgi and plasma-membrane-to-endosome directions, respectively, were not significant candidate genes (Figure S1B). Thus the screen results suggest that C. trachomatis invasion specifically depends on COPI-dependent vesicular trafficking and not vesicular trafficking in general.
To confirm that the COPI complex plays a role in enabling C. trachomatis invasion, HT29 Cas9-derived cells in which each of the eight COPI complex structural components were targeted with appropriate sgRNAs were created. In addition, HT29 Cas9 cells with sgRNAs targeting nucleotide exchange factor (GBF1) and GTPase-activating proteins (ARFGAPs) that regulate Arf-triggered COPI coat assembly were generated. In infection conditions that recapitulated those of the screen, most of these cell lines exhibited modest yet consistent reductions in the percent of cells infected relative to wild-type (WT) HT29 or cells mutated with a control sgRNA and/or in the mean fluorescence intensity in infected cells (Figure 2). However, immunoblotting did not reveal obvious reductions in the amounts of the targeted proteins. Our inability to generate cells fully deficient in COPI components likely reflects the fact that COPI function is thought to be critical for cell viability/proliferation; it was deemed to be essential for growth in several cell lines in a CRISPR screen to identify human essential genes (Wang et al., 2015). COPI-targeting guides may have been enriched in the screen because CRISPR mutagenesis yielded hypomorphic COPI alleles that compensated for reduced cell viability through protection from C. trachomatis invasion.
Figure 2.

Validation of COPI Candidate Genes Using Mutant Cell Lines
HT29 Cas9 cells lines expressing sgRNAs targeting the indicated COPI-associated genes (blue bars) were infected with fluorescent C. trachomatis and harvested 21 hpi for flow cytometry. Cells that exhibited fluorescence greater than that observed in the 99th percentile of uninfected control cells were scored as invaded. The difference in invasion relative to infection of wild-type HT29 Cas9 (calculated as follows: (% WT invasion − % mutant invasion)/% WT invasion) is presented. GMFI is the geometric mean of the fluorescence intensity of the total (ungated) population of cells. The sgCTRL cell has an sgRNA targeting an intergenic region. Error bars indicate SEM (n = 4). The p values (****p < 0.001, ***p < 0.005, **p < 0.01, *p < 0.1) are based on a Student's t test (two-tailed).
COPI Promotes Cell Surface Heparan Sulfate Presentation and C. trachomatis Attachment
To begin to address how COPI facilitates C. trachomatis invasion, we used small interfering RNA (siRNA)-mediated gene silencing to transiently knockdown (KD) COPA in HeLa cells, thereby circumventing our inability to generate stable COPI knockout cells. This approach yielded marked reduction in α-COP (encoded by COPA) at 48 hr post transfection (Figure 3A). Initially, we tested whether COPA KD had an impact on bacterial burden assayed at an early time post infection. There was a marked reduction in bacterial burden quantified with qPCR compared with control cells transfected with a non-targeting siRNA (siCTRL) at 6 hpi (Figure 3B). As this time point represents an early stage of infection, we hypothesized that COPI KD compromised either bacterial attachment to the host cell surface and/or an early step in internalization into host cells. Control and COPA KD HeLa cells were infected with C. trachomatis at a high MOI for 30 min, and unbound bacteria were washed off to test whether C. trachomatis attachment to COPA KD cells was impaired. Counting of attached bacteria, by staining of unpermeabilized cells with a fluorescently labeled anti-EB antibody, revealed a marked reduction in C. trachomatis attachment to the COPA KD cells (Figures 3C and 3D), suggesting that COPI facilitates C. trachomatis binding to host cells.
Figure 3.

Reduction in C. trachomatis Attachment and Entry into HeLa Cells with Diminished COPI
(A) Western blot of COPA protein in lysates of HeLa 48 hr post transfection with nontargeting (siCTRL) or targeting (siCOPA) siRNA. Beta-tubulin is a loading control.
(B) Quantitation of C. trachomatis burden (measured by qPCR) 6 hpi (MOI 10) in COPA knockdown cells relative to in cells targeted with siCTRL. Two different COPA siRNAs were used. Each group represents four replicates. Error bars indicate SEM. The p values (***p < 0.005) are based on a Student's t test (two-tailed).
(C) Immunofluorescence-microscopy-based detection of C. trachomatis attachment to siRNA-treated cells. C. trachomatis bound to non-permeabilized HeLa cells was detected with an antibody against EBs; cell boundaries are shown with dotted white lines, and nuclei are shown with solid white lines. Scale bar, 10 μm.
(D) Attachment assay. The number of bacteria bound to each cell in (C) was counted manually across four different microscopy fields; 25 cells were counted per group. The p values (****p < 0.001) are based on a Student's t test (two-tailed).
To corroborate and extend these observations, we took advantage of a previously characterized Chinese hamster ovary (CHO) cell line, ldlF, which bears a temperature-sensitive COPE allele (Guo et al., 1994, Guo et al., 1996). Depletion of COPE was achieved by a temperature shift from 34°C (permissive) to 40°C (non-permissive) for 12 hr (Cureton et al., 2012), which rendered ɛ-COP protein undetectable (Figure 4A). There was a similar burden of C. trachomatis 6 hpi in ldlF or WT CHO cells maintained at 34°C (Figure 4B). In contrast, there was a significantly lower burden of C. trachomatis in ldlF cells grown at 40°C compared with WT cells (Figure 4B). The temperature shift to 40°C did not alter the bacterial burden found in WT cells, suggesting that elevated temperature per se was not deleterious for C. trachomatis infection, at least in the time frame used for the experiment. In addition, visualization of adherent bacteria 30 min post infection revealed that pathogen attachment to ldlF cells and WT cells was similar at 34°C, but at 40°C, with depletion of COPE, far fewer bacteria were bound to ldlF than to WT cells (Figures 4C and 4D). The congruent observations obtained with the temperature-sensitive COPE in ldlF cells and the siRNA-mediated COPA KD in HeLa cells support the idea that at least one mechanism by which COPI promotes C. trachomatis infection is by enabling pathogen attachment.
Figure 4.

Reduction in C. trachomatis Attachment and Entry in CHO Cells Depleted in COPI
(A) Western blot of COPE protein (ɛ-COP) in lysates of WT and ldlF CHO cells incubated at 40°C for 12 hr.
(B) Quantitation of C. trachomatis burden (measured by qPCR) at 6 hpi. CHO cells were incubated at 34°C or 40°C for 12 hr before infection at MOI 10. Percentages are in reference to the WT burden measured at 34°C. There were four replicates per group. Error bars indicate SEM. The p values (***p < 0.005; n.s., not significant, p > 0.1) are based on a Student's t test (two-tailed).
(C) Immunofluorescence-microscopy-based detection of C. trachomatis attachment to COPI-undepleted or COPI-depleted CHO cells at 30 min post infection. WT or ldlF cells were incubated at 34°C or 40°C for 12 hr before infection. C. trachomatis bound to CHO cells was detected with an antibody against EBs. Cell boundaries are shown with dotted white lines, and nuclei are shown with solid white lines. Scale bar, 10 μm.
(D) Attachment assay. The number of bacteria bound to each cell in (C) was counted manually across four different microscopy fields; 25 cells were counted per group. Error bars indicate SEM. The p values (****p < 0.001; n.s., not significant, p > 0.1) are based on a Student's t test (two-tailed).
As the presence of heparan sulfate on cell surface proteoglycans is critical for C. trachomatis L2 to attach to host cells (Rosmarin et al., 2012), we tested whether COPI depletion decreased the abundance of cell surface heparan sulfate. Flow cytometric analyses of COPA KD HeLa cells stained with an antibody to heparan sulfate revealed reduced surface-localized heparan sulfate compared with control cells (siCTRL, Figure 5A). Also, there was reduced surface heparan sulfate in ldlF compared with WT CHO cells grown at 40°C but not at 34°C (Figure 5B). In contrast, heparan sulfate was undetectable in siEXT2 HeLa cells and pgsD-677 CHO cells, which have mutated EXT1/2 and thus cannot polymerize heparan sulfate (Lidholt et al., 1992). Collectively, these observations suggest that COPI vesicular trafficking in mammalian cells is required for optimal cell surface presentation of heparan sulfate. Therefore, the reduced capacity of C. trachomatis to bind to COPI-depleted cells (Figures 3C, 3D, 4C, and 4D) is likely a consequence of their lack of surface heparan sulfate.
Figure 5.

Depletion of COPI Reduces Cell Surface Heparan Sulfate
(A) Flow cytometric detection of surface heparan sulfate in HeLa cells treated with a control siRNA (siCTRL) or siRNAs targeting COPA or EXT2. Unstained cells were only incubated with secondary antibody. The GMFI (geometric mean fluorescence intensity) values for each of the conditions are shown. At least 10,000 cells were analyzed per plot. Data are representative of five independent experiments.
(B) Flow cytometric detection of surface heparan sulfate in wt, ldlF, and pgsD-677 CHO cell lines incubated at the permissive (34°C) or non-permissive (40°C) temperatures. Unstained cells were only incubated with secondary antibody. The GMFI (geometric mean fluorescence intensity) values for each of the conditions are shown. At least 10,000 cells were analyzed per plot. Data are representative of five independent experiments.
We tested whether COPI-deficient cells may be impeded in heparan sulfate biosynthesis due to downregulation or mislocalization of heparan sulfate biosynthetic enzymes. In HeLa cells, immunofluorescence of EXT1 and EXT2 revealed a predominantly ER localization, as evidenced by the colocalizing calnexin signal (Figure S2). In COPA-depleted cells, neither EXT1 nor EXT2 appeared to be significantly altered in signal intensity or subcellular localization. We hypothesize that COPI may regulate heparan sulfate biosynthesis at later steps, or may regulate the trafficking of heparan-sulfated GAGs through the Golgi cisternae and translocation to the cell surface.
As distinct vesicular trafficking pathways are governed by COPI and COPII, we tested whether COPII was required for heparan sulfate biosynthesis and/or C. trachomatis infection. HeLa cells were depleted of Sec13, an indispensable outer coat subunit of COPII (reviewed in Tang et al., 2005) via siRNA (Figure S3A). Analysis by flow cytometry demonstrated no defects in heparan sulfate cell surface expression, unlike COPI- or EXT2-depleted cells (Figure S3B versus Figure 5). In addition, infection with C. trachomatis L2, and qPCR quantitation of bacterial burdens at 6 hpi, revealed no significant differences (Figure S3C). Therefore COPI retrograde Golgi-to-ER, but not COPII anterograde ER-to-Golgi, trafficking appears necessary for efficient heparan sulfate biosynthesis and early infection.
COPI Promotes C. trachomatis Invasion at Steps beyond Attachment, Including T3SS Function
To begin to address whether COPI function is important for steps in C. trachomatis infection beyond heparan sulfate-mediated attachment, we used C. trachomatis serovar E, taking advantage of the fact that serovar E, unlike L2, attaches to host cells in a heparan sulfate-independent manner (Taraktchoglou et al., 2001). WT, ldlF, and pgsD-677 CHO cells were incubated at 40°C, infected with either serovar E or L2, and then assessed for bacterial burden via qPCR at 6 hpi. As expected, ldlF and pgsD-677 cells had significantly reduced burdens of serovar L2 relative to WT cells (Figure 6A). In contrast, pgsD-677 and WT cells had a similar burden of serovar E at 6 hpi, confirming that serovar E invasion is independent of heparan sulfate. However, ldlF cells exhibited a significantly reduced bacterial burden relative to both pgsD-677 and WT CHO cells following infection with serovar E C. trachomatis, although this reduction was less marked than seen following serovar L2 infection (Figure 6A). These results suggest that reduction in surface heparan sulfate only partially accounts for the resistance of COPI-deficient cells to C. trachomatis infection, and thus that COPI may also contribute to steps beyond pathogen attachment, such as pathogen T3 secretion.
Figure 6.

COPI Deficiency Reduces C. trachomatis Translocation of a T3SS Effector
(A) Comparison of C. trachomatis serovar E and serovar L2 infection of WT, ldlF, or pgsD-677 (pgsD) CHO cells. CHO cells were incubated at 40°C for 12 hr, then infected with serovar L2 or E at MOI 10, and the bacterial burden was assessed by qPCR at 6 hpi. Burdens are expressed relative to that of WT CHO cells infected with serovar L2 (set to 100%). Error bars indicate SEM (n = 4). The p values (***p < 0.005, **p < 0.01, *p < 0.1) are based on a Student's t test (two-tailed).
(B) Influence of golgicide A treatment on TarP translocation. HeLa cells were infected with a TarP-BlaM fusion or WT strain of C. trachomatis serovar L2. At 2 hpi, cells were treated with 1 μM CCF2-AM and 2.5 mM probenecid to load the beta-lactamase substrate, and then harvested, fixed, and analyzed by flow cytometry. Beta-lactamase activity (in arbitrary units) is reported as the GMFI change observed in the 535-nm emission channel (monitoring cleaved CCF2-AM), normalized to uninfected control (arbitrarily set to zero). At least 100,000 cells were analyzed. The error bars indicate SEM (n = 3). The p values shown are based on a Student's t test (two-tailed).
To investigate steps beyond C. trachomatis attachment, we used golgicide A treatment to rapidly inactivate COPI function while sparing heparan sulfate presentation. Golgicide A specifically inhibits GBF1, leading to rapid dissociation of COPI from cell membranes in treated cells (Sáenz et al., 2009), but, unlike brefeldin A, it does not inhibit the brefeldin A-inhibited guanine nucleotide exchange factors (BIG1/2), which participate in clathrin coat assembly (Pacheco-Rodriguez et al., 2002). Importantly, although the drug leads to rapid inactivation of COPI (e.g., the predominantly Golgi signal observed in a cell line expressing ɛ-COP-GFP became undetectable within 2 min of golgicide application; Figure S4A), cell surface heparan sulfate levels remained near WT levels after 4 hr of drug incubation (Figure S4B), providing a time window in which the effects of COPI inactivation on bacterial processes other than adhesion could be studied in the absence of an attachment defect (Figure S4C).
Binding of C. trachomatis EBs to the host cell surface is followed by activation of the pathogen T3SS, which translocates a pre-synthesized pool of effectors that are required for entry and early inclusion formation. One of the best characterized C. trachomatis effectors is TarP, which is secreted within minutes of host cell contact (Clifton et al., 2004). TarP possesses intrinsic actin-bundling activity through an F-actin nucleation domain (Jewett et al., 2006), and this activity is critical for EB invasion (Parrett et al., 2016). We used a C. trachomatis serovar L2 strain (Mueller and Fields, 2015) containing a TarP-beta-lactamase fusion (TarP-BlaM) to monitor TarP translocation during the first 2.5 hr of infection in golgicide A-treated versus control cells. Beta-lactamase activity was monitored by cleavage of a fluorescence resonance energy transfer substrate loaded into live cells, which were subsequently analyzed by flow cytometry. In this protocol, golgicide A treatment did not alter the bacterial burdens of infected cells (Figure S4D). Flow cytometry analyses revealed robust beta-lactamase activity in control cells infected with the strain expressing TarP-BlaM, but not in control cells left uninfected or infected with WT C. trachomatis (not bearing a TarP-BlaM fusion; Figure 6B). Finally, pretreatment of cells with golgicide A before infection with TarP-BlaM-expressing bacteria resulted in a dose-dependent reduction in beta-lactamase activity (Figure 6B). Collectively, these findings suggest that intact COPI function is necessary for efficient C. trachomatis T3SS translocation of effectors, independent of bacterial attachment. As TarP and other early injected effectors exist as pre-synthesized pools within the EB, we speculate that secretion may be impeded at steps such as T3SS translocon (pore) formation and/or needle stabilization, although future work is required to investigate these possibilities. The reduced T3SS translocation of TarP, and potentially other T3SS effectors that contribute to early stages of infection, could account for the reduced infection of COPI-deficient cells by serovar E (heparan sulfate-independent) C. trachomatis (Figure 6A).
Discussion
Here we undertook an unbiased FACS-based CRISPR screen of the human protein coding genome to identify key host molecules that promote C. trachomatis invasion. More than half of the top candidate genes in the screen encode factors required for heparan sulfate synthesis and surface presentation. As this GAG has previously been shown to be an important receptor for the pathogen, these hits validate our approach. Our screen also yielded components of the coatomer COPI, a central regulator of vesicular traffic, whose roles in C. trachomatis invasion into host cells have not been examined. Although Derré et al. (2007) identified COPI components in an RNAi microscopy-based screen for C. caviae replication in Drosophila S2 cells, they did not further explore the mechanistic basis of this hit. Here, we discovered a link between the COPI coatomer and the biogenesis of heparan sulfate, which likely contributes to COPI's critical role in C. trachomatis attachment. Furthermore, our data suggest that COPI promotes C. trachomatis invasion at steps beyond heparan sulfate-dependent attachment and show that golgicide A-mediated acute inactivation of COPI inhibits T3SS injection. Thus our study suggests multiple roles for COPI during C. trachomatis invasion into host cells.
COPI function has not previously been linked to biogenesis and/or cell surface presentation of heparan sulfate. COPI genes were not identified as candidate genes in a gene-trap-based screen of haploid leukemia cells that relied on FACS isolation of heparan sulfate-deficient mutants (Jae et al., 2013). Although this screen yielded several known heparan sulfate biosynthesis enzymes, it is likely that COPI loci were not identified because inactivation of the single copies of these genes in the haploid cells would have rendered the mutants nonviable. The hypertriploidy of the HT29 cells used in our screen (Kawai et al., 2002), together with the propensity of non-homologous end joining to result in diverse variants at the CRISPR cut site (Donovan et al., 2017), may have allowed enrichment of rare hypomorphic COPI alleles in our screen.
COPI deficiency may attenuate synthesis and/or surface presentation of heparan sulfate by several mechanisms. Previous work has demonstrated profound alterations to Golgi morphology upon COPI disruption in temperature-shifted ldlF CHO cells (Llorente et al., 2003). The dispersal of the Golgi found in COPI-depleted cells resembles that in cells depleted of COG proteins, which regulate intra-Golgi vesicular traffic (Pokrovskaya et al., 2011). Furthermore, in COG-deficient cells, there is mislocalization of several N-glycosylation enzymes that normally reside in the Golgi, which was proposed to explain the decrease in cell surface N-linked glycoconjugates and sialylated glycans in these cells. Diminished Golgi integrity in COPI-deficient cells may lead to a similar mislocalization and concomitant reduction in the activity of the O-glycosylation enzymes required in the biogenesis of heparan-sulfated GAGs. However, we found that EXT1 and EXT2, the copolymerases involved in the initiation of heparan sulfate biosynthesis, were not mislocalized by COPI KD in HeLa cells. We cannot rule out that enzymes downstream of EXT1/EXT2 are altered by COPI disruption, but none of the glycosyltransferases implicated in heparan sulfate biosynthesis contain the C-terminal KKXX or KXKXX motifs that are canonically selected by the COPI coatomer for retrograde sorting from the Golgi (Jackson et al., 2012), making it unlikely that COPI directly regulates heparan sulfate biosynthetic machinery. Rather, indirect effects of the disruption of Golgi, Golgi-to-ER transit, or otherwise unknown vesicular dynamics are more likely to account for the defect in heparan sulfate translocation to the cell surface upon COPI abrogation. Regardless of the mechanism(s) by which COPI influences heparan sulfate accumulation, our discovery that COPI depletion impedes cell surface heparan sulfate presentation implies that the myriad pathogens (bacterial, viral, and parasitic; reviewed in Bartlett and Park, 2011) that rely on heparan sulfate for cell attachment also rely on COPI for infection.
Although genes contributing to heparan sulfate biogenesis constituted the best hits in our screen, heparan sulfate-null mutants are not completely resistant to C. trachomatis infection (e.g., pgsD-677 cells in Figure 6A; HS-null clones in Rosmarin et al. (2012)), suggesting that C. trachomatis can use heparan sulfate-independent routes of entry. Several proteins have been proposed to serve as C. trachomatis receptors, including cystic fibrosis transmembrane conductance regulator (CFTR) (Chukwuemeka Ajonuma et al., 2010), mannose-binding protein (Swanson et al., 1998), fibroblast growth factor receptor (Kim et al., 2011), and protein disulfide isomerase (PDI) (Abromaitis and Stephens, 2009). However, our top candidate genes did not include potential cell surface receptors other than heparan sulfate. As heparan sulfate is a ubiquitous proteoglycan modification found on a variety of proteins, our findings do not support the idea that there is a discrete C. trachomatis receptor protein. It is possible that such host factors exist, but they did not score as candidate genes in our screen owing to functional redundancy or essentiality.
Our findings revealed that COPI also promotes steps in C. trachomatis entry into host cells that are downstream of attachment to host cells. Treatment of host cells with golgicide A, which acutely disrupts COPI while leaving surface heparan sulfate intact, led to a reduction in TarP translocation. Studies of T3SS function in other pathogens have yielded results that may help to explain the defective T3SS function in COPI-deficient cells. In Yersinia pseudotuberculosis, Sheahan and Isberg (2015) observed a reduction in translocon-mediated pore formation in COPI-depleted HeLa cells, suggesting that COPI promotes translocon insertion into and/or stability within the host cell membrane. Furthermore, like C. trachomatis, S. typhimurium invasion and effector translocation were found to depend on intact COPI (Misselwitz et al., 2011). This defect was attributed at least in part to the mislocalization of cholesterol and sphingolipids in COPI-depleted cells (Misselwitz et al., 2011). As several studies have linked membrane lipid composition to T3SS translocon insertion and/or effector translocation (van der Goot et al., 2004, Hayward et al., 2005, Jamison and Hackstadt, 2008), we hypothesize that alterations in membrane lipid composition in COPI-depleted cells may underlie the defective T3SS function in C. trachomatis as well as in Y. pseudotuberculosis. Regardless of the mechanism(s) by which COPI contributes to C. trachomatis T3SS function, it is remarkable that screens employing three diverse pathogens all revealed that COPI is implicated in T3SS activity, highlighting the conservation of host factors required for at least some T3SS.
Factors downstream of effector translocation may also contribute to diminished C. trachomatis invasion of COPI-depleted cells. In support of this possibility, a comprehensive proteomic screen to define effector targets revealed that CT223, a putative T3SS effector, interacted with COPG (Mirrashidi et al., 2015), suggesting that C. trachomatis may directly manipulate COPI. Additional studies are required to define how COPI promotes effector translocation and the significance of the CT223-COPG interaction.
Besides the invasive bacterial pathogens C. trachomatis and S. typhimurium, several human viruses also depend on host COPI for their entry and/or proliferation. These viruses include influenza (König et al., 2010), vesicular stomatitis virus (VSV) (Cureton et al., 2012), and Semliki Forest virus (Vonderheit and Helenius, 2005). In the case of VSV, COPI depletion is also thought to affect several steps in the viral life cycle, including attachment, endosomal trafficking, and intracellular replication (Cureton et al., 2012). The common reliance of C. trachomatis, S. typhimurium, and many epidemiologically important viruses on COPI for infection suggests that development of therapeutics that inactivate COPI could have broad application. However, developing an agent that would avoid the cellular toxicity of COPI inactivation would be challenging.
The pleiotropic effects of COPI on cell physiology make dissection of COPI's contributions to discrete steps in pathogen infection complex. Acute but transient perturbations are necessary for investigation of essential gene products such as COPI. In this regard, we benefited from the availability of ldl CHO cells and golgicide A for our studies; however, application of new protein perturbation strategies such as auxin-inducible degradation (Natsume et al., 2016) and mislocalization (Robinson et al., 2010) will facilitate future investigations of the roles of essential genes in pathogen infection. Also, our findings illustrate the utility of FACS-based CRISPR screens for revealing molecular mechanisms of pathogen invasion into host cells. Similar screens should be applicable to many other invasive microbes. Finally, our findings show once again the utility of using intracellular pathogens as probes to elucidate host cell biology.
Limitations of the Study
There are two principal limitations of this study. First, the aim of our study was to identify host factors required for C. trachomatis L2 invasion (entry and internalization), but our screen may have also yielded host factors required for C. trachomatis proliferation after the invasion process. We designed our screen to take advantage of the power of FACS to identify and sort uninvaded (non-fluorescent) cells. FACS was carried out 21 hr post C. trachomatis infection of the host cell library. This time point was chosen because by 21 hr it was possible to detect a sufficiently robust fluorescent signal from invaded cells for cell sorting. However, by this time, the pathogen is expected to have attached to the surface, internalized, and begun the early steps of EB-to-RB conversion. Thus, besides identifying factors important for attachment and entry, the screen could have yielded host factors that function “downstream” of cell entry, such as those that enable RB replication and inclusion survival. Second, Cas9-based CRISPR knockout screens cannot efficiently interrogate loci encoding essential proteins because it is not possible to generate null mutations in such genes. Additional essential gene products (besides components of COPI) maybe be required for C. trachomatis invasion, and such loci would have been missed in our screen. The use of CRISPRi, which relies on guide RNA-targeted dCas9-mediated inhibition of transcription, rather than gene interruption, should be a potent way to identify essential host factors required for C. trachomatis invasion in future studies.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Brigid Davis and Waldor laboratory members for providing helpful comments on the project and manuscript. We are grateful to Monty Krieger, Ken Fields, Sean Whelan, and Lindsey Robinson for reagents and advice. We thank Chad Araneo for his technical assistance with FACS. J.S.P., HHMI Medical Research Fellowship; J.E.L., T32 AI-007061; C.J.B., FONDECYT 11160901, REDI170269, and an HHMI-Gulbenkian International Research Scholar; M.N.S., AI39558, AI062827, and AI113187; M.K.W., AI-042347 and HHMI.
Author Contributions
J.S.P., J.D.H., J.E.L., M.N.S., and M.K.W. conceived of the project and designed the experiments. C.J.B. and J.G.D. prepared the CRISPR library, and J.S.P. and G.Y. performed the screen, sequencing, and analysis. J.S.P., J.D.H., and J.E.L. performed the experiments. J.S.P., J.D.H., J.E.L., and M.K.W. wrote the manuscript, and all authors edited the paper.
Declaration of Interests
The authors declare no competing interests.
Published: January 25, 2019
Footnotes
Supplemental Information includes Transparent Methods, four figures, and one table and can be found with this article online at https://doi.org/10.1016/j.isci.2018.12.011.
Supplemental Information
sgRNAs refers to the number of sgRNAs targeting that gene present in the Avana library (Blondel et al., 2016); score refers to the MAGeCK positive selection score; FDR, false discovery rate; log2 FC, the log2-transformed fold-change average of the sgRNA abundance relative to the control (uninfected) condition.
References
- Abromaitis S., Stephens R.S. Attachment and entry of Chlamydia have distinct requirements for host protein disulfide isomerase. PLoS Pathog. 2009;5:e1000357. doi: 10.1371/journal.ppat.1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeberhard L., Banhart S., Fischer M., Jehmlich N., Rose L., Koch S., Laue M., Renard B.Y., Schmidt F., Heuer D. The proteome of the isolated Chlamydia trachomatis containing vacuole reveals a complex trafficking platform enriched for retromer components. PLoS Pathog. 2015;11:e1004883. doi: 10.1371/journal.ppat.1004883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett A.H., Park P.W. Heparan sulfate proteoglycans in infection. In: Pavao M.S.G., editor. Glycans in Diseases and Therapeutics. Springer-Verlag; 2011. pp. 31–62. [Google Scholar]
- Béthune J., Wieland F., Moelleken J. COPI-mediated transport. J. Membr. Biol. 2006;211:65–79. doi: 10.1007/s00232-006-0859-7. [DOI] [PubMed] [Google Scholar]
- Blondel C.J., Park J.S., Hubbard T.P., Pacheco A.R., Kuehl C.J., Walsh M.J., Davis B.M., Gewurz B.E., Doench J.G., Waldor M.K. CRISPR/Cas9 screens reveal requirements for host cell sulfation and fucosylation in bacterial type III secretion system-mediated cytotoxicity. Cell Host Microbe. 2016;20:226–237. doi: 10.1016/j.chom.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham R.C., Rey-Ladino J. Immunology of Chlamydia infection: implications for a Chlamydia trachomatis vaccine. Nat. Rev. Immunol. 2005;5:149–161. doi: 10.1038/nri1551. [DOI] [PubMed] [Google Scholar]
- Carabeo R.A., Grieshaber S.S., Fischer E., Hackstadt T. Chlamydia trachomatis induces remodeling of the actin cytoskeleton during attachment and entry into HeLa cells. Infect. Immun. 2002;70:3793–3803. doi: 10.1128/IAI.70.7.3793-3803.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo R.A., Grieshaber S.S., Hasenkrug A., Dooley C., Hackstadt T. Requirement for the Rac GTPase in Chlamydia trachomatis invasion of non-phagocytic cells. Traffic. 2004;5:418–425. doi: 10.1111/j.1398-9219.2004.00184.x. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Chlamydia. 2016, STD Surveillance Report, https://www.cdc.gov/std/stats16/CDC_2016_STDS_Report-for508WebSep21_2017_1644.pdf.
- Chukwuemeka Ajonuma L., Lam Fok K., Sze Ho L., Kay Sheung Chan P., Chow P.H., Ling Tsang L., Hau Yan Wong C., Chen J., Li S., Kenneth Rowlands D. CFTR is required for cellular entry and internalization of Chlamydia trachomatis. Cell Biol. Int. 2010;34:593–600. doi: 10.1042/CBI20090227. [DOI] [PubMed] [Google Scholar]
- Clausen J.D., Christiansen G., Holst H.U., Birkelund S. Chlamydia trachomatis utilizes the host cell microtubule network during early events of infection. Mol. Microbiol. 1997;25:441–449. doi: 10.1046/j.1365-2958.1997.4591832.x. [DOI] [PubMed] [Google Scholar]
- Clifton D.R., Fields K.A., Grieshaber S.S., Dooley C.A., Fischer E.R., Mead D.J., Carabeo R.A., Hackstadt T. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc. Natl. Acad. Sci. U S A. 2004;101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P., Letourneur F. Coatomer interaction with di-lysine endoplasmic reticulum retention motifs. Science. 1994;263:1629–1631. doi: 10.1126/science.8128252. [DOI] [PubMed] [Google Scholar]
- Cureton D.K., Burdeinick-Kerr R., Whelan S.P.J. Genetic inactivation of COPI coatomer separately inhibits vesicular stomatitis virus entry and gene expression. J. Virol. 2012;86:655–666. doi: 10.1128/JVI.05810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani M.T., Gambarte Tudela J., Capmany A. Targeting eukaryotic Rab proteins: a smart strategy for chlamydial survival and replication. Cell. Microbiol. 2014;16:1329–1338. doi: 10.1111/cmi.12325. [DOI] [PubMed] [Google Scholar]
- Derré I., Pypaert M., Dautry-Varsat A., Agaisse H. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog. 2007;3:1446–1458. doi: 10.1371/journal.ppat.0030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan K.F., Hegde M., Sullender M., Vaimberg E.W., Johannessen C.M., Root D.E., Doench J.G. Creation of novel protein variants with CRISPR/Cas9-mediated mutagenesis: turning a screening by-product into a discovery tool. PLoS One. 2017;12:e0170445. doi: 10.1371/journal.pone.0170445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell C.A., Ceesay K., Kim J.H., Kalman D., Engel J. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 2008;9:e1000021. doi: 10.1371/journal.ppat.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell C., Mirrashidi K., Engel J. Chlamydia cell biology and pathogenesis. Nat. Rev. Microbiol. 2016;14:385–400. doi: 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor E.C., Emr S.D. COPI-independent anterograde transport: cargo-selective ER to Golgi protein transport in yeast COPI mutants. J. Cell Biol. 1997;136:789–802. doi: 10.1083/jcb.136.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Goot F.G., van Nhieu G.T., Allaoui A., Sansonetti P., Lafont F. Rafts can trigger contact-mediated secretion of bacterial effectors via a lipid-based mechanism. J. Biol. Chem. 2004;279:47792–47798. doi: 10.1074/jbc.M406824200. [DOI] [PubMed] [Google Scholar]
- Guo Q., Vasile E., Krieger M. Disruptions in Golgi structure and membrane traffic in a conditional lethal mammalian cell mutant are corrected by epsilon-COP. J. Cell Biol. 1994;125:1213–1224. doi: 10.1083/jcb.125.6.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q., Penman M., Trigatti B.L., Krieger M. A single point mutation in epsilon-COP results in temperature-sensitive, lethal defects in membrane transport in a Chinese hamster ovary cell mutant. J. Biol. Chem. 1996;271:11191–11196. doi: 10.1074/jbc.271.19.11191. [DOI] [PubMed] [Google Scholar]
- Gurumurthy R.K., Maurer A.P., Machuy N., Hess S., Pleissner K.P., Schuchhardt J., Rudel T., Meyer T.F. A Loss-of-Function screen reveals Ras- and Raf-independent MEK-ERK signaling during Chlamydia trachomatis infection. Sci. Signal. 2010;3:ra21. doi: 10.1126/scisignal.2000651. [DOI] [PubMed] [Google Scholar]
- Haggerty C.L., Gottlieb S.L., Taylor B.D., Low N., Xu F., Ness R.B. Risk of sequelae after Chlamydia trachomatis genital infection in women. J. Infect. Dis. 2010;201:134–155. doi: 10.1086/652395. [DOI] [PubMed] [Google Scholar]
- Hartenian E., Doench J.G. Genetic screens and functional genomics using CRISPR/Cas9 technology. FEBS J. 2015;282:1383–1393. doi: 10.1111/febs.13248. [DOI] [PubMed] [Google Scholar]
- Hayward R.D., Cain R.J., McGhie E.J., Phillips N., Garner M.J., Koronakis V. Cholesterol binding by the bacterial type III translocon is essential for virulence effector delivery into mammalian cells. Mol. Microbiol. 2005;56:590–603. doi: 10.1111/j.1365-2958.2005.04568.x. [DOI] [PubMed] [Google Scholar]
- Hu V.H., Harding-Esch E.M., Burton M.J., Bailey R.L., Kadimpeul J., Mabey D.C.W. Epidemiology and control of trachoma: systematic review. Trop. Med. Int. Health. 2010;15:673–691. doi: 10.1111/j.1365-3156.2010.02521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughson F.M. Copy coats: COPI mimics clathrin and COPII. Cell. 2010;142:19–21. doi: 10.1016/j.cell.2010.06.031. [DOI] [PubMed] [Google Scholar]
- Hybiske K., Stephens R.S. Mechanisms of Chlamydia trachomatis entry into nonphagocytic cells. Infect. Immun. 2007;75:3925–3934. doi: 10.1128/IAI.00106-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L.P., Lewis M., Kent H.M., Edeling M.A., Evans P.R., Duden R., Owen D.J. Molecular basis for recognition of dilysine trafficking motifs by COPI. Dev. Cell. 2012;23:1255–1262. doi: 10.1016/j.devcel.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jae L.T., Raaben M., Riemersma M., van Beusekom E., Blomen V.A., Velds A., Kerkhoven R.M., Carette J.E., Topaloglu H., Meinecke P. Deciphering the glycosylome of Dystroglycanopathies using haploid screens for lassa virus entry. Science. 2013;340:479–483. doi: 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamison W.P., Hackstadt T. Induction of type III secretion by cell-free Chlamydia trachomatis elementary bodies. Microb. Pathog. 2008;45:435–440. doi: 10.1016/j.micpath.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett T.J., Fischer E.R., Mead D.J., Hackstadt T. Chlamydial TARP is a bacterial nucleator of actin. Proc. Natl. Acad. Sci. U S A. 2006;103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C.M., Fisher D.J. Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II Intron. PLoS One. 2013;8:e83989. doi: 10.1371/journal.pone.0083989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R.M., Gérard H.C., Mishra M.K., Mao G., Wang S., Hali M., Whittum-Hudson J.A., Hudson A.P. Dendrimer-enabled transformation of Chlamydia trachomatis. Microb. Pathog. 2013;65:29–35. doi: 10.1016/j.micpath.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Kawai K., Viars C., Arden K., Tarin D., Urquidi V., Goodison S. Comprehensive karyotyping of the HT-29 colon adenocarcinoma cell line. Genes Chromosomes Cancer. 2002;34:1–8. doi: 10.1002/gcc.10003. [DOI] [PubMed] [Google Scholar]
- Kawamoto K., Yoshida Y., Tamaki H., Torii S., Shinotsuka C., Yamashina S., Nakayama K. GBF1, a guanine nucleotide exchange factor for ADP-ribosylation factors, is localized to the cis-Golgi and involved in membrane association of the COPI coat. Traffic. 2002;3:483–495. doi: 10.1034/j.1600-0854.2002.30705.x. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Jiang S., Elwell C.A., Engel J.N. Chlamydia trachomatis co-opts the FGF2 signaling pathway to enhance infection. PLoS Pathog. 2011;7:e1002285. doi: 10.1371/journal.ppat.1002285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokes M., Dunn J.D., Granek J.A., Nguyen B.D., Barker J.R., Valdivia R.H., Bastidas R.J. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe. 2015;17:716–725. doi: 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König R., Stertz S., Zhou Y., Inoue A., Hoffmann H.-H., Bhattacharyya S., Alamares J.G., Tscherne D.M., Ortigoza M.B., Liang Y. Human host factors required for influenza virus replication. Nature. 2010;463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur F., Gaynor E.C., Hennecke S., Démollière C., Duden R., Emr S.D., Riezman H., Cosson P. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Li W., Xu H., Xiao T., Cong L., Love M.I., Zhang F., Irizarry R.A., Liu J.S., Brown M., Liu X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014;15:554. doi: 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidholt K., Weinke J.L., Kiser C.S., Lugemwa F.N., Bame K.J., Cheifetz S., Massagué J., Lindahl U., Esko J.D. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc. Natl. Acad. Sci. U S A. 1992;89:2267–2271. doi: 10.1073/pnas.89.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente A., Lauvrak S.U., van Deurs B., Sandvig K. Induction of direct endosome to endoplasmic reticulum transport in Chinese hamster ovary (CHO) cells (LdlF) with a temperature-sensitive defect in epsilon-coatomer protein (epsilon-COP) J. Biol. Chem. 2003;278:35850–35855. doi: 10.1074/jbc.M303425200. [DOI] [PubMed] [Google Scholar]
- Mariotti S.P., Pascolini D., Rose-Nussbaumer J. Trachoma: global magnitude of a preventable cause of blindness. Br. J. Ophthalmol. 2009;93:563–568. doi: 10.1136/bjo.2008.148494. [DOI] [PubMed] [Google Scholar]
- Matsuura W., Yamazaki T., Yamaguchi-Iwai Y., Masuda S., Nagao M., Andrews G.K., Kambe T. SLC39A9 (ZIP9) regulates zinc homeostasis in the secretory pathway: characterization of the ZIP subfamily I protein in vertebrate cells. Biosci. Biotechnol. Biochem. 2009;73:1142–1148. doi: 10.1271/bbb.80910. [DOI] [PubMed] [Google Scholar]
- Mirrashidi K.M., Elwell C.A., Verschueren E., Johnson J.R., Frando A., Von Dollen J., Rosenberg O., Gulbahce N., Jang G., Johnson T. Global mapping of the inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe. 2015;18:109–121. doi: 10.1016/j.chom.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misselwitz B., Dilling S., Vonaesch P., Sacher R., Snijder B., Schlumberger M., Rout S., Stark M., von Mering C., Pelkmans L. RNAi screen of Salmonella invasion shows role of COPI in membrane targeting of cholesterol and Cdc42. Mol. Syst. Biol. 2011;7:474. doi: 10.1038/msb.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore E.R., Mead D.J., Dooley C.A., Sager J., Hackstadt T. The trans-Golgi SNARE syntaxin 6 is recruited to the chlamydial inclusion membrane. Microbiology. 2011;157:830–838. doi: 10.1099/mic.0.045856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller K.E., Fields K.A. Application of β-lactamase reporter fusions as an indicator of effector protein secretion during infections with the obligate intracellular pathogen Chlamydia trachomatis. PLoS One. 2015;10:e0135295. doi: 10.1371/journal.pone.0135295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller K.E., Plano G.V., Fields K.A. New frontiers in type III secretion biology: the Chlamydia perspective. Infect. Immun. 2014;82:2–9. doi: 10.1128/IAI.00917-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller K.E., Wolf K., Fields K.A. Gene deletion by fluorescence-reported allelic exchange mutagenesis in Chlamydia trachomatis. MBio. 2016;7 doi: 10.1128/mBio.01817-15. e01817–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T., Kiyomitsu T., Saga Y., Kanemaki M.T. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 2016;15:210–218. doi: 10.1016/j.celrep.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Nickel W., Brügger B., Wieland F.T. Vesicular transport: the core machinery of COPI recruitment and budding. J. Cell Sci. 2002;115:3235–3240. doi: 10.1242/jcs.115.16.3235. [DOI] [PubMed] [Google Scholar]
- Pacheco-Rodriguez G., Moss J., Vaughan M. BIG1 and BIG2: brefeldin A-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors. Methods Enzymol. 2002;345:397–404. doi: 10.1016/s0076-6879(02)45032-x. [DOI] [PubMed] [Google Scholar]
- Parrett C.J., Lenoci R.V., Nguyen B., Russell L., Jewett T.J. Targeted disruption of Chlamydia trachomatis invasion by in trans expression of dominant negative tarp effectors. Front. Cell. Infect. Microbiol. 2016;6:84. doi: 10.3389/fcimb.2016.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepperkok R., Scheel J., Horstmann H., Hauri H.P., Griffiths G., Kreis T.E. Beta-COP is essential for biosynthetic membrane transport from the endoplasmic reticulum to the Golgi complex in vivo. Cell. 1993;74:71–82. doi: 10.1016/0092-8674(93)90295-2. [DOI] [PubMed] [Google Scholar]
- Pokrovskaya I.D., Willett R., Smith R.D., Morelle W., Kudlyk T., Lupashin V. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011;21:1554–1569. doi: 10.1093/glycob/cwr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portaliou A.G., Tsolis K.C., Loos M.S., Zorzini V., Economou A. Type III secretion: building and operating a remarkable nanomachine. Trends Biochem. Sci. 2016;41:175–189. doi: 10.1016/j.tibs.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Robinson M.S., Sahlender D.A., Foster S.D. Rapid inactivation of proteins by rapamycin-Induced rerouting to mitochondria. Dev. Cell. 2010;18:324–331. doi: 10.1016/j.devcel.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosmarin D.M., Carette J.E., Olive A.J., Starnbach M.N., Brummelkamp T.R., Ploegh H.L. Attachment of Chlamydia trachomatis L2 to host cells requires sulfation. Proc. Natl. Acad. Sci. U S A. 2012;109:10059–10064. doi: 10.1073/pnas.1120244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz J.B., Sun W.J., Chang J.W., Li J., Bursulaya B., Gray N.S., Haslam D.B. Golgicide A reveals essential roles for GBF1 in Golgi assembly and function. Nat. Chem. Biol. 2009;5:157–165. doi: 10.1038/nchembio.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka H.A., Thompson J.W., Chen Y.-S., Kumar Y., Dubois L.G., Moseley M.A., Valdivia R.H. Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol. Microbiol. 2011;82:1185–1203. doi: 10.1111/j.1365-2958.2011.07877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore M.A., Fischer E.R., Hackstadt T. Restricted fusion of Chlamydia trachomatis vesicles with endocytic compartments during the initial stages of infection. Infect. Immun. 2003;71:973–984. doi: 10.1128/IAI.71.2.973-984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyunin A.S., Iles L.R., Bartholomeusz G., Mukhopadhyay S. Genome-wide siRNA screen identifies UNC50 as a regulator of Shiga toxin 2 trafficking. J. Cell Biol. 2017;216:3249–3262. doi: 10.1083/jcb.201704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G. Genome-Scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan K.-L., Isberg R.R. Identification of mammalian proteins that collaborate with type III secretion system function: involvement of a chemokine receptor in supporting translocon activity. MBio. 2015;6 doi: 10.1128/mBio.02023-14. e02023–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H., Raymond L., Rockey D.D., Fischer E., Hackstadt T., Caldwell H.D. A recombinant Chlamydia trachomatis major outer membrane protein binds to heparan sulfate receptors on epithelial cells. Proc. Natl. Acad. Sci. U S A. 1996;93:11143–11148. doi: 10.1073/pnas.93.20.11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson A.F., Ezekowitz R.A., Lee A., Kuo C.C. Human mannose-binding protein inhibits infection of HeLa cells by Chlamydia trachomatis. Infect. Immun. 1998;66:1607–1612. doi: 10.1128/iai.66.4.1607-1612.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B.L., Wang Y., Ong Y.S., Hong W. COPII and exit from the endoplasmic reticulum. Biochim. Biophys. Acta. 2005;1744:293–303. doi: 10.1016/j.bbamcr.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Taraktchoglou M., Pacey A.A., Turnbull J.E., Eley A. Infectivity of Chlamydia trachomatis serovar LGV but not E is dependent on host cell heparan sulfate. Infect. Immun. 2001;69:968–976. doi: 10.1128/IAI.69.2.968-976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderheit A., Helenius A. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biol. 2005;3:e233. doi: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kahane S., Cutcliffe L.T., Skilton R.J., Lambden P.R., Clarke I.N. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011;7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters M.G., Griff I.C., Rothman J.E. Proteins involved in vesicular transport and membrane fusion. Curr. Opin. Cell Biol. 1991;3:615–620. doi: 10.1016/0955-0674(91)90031-s. [DOI] [PubMed] [Google Scholar]
- Whitney J.A., Gomez M., Sheff D., Kreis T.E., Mellman I. Cytoplasmic coat proteins involved in endosome function. Cell. 1995;83:703–713. doi: 10.1016/0092-8674(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Wilfling F., Thiam A.R., Olarte M.-J., Wang J., Beck R., Gould T.J., Allgeyer E.S., Pincet F., Bewersdorf J., Farese R.V. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife. 2014;3:e01607. doi: 10.7554/eLife.01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2011). Prevalence and incidence of selected sexually transmitted infections, http://apps.who.int/iris/bitstream/handle/10665/75181/9789241503839_eng.pdf?sequence=1&isAllowed=y.
- Yabushita H., Noguchi Y., Habuchi H., Ashikari S., Nakabe K., Fujita M., Noguchi M., Esko J.D., Kimata K. Effects of chemically modified heparin on Chlamydia trachomatis serovar L2 infection of eukaryotic cells in culture. Glycobiology. 2002;12:345–351. doi: 10.1093/glycob/12.5.345. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
sgRNAs refers to the number of sgRNAs targeting that gene present in the Avana library (Blondel et al., 2016); score refers to the MAGeCK positive selection score; FDR, false discovery rate; log2 FC, the log2-transformed fold-change average of the sgRNA abundance relative to the control (uninfected) condition.