

Abstract
This scientific commentary refers to ‘Longitudinal multimodal MRI as prognostic and diagnostic biomarker in presymptomatic familial frontotemporal dementia’, by Jiskoot et al. (doi:10.1093/brain/awy288).
This scientific commentary refers to ‘Longitudinal multimodal MRI as prognostic and diagnostic biomarker in presymptomatic familial frontotemporal dementia’, by Jiskoot et al. (doi:10.1093/brain/awy288).
Investigators are increasingly using clinical, neuropsychological, biofluid and imaging measures in patients with familial frontotemporal lobar degeneration (FTLD) to improve understanding of the neuropathological substrate of evolving FTLD pathology and to plan for future clinical trials. Molecular alterations are predicted to lead to changes in grey and/or white matter indices on brain MRI, and these MRI changes are predicted to precede the onset of neuropsychological abnormalities and the clinical features of overt symptomatic FTLD (Fig. 1A). In this issue of Brain, Jiskoot and co-workers test these predictions by investigating longitudinal MRI changes in members of familial FTLD kindreds who were asymptomatic at baseline (Jiskoot et al., 2019).
Figure 1.
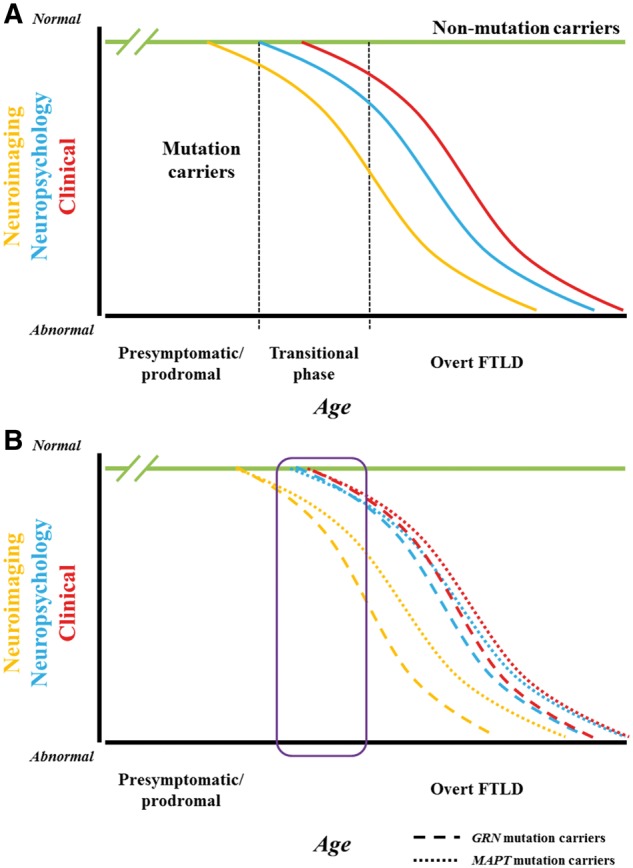
Implications of research findings on the hypothesized framework of evolving familial FTLD. (A) Hypothesized framework of evolving familial FTLD. Compared to non-mutation carrier relatives in familial FTLD kindreds (green line), one would expect that among mutation carriers, molecular alterations would lead to changes in grey and/or white matter indices on brain MRI (orange curve), and these MRI changes would precede the onset of neuropsychological abnormalities (blue curve) and overt clinical features of symptomatic FTLD (red curve). Presumably the curves would be sinusoidal—similar to other neurodegenerative disorders such as Alzheimer’s disease—with a slow initial slope, then an acceleration phase, then a deceleration phase, followed by a terminal gradual change. Inherent in this framework would be a transitional phase (represented by the column bounded by hashed vertical lines) with variable degrees of neuropsychological abnormalities (particularly in social cognition, executive functioning and language functioning) plus subtle clinical changes (such as mild apathy, disinhibition, altered food preferences, reduced empathy, declines in executive functioning and receptive or expressive language functioning) prior to the development of an overt FTLD phenotype such as behavioural variant frontotemporal dementia or primary progressive aphasia. (B) Implications of Jiskoot et al. data on the familial FTLD framework. The data in this manuscript relate to eight individuals—five MAPT mutation carriers who all converted to behavioural variant frontotemporal dementia and three GRN mutation carriers who all converted to non-fluent variant primary progressive aphasia—who were assessed over a 2–4 year timespan (roughly encompassed by the purple rectangle). The data suggest that indices of grey and white matter integrity changed prior to the onset of overt FTLD, with subtle neuropsychological changes around the time of conversion. Plus, the slopes of the imaging and neuropsychological/clinical changes in the GRN mutation carriers appeared to be steeper than those in the MAPT mutation carriers. With only three timepoints in six of the cases and two time points in the other two, it is difficult to determine if an initial slow slope evolved to a more rapid acceleration phase, as implied by these curves. Furthermore, while the period of assessment likely included the transitional phase between normal neurological functioning and overt FTLD for at least some of these cases, it is challenging to characterize this further based on the available data. Regardless, the longitudinal data in this paper provide intriguing preliminary support for the hypothetical familial FTLD model described above.
The investigators analysed grey and white matter indices on brain MRI in the prodromal phase of familial FTLD in kindreds with mutations in the genes encoding microtubule-associated protein tau (MAPT) or progranulin (GRN). The team compared findings on white matter integrity (using fractional anisotropy), diffusion tensor imaging (DTI) and grey matter volume in persons who evolved from asymptomatic to overt FTLD (converters; n = 8) to those in asymptomatic mutation carriers (non-converters; n = 35) and non-carriers (n = 30) from the same families. They found lower fractional anisotropy values in white matter regions (genu corpus callosum, forceps minor, uncinate fasciculus, and superior longitudinal fasciculus) and smaller grey matter volumes (prefrontal, temporal, cingulate, and insular cortex) over time in converters, which were present from 2 years before overt FTLD. Furthermore, they found that (i) MRI changes evolved more rapidly in GRN compared to MAPT converters; (ii) MAPT converters showed more decline in the uncinate fasciculus than GRN converters; and (iii) fractional anisotropy in corpus callosum declined more in GRN than MAPT converters. Subtle changes on neuropsychological measures were also suggested in the late presymptomatic phase. Jiskoot et al. concluded that there is evolving frontotemporal pathology near symptom onset and that these findings highlight the value of multimodal MRI as a prognostic biomarker in familial FTLD. While there are only eight converters in this analysis and sweeping conclusions are not appropriate, one could map the findings into the familial FTLD framework as shown in Fig. 1B.
These data extend what has been suggested on the basis of cross-sectional and longitudinal analyses (Rosen et al., 2002a, b; Knopman et al., 2009; Whitwell et al., 2010, 2012, 2015; Rohrer et al., 2015). Indeed, these results support the utility of longitudinal MRI in characterizing familial FTLD in the presymptomatic phase. Prior data, particularly from the Genetic Frontotemporal Dementia Initiative (GENFI) consortium (Rohrer et al., 2015), had suggested that MRI changes precede the onset of FTLD symptoms, possibly by as much as 10 years. That analysis used cross-sectional data and estimated ages of onset for asymptomatic individuals based on familial age of onset, but it is well accepted in the field that the highly variable age of onset in familial FTLD makes predictions of symptom onset tenuous. The findings by Jiskoot et al. are therefore notable because they confirm through prospective longitudinal analysis that brain volume and white matter integrity start to decline before overt symptom onset. However, their analysis suggested that these changes may begin about 2 years before symptom onset (they did not detect much difference 4 years prior to symptom onset). While general conclusions should not be made based on such a small sample, the findings clearly highlight the value of prospective longitudinal assessment for addressing this issue. Furthermore, while there are ample data indicating differences in the topography of degeneration across symptomatic MAPT, GRN and chromosome 9 open reading frame 72 (C9orf72) mutation carriers (Whitwell et al., 2012), this is one of the first studies to show topographic differences between genetic groups using longitudinal data.
Jiskoot et al. performed their study using a 3 T MRI scanner, available at many clinical and research centres, and using standard image analysis packages. These aspects of the study make the results more generalizable, but it is important to note that many centres still use 1.5 T scanners, and prior studies have indicated that field strength affects the power to detect longitudinal changes (Pankov et al., 2015). Furthermore, all subjects were scanned in the same machine. Although there have been great advances in methods for harmonizing acquisition procedures across centres and scanners (Jack et al., 2015), differences in scanner platforms and software still influence image characteristics, introducing noise into multicentre studies and decreasing sensitivity to detect change. Also, harmonization procedures are most advanced for T1-weighted MRI that quantifies volumetric changes, whereas harmonization for other types of imaging such as DTI is less well developed. All of these considerations underscore the importance of replicating and expanding these findings in a larger, multisite setting. If more advanced imaging techniques such as DTI really bring unique value for prediction, then additional technical developments will be needed to make this procedure perform optimally in a multisite setting.
The issue of a transitional stage between normal neurological functioning and overt FTLD also warrants comment. Jiskoot et al. analysed neuropsychological and imaging data using the onset of an overt FTLD syndrome—behavioural variant FTD (bvFTD) or a subtype of primary progressive aphasia (PPA)—as representing ‘symptom onset’ and hence conversion. By inference, the imaging changes identified were occurring during a transitional stage in at least some of the cases. The authors acknowledge the difficulty of identifying the exact timing of conversion, and discuss the concept and variable nomenclature of a transitional stage—similar to mild cognitive impairment in the evolution to Alzheimer’s disease dementia—in FTLD. However, they did not characterize the clinical features of this transitional stage. This is an important issue because clinicians and researchers will need guidance for when to pursue longitudinal imaging to look for the types of changes identified by Jiskoot et al. Data presented in the paper show a trend of mildly increased scores on the Neuropsychiatric Inventory Questionnaire, which captures some of the key neuropsychiatric FTLD features, in the non-converters compared to the non-carriers. Further studies may ultimately demonstrate that neuropsychiatric changes, or other clinical changes, are an early indicator of transition, and lead to a uniform set of operational criteria and terminology for this phase. Additional indicators of transition may ultimately come from protein biomarkers, hopefully measurable from peripheral blood (Rojas et al., 2016).
The authors also plot clinical, neuropsychological and imaging data for the eight converters at 4 years prior to conversion (when data were available), 2 years prior to conversion and at conversion. The Z-scores for the clinical and neuropsychological data are near 0 for almost every case at the evaluation 2 years prior to conversion. This suggests either that these measures are poorly sensitive to evolving FTLD, or that evaluating familial FTLD subjects every 2 years is insufficient to capture evolving features when nearing phenoconversion.
The findings from Jiskoot et al. and other reports, underscore the importance of these imaging modalities in familial FTLD clinical trials. Comprehensive prospective assessments consisting of longitudinal clinical, neuropsychological, biofluid and neuroimaging studies of a large number of patients with familial FTLD will be required to predict phenoconversion (that is, when and to what major phenotype—bvFTD versus PPA versus other), to identify the optimal outcome measures, and to estimate sample sizes for clinical trials. The success of future trials of disease-modifying therapies for familial FTLD—presumably using agents that affect tau pathophysiology in MAPT mutation carriers, progranulin ± TDP-43-related pathophysiology in GRN mutation carriers, and chromosome 9 repeat-associated non-standard (RAN) translation products ± TDP-43-related pathophysiology in C9orf72 mutation carriers—will likely require the identification of familial FTLD mutation carriers who have undergone at least two longitudinal evaluations prior to commencing therapy in order to assess whether the slope of change on MRI indices is altered during a clinical trial. Furthermore, regulators will likely require that sufficient natural history data have been acquired to determine which neuropsychological and clinical measures, and clinical outcomes, can best show that any intervention has delayed the onset of symptomatic FTLD. There are several prospective multisite studies that involve familial FTLD kindreds in progress (e.g. GENFI, ARTFL, LEFFTDS), and willingness among members of familial FTLD kindreds to participate in natural history studies is high. In addition, several interventions that may impact FTLD-related proteinopathies are nearing clinical trial readiness. The future thus appears bright for disease-modifying familial FTLD trials. The data by Jiskoot et al. underscore the importance of multimodal MRI, as well as a comprehensive battery of clinical and neuropsychological measures, in ongoing observational and future clinical trials.
Funding
This work is supported by the NIH (U01 AG045390 and U54 NS092089) and the Bluefield Project to Cure FTD.
Competing interests
B.F.B. has served as an investigator for clinical trials sponsored by GE Healthcare, Axovant and Biogen. He receives royalties from the publication of a book entitled Behavioral Neurology Of Dementia (Cambridge Medicine, 2009, 2017). He serves on the Scientific Advisory Board of the Tau Consortium. H.R. has served as a consultant for Inonis and Wave pharmaceuticals and received research support from Biogen pharmaceuticals and the Bluefield Project to Cure FTD.
References
- Jack CR Jr, Barnes J, Bernstein MA, Borowski BJ, Brewer J, Clegg S et al. Magnetic resonance imaging in Alzheimer’s Disease Neuroimaging Initiative 2. Alzheimers Dement 2015; 11(7): 740–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiskoot L, Panman J, Meeter L, Dopper E, Donker Kaat L, Franzen S et al. Longitudinal multimodal imaging as prognostic and diagnostic biomarker in presymptomatic FTD. Brain 2019; 142: 193–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Jack CR Jr, Kramer JH, Boeve BF, Caselli RJ, Graff-Radford NR et al. Brain and ventricular volumetric changes in frontotemporal lobar degeneration over 1 year. Neurology 2009; 72: 1843–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov A, Binney RJ, Staffaroni AM, Kornak J, Attygalle S, Schuff N et al. Data-driven regions of interest for longitudinal change in frontotemporal lobar degeneration. Neuroimage Clin 2015; 12: 332–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002a; 58: 198–208. [DOI] [PubMed] [Google Scholar]
- Rosen HJ, Kramer JH, Gorno-Tempini ML, Schuff N, Weiner M, Miller BL. Patterns of cerebral atrophy in primary progressive aphasia. Am J Geriatr Psychiatry 2002b; 10: 89–97. [PubMed] [Google Scholar]
- Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 2015; 14: 253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, Karydas A, Bang J, Tsai RM, Blennow K, Liman V et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol 2016; 3(3): 216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Avula R, Senjem ML, Kantarci K, Weigand SD, Samikoglu A et al. Gray and white matter water diffusion in the syndromic variants of frontotemporal dementia. Neurology 2010; 74: 1279–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 2012; 135: 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Boeve BF, Weigand SD, Senjem ML, Gunter JL, Baker MC et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol 2015; 22: 745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]