

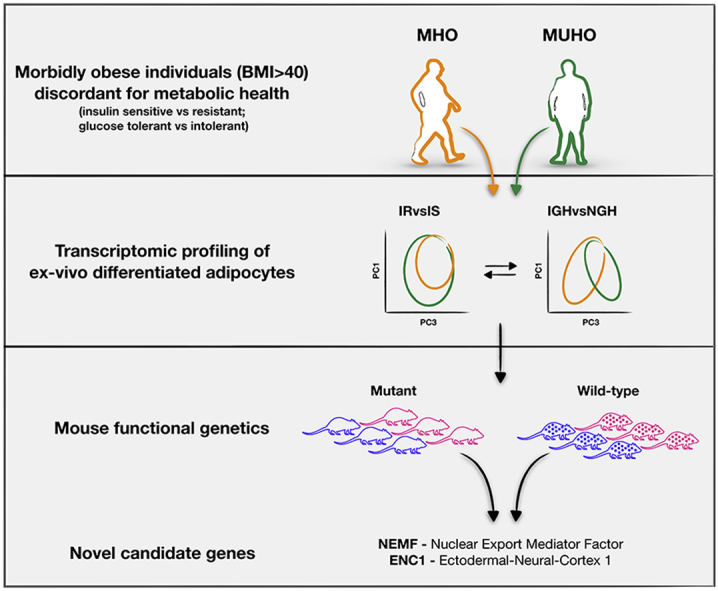
Abstract
Objective
Although debated, metabolic health characterizes 10–25% of obese individuals and reduces risk of developing life-threatening co-morbidities. Adipose tissue is a recognized endocrine organ important for the maintenance of whole-body metabolic health. Adipocyte transcriptional signatures of healthy and unhealthy obesity are largely unknown.
Methods
Here, we used a small cohort of highly characterized obese individuals discordant for metabolic health, characterized their adipocytes transcriptional signatures, and cross-referenced them to mouse phenotypic and human GWAs databases.
Results and conclusions
Our study showed that glucose intolerance and insulin resistance co-operate to remodel adipocyte transcriptome. We also identified the Nuclear Export Mediator Factor (NEMF) and the Ectoderm-Neural Cortex 1 (ENC1) as novel potential targets in the management of metabolic health in human obesity.
Keywords: Obesity, Glucose tolerance, Insulin sensitivity, Transcriptomics, Mouse genetics, Systemic phenotyping
Graphical abstract
Highlights
-
•
Healthy obese individuals maintain glucose tolerance and insulin sensitivity.
-
•
Glucose intolerance primarily remodels adipocyte transcriptome in human obesity.
-
•
Insulin resistance co-operates with glucose tolerance to remodel adipocyte transcriptome.
-
•
NEMF and ENC1 are potential targets to manage metabolic health in human obesity.
1. Introduction
Obesity, characterized by an excess of body fat, and global body fat dysfunction, has reached epidemic proportions worldwide. Almost 300 million people are clinically obese (BMI > 30 kg/m2) and, therefore, at high risk of developing life-threatening co-morbidities, such as diabetes, cardiovascular disease, stroke, and cancer. Of note, 10–25% of obese people stay healthy. These individuals remain glucose tolerant and insulin sensitive, with normal blood pressure and a favorable lipid profile [1]. A recent study demonstrated that the majority of obese individuals slip into poor health and chronic illness over time [2]. Conversely, other studies suggest that, while healthy obese individuals have higher risk of developing co-morbidities when compared to a lean population [3] they are still protected as the onset of co-morbidities is delayed compared to an unhealthy obese population [1]. Thus, maintaining metabolic health in obesity is important for long-term disease management.
Despite the current clinical definition of healthy obesity [1] and the recently discovered metabolomic signatures [4], the molecular underpinnings of metabolic health in obesity is largely unknown. Here, we analyzed primary subcutaneous adipocytes from morbidly obese individuals (BMI > 40) [4] discordant for metabolic health (Tables 1 and S3). We discovered that instead of insulin resistance, impaired glucose homeostasis is a major determinant of adipocyte transcriptional response. Insulin resistance, instead, appears to be a modifier of this response. Interrogating systemic mouse phenotypic data [5] and human GWAS catalogs, we identified 19 genetic associations with phenotypes of glucose homeostasis and/or body weight and composition. In particular, we describe the Nuclear Export Mediator Factor (NEMF) and the Ectoderm-Neural Cortex protein 1 (ENC1) as novel potential targets in the management of metabolic health in human obesity.
Table 1.
Characteristics of individuals with obesity classified as metabolically healthy (MHO) or unhealthy (MUHO).
| MHO | MUHO | p | |
|---|---|---|---|
| N (women/men) | 7/4 | 7/4 | 1.0a |
| Age (y) | 43 ± 10 | 39 ± 13 | 0.3b |
| BMI (kg/m2) | 52.5 ± 8.5 | 51.6 ± 6.7 | 1.0c |
| Body fat (%) | 49.5 ± 12.3 | 51.5 ± 11.7 | 0.7c |
| AUC0-120 glucose (mmol/L) | 16.2 ± 2.4 | 18.6 ± 3.5 | 0.0044c |
| HbA1c (%) | 5.6 ± 0.4 | 5.9 ± 0.3 | 0.05c |
| ISI-OGTT (x106 L/kg/min) | 7.10 ± 2.14 | 2.66 ± 0.70 | 0.0000003c |
| AUC0-30 C-peptide/glucose (pmol/mmol) | 208 ± 73 | 329 ± 101 | 0.6d |
| Leukocytes (μL−1) | 7,056 ± 1,782 | 10,086 ± 3,192 | 0.0127c |
| CRP (mg/dL) | 0.61 ± 0.51 | 1.30 ± 1.03 | 0.2c |
Data represents number or mean ± SD.
AUC – area under the curve; BMI – body mass index; CRP – C-reactive protein; HbA1c – hemoglobin A1c; ISI – insulin sensitivity index; MHO – metabolically healthy obese; MUHO – metabolically unhealthy obese; OGTT – oral glucose tolerance test.
χ2-test.
t-test.
Adjusted for age (multiple linear regression).
Adjusted for age and ISI-OGTT (multiple linear regression).
2. Results
2.1. Insulin sensitivity does not rewire adipocyte transcriptome in human obesity
Adipose tissue is a recognized endocrine organ critical for the maintenance of whole-body metabolic homeostasis [6]. We obtained subcutaneous adipose tissue biopsies during bariatric surgery of morbidly obese individuals discordant for metabolic health [7]. Accessible, frozen, primary pre-adipocytes were differentiated ex-vivo into mature adipocytes that were used for RNA-sequencing analyses. Total RNA was prepared from mature adipocytes and, after quality check, ribosomal RNA-depleted libraries were generated for single-end Illumina sequencing (see materials and methods section for more details). Although the use of ex-vivo differentiated adipocytes constitutes a limitation of the current study as the observed differences might be biased by intrinsic and individual differences in adipogenesis potential, we aimed to experimentally control for that. We therefore pulled out the 200 human adipogenesis-relevant genes from the Geneset Enrichment Analysis (GSEA) database (GSEA_Hallmark_human_Adipogenesis) and performed unsupervised clustering and principal component analysis to identify any association between metabolic status and adipogenesis efficiency. As shown in Supplementary Figure 1 (Fig. S1) preadipocytes from insulin sensitive or resistant individuals maintain the same differentiation potential, as highlighted by no differential clustering of the two metabolic conditions (Fig. S1A,S1B) as well as by the same expression of key markers of mature adipocytes such as PPARG (Peroxisome Proliferator Activator Receptor Gamma – Fig. S1C), FABP4 (Fatty Acid Binding Protein 4 – Fig. S1D) and PLIN2 (Perilipin 2 – Fig. S1E).
Since gender specificity is a common feature of metabolic phenotypes in humans, as well as in commonly used model organisms, we checked whether gender was significantly rewiring adipocytes transcriptome. As shown in Supplementary Figure 1, less than 10 genes are differentially expressed between males and females (Fig. S1F), and the vast majority of them are associated to sex chromosomes, thus confirming that gender is not a determinant of adipocyte transcriptional response.
Once controlled for adipogenesis potential and gender effect, we studied the adipocyte transcriptional signatures of metabolically healthy and unhealthy obese individuals. Since metabolic health is primarily defined by maintenance of whole-body insulin sensitivity, we compared adipocytes from insulin sensitive and resistant individuals (Table 1). Interestingly and unexpected, loss of insulin sensitivity did not affect adipocytes transcriptional programs as both principal component (Figure 1A) and differential expression (Figure 1B) analyses revealed a complete overlap between adipocytes from insulin sensitive and resistant individuals. These findings are in line with previously published work [8] showing that adipose tissue transcriptional response to insulin is determined primarily by obesity rather than insulin sensitivity.
Figure 1.

Glucose tolerance affects adipocyte transcriptional programs in human obesity. A-B. PCA (A) and Volcano plot (B) representation of RNA-Seq data from adipocytes of Insulin Sensitive (IS) and Resistant (IR) donors. C. PCA plot representation of RNA-Seq data from adipocytes of donors with Normal (NGT) or impaired (IGF and/or IGT) Glucose Tolerance. D-E. PCA plot (D) and Volcano Plot (E) representation of RNA-Seq data from adipocytes of donors with Normal (NGH) or Impaired (IGH) glucose homeostasis. Differentially Expressed Genes (DEGs) are represented as green (down-regulated) or red (up-regulated) dots. F-G. KEGG Pathway Analysis (F) of DEGs in IGH vs NGH samples and Heatmap representation (G) of those clustered in enriched pathways (colored dots indicate the respective KEGG terms).
2.2. Glucose homeostasis rewires adipocyte transcriptome in human obesity
Together with insulin sensitivity, metabolic health is characterized by maintenance of whole body glucose homeostasis. Hyperglycemia, independent from insulin resistance, has a strong impact on adipose tissue function as it activates several pathways important for adipocyte cellular homeostasis (e. g., ER stress response, mRNA processing and protein translation) and is critical in directing adipose tissue growth towards the unhealthy hypertrophic state (reviewed in [9]). Mechanistically, glucose has been shown to activate several downstream signaling pathways, which culminate in rewiring of cellular transcriptional responses [10], [11].
To test the effect of glucose intolerance on adipocyte transcriptional programs, we re-grouped the obese individuals based on their glucose tolerance (Table 2) and re-ran the bioinformatic analysis. Deterioration of glucose tolerance is stepwise and proceeds from Normal Glucose Tolerance (NGT – fasting glycemia ≤5.5 mmol/L and 2hr after OGTT ≤7.7 mmol/L) to Impaired Fasting Glycemia (IFG – fasting glycemia 5.6–6.9 mmol/L and 2hr after OGTT ≤7.7 mmol/L), Impaired Glucose Tolerance (IGT - fasting glycemia ≤5.5 mmol/L and 2hr after OGTT 7.8–11 mmol/L) and a more severe derangement of glucose homeostasis characterized by both IFG and IGT (IFG + IGT). Interestingly, the number of differentially expressed genes (DEGs) between adipocytes from tolerant and intolerant individuals substantially increased with decreasing glucose tolerance (Fig. S2A–C and Table S1) and, while enriched for lipid metabolism in both IFG and IGT samples, they clustered to pathways critical for proper metabolic control (e. g. Insulin, PI3K/Akt, FoxO) in IFG + IGT samples (Fig. S2D–F and Table S1). Also, a principal component analysis run on all-expressed genes (fpkm > 0.3) indicated that adipocytes from intolerant individuals cluster together and away from those of tolerant individuals (Figure 1C).
Table 2.
Characteristics of individuals with obesity regrouped based on their glucose homeostasis (NGH or IGH) and insulin sensitivity (IS or IR).
| NGH_IS | NGH_IR | IGH_IS | IGH_IR | p | |
|---|---|---|---|---|---|
| N (women/men) | 3/0 | 2/3 | 4/4 | 5/1 | 0.2a |
| Age (y) | 39 ± 16 | 29 ± 6 | 44 ± 8 | 48 ± 11 | 0.0142b |
| BMI (kg/m2) | 52.8 ± 5.1 | 47.7 ± 3.1 | 52.4 ± 9.7 | 54.8 ± 7.4 | 0.4c |
| Body fat (%) | 60.1 ± 2.1 | 48.2 ± 12.2 | 44.9 ± 12.0 | 54.1 ± 11.7 | 0.5c |
| Fasting glucose (mmol/L) | 5.37 ± 0.14 | 5.16 ± 0.17 | 5.54 ± 0.51 | 6.16 ± 0.39 | 0.0070d |
| 2-hr glucose (mmol/L) | 6.89 ± 0.39 | 5.92 ± 0.79 | 7.31 ± 1.45 | 9.05 ± 1.27 | 0.0002d |
| HbA1c (%) | 5.87 ± 0.23 | 6.00 ± 0.38 | 5.49 ± 0.42 | 5.90 ± 0.14 | 0.07d |
| ISI-OGTT (x1019 L2/mol2) | 6.62 ± 1.44 | 2.69 ± 0.48 | 7.28 ± 2.42 | 2.63 ± 0.90 | 0.000015d |
| Insulinogenic index (×10−9) | 214 ± 105 | 351 ± 83 | 195 ± 163 | 253 ± 150 | 0.6e |
| Leukocytes (μL−1) | 7,540 ± 1,607 | 12,390 ± 3,378 | 6,875 ± 1,913 | 8,167 ± 1,230 | 0.06d |
| CRP (mg/dL) | 0.65 ± 0.60 | 0.97 ± 0,93 | 0.60 ± 0.52 | 1.48 ± 1.10 | 0.7d |
| AST (U/L) | 21 ± 4 | 25 ± 11 | 23 ± 6 | 25 ± 10 | 1.0d |
| ALT (U/L) | 23 ± 9 | 46 ± 25 | 25 ± 10 | 32 ± 15 | 0.6d |
| Fetuin-A (μg/mL) | 281 ± 60 | 275 ± 55 | 275 ± 37 | 256 ± 38 | 0.7d |
Data represent numbers or means ± SD.
ALT – alanine transaminase; AST – aspartate transaminase; BMI – body mass index; CRP – C-reactive protein; HbA1c – hemoglobin A1c; IGH – impaired glucose homeostasis; IR – insulin-resistant; IS – insulin-sensitive; ISI – insulin sensitivity index; NGH – normal glucose homeostasis; OGTT – oral glucose tolerance test.
χ2-test.
Multiple linear regression (adjusted for gender).
Multiple linear regression (adjusted for gender and age).
Multiple linear regression (adjusted for gender, age, and BMI).
Multiple linear regression (adjusted for gender, age, BMI, and insulin sensitivity).
Thus, hyperglycemia rewires adipocyte transcription and, while different adipocyte transcriptional programs reflect the degree of glucose intolerance, intolerant donors constitute a “single” group whose health determinant is glucose homeostasis.
Stemming from these findings, we grouped the adipocytes samples as coming from individuals with normal (NGH) or impaired glucose homeostasis (IGH – which includes IFG, IGT and IFG + IGT individuals – Table 1) and re-ran the bioinformatic pipeline reasoning that this would deepen our knowledge on the biological consequences of impaired glucose homeostasis in adipocytes. Indeed, both principal component (Figure 1D) and differential expression (Figure 1E) analyses showed that glucose homeostasis highlights two transcriptionally different groups defined by more than 1000 DEGs (Figure 1D–E), which cluster to pathways important for longevity and metabolic control, such as the Insulin, the MAPK and the AMPK signaling pathways (Figure 1F–G and Table S1). Critically, the NGH group was significantly younger than the IGH, and therefore we ran a control analysis to make sure that the observed differences in gene expression were not age-dependent. Briefly, we stratified the obese individuals by age and looked at the expression of the differentially expressed genes between NGH and IGH individuals. As shown in supplementary figure 3 (Fig. S3A), differential gene expression is age-independent as expression patterns of young IGH (Ad20 and Ad03) or old NGH (Ad26) individuals still reflect their metabolic status rather than the age group.
Thus, impaired glucose homeostasis alters the transcription of critical metabolic pathways in ex-vivo differentiated adipocytes from morbidly obese individuals.
Unfortunately, data on adipose tissue morphology and immune cell infiltration – important parameters to define a metabolically healthy adipose tissue – are not available from this cohort and the exact relationship between these parameters and the adipocyte transcriptional response remain a limitation of the study. Nevertheless, the same individuals have been previously characterized for adipose tissue insulin sensitivity and systemic inflammation [4]; here, we have deepened our adipocyte characterization by integrating individuals’ glucose homeostasis and insulin sensitivity in the analysis to understand the relationship between the two and their impact on adipocyte transcriptional response.
2.3. Insulin sensitivity cooperates with glucose intolerance to modify adipocytes transcriptional programs in human obesity
Glucose intolerance and insulin resistance are independent and additive risk factors for the development of diabetes, cardiovascular disease and premature death. Insulin resistance occurs independently from glucose tolerance also in the study cohort, with both NGH and IGH individuals being insulin sensitive or resistant (Figure 2A). Interestingly enough, insulin sensitivity and glucose tolerance interact in regulating adipocytes transcription. While the magnitudes of transcriptional changes between IGH and NGH individuals being insulin sensitive or resistant are similar (IS_IGH_NGH = 175 DEGs; IR_IGH_NGH = 226 DEGs – Figure 2B,C), the biological functions and the underlying transcriptional regulatory mechanisms are very divergent. While glucose intolerance drives differential expression of genes involved in tissue remodeling (Figure 2D and Table S1) and targets of transcription factors important for development (RNF2, SUZ12, JARID2, EZH2), adipogenesis (CLOCK, PAX3), and stemness (ZFP281, GATA2) (Figure 2F upper left panel, Fig. S4A and Table S1) in adipocytes from insulin sensitive individuals; in those from insulin resistant individuals, DEGs are involved in the establishment and maintenance of inflammation (Figure 2E and Table S1) and are targets of transcription factors important for inflammatory related processes such as Epithelial-Mesenchymal Transition (SALL4, SMAD), Apoptosis (ZNF217), Inflammation (CEBPD, ESR1, SMAD) and insulin resistance (NRF2) (Figure 2F lower left panel, Fig. S4C and Table S1).
Figure 2.

Insulin sensitivity is a modifier of adipocyte transcriptional programs imposed by glucose tolerance. A. ISI Matsuda Index in NGH or IGH donors. Horizontal bars indicate the median. Significance explained in Table 1. B-C. Volcano plot representation of RNA-Seq data from adipocytes of NGH vs IGH donors featuring insulin sensitivity (B), or resistance (C). D-E. KEGG pathway analysis of DEGs in IS_IGH_NGH (D) and IR_IGH_NGH (E). F. Enrichr-based grid representation of transcriptional regulatory clusters. Left part: ChEA-based enriched transcription factors in DEGs from IS_IGH_NGH (top) or IR_IGH_NGH (bottom). p-value < 0.05 is considered statistically significant. Cluster 1 – RNF2, JARID2, PAX3; Cluster 2 – EZH2, p53, GATA2; Cluster 3 – SUZ12, SMAD, ZNF217, ESR1, CEBPD, NRF2. Right part: ENCODE-based enriched histone PTMs on DEGs from IS_IGH_NGH (top) and IR_IGH_NGH (bottom).
Interestingly, a consistent, though not significant clustering to Polycomb target genes (Figure 2F right panels and S4B and D – H3K27me3) indicates, in line with recently published findings [12], [13], [14], deregulation of Polycomb domains on chromatin as a common feature of metabolic dysfunction also in human adipocytes.
These findings, although obtained in a simplified system such as ex-vivo differentiated and cultured adipocytes, represent a good example of the complex interplay between hyperglycemia and insulin resistance in the maintenance of metabolic health in human obesity. Highly divergent transcriptional regulatory mechanisms, although converging into the activation of common downstream pathways, indicate that different intracellular molecular machineries, aimed at fine-tuning cellular responses potentially critical for organismal health, integrate extracellular metabolic signals. Intriguingly, activation of downstream pathways which are commonly associated to loss of metabolic health in intact adipose tissue in vivo, such as those involved in inflammation and tissue remodeling, suggest – on one side – their adipocyte origin in vivo and – on the other side – that ex-vivo differentiated adipocytes reflects, at least to a certain extent, in vivo physiology.
2.4. NEMF and ENC1 emerge as novel potential target in organismal metabolic homeostasis
In keeping with the divergence in the transcriptional regulatory mechanisms, the number of co-DEGs between the insulin sensitive and resistant sample sets is restricted to roughly 5% of all DEGs (Figure 3A). To further understand the role of these genes in metabolic control, we turned to mouse and human genetics and interrogated public databases for genetic associations with relevant phenotypes. We started by harnessing the resource of the International Mouse Phenotyping Consortium (IMPC) [5], [15], which has already provided the community with several thousands disease models [15], [16] and identified genes important for hearing [17], sexual dimorphism [18], and metabolic control [19]. To date, the IMPC has generated and phenotyped ∼25% (96 genes) of the 401 genes differentially expressed in adipocytes from the different subgroups of insulin sensitive and resistant individuals, and ∼20% of those (21 genes) have significant associations with Mouse Phenotype (MP) terms related to glucose homeostasis and/or body composition (Figure 3B,C and Table S2).
Figure 3.

Combined mouse and human genetics identifies novel determinant of metabolic health in human obesity. A. Venn diagram representation of co-differentially expressed genes in IS_IGH_NGH and IR_IGH_NGH. B-C. Heatmap representation of IS_IGH_NGH (B) and IR_IGH_NGH (C) DEGs with significant MP term associations (red – glucose homeostasis; blue – body weight and composition). D-I. 5–95 percentile plots of representative genes and MP terms (* = p < 0.01 calculated with one-way ANOVA with multiple comparisons using GraphPad Prism 6). Plain colored boxes represent mutant animals; dotted colored boxes represent control animals. Males in blue, females in pink.
Supporting the robustness and sensitivity of our approach, we identified, among the 21 genes with significant MP term associations, two with known function in metabolic control, namely the leptin gene (LEP) [20], [21] and the Polycomb repressive complex 1 subunit BMI1 (B Lymphoma Mo-MLV Insertion Region 1) [14]. Among the remaining 19 genes with significant MP term associations, we focused on the 6 associated with phenotypes related to whole body glucose homeostasis. Of those, two are novel potential targets for the management of glucose homeostasis in obesity, namely the Nuclear Export Mediator Factor (NEMF) and the Ectodermal-Neural Cortex 1 (ENC1). NEMF and ENC1 are up-regulated in adipocytes from glucose intolerant donors either insulin sensitive (NEMF – Figure 3B) or resistant (ENC1 – Figure 3C) and, in both cases, genetic ablation in the mouse results in reduced non-fasted glycemia (Figure 3D,F). ENC1 knockout mice also show a favorable switch in body composition with reduced fat mass (Figure 3F). Similar phenotypes also result from the manipulation of the Engulfment and cell Motility 1 (ELMO1) gene, which is up-regulated in adipocytes from glucose intolerant and insulin resistant donors (Figure 3C) and whose ablation leads to male-specific reduction in fasted glycemia (Figure 3E). Of note, none of these three genes has a known function in adipocyte biology or metabolic control. Conversely, they have central roles in cellular physiology, namely quality control of protein translation (NEMF) [22] and cellular morphology (ENC1 and ELMO1) [23], [24]. Intriguingly, these cellular functions are impaired in the transition from hyperplastic to hypertrophic adipocytes, associated to loss of endocrine function and of whole-body metabolic homeostasis [25].
The remaining three genes – the Aspartoacylase (ASPA), the Frizzled Receptor 3 (FZD3) and the Cytochrome P450 Family 7 Subfamily B Member 1 (CYP7B1) – are up-regulated in adipocytes from glucose intolerant donors either insulin sensitive (ASPA – Figure 3B) or resistant (FZD3 and CYP7B1 – Figure 3C) and lead to impaired glucose homeostasis when knocked-out in mice (Figure 3G–I), namely elevated fasting glycemia (ASPA – Figure 3H) or impaired glucose tolerance (CYP7B1 and FZD3 – Figure 3G,I). ASPA, FZD3 and CYP7B1 are all involved in different aspects of lipid metabolism and adipose tissue biology.
CYP7B1, also known as Oxysterol 7-Alpha-Hydroxylase, functions in hepatic and extrahepatic tissues to metabolize cholesterol to oxysterols and bile acids, which are important for metabolic physiology [26].
ASPA and FZD3 are expressed in adipose tissue and function in adipogenesis, lipid metabolism and organismal development. ASPA de-acetylates N-Acetyl-l-Aspartic Acid (NAA) into aspartate and acetate and is critical for the formation of myelin sheaths [27]. Loss-of-function mutations result in NAA accumulation and Canavan disease in humans [28]. The role of ASPA in adipocyte biology and metabolic health is unclear, although preliminary observations have linked ASPA to lipid turnover, histone acetylation and function of brown adipocytes [29], [30]. FZD3 is a member of the Frizzled gene family and encodes a seven-transmembrane domain protein, receptor of the Wnt family of signaling proteins. While most of the frizzled receptors activate the canonical wnt/beta-catenin signaling, FZD3 is a Wnt5A receptor and activator of non-canonical Wnt signaling pathway in inflammatory conditions [31], axon guidance, and neurogenesis [32]. In keeping with a critical function in neuronal development, FZD3 knockout mice show prenatal lethality (Table S2). Of note, the reported phenotypes are just the most relevant to this manuscript. Complete phenotypic information is available from the IMPC database and links to individual mouse lines are reported in the Supplementary Table S2.
SNP analysis did not reveal genome-wide significant associations with human metabolic syndrome for any of the 6 genes with significant annotations to MP terms (Table S2), indicating that the observed alteration in their expression results from alternative mechanisms, such as gene/environment interaction and epigenetic phenomena.
3. Conclusions
Taken together, our findings show that glucose intolerance and insulin resistance cooperatively regulate adipocyte transcription in human obesity, with glucose intolerance (and thus hyperglycemia) being – most likely – the most potent signal. Our data not only validates genes previously associated to metabolic health (e. g. LEP and BMI1), but also identifies six novel candidates functionally validated using mouse genetics and systemic phenotyping. Among those, NEMF and ENC1 are upregulated in unhealthy obese individuals and, when knocked out in mice, show consistent amelioration of metabolic homeostasis. Therefore, NEMF and ENC1 appear as two potential novel targets for the management of glucose homeostasis in human obesity.
4. Materials and methods
4.1. Characteristics and stratification of adipose tissue donors
Twenty-two, age-matched Caucasian morbidly obese individuals, who underwent sleeve gastrectomy and concomitant abdominal subcutaneous fat biopsy, were matched for gender and BMI and grouped into two equally sized subgroups discordant for whole-body insulin sensitivity (Table 1). The reason for choosing morbidly obese individuals, although they do not fully represent the more common population of “mild” obese individuals is at least two fold: (1) “Mild” obese individuals unlikely undergo bariatric surgery and therefore, it would have been more complicated to get access to biopsies; and (2) A population of morbidly obese individuals is more homogeneous (in terms of obesity development) and we expected adipose tissue phenotypes to mostly reflect their metabolic health status, rather than their obesity stage. Overt diabetes, other severe diseases, and/or glucose-tolerance-affecting medication were exclusion criteria. From all 22 obese individuals, information about comorbidities is available (Table S3). To estimate whether these comorbidities could have affected our results, we tested whether they were unevenly distributed between the groups (between IS and IR as well as between NGH_IS, NGH_IR, IGH_IS, and IGH_IR). We only tested the most frequent comorbidities, i.e., musculoskeletal problems (N = 11) and arterial hypertension (N = 10), because tests with less frequent comorbidities, e. g., depression (N = 6) or sleep apnea (N = 5), would have been prone to false-positive results. Using χ2-tests, we could not detect uneven distribution of musculoskeletal problems or arterial hypertension between the groups (p ≥ 0.2, all). All participants underwent physical examination and routine laboratory tests. For determination of glucose tolerance and insulin sensitivity, they underwent a 5-point 75-g oral glucose tolerance test with glucose, insulin, and C-peptide measurements, as reported earlier [33]. Whole-body insulin sensitivity was calculated according to Matsuda and DeFronzo [34]: insulin sensitivity (in 1019 L2/mol2) = 10,000/(fasting glucose x fasting insulin x mean glucose x mean insulin)1/2. Body fat content (in %) was measured by bioelectrical impedance (BIA-101, RJL Systems, Detroit, MI, USA).
All participants gave written informed consent to the study. The study adhered to the ethical principles of the Declaration of Helsinki, and the ethics committee of the University of Tübingen approved the study protocol.
4.2. Preadipocyte isolation and differentiation
From all donors, paired samples of subcutaneous and visceral preadipocytes were prepared from adipose tissue biopsies [7] and properly frozen and biobanked. While frozen visceral preadipocytes are hard to differentiate ex-vivo, we could obtain good and homogeneous differentiation efficiency from subcutaneous preadipocytes.
Briefly, isolated cells were grown in α-MEM/Ham's nutrient mixture F12 (1:1), 20% fetal calf serum, 1% chicken embryo extract (Sera Laboratories, Haywards Heath, UK), 100 IU/mL penicillin, 0.1 mg/mL streptomycin, 0.5 mg/mL fungizone, 2 mmol/L glutamine (= growth medium). Second-pass cells were stored in liquid nitrogen (in 90% fetal calf serum, 10% DMSO). Stored cells were thawed and grown to confluence in growth medium. Thereafter, adipocyte differentiation was started by shifting the cultures to DMEM/Ham's nutrient mixture F12 (1:1) containing 5% fetal calf serum, 17 μmol/L pantothenate, 1 μmol/L biotin, 2 μg/mL apo-transferrin, 1 μmol/L human insulin, 1 μmol/L dexamethasone, 100 IU/mL penicillin, 0.1 mg/mL streptomycin, 0.5 mg/mL fungizone, 2 mmol/L glutamine (= differentiation medium) supplemented with 0.5 mmol/L 3-isobutyl-1-methyl-xanthine, 2 nmol/L triiodo-thyronine, 50 μmol/L indomethacin, 10 μmol/L Troglitazone and maintaining them in these conditions for seven days. Finally, the cells were allowed to terminally differentiate for another 11 days by culturing them in differentiation medium alone. Culture media and supplements were purchased from Lonza (Basel, Switzerland) and Biochrom (Berlin, Germany).
4.3. RNA-sequencing and data analysis
Total RNA was prepared from differentiated adipocytes using the RNeasy mini kit (QIAGEN) according to the manufacturer's instructions. RNA concentration and integrity was then controlled on a Bioanalyzer system (Agilent) and only RNA samples with RIN (RNA Integrity Number) values > 7 were used for downstream applications. Library construction and sequencing was outsourced to IGA Technology Services Srl. Libraries were constructed using the Nextera Library Prep Kit (Illumina) according to the manufacturer's instructions and sequenced on an Illumina HiSeq 2500 at 75bp single-ended, with a minimum output of 40 million reads per sample. Read mapping and differential expression analysis was performed using the A.I.R (Artificial Intelligence RNA-Seq) software from Sequentia Biotech with the following pipeline: BBDuk (reads trimming – http://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk-guide/), STAR (reads mapping to the human genome GRCh38 [ENSEMBL] – https://github.com/alexdobin/STAR), featureCounts (gene expression quantification – http://bioinf.wehi.edu.au/featureCounts/), and EdgeR (differential gene expression – https://www.bioconductor.org/packages/devel/bioc/html/edgeR.html). Heatmap and PCA analyses were performed with the web-application ClustVis using default parameters [35]. Knowledge-based analysis, including KEGG, ChEA, and ENCODE Histone PTMs was performed using the web-based application Enrichr (http://amp.pharm.mssm.edu/Enrichr/) [36].
4.4. IMPC data extrapolation and analysis
Mouse phenotypic information was extrapolated from the database of the International Mouse Phenotyping Consortium (IMPC – http://www.mousephenotype.org) using the batch query tool on differentially expressed genes in adipocytes from NGH and IGH donors being either insulin sensitive or resistant. Raw data from mouse lines with relevant MP term associations were downloaded from the database, re-analyzed and re-plotted using GraphPad Prism 6.
4.5. SNP analysis
SNP analysis was performed as previously described [19] with slight modifications. Differentially expressed genes associated to significant MP terms were used for analysis. We searched for SNPs in the Type 2 Diabetes Knowledge Portal (http://www.type2diabetesgenetics.org) and SNiPA (https://snipa.helmholtz-muenchen.de/snipa3/index.php) databases and used the cross-phenotype meta-analysis (CPMA) method to evaluate their significance across 16 metabolic phenotype GWAs from various consortia, as described [19].
Authors’ contributions
H. S. and R. T. conceived the study, analyzed the data and wrote the manuscript; R. G., L. B., J. D., M. L., F. S., A. K., A. B., and H. G. performed the experiments and analyzed the data; M. H. A., H. G., and H. H. read and commented on the manuscript.
Acknowledgements
This work has been supported by the grant of the German Diabetes Research Center (DZD - Germany): “Healthy and unhealthy obesity under the lens: dissecting epigenetic mechanisms of phenotypic variation” to RT and HS. The authors thank the Helmholtz Association and the German Diabetes Research Center for funding the positions of R. G., J. D., M. L., F. S., L. B., and R. T.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2018.09.004.
Contributor Information
H. Staiger, Email: Harald.Staiger@med.uni-tuebingen.de.
R. Teperino, Email: Raffaele.teperino@helmholtz-muenchen.de.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
figs1.

Ex-vivo differentiated preadipocytes maintain differentiation potential and their transcriptome is gender independent – relates toFigure 1. A-B. Unsupervised clustering by heatmap (A) and PCA (B) of human adipogenesis-relevant genes. C-E. Expression levels of PPARG (C), FABP4 (D) and PLIN2 (E) in mature adipocytes from insulin sensitive and resistant individuals. F. Volcano plot representation of RNA-Seq data from adipocytes of male vs female individual donors.
figs2.

Step-wise deterioration of glucose tolerance is reflected in adipocyte transcriptional alterations – relates toFigure 1. A-C. Volcano plot representation of RNA-Seq data from adipocytes of obese NGH (n = 8) vs IFG (n = 4) (A), IGT (n = 3) (B) or IFG + IGT (n = 6) (C) individuals. D-F. KEGG Pathway Analysis of DEGs in NGH vs IFG (D), IGT (E) or IFG + IGT (F) samples.
figs3.

Observed transcriptional differences in adipocytes from NGH and IGH individuals do not depend on donors' age – relates toFigure 1. A. Heatmap representation of 1449 DEGs from NGHvsIGH individuals stratified by age. Black lines are used to highlight the samples Ad20, Ad03 and Ad26.
figs4.

Insulin sensitivity is a modifier of adipocyte transcriptional programs imposed by glucose tolerance – relates toFigure 2. A-D. Volcano plot-based representation of ChEA (A-C) and ENCODE (B-D) dataset analysis in IS_IGH_NGH (A-B) and IR_IGH_NGH (C-D) sample sets.
References
- 1.Stefan N., Häring H.-U., Schulze M.B. Obesity 3 Metabolically healthy obesity: the low-hanging fruit in obesity treatment? The Lancet Diabetes & Endocrinology. 2017:1–10. doi: 10.1016/S2213-8587(17)30292-9. [DOI] [PubMed] [Google Scholar]
- 2.Bell J.A., Kivimaki M., Hamer M. Metabolically healthy obesity and risk of incident type 2 diabetes: a meta-analysis of prospective cohort studies. Obesity Reviews. 2014;15:504–515. doi: 10.1111/obr.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caleyachetty R., Thomas N.G., Toulis K.A., Nuredin M., Gokhale K.M., Balachandran K. Metabolically healthy obese and incident cardiovascular disease events among 3.5 million men and women. Journal of the American College of Cardiology. 2017;70:1429–1437. doi: 10.1016/j.jacc.2017.07.763. [DOI] [PubMed] [Google Scholar]
- 4.Böhm A., Halama A., Meile T., Zdichavsky M., Lehmann R., Weigert C. Metabolic signatures of cultured human adipocytes from metabolically healthy versus unhealthy obese individuals. PLoS One. 2014;9 doi: 10.1371/journal.pone.0093148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown S.D.M., Moore M.W. The International Mouse Phenotyping Consortium: past and future perspectives on mouse phenotyping. Mammalian Genome. 2012;23:632–640. doi: 10.1007/s00335-012-9427-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosen E.D., Spiegelman B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature Publishing Group. 2006;444:847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berti L., Irmler M., Zdichavsky M., Meile T., Böhm A., Stefan N. Fibroblast growth factor 21 is elevated in metabolically unhealthy obesity and affects lipid deposition, adipogenesis, and adipokine secretion of human abdominal subcutaneous adipocytes. Molecular Metabolism. 2015;4:519–527. doi: 10.1016/j.molmet.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rydén M., Hrydziuszko O., Mileti E., Raman A., Bornholdt J., Boyd M. The adipose transcriptional response to insulin is determined by obesity, not insulin sensitivity. Cell Reports. 2016;16:2317–2326. doi: 10.1016/j.celrep.2016.07.070. [DOI] [PubMed] [Google Scholar]
- 9.Sorisky A. Effect of high glucose levels on white adipose cells and Adipokines—fuel for the fire. International Journal of Molecular Sciences. 2017;18:944–946. doi: 10.3390/ijms18050944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meugnier E., Faraj M., Rome S., Beauregard G., Michaut A., Pelloux V. Acute hyperglycemia induces a global downregulation of gene expression in adipose tissue and skeletal muscle of healthy subjects. Diabetes. 2007;56:992–999. doi: 10.2337/db06-1242. [DOI] [PubMed] [Google Scholar]
- 11.Meugnier E., Rome S., Vidal H. Regulation of gene expression by glucose. Current Opinion in Clinical Nutrition and Metabolic Care. 2007;10:518–522. doi: 10.1097/MCO.0b013e3281298fef. [DOI] [PubMed] [Google Scholar]
- 12.Chen H., Gu X., Su I.-H., Bottino R., Contreras J.L., Tarakhovsky A. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes & Development. 2009;23:975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu T.T.-H., Heyne S., Dror E., Casas E., Leonhardt L., Boenke T. The polycomb-dependent epigenome controls β cell dysfunction, dedifferentiation, and diabetes. Cell Metabolism. 2018;27:1294–1308. doi: 10.1016/j.cmet.2018.04.013. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cannon C.E., Titchenell P.M., Groff D.N., El Ouaamari A., Kulkarni R.N., Birnbaum M.J. The Polycomb protein, Bmi1, regulates insulin sensitivity. Molecular Metabolism. 2014;3:794–802. doi: 10.1016/j.molmet.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Angelis M.H., Nicholson G., Selloum M., White J.K., Morgan H., Ramirez-Solis R. Analysis of mammalian gene function through broad-based phenotypic screens across a consortium of mouse clinics. Nature Genetics. 2015;47:969–978. doi: 10.1038/ng.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meehan T.F., Conte N., West D.B., Jacobsen J.O., Mason J., Warren J. Disease model discovery from 3,328 gene knockouts by the international mouse phenotyping consortium. Nature Publishing Group. 2017;49:1231–1238. doi: 10.1038/ng.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bowl M.R., Simon M.M., Ingham N.J., Greenaway S., Santos L., Cater H. A large scale hearing loss screen reveals an extensive unexplored genetic landscape for auditory dysfunction. Nature Communications. 2017;2017:1–10. doi: 10.1038/s41467-017-00595-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mason J., Beaudet A.L., Benjamini Y., Bower L., Braun R.E., Brown S.D.M. Prevalence of sexual dimorphism in mammalian phenotypic traits. Nature Communications. 2017;8:1–12. doi: 10.1038/ncomms15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rozman J., Rathkolb B., Oestereicher M.A., Schütt C., Ravindranath A.C., Leuchtenberger S. Identification of genetic elements in metabolism by high-throughput mouse phenotyping. Nature Communications. 2018:1–16. doi: 10.1038/s41467-017-01995-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman J.M., Halaas J.L. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 21.Kamohara S., Burcelin R., Halaas J.L., Friedman J.M., Charron M.J. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–377. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 22.Shao S., Brown A., Santhanam B., Hegde R.S. Structure and assembly pathway of the ribosome quality control complex. Molecular Cell. 2015;57:433–444. doi: 10.1016/j.molcel.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez M.C., Andres-Barquin P.J., Martinez S., Bulfone A., Rubenstein J.L., Israel M.A. ENC-1: a novel mammalian kelch-related gene specifically expressed in the nervous system encodes an actin-binding protein. Journal of Neuroscience. 1997;17:3038–3051. doi: 10.1523/JNEUROSCI.17-09-03038.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevenson C., de la Rosa G., Anderson C.S., Murphy P.S., Capece T., Kim M. Essential role of Elmo1 in Dock2-dependent lymphocyte migration. The Journal of Immunology. 2014;192:6062–6070. doi: 10.4049/jimmunol.1303348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun K., Kusminski C.M., Scherer P.E. Adipose tissue remodeling and obesity. Journal of Clinical Investigation. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiological Reviews. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 27.Bogner-Strauss J.G. N-acetylaspartate metabolism outside the brain: Lipogenesis, histone acetylation, and cancer. Frontiers in Endocrinology. 2017;8:804–805. doi: 10.3389/fendo.2017.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Namboodiri A.M.A., Peethambaran A., Mathew R., Sambhu P.A., Hershfield J., Moffett J.R. Canavan disease and the role of N-acetylaspartate in myelin synthesis. Molecular and Cellular Endocrinology. 2006;252:216–223. doi: 10.1016/j.mce.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 29.Pessentheiner A.R., Pelzmann H.J., Walenta E., Schweiger M., Groschner L.N., Graier W.F. NAT8L ( N-Acetyltransferase 8-Like) accelerates lipid turnover and increases energy expenditure in Brown adipocytes. Journal of Biological Chemistry. 2013;288:36040–36051. doi: 10.1074/jbc.M113.491324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prokesch A., Pelzmann H.J., Pessentheiner A.R., Huber K., Madreiter-Sokolowski C.T., Drougard A. N-acetylaspartate catabolism determines cytosolic acetyl-CoA levels and histone acetylation in brown adipocytes. Scientific Reports. 2016;6:1–12. doi: 10.1038/srep23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuriaga M.A., Fuster J.J., Farb M.G., MacLauchlan S., Breton-Romero R., Karki S. Activation of non-canonical WNT signaling in human visceral adipose tissue contributes to local and systemic inflammation. Scientific Reports. 2017:1–10. doi: 10.1038/s41598-017-17509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morello F., Prasad A.A., Rehberg K., Vieira de Sá R., Antón-Bolaños N., Leyva-Diaz E. Frizzled3 controls axonal polarity and intermediate target entry during striatal pathway development. Journal of Neuroscience. 2015;35:14205–14219. doi: 10.1523/JNEUROSCI.1840-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Böhm A., Wagner R., Machicao F., Holst J.J., Gallwitz B., Stefan N. DPP4 gene variation affects GLP-1 secretion, insulin secretion, and glucose tolerance in humans with high body adiposity. PLoS One. 2017;12 doi: 10.1371/journal.pone.0181880. e0181880–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuda M., DeFronzo R.A. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- 35.Metsalu T., Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Research. 2015;43:W566–W570. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.