

Abstract
Heparan sulfate (HS) proteoglycans on immune cells have the ability to bind to and regulate the bioactivity more than 400 bioactive protein ligands, including many chemokines, cytokines, and growth factors. This makes them important regulators of the phenotype and behavior of immune cells. Here we review how HS biosynthesis in macrophages is regulated during polarization and in chronic inflammatory diseases such as rheumatoid arthritis, atherosclerosis, asthma, chronic obstructive pulmonary disease and obesity, by analyzing published micro-array data and mechanistic studies in this area. We describe that macrophage expression of many HS biosynthesis and core proteins is strongly regulated by macrophage polarization, and that these expression patterns are recapitulated in chronic inflammation. Such changes in HS biosynthetic enzyme expression are likely to have a significant impact on the phenotype of macrophages in chronic inflammatory diseases by altering their interactions with chemokines, cytokines, and growth factors.
Keywords: heparan sulfate, inflammation, macrophages, proteoglycans, rheumatoid arthritis
Macrophages Perpetuate Chronic Inflammation
Inflammatory responses are essential for combating infection and promoting tissue repair and are normally quickly resolved. During chronic inflammation, unchecked inflammation persists for months to years and can contribute to the development of various diseases, such as rheumatoid arthritis (RA), cardiovascular disease (CVD) and diabetes. Incidence of many chronic inflammatory diseases is on the rise, increasing the need to understand the molecular events that initiate and drive disease pathogenesis, to identify novel targets for intervention. For example, RA is a chronic inflammatory disease in which joint inflammation causes pain and loss of joint function. RA affects approximately 1% of the population and while its treatment was revolutionized in the 1990s by development of anti-tumor necrosis factor (TNF)-targeted biologic therapies,1 30% to 40% of patients exhibit only partial or temporary responses to treatment.2 Novel approaches are thus required to improve treatment of RA and other chronic inflammatory conditions.
Chronic inflammatory conditions such as RA are characterized by elevated levels of pro-inflammatory cytokines (e.g., TNF, interleukin-1 [IL-1]) and influx of many immune cell types into affected tissues.3 Macrophages are a critical component of this elevated inflammatory response, and in RA, macrophage abundance in tissue correlates with joint erosion.4 Macrophages respond to stimuli (such as danger signals) in their environment by adopting a range of activation signatures that define and regulate their biological responses. In vitro, two extreme activation signatures (also known as polarization states) are defined: namely, classically activated (pro-inflammatory; previously known as “M1”) and alternatively activated (reparative; previously known as “M2”) macrophages (Fig. 1A). Interferon-γ (IFNγ) and lipopolysaccharide (LPS) promote M1 polarization and the expression of pro-inflammatory cytokines (e.g., IL-1, IL-6, and TNF), whereas IL-4 and IL-13 result in M2 polarization and the secretion of anti-inflammatory cytokines like IL-10. In vivo, macrophages are thought to adopt a range of activation states between these two extremes, fine-tuned by their local environment.5,6
Figure 1.

Several macrophage-modulating bioactive proteins bind to HS. (A). In vitro, two extreme activation signatures are defined for macrophages. Interferon-γ (IFNγ) and lipopolysaccharide (LPS) or tumor necrosis factor (TNF) are responsible for classical activation (previously termed “M1”), which is characterized by nitric oxide synthase (iNOS), cluster of differentiation 80 (CD80) and major histocompatibility complex-II (MHC-II) expression and secretion of pro-inflammatory cytokines. IL-4 and IL-13 result in alternative macrophage activation (previously termed “M2”), which is associated with arginase (Arg-1) and mannose receptor (MR or CD206) expression and secretion of IL-10 and TGFβ. In vivo, heterogeneous activation states are present, depending on stimuli in their local environment.5,6 Granulocyte-macrophage colony-stimulating factor (GM-CSF), various chemokines7 and the complement factors C3a, C5a, and C5b-98 also promote M1-like differentiation, while immune complexes (IC), IL-10, TGFβ, glucocorticoids,6 the complement factors C1q and C3b,8 tenascin C,9 Indian hedgehog (IHH) signaling,10 and apolipoprotein E (ApoE)11 contribute to M2-like activation. Many additional factors are also likely to affect macrophage polarization. (B). Many HS-binding proteins (as reviewed by Ori et al.12) are known to modulate macrophage function. In addition to the well-known cytokines and chemokines,7 proteins such as complement factors can modulate macrophage activation.8 MMPs are involved in macrophage infiltration13–15 and MMP-7 suppress M1 polarization, while SHH has been shown to act as a macrophage chemoattractant.16 Abbreviations: HS, heparan sulfate; TGFβ, transforming growth factor beta; IL, interleukin; MMPs, matrix metalloproteinases.
In active RA, as in many other chronic inflammatory diseases, macrophages are thought to adopt a persistently activated state, resembling an M1 signature, associated with secretion of pro-inflammatory cytokines (e.g., TNF, IL-6, IL-12, and IL-23).17 In several other chronic inflammatory conditions, a similar M1 signature has been demonstrated in macrophages isolated from affected tissues.18 For example, macrophages from patients with atherosclerosis18 and obesity18 also exhibit an M1-like activation signature. Certain other chronic inflammatory conditions, such as asthma19 and chronic obstructive pulmonary disease (COPD),20 are associated with a more M2-like profile.
Macrophage phenotype is determined in response to molecules that the cells interact with in their environment. These include, for example, bacterial components (that serve as pathogen-associated molecular patterns or PAMPs), tissue components (such as extracellular matrix breakdown products that act as damage-associated molecular patterns or DAMPs), as well as cytokines, chemokines, and growth factors (e.g., granulocyte-macrophage colony-stimulating factor [GM-CSF], IFNβ, IFNγ, IL-4, IL-10, IL-13) released by other cells.9,21–23 The biological activity of cytokines, chemokines, and growth factors in particular is highly regulated by their interaction with heparan sulfate (HS) proteoglycans, which are structurally diverse molecules ubiquitously expressed on cell surfaces and in the extracellular matrix. HS proteoglycans consist of a core protein that carries one or more linear HS polysaccharide chains consisting of alternating N-acetylglucosamine (GlcNAc) and hexuronic acid monomers, decorated with variable patterns and levels of sulfation.24
Heparan Sulfate Proteoglycans Modulate the Activity of Inflammatory Mediators
As comprehensively reviewed by Ori et al.,12 HS has been shown to bind to more than 400 proteins, including a multitude of chemokines, cytokines, and growth factors. Several of these are known to affect macrophage activation and behavior (Fig. 1B), and in turn, their activity is modulated by binding to HS. Binding of proteins to HS can establish gradients (e.g., concentrate them on the cell surface), protect them from proteolysis, modulate their bioactivity (e.g., oligomerize chemokines), and affect receptor binding.25,26 For example, IFNγ, which is critical for polarization of macrophages toward a pro-inflammatory phenotype, binds to HS and this interaction inhibits IFNγ binding to its receptor.27 Paradoxically, HS binding has been shown to increase the activity of IFNγ by promoting its local accumulation and inhibiting its rapid proteolytic degradation.28 Many other HS-binding proteins are also likely to or may affect macrophage function, for example, Indian hedgehog (IHH) promotes alternative activation of macrophages.10
Proteins bind to HS via positively charged amino acid residues (Arg, Lys) that interact with the negatively charged sulfate groups on HS.29 Changes in HS structure change its binding to protein ligands, and consequently affect tissue homeostasis.30 HS structure is dynamically regulated and has been shown to change with age,31–34 during development,35 and in many pathological contexts.36 The structure of HS is thought to be determined by the relative abundance of the 25 different HS biosynthetic enzymes (Fig. 2),37,38 which collectively create a complex sulfation pattern that determines affinity for protein ligands.39 However, little is known about the transcriptional and translational regulation of these HS biosynthetic enzymes. To date a systematic examination of how HS biosynthesis is regulated in chronic inflammation is lacking, and it is not known whether HS biosynthesis has an impact on the development of diseases such as RA.
Figure 2.

Enzymes responsible for HS biosynthesis. HS biosynthesis is initiated in the Golgi by the formation of a tetrasaccharide linker region (on serine residues of the core protein) consisting of a xylose, 2 galactose, and a glucuronic acid (GlcA) residue. Subsequently, HS chains are synthesized and modified in a template-independent manner by up to 25 different enzymes. First, the exostosis (EXT) and exostosin-like (EXTL) glycosyltransferase enzyme family elongate the HS chain by alternating the addition of N-acetyl glucosamine (GlcNAc) and GlcA. Next, the N-deacetylase/N-sulfotransferase (NDST) family starts the modification of the HS backbone by deacetylating GlcNAc, followed by N-sulfation. Afterwards, glucuronic acid epimerase (GLCE) converts some GlcA residues into iduronic acid (IdoA) and a 2-O-sulfotransferase (HS2ST1) can then sulfate IdoA. Alternatively, glucosamine can be sulfated by 6-O-sulfotransferases (HS6ST1-3) and 3-O-sulfotransferases (HS3ST1-6). As these enzymes do not completely sulfate the HS chain, they generate complex sulfation patterns that form structurally diverse protein-binding sites. 6- and 3-O-sulfotransferases are thought to be particularly important, since these 10 enzymes have slightly different substrate specificities and are highly evolutionarily conserved. Even after HS biosynthesis in the Golgi system, HS can be extracellularly modified by the endosulfatases sulfatase (SULF) 1 and 2, which can specifically remove sulfate groups from the 6-O position. Moreover, heparanase can degrade the HS chain into shorter oligosaccharides. Together, these biosynthetic enzymes account for the structural diversity of HS.37,38 Abbreviation: HS, heparan sulfate.
Macrophage HS Is Regulated Upon Polarization
Using published microarray data, we analyzed expression of HS biosynthetic enzymes and core proteins in macrophages upon in vitro polarization toward an M1 or an M2 phenotype.40–45 This meta-analysis showed that HS sulfotransferase expression was largely downregulated in M1 compared with M2 macrophages, for both human and murine macrophages (Fig. 3). In line with this change in expression of HS biosynthetic enzymes, human M1 macrophages have been shown to contain half the amount of HS and have less 2-O and potentially less 3-O sulfation than M2 macrophages.46 However, 3-O sulfation cannot be directly quantified due to the lack of commercial standards.
Figure 3.
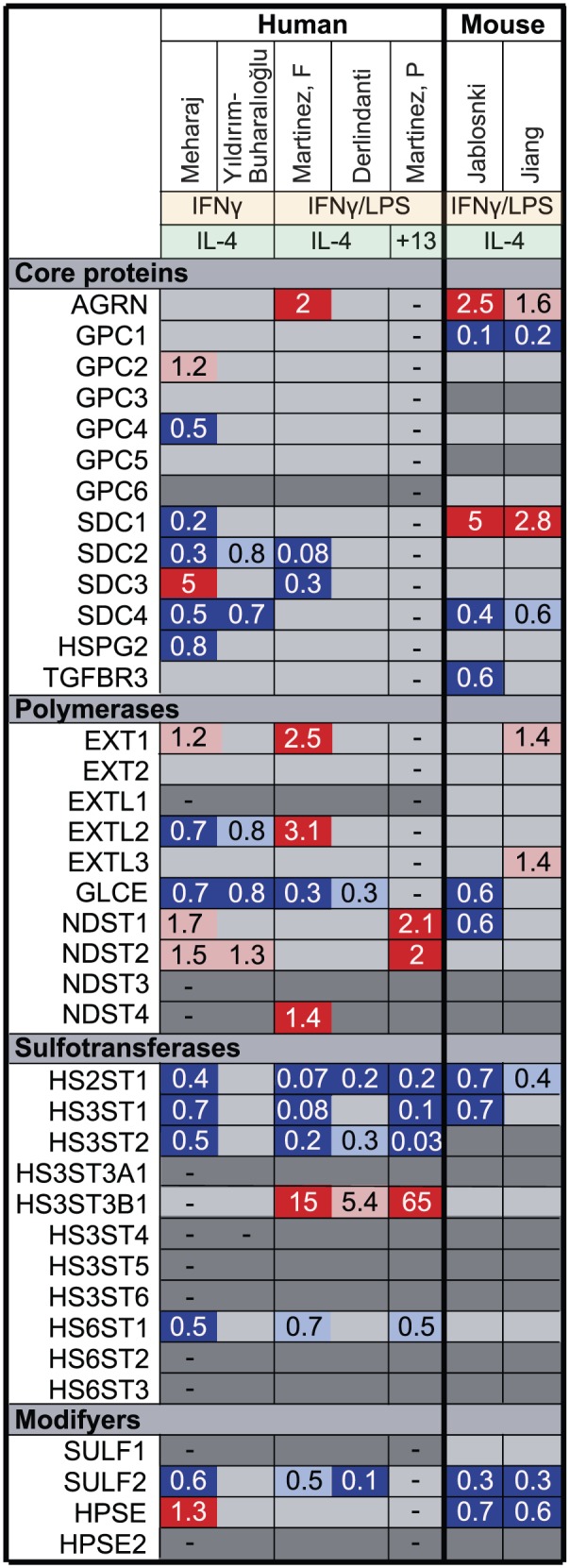
Fold change in the expression of HS biosynthetic enzymes and core proteins in in vitro–polarized M1 compared with M2 macrophages. Using microarray data, we analyzed the expression of HS core protein and biosynthetic enzymes upon macrophage polarization in vitro with either IFNγ/LPS (M1) or IL-4/IL-13 (M2). Genes marked in dark gray show minimal expression in macrophages in our experience. Genes upregulated in M1 compared with M2 macrophages are marked in red (p<0.05) or pink (p>0.05), genes downregulated in M1 macrophages are marked in dark blue (p<0.05) or light blue (p>0.05) and genes that were not measured are marked with a dash (–). Mehraj,40 Martinez F,41 Derlindanti,42 Yıldırım-Buharalıoğlu43 compared human M1 and M2 macrophages by microarray. Jablonski44 and Jiang45 compared M1 and M2 macrophages by microarray from C57BL/6 or BALB/c mice, respectively. Martinez P46 performed individual quantitative polymerase chain reactions (qPCRs) for NDSTs and sulfotransferases in human M1 and M2 macrophages. Abbreviations: HS, heparan sulfate; IFNγ, interferon-γ; LPS, lipopolysaccharide; IL, interleukin; NDST, N-deacetylase/N-sulfotransferase.
Macrophage HS Is Regulated in Chronic Inflammation
Similarly, we analyzed published microarrays examining expression of HS-associated genes in macrophages isolated from patients with RA47–49 (Fig. 4). This revealed that RA macrophages broadly resemble M1 macrophages in terms of HS biosynthesis gene expression, in line with studies indicating that RA macrophages exhibit an elevated chronic inflammatory state, with increased release of TNF.17 In contrast, macrophages from human chronic lung diseases (COPD and asthma) exhibited a more M2-like phenotype in terms of the HS biosynthesis gene expression profile, in line with reports that M2 macrophages predominate in these diseases.50–52
Figure 4.

Fold change in the expression of HS biosynthetic enzymes and core proteins in chronic inflammatory diseases. We analyzed microarray data on expression of HS core protein and biosynthetic enzymes in macrophages isolated from chronically inflamed tissues from more M1- (yellow) or M2-associated (green) diseases by FACS or laser-capture microdissection. Genes which were increased in disease compared with control are marked in red (p<0.05) or pink (p>0.05) and downregulated genes are marked in dark blue (p<0.05) or light blue (p>0.05). Kang,47 Yarilina,48 and You49 compared freshly isolated CD14+ cells from human RA synovial fluid to CD14+ monocyte-derived macrophages (MDM) from healthy donors. Hägg53 compared MDMs isolated from blood of patients with subclinical atherosclerosis and a family history of CVD after exposure to oxidized LDL (oxLDL) to mimic the atherosclerotic environment with those from healthy donors. Woodruff,52 Shaykhiev,50 and Kazeros51 compared macrophages isolated from broncheoalveolar lavage from smokers, patients with asthma or COPD to those of healthy donors. Goo54 compared microdissected macrophages from the aorta of ApoE–/– mice on a high fat western-type diet (WTD) for 14 weeks to a normal diet. Feig55 compared microdissected macrophages in a progression (Pr) or regression (Re) model of atherosclerosis, which promoted M1 or M2 polarization, respectively. Berisha56 compared bone marrow-derived macrophages (BMDM) from male (M) or female (F) atherosclerosis-resistant ApoE–/– AKR mice to BMDM from atherosclerosis-prone ApoE–/– DBA/2 mice after loading with acetylated LDL (acLDL). Aurora57 compared CD11b+Ly6G– macrophages from the heart of mice 3 days after experimental myocardial infarction at postnatal day 14 (P14; nonregenerating) to P1 (regenerating). Xu58 compared CD11c+ and CD11c– (generally considered to represent M1 and M2 macrophages) CD11b+F4/80+ macrophages from the perigonodal cells of leptin-deficient obese mice to wild-type mice. Although HS-associated genes were differentially expressed upon obesity in CD11c+ macrophages, no differences were observed in CD11c– macrophages. Prieur59 compared CD11b+ macrophages from gonadal white adipose tissue of leptin-deficient obese mice after 5 (5W) or 16 weeks (16W) of WTD to wild-type mice. They demonstrated that macrophages have a more M2-like phenotype after 5 weeks and more M1-like after 16 weeks of diet. Abbreviations: HS, heparan sulfate; RA, rheumatoid arthritis; CVD, cardiovascular disease; COPD, chronic obstructive pulmonary disease; ApoE, apolipoprotein E.
Here, we examine the expression of individual HS biosynthesis enzymes and core proteins in macrophages upon polarization, in RA, as well as in other chronic inflammatory conditions. This gives insight as to the contribution of these genes to chronic inflammation.
Polymerases
EXT1 and EXT2
Exostosin glycosyltransferase 1 (EXT1) and EXT2 act as a heterodimer to synthesize the HS backbone by alternating addition of glucuronic acid (GlcA) and GlcNAc to the growing HS chain.60 They are thus critical for HS biosynthesis, and mutation of either enzyme causes hereditary multiple exostoses (HME), characterized by the formation of multiple tumors on the long bones due to abnormal HS-dependent growth factor signaling.61 EXT1 has a strong polymerizing activity, whereas EXT2 is thought to act as a chaperone bringing biosynthetic enzymes to the correct position in the Golgi.60 It has been hypothesized that EXT1 is the rate-limiting enzyme and that EXT2 is normally synthesized in excess.62 This is supported by the predominance of EXT1 mutations in HME and the observation that EXT1 mutations correlate with more severe disease than EXT2.63 A study by Bao et al.64 indicates that EXT expression is important in immune responses, as Ext1 deficiency in endothelial cells in mice has been shown to impair lymphocyte recruitment in a contact hypersensitivity model due to impaired chemokine presentation, but little is known about its role in macrophages.
Expression of EXT1 was increased by treatment of human macrophages with IFNγ and LPS,41 and expression of the enzyme increased 2-fold in in vitro–polarized M1 macrophages (Fig. 3).40,41 Increased expression of EXT1 in pro-inflammatory environments may imply that HS chain elongation is stimulated by pro-inflammatory signals. In support of this, HS chain length is also increased by pro-inflammatory cytokines such TNF and decreased by M2-like stimuli such as TGFβ65 in other cell types, such as human umbilical endothelial cells (HUVEC). EXT1 expression was increased 3-fold in RA macrophages47,48 and also in a murine atherosclerosis model,56 in line with the more M1-like phenotype of macrophages in these diseases. In contrast, EXT1 was largely not regulated in M2-like macrophages from patients with chronic lung diseases such as asthma and COPD,50,52 and was downregulated in macrophages in murine obesity models.58,59
However, EXT2 appeared to be unregulated or decreased by M1 polarization40–43 and in RA.47 Changes in EXT2 expression may have more limited effects on HS structure as EXT2 is normally produced in excess.62
EXTL1-3
The EXT like (EXTL) family of genes share sequence homology with the EXTs, but their role in HS biosynthesis is poorly understood.66 EXTL1 is not expressed by human macrophages in vitro, while EXTL3 is expressed but not regulated by polarization.
Among the family, EXTL2 appears to be most regulated in macrophages and its expression is dependent on the polarization protocol. A study in which macrophages were polarized in vitro with IFNγ and LPS41 found an increase in EXTL2 expression, while studies that used only IFNγ40,43 found downregulation of the gene. This suggests that EXTL2 expression is increased by LPS and decreased by IFNγ. This IFNγ polarization profile mirrors the decrease of EXTL2 in RA.47 However, Extl2 expression was increased in murine models of other M1 chronic inflammatory diseases,52,55–58 implying that the regulation of EXTL2 expression is complex.
Busse et al. hypothesized that EXTL2 initiates HS side chain formation by catalyzing the addition of the first GlcNAc in the Golgi,62 but subsequent studies indicated that decreased expression of EXTL2 resulted in longer HS side chains.67 Nadanaka et al. proposed that the addition of the first GlcNAc to a phosphorylated tetrasaccharide linker (see “Tetrasaccharide linker region” below for more detail) by EXTL2 is involved in chain termination and quality control of HS production by avoiding over- and abnormal HS production.68 It has been demonstrated that EXTL2 deficiency results in HS overproduction and structural changes, which affected cell signaling and liver and blood vessel remodeling upon injury.69,70 Further studies are required to understand how inflammation changes expression of the EXTL family and how this affects HS chain length, amount, quality, and ultimately macrophage signaling.
N-Deacetylase/N-Sulfotransferase
The N-deacetylase/N-sulfotransferase (NDST) family consists of four isoforms that perform the first HS modification step.39 They deacetylate GlcNAc and subsequently N-sulfate glucosamine residues. NDST1 and two are ubiquitously expressed, while NDST3 and four are mainly expressed in the brain and fetal tissues,71,72 but not in macrophages.
NDST1 and NDST2 are upregulated in human IFNγ-polarized M1 macrophages in vitro,40,43 indicating a pro-inflammatory role. Other studies reported that inflammatory mediators such as LPS or IFNγ+TNF also increase NDST1 and/or NDST2 expression in microvascular endothelial cells.73,74 NDST1 and NDST2 levels were also increased 1.6-fold in RA macrophages.47 In contrast, NDST expression is overall not regulated in macrophages from patients with more M2-driven diseases, such as asthma.50,52
NDST expression seems to be differentially regulated in human and murine macrophages. For example, Ndst1 levels decreased in mouse M1 macrophages in vitro44 and Ndst1 and Ndst2 were also decreased in murine M1-driven diseases, such as CVD.56,57 In line, macrophage-specific inactivation of Ndst1 (LysMCre+Ndst1f/f) in mice diminished sulfation and accelerated atherosclerosis development and diet-induced obesity. The authors proposed that HS proteoglycan sulfation is required for the suppression of basal macrophage activation through interferon-β (IFNβ) sequestration,75,76 but many other factors that bind HS might be involved in the observed phenotype of these mice. In contrast to the detrimental effect of Ndst1 deficiency in M1-diseases, deficiency of Ndst1 in the endothelium and leukocytes (TekCre+Ndst1f/f) was found to be protective against allergen-induced airway remodeling (an M2-driven disease), by reducing the recruitment of inflammatory cells and the expression of IL-13, vascular endothelial growth factor (VEGF), TGFβ, and fibroblast growth factor-2 (FGF-2).77 However, from this study it cannot be concluded whether the protective effect depends on Ndst1 deficiency in the endothelium or leukocytes. Overall, this indicates that the role of NDST enzymes is highly complex and that differences in regulation of their expression between mouse and human should be taken into account when performing murine studies.
D-Glucuronyl C5-Epimerase
D-Glucuronyl C5-epimerase (GLCE) converts GlcA to iduronic acid (IdoA) in an irreversible reaction that increases the flexibility of the HS chain and so modulates ligand binding.78,79 GLCE expression was found to be downregulated in human40–43 and mouse44 M1 macrophages, indicating an anti-inflammatory role for this enzyme. In support, GLCE expression was also decreased in RA macrophages.47 Currently, the functional effects of GLCE in macrophages are unknown. Glce–/– mice displayed neonatal lethality with defects in kidney, lung, and skeletal development80; lymphoid organ development81; and B-cell survival and maturation.82 It is highly probable that GLCE also affects binding and signaling in macrophages, as all the characterized protein binding HS-sites contain at least one IdoA.39
Heparan Sulfate Sulfotransferases
Following epimerization of GlcA into IdoA, one HS 2-O sulfotransferase (HS2ST1) enzyme can O-sulfate IdoA at the carbon position 2 (Fig. 2). Finally, 3-O sulfotransferases (seven isoforms) and 6-O sulfotransferases (three isoforms) can O-sulfate at carbons 3 and 6 of glucosamine.37,38 Martinez et al. reported that M1 macrophages have decreased 2-O- and 3-O-, but not 6-O-sulfation, as determined by reverse phase high-performance liquid chromatography (RP-HPLC) and glycosaminoglycan (GAG) specific antibodies.46 The expression of HS2ST1, HS3ST1, HS3ST2, and HS6ST1 is dramatically downregulated up to 12.5-fold in human and murine M1 macrophages in vitro,40–42,44,45 strongly suggesting an anti-inflammatory role of O-sulfation. In line with these studies, HS2ST1, HS3ST1, HS3ST2, and HS6ST1 are also decreased up to 100-fold in macrophages from M1-driven diseases such as RA,47,48 while their expression is increased in macrophages from more M2-associated diseases, such as asthma and COPD.50,52
In contrast to the other sulfotransferases, HS3ST3B1 is increased in M1 polarized macrophages up to 15-fold in vitro,41,42 implying an inflammatory role of this enzyme. In agreement, HS3ST3B1 is also upregulated 5-fold in RA macrophages,47 while it is decreased up to 3-fold in macrophages in more M2-driven diseases such as COPD.50
Sulfotransferases are also downregulated in murine M1 macrophages in vitro,44 but the effects in murine disease models are more heterogeneous. Hs3st1 and Hs3st2 are indeed downregulated in CVD and obesity,54,59 while Hs2st1 is slightly increased in obesity59 and Hs6st1 is increased in CVD and obesity.56,58,59 This illustrates that sulfotransferase expression is also affected by factors other than those (IFNγ, LPS, IL-4, and IL-13) which were used for the in vitro studies. This is supported by studies showing that TNF (but not IL-4, IL-6, and IFNγ) as well as TLR2 (peptidoglycan) and TLR4 (LPS) ligands increase HS3ST3B1 expression in human monocytes.83 HS3ST1 is also upregulated by IL-4+IL-13 in the intestinal epithelium,84 whereas HS3ST2 is regulated by the circadium rhythm and increased by light85,86 and HS3ST3A1 is increased by IL-6 in the canine urothelium.87 HS6T1 expression is decreased by TGFβ lung fibroblasts,88 but not affected by TNF, IFNγ, or TGFβ in renal epithelial cells.89 Conversely, 6-O sulfation in HUVECs is reported to be decreased by stimulation with TNF and TGFβ and slightly increased upon IL-1α stimulation.65 These studies indicate that sulfotransferases are highly regulated by inflammatory signals, but a systematic study of their regulation in macrophages and other immune cells is lacking.
Modifyers
HS can be postsynthetically modified by the extracellular 6-O endosulfatases (SULF1 and SULF2) and the endoglycosidase heparanase (HPSE).90
Sulfatases
Sulfatases act extracellularly to remove 6-O sulfate groups and so modulate ligand binding to HS. Moreover, it has been suggested that SULFs can also translocate to the nucleus where they might be involved in cell-cycle regulation.91 SULF1 is not expressed by macrophages in vitro, while SULF2 is expressed and downregulated upon M1 polarization in vitro of human and murine macrophages.40,41,44,45 It is overall not regulated in M2-driven diseases such as asthma.52 SULF2 appears to be variably regulated in M1-associated diseases, as its expression is decreased in obesity59 and in one CVD56 study, but increased in RA47 and another CVD57 study. This suggests that factors other than IFNγ, LPS, IL-4, and IL-13 affect SULF2 expression in vivo. This is supported by studies in other cell types. For example, SULF2 expression is increased by TGFβ in renal epithelial cells,89 by TNF in nucleus pulposis cells92 and by IL-1α in HeLa cells.93 Dhoot et al. demonstrated that SULF2 can be expressed as an alternatively spliced catalytically inactive variant and found that various tumors express this at high levels.94–96 Mechanistically, the inactive variant titrates out the active variant and results in an increase in 6-O sulfation and receptor tyrosine kinase activation. It would thus also be important to check which splice variants of SULF2 are expressed in macrophages and other immune cell types during chronic inflammation.
Heparanase
The HPSE family consists of two isoforms: HPSE1 and HPSE2. HPSE1 degrades HS chains by specifically cleaving the bond between GlcA and GlcNAc in HS.97 Although HPSE2 shares 40% similarity with HPSE1, it is unable to cleave HS chains.97 HPSE1 expression is increased in various inflammatory conditions, such as inflammatory bowel disease (IBD), Crohn’s disease, ulcerative colitis, and also in the synovial fluid of patients with RA.98,99 Inflammatory cytokines such as TNF, IFNγ, and IL-1β increase HPSE secretion from U937 macrophages and endothelial cells.100–102 HPSE1 is thought to increase inflammation through three different mechanisms. First, inflammation is increased in a positive feedback loop since HPSE stimulates pro-inflammatory cytokine secretion from macrophages by generating HS oligomers that signal via TLR4.103 Second, increased HPSE1 expression might also indirectly enhance inflammation by accelerating HS turnover and promoting reshaping of the HS structure. In a tumor model, HPSE1 has been shown to increase levels of sulfation and consequently augment growth factor signaling.104 Finally, HPSE1 might change the free concentration of inflammatory mediators in the microenvironment by promoting their release from HS.97
Despite the evidence for an important inflammatory role of HPSE1 in chronic diseases, one microarray study showed a decrease in HPSE1 expression in RA macrophages.47 It is unclear whether macrophage polarization in vitro affects HPSE1 expression, as one study on human macrophages reported increased expression40 upon polarization with IFNγ, while murine studies reported decreased expression.44,45 We cannot rule out differences in HPSE1 regulation in mouse and human. In addition, HPSE1 expression in other diseases does not correlate with the macrophage polarization state. For example, its expression was decreased in smoking and COPD50,52 and upregulated by oxLDL53 in human studies. In murine studies, it was decreased in CVD56 but increased in obesity.59 It is tempting to speculate that feedback mechanisms might be in place to limit the inflammatory effects of HSPE1.
Expression of HPSE2 was also increased in RA,47 although its expression is undetectable in in vitro–polarized macrophages. Specific environmental cues may upregulate HPSE2 in RA. Nevertheless, the functional significance of HPSE2 is unknown.
Core Proteins
HS proteoglycan core proteins carry the HS chains, and are also known to have biological functions themselves in some cases. They can be grouped into families based on their structure and location. The transmembrane syndecans (SDC; four members), the glycosyl phosphoinositol (GPI)-anchored glypicans (GPC; six members), and TGFβ receptor 3 (TGFβR3) are expressed on the cell surface, while agrin (AGRN), and perlecan (HSPG2) are present in the extracellular matrix.
Agrin and Perlecan
Perlecan does not appear to be regulated by macrophage polarization in vitro or by chronic inflammation, while AGRN expression is increased 2- to 2.5-fold in murine44,45 and human41 macrophages upon M1 polarization in vitro. Inflammatory stimuli, such as IL-1β, have also been shown to increase agrin secretion from other cell types.65
In contrast, AGRN is significantly downregulated in RA (by to 2-fold48) and also in other chronic inflammatory conditions (i.e., asthma,52 COPD,50 and obesity59), but increased in CVD.57 Other factors are likely to regulate AGRN expression in these contexts, or a negative feedback loop may be responsible for AGRN downregulation. Transgenic mice studies indicate that AGRN is vital for macrophage function. Agrn–/– mice demonstrate neonatal lethality due to neuromuscular synapse dysfunction.105 This phenotype can be partially rescued by selective expression of Musk in skeletal muscle (Musk-L; Agrn–/–).106 Such mice displayed reduced differentiation and viability of monocytes, as well as a reduction in subsequent differentiation into macrophages and impaired biological function, with reduced phagocytosis and cytoskeletal remodeling.106
Glypicans
Among the glypicans, expression of GPC3 and GPC4 is regulated in macrophages upon chronic inflammation, whereas the other family members are overall not affected. For example, GPC3 is downregulated in RA and other chronic inflammatory M1- and M2-driven conditions,47,52,57,59 but is not differentially regulated by macrophage polarization.40–45 Therefore, mediators other than IFNγ, LPS, and IL-4 might be important for GPC3 regulation. GPC3 is likely to be important for the generation of macrophages, as Gpc3–/– mice have impaired hematopoiesis of monocyte/macrophage progenitors,107 while peripheral cell numbers are unaffected. In addition, defective hematopoiesis has not been reported in patients with loss of function mutations in GPC3, which causes Simpson-Golabi-Behmel Syndrome. It could be hypothesized that compensatory mechanisms maintain peripheral cell numbers, but it would be interesting to address this during chronic inflammation.
In contrast, GPC4 is strongly upregulated by up to 33-fold in RA47,48 and other M1-driven conditions such as obesity.59 As GPC4 is decreased upon M1 macrophage polarization in vitro,40 factors other than IFNγ are likely to be responsible for the upregulation of GPC4 during chronic inflammation. GPC4 expression has previously been shown to increase in the synovial lining layer and blood vessels in RA,108 but its function is currently unknown. We speculate that GPC4 contributes to inflammation in RA, since GPC4 has been shown to bind and enhance Wnt3a and Wnt5a signaling,109 which is increased in RA110 and promotes pro-inflammatory cytokine release by macrophages (e.g., IL-6 and TNF).111 However, GPC4 is shed in obesity and levels of shed GPC4 correlate with BMI and insulin resistance.112 This raises the question of whether GPC4 is shed in RA and why GPC4 is not upregulated in other chronic inflammatory conditions.
Syndecans
Sdc1 is expressed at high levels in M2 macrophages and not in M1 macrophages,113 and is thought to exert an anti-inflammatory effect by sequestering inflammatory mediators on its glycan chains.114 In addition, Angsana et al. showed that macrophage motility and consequently resolution of inflammation is impaired in Sdc1–/– mice.113
Among the syndecan family, only SDC2 and SDC3 are differentially expressed in RA. SDC2 expression was found to increase slightly in RA,49 CVD,54,55 and obesity59 models, but was also increased in M2-driven diseases, such as asthma and COPD.50–52 Previous studies indicated that macrophages increase SDC2 expression upon IL-1α, TNF, and LPS treatment115 and IL-1β, IL-6, and TGFβ have also been shown to promote SDC2 expression in other cell types.116–118 In contrast, SDC2 was downregulated in human macrophages upon IFNγ-dependent M1 polarization in vitro.40,41,43
SDC2 is also important for the response of macrophages to growth factors and cytokines through its core protein. For example, the core protein binds TGFβ, and SDC2 upregulates TGFβR expression and enhances its signaling.119 Finally, SDC2 expression is increased in many cancer types, where it is important for cancer cell adhesion and migration.120 SDC2 may also modulate immune cell adhesion and migration. Shedding of SDC2 is also increased by inflammatory signals, such as TNF,121 potentially via ADAM17 activation.122 Such shedding may enable secondary downregulation of SDC2 biological activity on the cell surface. Moreover, levels of matrix metalloproteinases (MMP) that shed SDC2, such as MMP-7 and MMP-14, are elevated in various chronic inflammatory diseases.123,124 Systemic study of SDC2 expression, shedding, and function in chronic inflammation is required to better understand the contribution of this proteoglycan.
Little is currently know about the role and regulation of SDC3 in inflammation. In contrast to SDC2, SDC3 is downregulated in macrophages from RA47,48 and models of M1-driven diseases such as CVD56 and obesity.59 SDC3 was upregulated in human macrophages by in vitro IFNγ polarization, but downregulated by IFNγ and LPS, suggesting that regulation may be complex. Its biological activity is also likely to be complex, as one study demonstrated that SDC3 is pro-inflammatory in the joint, but protective in the skin.125
Tetrasaccharide Linker Region
Synthesis of HS as well as chondroitin (CS) and closely related dermatan sulfate (DS) starts at the tetrasaccharide linker that attaches the sugar chain to the core protein (reviewed by Mikami et al.126), so HS biosynthesis in macrophages during inflammation might also be regulated at the level of this tetrasaccharide linker. Modifications of the linker region determine whether the linker will continue to be synthesized as either HS or CS/DS chains and can therefore act as a switch between HS and CS/DS synthesis.127
The tetrasaccharide linker is formed by the action of four transferase enzymes, namely, xylosyltransferase (XYLT1 and XYLT2), β1,4-galactosyltransferase-I (GalT-I), β1,3-galactosyltransferase-II (GalT-II), and β1,3-glucoronyltransferase-I (GlcAT-I) that sequentially add a xylose, two galactose (Gal), and a GlcA residue.128 Of these enzymes, XYLT1 has been found to be most drastically regulated by macrophage polarization, with a decrease of up to a 20-fold in M1 macrophages 40–43,45 (Fig. 5) and a 3-fold decrease in expression in RA macrophages.47 This suggests that a decrease in linker formation might be an important contributor to the decrease in HS content in M1 macrophages observed by Martinez et al.46 However, fibroblasts from patients with mutations in XYLT1 produce less CS, but not less HS, suggesting that XYLT1 might be more associated with the formation of CS chains.129
Figure 5.

Fold change in the expression of enzymes involved in the formation of the tetrasaccharide linker and CS biosynthetic enzymes and core proteins upon macrophage polarization and RA. Using microarray data, we analyzed the expression of HS core protein and biosynthetic enzymes upon macrophage polarization in vitro with either IFNγ/LPS (M1) or IL-4/IL-13 (M2) or in RA. Genes marked in dark gray show minimal expression in macrophages in our experience. Genes upregulated in M1/RA compared with M2/healthy macrophages are marked in red (p<0.05) or pink (p>0.05), genes downregulated in M1/RA macrophages are marked in dark blue (p<0.05) or light blue (p>0.05) and genes that were not measured are marked with a dash (–). Mehraj,40 Martinez F,41 Derlindanti,42 Yıldırım-Buharalıoğlu43 compared human M1 and M2 macrophages by microarray. Jablonski44 and Jiang45 compared M1 and M2 macrophages by microarray from C57BL/6 or BALB/c mice, respectively. Martinez P46 performed individual quantative polymerase chain reactions (qPCRs) for NDSTs and sulfotransferases in human M1 and M2 macrophages. Kang,47 Yarilina,48 and You49 compared CD14+ cells freshly isolated from human RA synovial fluid to CD14+ monocyte-derived macrophages (MDM) from healthy donors. CS, chondroitin; RA, rheumatoid arthritis; HS, heparan sulfate; IFNγ, interferon-γ; LPS, lipopolysaccharide; IL, interleukin; NDST, N-deacetylase/N-sulfotransferase.
The expression of XYLT2, GalT-I, and GalT-II is also downregulated in in vitro IFNγ−polarized macrophages43 and macrophages from RA patients.47 In contrast, expression of these enzymes is increased upon in vitro stimulation with IFNγ in combination with LPS.41,44
All four of the sugars in the linker region can be modified to regulate the HS-CS/DS switch. The xylose subunit can be transiently phosphorylated by FAM20B at C2. This phosphorylation substantially increases addition of the third and fourth sugar units by GalT-II and GlcAT-I, and is thus a prerequisite for the completion of the linker region.130,131 Once the linker has been synthesized, this xylose residue must be dephosphorylated for further HS/CS polymerization. If this does not occur, EXTL2 can add a fifth (GlcNac) unit, leading to chain termination.69 This series of events is poorly understood. For example, while FAM20B overexpression increases both HS and CS synthesis in vitro,132 expression of the enzyme was increased in RA47 as well as M140,41,45 macrophages, which have lower HS content.46 In addition, the enzyme responsible for dephosphorylating the xylose residue is unknown.
The second and third residues of the linker region can be respectively 6-O, and 6-O and 4-O sulfated, with these modifications thought to switch synthesis toward CS/DS, but little is known about the enzymes involved.125
Finally, the fourth sugar (GlcA) can be 3-O-sulfated by human natural killer 1 sulfotransferase (HNK1ST), promoting chain termination.126 Expression of HNK1ST is not regulated by macrophage polarization.40–45
Several of the enzymes responsible for synthesis of the tetrasaccharide linker region are thus regulated by macrophage polarization and in RA, suggesting they may contribute to changes in HS synthesis in acute and chronic inflammation. Further investigation is required to evaluate the role(s) of these enzymes and whether their expression and activity may be coordinately regulated.
Chondroitin and Dermatan Sulfate
HS only accounts for 37% to 45% of the total GAG content in primary human M1 and M2 macrophages, while CS/DS makes up to 60% to 77% of the GAG content in human macrophages46 and the murine macrophage cell line J774A.1.133 CS/DS differ from HS in that their backbone consists of GlcA and GalNAc monomers instead of GlcA and GlcNAc, and DS is distinguished from CS by the presence of IdoA instead of GlcA.126 CS/DS can also be sulfated by various sulfotransferases and while CS binding to bioactive proteins is less well studied than for HS, the available data suggest that CS can bind to and modulate the activity of mediators in a similar fashion to HS. For example, CS is known to influence Wnt3a signaling.134,135 Our microarray analysis indicates that all of the CS sulfotransferases are downregulated in IFNγ−polarized macrophages40,43 (Fig. 5) and overall that their expression is also decreased in RA macrophages.47,48 Therefore, it is likely that changes in CS also contribute to altered binding of mediators in acute and chronic inflammation. This under-studied area might prove to be important for understanding macrophage behavior during inflammation.
Future Directions
Macrophage HS biosynthesis and core protein expression is dramatically affected by polarization in vitro and upon chronic inflammation, indicating that HS is likely to be important for the function of macrophages in inflammation. Although there are many similarities between HS expression profile in chronic inflammatory diseases and following in vitro polarization, these patterns do not fully mirror each other, suggesting that other factors in the microenvironment fine-tune expression of these genes. The importance of microenvironment is further supported by studies showing that HS expression is not significantly different in in vitro monocyte-derived macrophages from the blood of patients with atherosclerosis or systemic lupus erythematosus and healthy donors.53,136–138 Therefore, to understand the biological function of HS in macrophages, it is necessary to study macrophages isolated directly from the diseased tissue. Unfortunately, there are currently no published data sets with the proper controls to analyze macrophage expression of HS-associated genes in human atherosclerosis, obesity, or IBD. In addition, differences in the macrophage isolation method (e.g., markers for fluorescence-activated cell sorting [FACS], microdissection) between studies might be partly responsible for the observed differences, since purification methods can affect the purity or subset of macrophages analyzed.
It is difficult to predict how changes in expression of HS biosynthesis enzymes will specifically affect HS structure. For example, although M1 macrophages express more EXT1 and NDST1/2 than M2 macrophages, they have lower amounts of HS and lower HS sulfation. This highlights the importance of validating changes in mRNA expression at the protein level, and of analyzing effects on HS structure directly. Limited availability of good antibodies for HS biosynthetic enzymes can make this challenging. Furthermore, Esko and Selleck hypothesized that HS biosynthetic enzymes might function as a physical complex, which they termed the GAGosome, in which the relative abundance of the biosynthetic enzymes affects their function and ultimately HS structure and modification.39 Various studies demonstrate that changes in the expression of one of the biosynthesis enzymes can affect the function of other HS biosynthesis enzymes, which supports the existence of the GAGosome.39 For example, one study showed that the relative abundance of EXT1, EXT2, and NDST1 determine the activity of NDST1.139 Overexpression of EXT2 increased expression and activity of NDST1, while EXT1 had the opposite effect. NDST1 might compete with EXT1 for binding to EXT2 and require EXT2 for transport to the Golgi to perform its activity. In another study, inactivation of Hs2st1 (TieCre+Hs2t1f/f mice) has been shown to not only decrease 2-O sulfation, but also to increase N-sulfation and 6-O sulfation and, as a result to alter cytokine and chemokine binding.140 Moreover, the catalytic activity of HS3ST enzymes has been reported to depend on 2-O sulfation.141 The complexity of these inter-relationships makes it difficult to predict how the changes in expression of HS-associated genes shown in Fig. 3 and Fig. 4 impact HS structure. Figure 6 depicts how we think the changes in expression of HS-associated genes in RA macrophages might affect HS structure. Studies directly examining macrophage HS structure in disease are likely to be challenging, but are clearly required.
Figure 6.

Schematic summary of changes in HS biosynthesis in macrophages from RA patients. Analysis of microarray studies demonstrated that macrophages from RA synovial fluid express more glypican-4. As EXT1 was also increased, we hypothesize that the HS chain length is increased in RA. NDST expression was also increased, suggesting increased N-sulfation, while decreased GLCE and 2-O sulfotransferase expression suggest decreased epimerization and 2-O sulfation. Decreased 6-O sulfatase expression combined with an increase in SULF2 point to a net decrease in 6-O sulfation. The pattern of 3-O sulfation might also be altered, as the HS3ST1 and two isoforms are decreased, while HS3ST3B1 was increased. Nevertheless, expression data alone cannot be used to predict HS structure and studies directly studying HS structure are required. Abbreviations: HS, heparan sulfate; RA, rheumatoid arthritis; NDST, N-deacetylase/N-sulfotransferase; GLCE, glucuronic acid epimerase; GlcA, glucuronic acid; IdoA, iduronic acid; GlcNAc, N-acetyl glucosamine; GlcNS, N-sulfated glucuronic acid; NS, N-sulfation; 2S, 2-O sulfation; 3S, 3-O sulfation; 6S, 6-O sulfation.
Most of the studies that address functional changes in HS gene expression in disease focus on the role of one heparan-binding protein, with FGF, hepatocyte growth factor (HGF), platlet-derived growth factor (PDGF), wingless/integrated (WNT), VEGF, and TGFβ commonly studied. In chronic diseases, it is likely that changes in HS structure affect more than one signaling pathway due to the heterogeneity and complexity of HS. Immune cells, such as macrophages (Fig. 1B), frequently depend on multiple signaling molecules for their function. Therefore, it would be beneficial to study differences in the whole macrophage heparan-binding proteome in chronic inflammatory diseases in an unbiased way (e.g., by quantitative mass spectrometry142). This approach might help to identify potential therapeutic targets to treat chronic inflammatory diseases. HS binding of these targets can be inhibited using small peptides that resemble their HS-binding site.143 This approach has been effective for IFNγ and chemokines.144,145
Finally, we should be careful in interpreting the effect of macrophage HS biosynthetic enzymes and core proteins in mouse models of chronic inflammatory diseases, since some of the genes (e.g., NDST1) appear to be differentially expressed in human and mouse macrophages.
In summary, changes in expression of HS core proteins and biosynthetic enzymes have the potential to drastically alter signals that macrophages receive and therefore to affect their function in chronic inflammation. Systematic investigation of how HS changes in chronic inflammation and which signaling pathways this affects will broaden our understanding of how macrophage behavior is regulated in chronic inflammation and disease.
Footnotes
Competing Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: MS performed micro-array meta-analysis and drafted and revised the review; LT critically revised the review. All authors have read and approved the final manuscript.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: M.S. is supported by a PhD scholarship from Arthritis Research UK (21245) and the work is further supported by Arthritis Research UK grants 20887, 20205, and 21621.
Contributor Information
Maarten Swart, Kennedy Institute of Rheumatology, University of Oxford, Oxford, United Kingdom.
Linda Troeberg, Kennedy Institute of Rheumatology, University of Oxford, Oxford, United Kingdom.
Literature Cited
- 1. Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti-TNF therapy: past, present and future. Int Immunol. 2015;27(1):55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bek S, Bojesen AB, Nielsen JV, Sode J, Bank S, Vogel U, Andersen V. Systematic review and meta-analysis: pharmacogenetics of anti-TNF treatment response in rheumatoid arthritis. Pharmacogenomics J. 2017;17(5):403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–19. [DOI] [PubMed] [Google Scholar]
- 4. Udalova IA, Mantovani A, Feldmann M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol. 2016;12(8):472–85. [DOI] [PubMed] [Google Scholar]
- 5. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege J-L, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dembic Z. Chemokines. In: The Cytokines of the Immune System. Elsevier; 2015:241–62. [Google Scholar]
- 8. Bohlson SS, O’Conner SD, Hulsebus HJ, Ho M-M, Fraser DA. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front Immunol. 2014;5:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abbadi D, Laroumanie F, Bizou M, Pozzo J, Daviaud D, Delage C, Calise D, Gaits-Iacovoni F, Dutaur M, Tortosa F, Renaud-Gabardos E, Douin-Echinard V, Prats A-C, Roncalli J, Parini A, Pizzinat N. Local production of tenascin-C acts as a trigger for monocyte/macrophage recruitment that provokes cardiac dysfunction. Cardiovasc Res. 2018;114(1):123–37. [DOI] [PubMed] [Google Scholar]
- 10. Pereira TA, Xie G, Choi SS, Syn W-K, Voieta I, Lu J, Chan IS, Swiderska M, Amaral KB, Antunes CM, Secor WE, Witek RP, Lambertucci JR, Pereira FL, Diehl AM. Macrophage-derived hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni. Liver Int. 2013;33(1):149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G, Bot M, Herz J, Robenek H, von Eckardstein A, Nofer J-R. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol. 2011;31(5):1160–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ori A, Wilkinson MC, Fernig DG. A systems biology approach for the investigation of the heparin/heparan sulfate interactome. J Biol Chem. 2011;286(22):19892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishida M, Okumura Y, Ozawa S, Shiraishi I, Itoi T, Hamaoka K. MMP-2 inhibition reduces renal macrophage infiltration with increased fibrosis in UUO. Biochem Biophys Res Commun. 2007;354(1):133–9. [DOI] [PubMed] [Google Scholar]
- 14. Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest. 2008;118(9):3012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klose A, Zigrino P, Mauch C. Monocyte/macrophage MMP-14 modulates cell infiltration and T-cell attraction in contact dermatitis but not in murine wound healing. Am J Pathol. 2013;182(3):755–64. [DOI] [PubMed] [Google Scholar]
- 16. Schumacher MA, Donnelly JM, Engevik AC, Xiao C, Yang L, Kenny S, Varro A, Hollande F, Samuelson LC, Zavros Y. Gastric sonic hedgehog acts as a macrophage chemoattractant during the immune response to helicobacter pylori. Gastroenterology. 2012;142(5):1150–59.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laria A, Lurati A, Marrazza M, Mazzocchi D, Re KA, Scarpellini M. The macrophages in rheumatic diseases. J Inflamm Res. 2016;9:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Girodet P-O, Nguyen D, Mancini JD, Hundal M, Zhou X, Israel E, Cernadas M. Alternative macrophage activation is increased in asthma. Am J Respir Cell Mol Biol. 2016;55(4):467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He S, Xie L, Lu J, Sun S. Characteristics and potential role of M2 macrophages in COPD. Int J Chron Obstruct Pulmon Dis. 2017;12:3029–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamidzadeh K, Christensen SM, Dalby E, Chandrasekaran P, Mosser DM. Macrophages and the recovery from acute and chronic inflammation. Annu Rev Physiol. 2017;79(1):567–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage M1-M2 polarization balance. Front Immunol. 2014;5:614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Couchman JR, Pataki CA. An introduction to proteoglycans and their localization. J Histochem Cytochem. 2012;60(12):885–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6(9):633–43. [DOI] [PubMed] [Google Scholar]
- 26. Simon Davis DA, Parish CR. Heparan sulfate: a ubiquitous glycosaminoglycan with multiple roles in immunity. Front Immunol. 2013;4:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sadir R, Forest E, Lortat-Jacob H. The heparan sulfate binding sequence of interferon-gamma increased the on rate of the interferon-gamma-interferon-gamma receptor complex formation. J Biol Chem. 1998;273(18):10919–25. [DOI] [PubMed] [Google Scholar]
- 28. Lortat-Jacob H. Interferon and heparan sulphate. Biochem Soc Trans. 2006;34(3):461–4. [DOI] [PubMed] [Google Scholar]
- 29. Billings PC, Pacifici M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: mechanisms and mysteries. Connect Tissue Res. 2015;56(4):272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stanford KI, Wang L, Castagnola J, Song D, Bishop JR, Brown JR, Lawrence R, Bai X, Habuchi H, Tanaka M, Cardoso WV, Kimata K, Esko JD. Heparan sulfate 2-O-sulfotransferase is required for triglyceride-rich lipoprotein clearance. J Biol Chem. 2010;285(1):286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huynh MB, Villares J, Sepúlveda Díaz JE, Christiaans S, Carpentier G, Ouidja MO, Sissoeff L, Raisman-Vozari R, Papy-Garcia D. Glycosaminoglycans from aged human hippocampus have altered capacities to regulate trophic factors activities but not Aβ42 peptide toxicity. Neurobiol Aging. 2012;33(5):1005.e11–22. [DOI] [PubMed] [Google Scholar]
- 32. Huynh MB, Morin C, Carpentier G, Garcia-Filipe S, Talhas-Perret S, Barbier-Chassefiere V, van Kuppevelt TH, Martelly I, Albanese P, Papy-Garcia D. Age-related changes in rat myocardium involve altered capacities of glycosaminoglycans to potentiate growth factor functions and heparan sulfate-altered sulfation. J Biol Chem. 2012;287(14):11363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghadiali RS, Guimond SE, Turnbull JE, Pisconti A. Dynamic changes in heparan sulfate during muscle differentiation and ageing regulate myoblast cell fate and FGF2 signalling. Matrix Biol. 2017;59:54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamada T, Kerever A, Yoshimura Y, Suzuki Y, Nonaka R, Higashi K, Toida T, Mercier F, Arikawa-Hirasawa E. Heparan sulfate alterations in extracellular matrix structures and fibroblast growth factor-2 signaling impairment in the aged neurogenic niche. J Neurochem. 2017;142(4):534–44. [DOI] [PubMed] [Google Scholar]
- 35. Häcker U, Nybakken K, Perrimon N. Developmental cell biology: heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6(7):530–41. [DOI] [PubMed] [Google Scholar]
- 36. Lindahl U, Kjellen L, Kjellén L. Pathophysiology of heparan sulphate: many diseases, few drugs. J Intern Med. 2013;273(6):555–71. [DOI] [PubMed] [Google Scholar]
- 37. Sugahara K, Kitagawa H. Heparin and heparan sulfate biosynthesis. IUBMB Life. 2002;54(4):163–75. [DOI] [PubMed] [Google Scholar]
- 38. Kreuger J, Kjellén L. Heparan sulfate biosynthesis. J Histochem Cytochem. 2012;60(12):898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71(1):435–71. [DOI] [PubMed] [Google Scholar]
- 40. Mehraj V, Textoris J, Ben Amara A, Ghigo E, Raoult D, Capo C, Mege J-L. Monocyte responses in the context of Q fever: from a static polarized model to a kinetic model of activation. J Infect Dis. 2013;208(6):942–51. [DOI] [PubMed] [Google Scholar]
- 41. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–11. [DOI] [PubMed] [Google Scholar]
- 42. Derlindati E, Dei Cas A, Montanini B, Spigoni V, Curella V, Aldigeri R, Ardigò D, Zavaroni I, Bonadonna RC. Transcriptomic analysis of human polarized macrophages: more than one role of alternative activation? PLoS ONE. 2015;10(3):e0119751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yıldırım-Buharalıoğlu G, Bond M, Sala-Newb G, Hindmarch C. Regulation of epigenetic modifiers, including KDM6B, by interferon-γ and interleukin-4 in human macrophages. Front Immunol. 2017;8:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado J, de D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. 2015;10(12):e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jiang L, Li X, Zhang Y, Zhang M, Tang Z, Lv K. Microarray and bioinformatics analyses of gene expression profiles in BALB/c murine macrophage polarization. Mol Med Rep. 2017;16(5):7382–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martinez P, Denys A, Delos M, Sikora AS, Carpentier M, Julien S, Pestel J, Allain F. Macrophage polarization alters the expression and sulfation pattern of glycosaminoglycans. Glycobiology. 2015;25(5):502–13. [DOI] [PubMed] [Google Scholar]
- 47. Kang K, Park SH, Chen J, Qiao Y, Giannopoulou E, Berg K, Hanidu A, Li J, Nabozny G, Kang K, Park-Min K-H, Ivashkiv LB. Interferon-γ represses M2 gene expression in human macrophages by disassembling enhancers bound by the transcription factor MAF. Immunity. 2017;47(2):235–50.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yarilina A, Park-Min K-H, Antoniv T, Hu X, Ivashkiv LB. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat Immunol. 2008;9(4):378–87. [DOI] [PubMed] [Google Scholar]
- 49. You S, Yoo S-A, Choi S, Kim J-Y, Park S-J, Ji JD, Kim T-H, Kim K-J, Cho C-S, Hwang D, Kim W-U. Identification of key regulators for the migration and invasion of rheumatoid synoviocytes through a systems approach. Proc Natl Acad Sci U S A. 2014;111(1):550–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey B-G, O’Connor TP, Crystal RG. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183(4):2867–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kazeros A, Harvey B-G, Carolan BJ, Vanni H, Krause A, Crystal RG. Overexpression of apoptotic cell removal receptor MERTK in alveolar macrophages of cigarette smokers. Am J Respir Cell Mol Biol. 2008;39(6):747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Woodruff PG, Koth LL, Yang YH, Rodriguez MW, Favoreto S, Dolganov GM, Paquet AC, Erle DJ. A distinctive alveolar macrophage activation state induced by cigarette smoking. Am J Respir Crit Care Med. 2005;172(11):1383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hägg DA, Jernås M, Wiklund O, Thelle DS, Fagerberg B, Eriksson P, Hamsten A, Olsson B, Carlsson B, Carlsson LMS, Svensson P-A. Expression profiling of macrophages from subjects with atherosclerosis to identify novel susceptibility genes. Int J Mol Med. 2008;21(6):697–704. [PubMed] [Google Scholar]
- 54. Goo Y, Son S, Yechoor V, Paul A. Transcriptional profiling of foam cells reveals induction of guanylate-binding proteins following western diet acceleration of atherosclerosis in the absence of global changes in inflammation. J Am Heart Assoc. 2016;5(4):e002663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Feig JE, Vengrenyuk Y, Reiser V, Wu C, Statnikov A, Aliferis CF, Garabedian MJ, Fisher EA, Puig O. Regression of atherosclerosis is characterized by broad changes in the plaque macrophage transcriptome. PLoS ONE. 2012;7(6):e39790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Berisha S, Hsu J, Robinet P, Smith J. Transcriptome analysis of genes regulated by cholesterol loading in two strains of mouse macrophages associates lysosome pathway and ER stress response with atherosclerosis susceptibility. PLoS ONE. 2013;8(5):e65003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124(3):1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, Ferrante AW. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18(6):816–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Prieur X, Mok CYL, Velagapudi VR, Núñez V, Fuentes L, Montaner D, Ishikawa K, Camacho A, Barbarroja N, O’Rahilly S, Sethi JK, Dopazo J, Orešič M, Ricote M, Vidal-Puig A. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. 2011;60(3):797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCormick C, Duncan G, Goutsos KT, Tufaro F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci U S A. 2000;97(2):668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pacifici M. The pathogenic roles of heparan sulfate deficiency in hereditary multiple exostoses. Matrix Biol. 2017, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Busse M, Feta A, Presto J, Wilén M, Grønning M, Kjellén L, Kusche-Gullberg M. Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain elongation. J Biol Chem. 2007;282(45):32802–10. [DOI] [PubMed] [Google Scholar]
- 63. Francannet C, Cohen-Tanugi A, Le Merrer M, Munnich A, Bonaventure J, Legeai-Mallet L. Genotype-phenotype correlation in hereditary multiple exostoses. J Med Genet. 2001;38(7):430–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bao X, Moseman EA, Saito H, Petryniak B, Petryanik B, Thiriot A, Hatakeyama S, Ito Y, Kawashima H, Yamaguchi Y, Lowe JB, von Andrian UH, Fukuda M. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity. 2010;33(5):817–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reine TM, Kusche-Gullberg M, Feta A, Jenssen T, Kolset SO. Heparan sulfate expression is affected by inflammatory stimuli in primary human endothelial cells. Glycoconj J. 2012;29(1):67–76. [DOI] [PubMed] [Google Scholar]
- 66. Busse-Wicher M, Wicher KB, Kusche-Gullberg M. The exostosin family: proteins with many functions. Matrix Biol. 2014;35:25–33. [DOI] [PubMed] [Google Scholar]
- 67. Katta K, Imran T, Busse-Wicher M, Grønning M, Czajkowski S, Kusche-Gullberg M. Reduced expression of EXTL2, a member of the exostosin (EXT) family of glycosyltransferases, in human embryonic kidney 293 cells results in longer heparan sulfate chains. J Biol Chem. 2015;290(21):13168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nadanaka S, Kitagawa H. EXTL2 controls liver regeneration and aortic calcification through xylose kinase-dependent regulation of glycosaminoglycan biosynthesis. Matrix Biol. 2014;35:18–24. [DOI] [PubMed] [Google Scholar]
- 69. Nadanaka S, Zhou S, Kagiyama S, Shoji N, Sugahara K, Sugihara K, Asano M, Kitagawa H. EXTL2, a member of the EXT family of tumor suppressors, controls glycosaminoglycan biosynthesis in a xylose kinase-dependent manner. J Biol Chem. 2013;288(13):9321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Purnomo E, Emoto N, Nugrahaningsih DAA, Nakayama K, Yagi K, Heiden S, Nadanaka S, Kitagawa H, Hirata K-I. Glycosaminoglycan overproduction in the aorta increases aortic calcification in murine chronic kidney disease. J Am Heart Assoc. 2013;2(5):e000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Aikawa J, Grobe K, Tsujimoto M, Esko JD. Multiple isozymes of heparan sulfate/heparin GlcNAc N-deacetylase/GlcN N-sulfotransferase. Structure and activity of the fourth member, NDST4. J Biol Chem. 2001;276(8):5876–82. [DOI] [PubMed] [Google Scholar]
- 72. Aikawa J, Esko JD. Molecular cloning and expression of a third member of the heparan sulfate/heparin GlcNAc N-deacetylase/ N-sulfotransferase family. J Biol Chem. 1999;274(5):2690–5. [DOI] [PubMed] [Google Scholar]
- 73. Krenn EC, Wille I, Gesslbauer B, Poteser M, van Kuppevelt TH, Kungl AJ. Glycanogenomics: a qPCR-approach to investigate biological glycan function. Biochem Biophys Res Commun. 2008;375(3):297–302. [DOI] [PubMed] [Google Scholar]
- 74. Carter NM, Ali S, Kirby JA. Endothelial inflammation: the role of differential expression of N-deacetylase/N-sulphotransferase enzymes in alteration of the immunological properties of heparan sulphate. J Cell Sci. 2003;116(Pt 17):3591–600. [DOI] [PubMed] [Google Scholar]
- 75. Gordts P, Foley EM, Lawrence R, Sinha R, Lameda-Diaz C, Deng L, Nock R, Glass CK, Erbilgin A, Lusis AJ, Witztum JL, Esko JD. Reducing macrophage proteoglycan sulfation increases atherosclerosis and obesity through enhanced type I interferon signaling. Cell Metab. 2014;20(5):813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gordts P, Esko JD. Heparan sulfate proteoglycans fine-tune macrophage inflammation via IFN-β. Cytokine. 2015;72(1):118–9. [DOI] [PubMed] [Google Scholar]
- 77. Ge XN, Ha SG, Rao A, Greenberg YG, Rushdi MN, Esko JD, Rao SP, Sriramarao P. Endothelial and leukocyte heparan sulfates regulate the development of allergen-induced airway remodeling in a mouse model. Glycobiology. 2014;24(8):715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hagner-McWhirter Å, Li J-P, Oscarson S, Lindahl U. Irreversible glucuronyl C5-epimerization in the biosynthesis of heparan sulfate. J Biol Chem. 2004;279(15):14631–8. [DOI] [PubMed] [Google Scholar]
- 79. Jia J, Maccarana M, Zhang X, Bespalov M, Lindahl U, Li J-P. Lack of L-iduronic acid in heparan sulfate affects interaction with growth factors and cell signaling. J Biol Chem. 2009;284(23):15942–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li J-P, Gong F, Hagner-McWhirter Å, Forsberg E, Åbrink M, Kisilevsky R, Zhang X, Lindahl U. Targeted disruption of a murine glucuronyl C5-epimerase gene results in heparan sulfate lacking L-iduronic acid and in neonatal lethality. J Biol Chem. 2003;278(31):28363–6. [DOI] [PubMed] [Google Scholar]
- 81. Reijmers RM, Vondenhoff MFR, Roozendaal R, Kuil A, Li J-P, Spaargaren M, Pals ST, Mebius RE. Impaired lymphoid organ development in mice lacking the heparan sulfate modifying enzyme glucuronyl C5-epimerase. J Immunol. 2010;184(7):3656–64. [DOI] [PubMed] [Google Scholar]
- 82. Reijmers RM, Groen RWJ, Kuil A, Weijer K, Kimberley FC, Medema JP, van Kuppevelt TH, Li J-P, Spaargaren M, Pals ST. Disruption of heparan sulfate proteoglycan conformation perturbs B-cell maturation and APRIL-mediated plasma cell survival. Blood. 2011;117(23):6162–71. [DOI] [PubMed] [Google Scholar]
- 83. Sikora A-S, Delos M, Martinez P, Carpentier M, Allain F, Denys A. Regulation of the expression of heparan sulfate 3-O-sulfotransferase 3B (HS3ST3B) by Inflammatory stimuli in human monocytes. J Cell Biochem. 2016;117(7):1529–42. [DOI] [PubMed] [Google Scholar]
- 84. Takeda K, Hashimoto K, Uchikawa R, Tegoshi T, Yamada M, Arizono N. Direct effects of IL-4/IL-13 and the nematode Nippostrongylus brasiliensis on intestinal epithelial cells in vitro. Parasite Immunol. 2010;32(6):420–29. [DOI] [PubMed] [Google Scholar]
- 85. Borjigin J, Deng J, Sun X, De Jesus M, Liu T, Wang MM. Diurnal pineal 3-O-sulphotransferase 2 expression controlled by beta-adrenergic repression. J Biol Chem. 2003;278(18):16315–9. [DOI] [PubMed] [Google Scholar]
- 86. Kuberan B, Lech M, Borjigin J, Rosenberg RD. Light-induced 3-O-sulfotransferase expression alters pineal heparan sulfate fine structure. A surprising link to circadian rhythm. J Biol Chem. 2004;279(7):5053–4. [DOI] [PubMed] [Google Scholar]
- 87. Wood MW, Breitschwerdt EB, Gookin JL. Autocrine effects of interleukin-6 mediate acute-phase proinflammatory and tissue-reparative transcriptional responses of canine bladder mucosa. Infect Immun. 2011;79(2):708–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lu J, Auduong L, White ES, Yue X. Up-regulation of heparan sulfate 6-O-sulfation in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;50(1):106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Alhasan AA, Spielhofer J, Kusche-Gullberg M, Kirby JA, Ali S. Role of 6-O-sulfated heparan sulfate in chronic renal fibrosis. J Biol Chem. 2014;289(29):20295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. El Masri R, Seffouh A, Lortat-Jacob H, Vivès RR. The “in and out” of glucosamine 6-O-sulfation: the 6th sense of heparan sulfate. Glycoconj J. 2017;34(3):285–98. [DOI] [PubMed] [Google Scholar]
- 91. Krishnakumar K, Chakravorty I, Foy W, Allen S, Justo T, Mukherjee A, Dhoot GK. Multi-tasking Sulf1/Sulf2 enzymes do not only facilitate extracellular cell signalling but also participate in cell cycle related nuclear events. Exp Cell Res. 2018;364(1):16–27. [DOI] [PubMed] [Google Scholar]
- 92. Johnson ZI, Doolittle AC, Snuggs JW, Shapiro IM, Le Maitre CL, Risbud M V. TNF-α promotes nuclear enrichment of the transcription factor TonEBP/NFAT5 to selectively control inflammatory but not osmoregulatory responses in nucleus pulposus cells. J Biol Chem. 2017;292(42):17561–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Macur K, Grzenkowicz-Wydra J, Konieczna L, Bigda J, Temporini C, Tengattini S, Bączek T. A proteomic-based approach to study the mechanism of cytotoxicity induced by interleukin-1α and cycloheximide. Chromatographia. 2018;88(1):47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gill RMS, Mehra V, Milford E, Dhoot GK. Short SULF1/SULF2 splice variants predominate in mammary tumours with a potential to facilitate receptor tyrosine kinase-mediated cell signalling. Histochem Cell Biol. 2016;146(4):431–44. [DOI] [PubMed] [Google Scholar]
- 95. Gill RMS, Michael A, Westley L, Kocher HM, Murphy JI, Dhoot GK. SULF1/SULF2 splice variants differentially regulate pancreatic tumour growth progression. Exp Cell Res. 2014;324(2):157–71. [DOI] [PubMed] [Google Scholar]
- 96. Graham K, Murphy JI, Dhoot GK. SULF1/SULF2 reactivation during liver damage and tumour growth. Histochem Cell Biol. 2016;146(1):85–97. [DOI] [PubMed] [Google Scholar]
- 97. Li J, Vlodavsky I. Heparin, heparan sulfate and heparanase in inflammatory reactions. Thromb Haemost. 2009;102(5):823–8. [DOI] [PubMed] [Google Scholar]
- 98. Waterman M, Ben-Izhak O, Eliakim R, Groisman G, Vlodavsky I, Ilan N. Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod Pathol. 2007;20(1):8–14. [DOI] [PubMed] [Google Scholar]
- 99. Li RW, Freeman C, Yu D, Hindmarsh EJ, Tymms KE, Parish CR, Smith PN. Dramatic regulation of heparanase activity and angiogenesis gene expression in synovium from patients with rheumatoid arthritis. Arthritis Rheum. 2008;58(6):1590–600. [DOI] [PubMed] [Google Scholar]
- 100. Secchi MF, Crescenzi M, Masola V, Russo FP, Floreani A, Onisto M. Heparanase and macrophage interplay in the onset of liver fibrosis. Sci Rep. 2017;7(1):14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Edovitsky E, Lerner I, Zcharia E, Peretz T, Vlodavsky I, Elkin M. Role of endothelial heparanase in delayed-type hypersensitivity. Blood. 2006;107(9):3609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen G, Wang D, Vikramadithyan R, Yagyu H, Saxena U, Pillarisetti S, Goldberg IJ. Inflammatory cytokines and fatty acids regulate endothelial cell heparanase expression. Biochemistry. 2004;43(17):4971–7. [DOI] [PubMed] [Google Scholar]
- 103. Goodall KJ, Poon IKH, Phipps S, Hulett MD. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS ONE. 2014;9(10):e109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Escobar Galvis ML, Jia J, Zhang X, Jastrebova N, Spillmann D, Gottfridsson E, van Kuppevelt TH, Zcharia E, Vlodavsky I, Lindahl U, Li J-P. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat Chem Biol. 2007;3(12):773–8. [DOI] [PubMed] [Google Scholar]
- 105. Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee K-F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410(6832):1057–64. [DOI] [PubMed] [Google Scholar]
- 106. Mazzon C, Anselmo A, Soldani C, Cibella J, Ploia C, Moalli F, Burden SJ, Dustin ML, Sarukhan A, Viola A. Agrin is required for survival and function of monocytic cells. Blood. 2012;119(23):5502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Viviano BL, Silverstein L, Pflederer C, Paine-Saunders S, Mills K, Saunders S. Altered hematopoiesis in glypican-3-deficient mice results in decreased osteoclast differentiation and a delay in endochondral ossification. Dev Biol. 2005;282(1):152–62. [DOI] [PubMed] [Google Scholar]
- 108. Patterson AM, Cartwright A, David G, Fitzgerald O, Bresnihan B, Ashton BA, Middleton J. Differential expression of syndecans and glypicans in chronically inflamed synovium. Ann Rheum Dis. 2007;67(5):592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sakane H, Yamamoto H, Matsumoto S, Sato A, Kikuchi A. Localization of glypican-4 in different membrane microdomains is involved in the regulation of Wnt signaling. J Cell Sci. 2012;125(2):449–60. [DOI] [PubMed] [Google Scholar]
- 110. Bhatt PM, Malgor R. Wnt5a: a player in the pathogenesis of atherosclerosis and other inflammatory disorders. Atherosclerosis. 2014;237(1):155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ouchi N, Higuchi A, Ohashi K, Oshima Y, Gokce N, Shibata R, Akasaki Y, Shimono A, Walsh K. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science. 2010;329(5990):454–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tamori Y, Kasuga M. Glypican-4 is a new comer of adipokines working as insulin sensitizer. J Diabetes Investig. 2013;4(3):250–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Angsana J, Chen J, Smith S, Xiao J, Wen J, Liu L, Haller CA, Chaikof EL. Syndecan-1 modulates the motility and resolution responses of macrophages. Arterioscler Thromb Vasc Biol. 2015;35(2):332–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Teng YH-F, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012;31(1):3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Clasper S, Vekemans S, Fiore M, Plebanski M, Wordsworth P, David G, Jackson DG. Inducible expression of the cell surface heparan sulfate proteoglycan syndecan-2 (fibroglycan) on human activated macrophages can regulate fibroblast growth factor action. J Biol Chem. 1999;274(34):24113–23. [DOI] [PubMed] [Google Scholar]
- 116. Choi S, Chung H, Hong H, Kim SY, Kim S-E, Seoh J-Y, Moon CM, Yang EG, Oh E-S. Inflammatory hypoxia induces syndecan-2 expression through IL-1β-mediated FOXO3a activation in colonic epithelia. FASEB J. 2017;31(4):1516–30. [DOI] [PubMed] [Google Scholar]
- 117. Worapamorn W, Tam SP, Li H, Haase HR, Bartold PM. Cytokine regulation of syndecan-1 and -2 gene expression in human periodontal fibroblasts and osteoblasts. J Periodontal Res. 2002;37(4):273–8. [DOI] [PubMed] [Google Scholar]
- 118. Sebestyén A, Gallai M, Knittel T, Ambrust T, Ramadori G, Kovalszky I. Cytokine regulation of syndecan expression in cells of liver origin. Cytokine. 2000;12(10):1557–60. [DOI] [PubMed] [Google Scholar]
- 119. Chen L, Klass C, Woods A. Syndecan-2 regulates transforming growth factor-β signaling. J Biol Chem. 2004;279(16):15715–8. [DOI] [PubMed] [Google Scholar]
- 120. Park H, Kim Y, Lim Y, Han I, Oh E-S. Syndecan-2 mediates adhesion and proliferation of colon carcinoma cells. J Biol Chem. 2002;277(33):29730–6. [DOI] [PubMed] [Google Scholar]
- 121. De Rossi G, Evans AR, Kay E, Woodfin A, McKay TR, Nourshargh S, Whiteford JR. Shed syndecan-2 inhibits angiogenesis. J Cell Sci. 2014;127(21):4788–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, Hoettecke N, Schmidt B, Sechi A, Uhlig S, Ludwig A. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem. 2010;285(1):555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Alameddine HS, Morgan JE. Matrix metalloproteinases and tissue inhibitor of metalloproteinases in inflammation and fibrosis of skeletal muscles. J Neuromuscul Dis. 2016;3(4):455–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. O’Sullivan S, Gilmer JF, Medina C. Matrix metalloproteinases in inflammatory bowel disease: an update. Mediators Inflamm. 2015;2015:964131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kehoe O, Kalia N, King S, Eustace A, Boyes C, Reizes O, Williams A, Patterson A, Middleton J. Syndecan-3 is selectively pro-inflammatory in the joint and contributes to antigen-induced arthritis in mice. Arthritis Res Ther. 2014;16(4):R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Mikami T, Kitagawa H. Biosynthesis and function of chondroitin sulfate. Biochim Biophys Acta. 2013;1830(10):4719–33. [DOI] [PubMed] [Google Scholar]
- 127. Prydz K. Determinants of glycosaminoglycan (GAG) structure. Biomolecules. 2015;5(3):2003–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Mizumoto S, Ikegawa S, Sugahara K. Human genetic disorders caused by mutations in genes encoding biosynthetic enzymes for sulfated glycosaminoglycans. J Biol Chem. 2013;288(16):10953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bui C, Huber C, Tuysuz B, Alanay Y, Bole-Feysot C, Leroy JG, Mortier G, Nitschke P, Munnich A, Cormier-Daire V. XYLT1 mutations in desbuquois dysplasia type 2. Am J Hum Genet. 2014;94(3):405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wen J, Xiao J, Rahdar M, Choudhury BP, Cui J, Taylor GS, Esko JD, Dixon JE. Xylose phosphorylation functions as a molecular switch to regulate proteoglycan biosynthesis. Proc Natl Acad Sci U S A. 2014;111(44):15723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tone Y, Pedersen LC, Yamamoto T, Izumikawa T, Kitagawa H, Nishihara J, Tamura J, Negishi M, Sugahara K. 2-O-phosphorylation of Xylose and 6-O-sulfation of galactose in the protein linkage region of glycosaminoglycans influence the glucuronyltransferase-I activity involved in the linkage region synthesis. J Biol Chem. 2008;283(24):16801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Koike T, Izumikawa T, Tamura J-I, Kitagawa H. FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem J. 2009;421(2):157–62. [DOI] [PubMed] [Google Scholar]
- 133. Kaplan M, Aviram M. Macrophage plasma membrane chondroitin sulfate proteoglycan binds oxidized low-density lipoprotein. Atherosclerosis. 2000;149(1):5–17. [DOI] [PubMed] [Google Scholar]
- 134. Nadanaka S, Ishida M, Ikegami M, Kitagawa H. Chondroitin 4-O-sulfotransferase-1 modulates Wnt-3a signaling through control of E disaccharide expression of chondroitin sulfate. J Biol Chem. 2008;283(40):27333–43. [DOI] [PubMed] [Google Scholar]
- 135. Sugahara K, Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol. 2003;13(5):612–20. [DOI] [PubMed] [Google Scholar]
- 136. Mosig S, Rennert K, Büttner P, Krause S, Lütjohann D, Soufi M, Heller R, Funke H. Monocytes of patients with familial hypercholesterolemia show alterations in cholesterol metabolism. BMC Med Genomics. 2008;1(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Schirmer SH, Fledderus JO, van der Laan AM, van der Pouw-Kraan TCTM, Moerland PD, Volger OL, Baggen JM, Böhm M, Piek JJ, Horrevoets AJG, van Royen N. Suppression of inflammatory signaling in monocytes from patients with coronary artery disease. J Mol Cell Cardiol. 2009;46(2):177–85. [DOI] [PubMed] [Google Scholar]
- 138. Korman BD, Huang C-C, Skamra C, Wu P, Koessler R, Yao D, Huang QQ, Pearce W, Sutton-Tyrrell K, Kondos G, Edmundowicz D, Pope R, Ramsey-Goldman R. Inflammatory expression profiles in monocyte-to-macrophage differentiation in patients with systemic lupus erythematosus and relationship with atherosclerosis. Arthritis Res Ther. 2014;16(4):R147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Presto J, Thuveson M, Carlsson P, Busse M, Wilen M, Eriksson I, Kusche-Gullberg M, Kjellen L. Heparan sulfate biosynthesis enzymes EXT1 and EXT2 affect NDST1 expression and heparan sulfate sulfation. Proc Natl Acad Sci U S A. 2008;105(12):4751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Axelsson J, Xu D, Kang BN, Nussbacher JK, Handel TM, Ley K, Sriramarao P, Esko JD. Inactivation of heparan sulfate 2-O-sulfotransferase accentuates neutrophil infiltration during acute inflammation in mice. Blood. 2012;120(8):1742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Thacker BE, Xu D, Lawrence R, Esko JD. Heparan sulfate 3-O-sulfation: a rare modification in search of a function. Matrix Biol. 2014;35:60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Gesslbauer B, Rek A, Falsone F, Rajkovic E, Kungl AJ. Proteoglycanomics: tools to unravel the biological function of glycosaminoglycans. Proteomics. 2007;7(16):2870–80. [DOI] [PubMed] [Google Scholar]
- 143. Farrugia BL, Lord MS, Melrose J, Whitelock JM. The role of heparan sulfate in inflammation, and the development of biomimetics as anti-inflammatory strategies. J Histochem Cytochem. 2018;66(4):321-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Fernandez-Botran R, Gorantla V, Sun X, Ren X, Perez-Abadia G, Crespo FA, Oliver R, Orhun HI, Quan EE, Maldonado C, Ray M, Barker JH. Targeting of glycosaminoglycan-cytokine interactions as a novel therapeutic approach in allotransplantation. Transplantation. 2002;74(5):623–9. [DOI] [PubMed] [Google Scholar]
- 145. Johnson Z, Kosco-Vilbois MH, Herren S, Cirillo R, Muzio V, Zaratin P, Carbonatto M, Mack M, Smailbegovic A, Rose M, Lever R, Page C, Wells TNC, Proudfoot AEI. Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J Immunol. 2004;173(9):5776–85. [DOI] [PubMed] [Google Scholar]