

Summary
TRIM25 is an E3 ubiquitin ligase that activates RIG-I to promote the anti-viral interferon response. The NS1 protein from all strains of influenza A virus bind TRIM25, although not all virus strains block the interferon response, suggesting alternative mechanisms for TRIM25 action. Here we present a nuclear role for TRIM25 in specifically restricting influenza A virus replication. TRIM25 inhibits viral RNA synthesis through a direct mechanism, independent of its ubiquitin ligase activity and the interferon pathway. This activity can be inhibited by the viral NS1 protein. TRIM25 inhibition of viral RNA synthesis results from its binding to viral ribonucleoproteins (vRNPs), the structures containing individual viral RNA segments, the viral polymerase, and multiple viral nucleoproteins. TRIM25 binding does not inhibit initiation of capped RNA-primed viral mRNA synthesis by the viral polymerase. Rather, the onset of RNA chain elongation is inhibited because TRIM25 prohibits the movement of RNA into the polymerase complex.
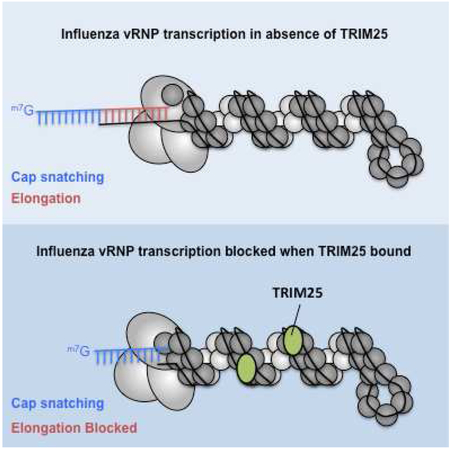
Graphical Abstract
eTOC blurb
Meyerson et al. identify an anti-influenza activity for TRIM25 in the nucleus. TRIM25 specifically targets influenza virus replication by binding viral RNA-containing ribonucleoproteins (vRNPs). TRIM25 binds vRNPs in nuclei of infected cells, acting as a molecular clamp that deprives the viral polymerase of its RNA template.
Introduction
TRIM25 promotes the interferon response to virus infection. Specifically, TRIM25 activates RIG-I, initiating the signaling pathway that leads to the activation of interferon regulatory factor 3 (IRF3) and transcription of interferon genes (Gack et al., 2008; 2007). TRIM25 is an E3 ubiquitin ligase and activates RIG-I either by ubiquitinating it directly and/or by synthesizing unanchored polyubiquitin chains that bind to RIG-I (Gack et al., 2007; 2008; Zeng et al., 2010).
Influenza A viruses cause an annual highly contagious respiratory disease in humans, and are responsible for pandemics that result in high mortality rates (Wright et al., 2013). The influenza A virus genome is comprised of eight segments of negative sense viral RNA. The smallest segment encodes the NS1 protein, a multifunctional nonstructural protein that antagonizes host antiviral responses and regulates other virus-host interactions (Krug and Sastre, 2013). One function of the NS1 protein is to bind the TRIM25 E3 ligase, inhibiting its multimerization and thus its ligase activity, an action that was postulated to block activation of the RIG-I pathway (Gack et al., 2009). However, not all human-circulating influenza A virus strains block RIG-I-mediated activation of IRF3 and interferon-β transcription (Kuo et al., 2016; 2010), although the NS1 proteins of all tested strains bind TRIM25 (Gack et al., 2009; Kuo et al., 2010; Rajsbaum et al., 2012). Notably, strong activation of IRF3 and interferon-β transcription still occurs in cells infected with viruses expressing NS1 proteins of human-circulating H3N2 and H2N2 viruses (Kuo et al., 2010; 2016). Consequently, binding of TRIM25 by the NS1 protein does not directly correlate to the suppression of interferon-β induction in influenza A virus-infected cells. These results led us to propose that the NS1 protein is antagonizing a different, unknown function of TRIM25.
In the present study, we identify this previously unknown TRIM25 function which is inhibited by the NS1 protein. We demonstrate that TRIM25 directly restricts influenza viral RNA synthesis in infected cells. Like restriction enzymes in bacteria, mammalian restriction factors block virus replication directly, without the need for extensive cellular signaling (Blanco-Melo et al., 2012). The restriction activity of TRIM25 is independent of both the ubiquitin ligase activity of TRIM25 and the interferon induction pathway. In fact, TRIM25 restriction of influenza viruses is so direct that it can be reconstituted with purified components in vitro. We show that inhibition of viral RNA synthesis results from TRIM25 binding to double-helical influenza viral ribonucleoproteins (vRNPs), the complexes that contain individual viral RNAs, the viral RNA polymerase, and multiple viral nucleoporin (NP) proteins that coat the viral RNA (Arranz et al., 2012; Resa-Infante et al., 2014; Velthuis and Fodor, 2016; Ye et al., 2006). We show that TRIM25 binds to vRNPs in in vitro assays, and also in the nucleus of virus-infected cells where viral RNA synthesis takes place (Herz et al., 1981). Glycerol gradient analysis and electron microscopy (EM) revealed that TRIM25 binds along the length of the vRNPs in an RNA-dependent manner, with no apparent preferential binding site. In vitro transcription assays indicated that TRIM25 binding onto vRNPs does not inhibit initiation of capped RNA-primed viral mRNA synthesis by the vRNP-associated polymerase. Rather, TRIM25 inhibits the onset of RNA chain elongation, most likely because TRIM25 acts as a molecular clamp on the vRNP-associated template RNA. In this model, TRIM25 simultaneously binds both RNA and protein components of the vRNP, locking the RNA in place and blocking its movement into the viral polymerase.
Results
TRIM25 inhibits influenza virus replication in infected cells
Mutations introduced into the multifunctional NS1 protein frequently result in the perturbation of more than one of its many functions (Krug and Sastre, 2013). For this reason, we took two alternative approaches to obviate NS1 antagonism so that we could study the activity of TRIM25 that NS1 is suppressing. In our first approach, we overexpressed TRIM25 to a level exceeding the amount of the NS1 protein produced in infected cells. To overexpress TRIM25, we used retroviral vectors to generate transgenic cell lines stably expressing either FLAG-tagged human TRIM25 or FLAG-tagged TRIM25 from three different nonhuman primate species (agile gibbon, orangutan, or talapoin). The latter represent versions of TRIM25 to which human influenza A viruses have not been exposed. These nonhuman primate TRIM25 proteins were found to be especially potent against human influenza A virus, and one of them (gibbon TRIM25) proved to be crucial for determining the mechanism of TRIM25 action against influenza virus. We found that, of all the cell backgrounds that we tested, only CRFK cells support stable high-level expression of these FLAG-TRIM25 proteins (Figure 1A), higher than the level of endogenous human TRIM25 expressed in human cells (Figure S1A). CRFK cells transduced with the empty vector, or with a vector expressing the HIV-1 restriction factor TRIM5α (Stremlau et al., 2004), were created as negative controls. Comparable amounts of human and nonhuman primate TRIM25 proteins were stably produced by the CRFK cell lines. The expressed TRIM25 is a doublet: the upper band is auto-ubiquitinated TRIM25 (Inn et al., 2011), as demonstrated below.
Figure 1. TRIM25 inhibits influenza viral replication in infected cells.

(A) CRFK cells engineered to stably express the indicated FLAG-tagged TRIM25 proteins, or FLAG-TRIM5α (human) as a negative control. Immunoblots were probed with mouse anti-FLAG antibody and mouse anti-βActin antibody. (B) The CRFK cells from panel A were infected with 0.2 pfu/cell of influenza A Udorn virus. Whole cell extracts collected at 8 hours post infection were analyzed by immunoblotting with an anti-Udorn goat polyclonal antibody recognizing HA, NP, and M1 (Chen et al., 2007). Quantitation of immunoblots was performed using ImageJ software, and the percent reduction in viral protein levels relative to the vector control are shown. The values for percent reduction correspond to the average change in the levels of HA, NP and M1. Each viral protein band was normalized using the actin control. (C) CRFK cell lines expressing human TRIM25 were pre-treated with the indicated amounts of wildtype (wt) NS1-expressing retrovirus, 38/41 mutant NS1-expressing retrovirus, or a control retrovirus (ctl). These cells were then infected with 0.2 pfu/cell of Udorn virus. A cell extract collected from mock infected cells was immunoblotted using rabbit anti-NS1 antibody as a reference for the amount of NS1 present at the time of infection. Extracts of cells collected at 8 hours post infection were immunoblotted using the anti-Udorn polyclonal antibody. Quantitation of immunoblots was performed as in panel B, with fold rescue relative to the sample receiving control retrovirus reported. Data presented in panels B and C are representative of two experimental replicates. (D) Virus replication after infection of the indicated CRFK cells with 0.2 pfu/cell of Udorn virus. Error bars represent standard error from three independent experiments. Asterisks indicate significant one-way ANOVA comparisons (*p < 0.01). For each time point, a post-hoc Tukey-Kramer test found all pairwise comparisons significant with p < 0.05. (E) qRT-PCR measurements of HA, NP, and NS1 mRNA and vRNA levels at 7 hours after infection of the indicated CRFK cell line with 0.2 pfu/cell of Udorn virus. Expression is normalized to GAPDH mRNA level. Error bars represent standard deviation of three independent experiments. Each mRNA/vRNA group passed a one-way ANOVA test with p < 0.01. Asterisks indicate significant (*p < 0.01) pairwise comparisons in a post-hoc Tukey-Kramer test. See also Figures S1-S3.
Each CRFK cell line was infected with 0.2 plaque-forming units (pfu)/cell of H3N2 influenza A/Udorn/72 (Udorn) virus, and virus replication was monitored by determining the levels of viral proteins at 8 hours post infection using immunoblots (Figure 1B). In cells expressing human TRIM25, viral protein synthesis was reduced in Udorn-infected cells by 35% compared to the negative controls. The reduction in viral protein levels was much greater in cells expressing any of the nonhuman primate TRIM25 molecules. For example, viral protein levels in cells expressing gibbon TRIM25 were reduced by 90% in Udorn-infected cells. This is despite the fact that human and gibbon TRIM25 differ in sequence at only 21 out of 630 amino acid positions (Figure S2). Similar results were obtained using the 2009 pandemic H1N1 influenza A/California/09 virus (Figure S1B). The production of influenza B virus proteins was also inhibited by human or gibbon TRIM25, but this was not the case for Sendai virus or a panel of retroviruses (Figure S1C-E), indicating that TRIM25 inhibition is directed against both influenza A and B viruses.
Inhibition of influenza A virus protein production was relieved by boosting NS1 protein levels prior to infection using a retrovirus expressing the NS1 protein (Figure 1C). The NS1 protein of influenza A/PR/8/34 (PR8) was used for these experiments because this NS1 protein does not bind CPSF30, which would have the effect of limiting the amount of NS1 mRNA via the inhibition of the 3’-end processing of its polymerase II-driven pre-mRNA (Krug and Sastre, 2013). We tested both wild-type NS1 and the R38A/K41A mutant NS1 protein, the latter of which does not bind TRIM25 (Gack et al., 2009). Wild-type NS1 protein led to a 14-fold increase in influenza A virus protein production in the CRFK cells expressing human TRIM25, whereas this level of the mutant NS1 protein led to only a 2-fold increase in viral protein production in these cells. These results indicate that the NS1 protein antagonizes the antiviral activity of the excess TRIM25 molecules produced in CRFK cells.
Relative to the control CRFK cells, the rate of virus replication was inhibited 10-fold in the CRFK cells expressing human TRIM25, and 100-fold in the CRFK cells expressing gibbon TRIM25 (Figure 1D). At 7 hours after infection (0.2 pfu/cell), we used quantitative RT-PCR to measure the levels of mRNAs and viral RNAs (vRNAs) derived from three viral genes: NP, NS1, and HA (hemagglutinin; Figure 1E). The levels of these virus-specific RNAs were lower in CRFK cells expressing either human or gibbon TRIM25 compared to the cells transduced with the empty plasmid. Gibbon TRIM25 inhibited virus-specific RNA synthesis more effectively than human TRIM25. For example, viral mRNA levels were reduced by 50-60% in CRFK cells expressing human TRIM25, and by 90% in CRFK cells expressing gibbon TRIM25. We conclude that TRIM25 inhibits influenza viral replication in infected cells, at the step of viral RNA synthesis or at some step upstream of RNA synthesis. We also conclude that gibbon TRIM25 is a more potent inhibitor of viral replication than human TRIM25. Because human and gibbon TRIM25 have similar binding affinities for the NS1 protein (Figure S3), it is unlikely that the greater inhibition observed with gibbon TRIM25 is due to differential inhibition by the NS1 protein, as verified by the minigenome assays described below.
Our second approach was to demonstrate that endogenous TRIM25 in human cells inhibits influenza replication. Accordingly, we deleted the TRIM25 gene of human A549 cells using CRISPR-Cas9 (Figure S4). To ensure that the effect of the deletion of TRIM25 does not simply manifest through failed activation of the interferon response via the RIG-I pathway, we also deleted the RIG-I gene in the background of both the control and TRIM25 deletion lines. Thus, wild-type A549 cells, RIG-I knockout A549 cells, and double (TRIM25 and RIG-I) knockout A549 cells were each infected with 0.002 pfu/cell of Udorn virus, and virus replication was monitored by determining the levels of viral proteins at 24 hours post infection using immunoblots (Figure 2A). The level of viral protein production in the cells in which both TRIM25 and RIG-I were knocked out (lane 3) was 2-3-fold higher than the level in the cells in which only RIG-I was knocked out (lane 2), demonstrating that endogenous TRIM25 inhibits viral protein production independently of the RIG-I pathway. To ensure that the effect we observed was TRIM25-specific, we stably complemented the RIG-I/TRIM25 double knockout A549 cell line with FLAG-tagged versions of human or gibbon TRIM25, or with an empty vector as a control (Figure 2B). It is important to note that stable over expression of FLAG-TRIM25 was tolerated in the RIG-I/TRIM25 double knockout A549 cells, whereas it was not in wild-type A549 cells. In the presence of human TRIM25, influenza protein production is reduced to levels observed in the wild-type A549 cells (Figure 2B). The cell line expressing gibbon TRIM25 shows an even greater reduction in viral protein production, in line with the experiments carried out in the CRFK cell line. These complementation experiments show that the effect we observe in the A549 knockout cells is TRIM25 specific.
Figure 2. Endogenous TRIM25 inhibits influenza RNA synthesis in infected cells.

(A) Wild-type, RIG-I knockout (KO), and RIG-I/TRIM25 (T25) double KO human A549 cells were infected with 0.002 pfu/cell of Udorn virus. Whole cell extracts collected at 24 hours post infection were immunoblotted with anti-TRIM25, anti-RIG-I, anti-IRF3, anti-pIRF3, or anti-βActin antibodies. (B) Wild-type RIG-I/TRIM25 double knockout cells, or the same cells stably expressing human (Hu) TRIM25, gibbon (Gi) TRIM25, or an empty vector control, were infected with 0.002 pfu/cell of Udorn virus. Whole cell extracts were collected and immunoblotted with the indicated antibodies. Data in panels A and B are representative of two experimental replicates. (C) qRT-PCR measurements of the indicated mRNA and vRNA levels at 24 hours after infection of the indicated A549 cells with 0.002 pfu/cell of Udorn virus, carried out as described in the legend to Figure 1E. Error bars represent standard deviation of three independent experiments. Each mRNA/vRNA group passed a one-way ANOVA test with p < 0.01. Asterisks indicate significant (*p < 0.01) pairwise comparisons in a post-hoc Tukey-Kramer test. ns = not significant. (D) Time course of virus replication after infection of the indicated A549 cells with 0.002 pfu/cell of Udorn virus. Error bars represent standard error from three independent experiments. Asterisks indicate significant one-way ANOVA comparisons (*p < 0.01). For each significant time point, a post-hoc Tukey-Kramer test found all comparisons with the double knockout cell line to be significant (p < 0.01), and each comparison between wild-type and RIG-I knockout cell lines was not significant. See also Figure S4.
We used quantitative RT-PCR to measure the levels of several mRNAs and viral RNAs (vRNAs) at the same time point (24 hours) after infection of wild-type, RIG-I knockout cells and RIG-I/TRIM25 double knockout cells (Figure 2C). The synthesis of viral mRNAs and vRNAs was increased 2-fold in the double knockout cells relative to wild-type and RIG-I knockout cells (Figure 2C), accounting for the 2-3-fold inhibition of viral protein synthesis, indicating that the inhibition of viral protein synthesis is due to the inhibition of viral RNA synthesis or a step upstream of RNA synthesis. As a consequence of the increased levels of viral RNA and protein synthesis, the rate of virus replication in the double knockout cells was increased 10-fold relative to virus replication in the wildtype and RIG-I knockout cells (Figure 2D). Based on the TRIM25 overexpression and CRISPR-Cas9 knockout results, we conclude that TRIM25 blocks influenza virus replication within cells.
TRIM25 restricts influenza virus independently of its ligase activity
Because TRIM25 activation of RIG-I and the interferon induction pathway requires its ligase activity (Gack et al., 2007; Zeng et al., 2010), we determined whether this is also the case for the TRIM25 antiviral activity that we describe here. Accordingly, we mutated human and gibbon TRIM25 at critical zinc-coordinating cysteines in the RING domain (C13A/C16A) (Reymond et al., 2001) to abolish ubiquitin ligase activity (Figure S5A, B), and established stable CRFK cell lines expressing these mutated TRIM25 proteins. As shown in Figure 3A (bottom panel), these mutations eliminate auto-ubiquitination of TRIM25 (T25), but do not impair the ability of TRIM25 to inhibit influenza virus replication (upper panel).
Figure 3. TRIM25-mediated inhibition of viral RNA synthesis is independent of its ligase activity.

(A) CRFK cell lines stably expressing human or gibbon TRIM25 (T25) or their C13A/C16A RING mutants were infected with 0.002 pfu/cell of Udorn virus. At 36 hours post infection virus production was measured by plaque assay. Error bars represent standard error from three independent experiments. These data pass a one-way ANOVA comparison with p < 0.01. (B) Minigenome assay was conducted in 293T cells lacking the RIG-I gene using plasmids expressing either human or gibbon TRIM25 containing C13A/C16A RING mutations that inactivate ligase activity. Influenza polymerase components (NP, PB1, PB2, PA) used in this assay were from Udorn virus. The Firefly/Renilla luciferase ratio of each sample is presented relative to an empty vector control. Error bars indicate the standard error of three independent replicates. Whole cell extract was prepared from an additional replicate and immunoblotted with anti-FLAG (TRIM25), anti-PB2, anti-NP, and anti-βActin antibodies. Significance between samples receiving no TRIM25 plasmid and samples receiving 2 micrograms of TRIM25 plasmid was calculated using a two-tailed, unpaired student’s t-test (*p < 0.01). (C) Minigenome assay was conducted in RIG-I knock-out 293T cells using plasmids expressing ligase-deficient human or gibbon TRIM25, and where indicated, plasmids expressing wild-type PR8 NS1 protein or PR8 NS1-R38A/R41A mutant protein. Samples were normalized to the vector sample with no NS1. Error bars represent the standard error from three independent experiments. Whole cell extract was prepared from an additional replicate and immunoblotted with the indicated antibodies. Significance between human TRIM25 samples or gibbon TRIM25 samples was calculated using a one-way ANOVA (p < 0.01) coupled with a post-hoc Tukey-Kramer test. Asterisks indicate significance for the Tukey-Kramer test (*p < 0.05). See also Figure S5.
To confirm that TRIM25 lacking ligase activity inhibits viral RNA synthesis, we carried out minigenome assays to measure viral RNA synthesis. Influenza virus RNA synthesis takes place in the context of vRNPs. Each influenza vRNP is comprised of one of the RNA genome segments, the viral polymerase complex comprised of the PA, PB1, and PB2 proteins, and multiple NP proteins that coat the RNA genome segment (Arranz et al., 2012; Resa-Infante et al., 2014; Velthuis and Fodor, 2016; Ye et al., 2006). In the minigenome assay, cells are transfected with plasmids that express all the protein components of the vRNP, and a plasmid that expresses in the negative sense a Firefly luciferase reporter gene flanked by the 5′- and 3′- terminal regions of an influenza vRNA. This reporter gene is expressed only in the context of an active influenza polymerase complex. A plasmid expressing the Renilla luciferase is co-transfected along with the other plasmids to control for transfection efficiencies, so that the Firefly/Renilla luciferase ratio measures viral RNA synthesis. To determine the effect of TRIM25 on viral RNA synthesis, we co-transfected along with these other plasmids increasing amounts of a plasmid expressing the ligase-deficient human or gibbon TRIM25 protein. To totally eliminate the RIG-I pathway in this analysis, we engineered 293T cells via CRISPR-Cas9 to disrupt RIG-I (Figure S5C, D). As shown in Figure 3B, human and gibbon TRIM25 both inhibit influenza RNA synthesis in this assay which, it should be noted, is conducted in the absence of both NS1 and RIG-I. These results show that neither the E3 ubiquitin ligase activity of TRIM25, nor interferon signaling, is required for the TRIM25-mediated inhibition of influenza A virus replication observed here.
The inhibition of viral RNA synthesis in the minigenome assay by gibbon or human TRIM25 was reversed when a plasmid encoding the wild-type NS1 protein was additionally transfected (Figure 3C). The reversal of inhibition was similar for gibbon and human TRIM25, consistent with our observation that both have similar binding affinities for the NS1 protein (Figure S3). In contrast, inhibition was not reversed by a plasmid expressing the mutant (R38A/K41A) NS1 protein that does not bind TRIM25, mirroring the infected cell results. These results demonstrate that the NS1 protein counteracts the inhibition of influenza viral RNA synthesis caused by the ligase-deficient TRIM25 protein.
TRIM25 binds influenza vRNPs in vitro and in the nucleus of virus-infected cells
TRIM25-mediated inhibition of viral RNA synthesis in minigenome assays indicated that TRIM25 targets one or more components of influenza vRNPs. To determine whether TRIM25 binds to vRNPs themselves, we isolated vRNPs from purified virions (Plotch et al., 1981), and purified baculovirus-expressed His-tagged human and gibbon TRIM25 (Figure S6). Each TRIM25 protein was incubated with vRNPs, followed by analysis by glycerol gradient centrifugation. To obtain the total TRIM25-associated vRNPs, the proteins in each glycerol gradient fraction (400 ¼l) were precipitated with trichloracetic acid (TCA). The precipitates were dissolved in a small volume (25 ¼l) of a SDS-containing buffer and then analyzed by immunoblotting for the presence of NP and TRIM25. Both human and gibbon TRIM25 co-migrated with vRNPs that were detected by NP immunoblots (Figure 4A), demonstrating that both TRIM25 proteins bind to vRNPs. Different amounts of free TRIM25 were observed at the top of the two glycerol gradients.
Figure 4. TRIM25 binds influenza vRNPs in vitro and in the nucleus of virus-infected cells.

(A) Purified, baculovirus-expressed His-tagged human or gibbon TRIM25 was incubated alone or with purified influenza vRNPs, followed by centrifugation on a glycerol gradient. Immunoblots were probed with anti-His (TRIM25) and anti-NP antibodies to identify the high molecular weight gradient fractions containing vRNPs. (B) Fractionation of wildtype (wt) or RIG-I knockout (ΔRIG-I) A549 cells. Whole cell extracts (W), cytoplasmic extracts (C), or nuclear extracts (N) from mock or Udorn-infected cells were immunoblotted with anti-TRIM25, anti-NP, anti-βActin (cytoplasmic marker), and anti-Mre11 (nuclear marker) antibodies. (C) Co-immunoprecipitation using TRIM25 antibody of endogenous TRIM25 and NP in nuclear extracts of Udorn-infected RIG-I knockout A549 cells. Mouse IgG was used for a control immunoprecipitation. Antibodies for immunoblotting were the same as in panel B. Data for all panels are representative of two experimental replicates. See also Figures S6, S7, and supplemental videos.
Transcription and viral RNA replication catalyzed by the polymerase in influenza vRNPs takes place in the nucleus of infected cells (Herz et al., 1981). While TRIM25 is predominately found in the cytoplasm, two recent studies found that a significant amount of TRIM25 is located in the nucleus (Choudhury et al., 2014; Zhang et al., 2015). To assess the validity of these recent studies, we fractionated A549 cells into cytoplasmic and nuclear fractions using our recently developed procedure (Zhao et al., 2016). This procedure yields a cytoplasmic fraction containing the cytoplasmic β-actin protein and lacking the nuclear Mre11 protein, and a nuclear fraction containing Mre11, but not β-actin (Figure 4B). TRIM25 was found in both the cytoplasm and nucleus of wild-type and RIG-I knockout A549 cells. We quantitated relative amounts of cytoplasmic and nuclear TRIM25 by measuring band intensities and taking into account volumes of lysis buffer used for the cytoplasmic and nuclear extracts. The amount of TRIM25 in the nucleus is approximately 5% of the amount in the cytoplasm in wild-type A549 cells, and approximately 8% in the RIG-I knockout A549 cells. We do not detect any difference in TRIM25 localization resulting from Udorn virus infection (Figure 4B). To verify the cell fractionation results, we carried out live imaging experiments. For this purpose, we generated RIG-I/TRIM25 double knockout A549 cells that stably express GFP-tagged human or gibbon TRIM25. Live imaging of these cells showed that TRIM25 foci are present in most nuclei (Movies S1, S2).
To determine whether nuclear TRIM25 is associated with vRNPs, nuclear extracts from virus-infected RIG-I knockout A549 cells were immunoprecipitated with antibody recognizing TRIM25. Both endogenous TRIM25, as well as NP, were recovered (Figure 4C). These results show that TRIM25 is associated with nuclear vRNPs, because immunoprecipitation of TRIM25 does not pull down individual components of vRNPs in co-transfection experiments, i.e., NP or viral polymerase proteins (Figure S7A). Because TRIM25 is responsible for the inhibition of viral RNA synthesis and virus replication in RIG-I knockout A549 cells (Figure 2C and 2D), we conclude that endogenous TRIM25 causes these inhibitions by binding to nuclear vRNPs, the vRNPs that catalyze viral RNA synthesis in infected cells (Herz et al., 1981). This mechanism of inhibition by endogenous TRIM25 mirrors the mechanism of TRIM25-mediated inhibition of viral RNA synthesis in vitro using purified vRNPs and TRIM25 proteins, as described below (Figures 5-7).
Figure 5. TRIM25 binds influenza vRNPs at various locations in a RNA-dependent manner.

(A) Human FLAG-TRIM25 was incubated with purified vRNPs, with or without RNase A digestion, followed by glycerol gradient centrifugation and TCA precipitation of each glycerol gradient fraction. Immunoblots of the TCA precipitates were probed with anti-FLAG antibody and anti-NP antibody to identify the high molecular weight gradient fractions containing vRNPs or RNA-free NP complexes. (B) Immunoprecipitation using FLAG antibody (to precipitate human (Hu) or gibbon (Gi) FLAG-TRIM25) in nuclear extracts of RIG-I knockout 293T cells transfected with all the minigenome components described in Figure 3, with or without RNase treatment. Data from panels A and B are representative of two experimental replicates. (C) Electron micrographs show purified influenza vRNPs mixed with Ni-NTA-nanogold labeled His-TRIM25 or a His-SUMO negative control. (D) Quantification of TRIM25 or SUMO molecules bound to vRNPs in EM images. Error bars represent twice the standard error (see methods for number of images and vRNPs counted). Data from both graphs passed a one-way ANOVA test with p<0.01 Asterisks indicate significant (*p<0.01) pairwise comparisons in a post-hoc Tukey-Kramer test. ns = not significant. See also Figure S7.
Figure 7. Model of the mechanism of TRIM25 inhibition.

Models for capped RNA-primed viral mRNA synthesis in the absence (panel A) and presence (panel B) of TRIM25. The relative sizes of NP (grey balls), TRIM25 (aqua) and the viral polymerase (brown) correspond to their relative molecular weights. The three protein subunits of the polymerase (PB1, PB2 and PA) make multiple interactions with each other, but these interactions are not shown in this model. Instead, the main body of the polymerase is represented as a brown “donut,” and entry and exit sites used during viral mRNA synthesis are depicted. The “donut hole” contains the catalytic site for nucleotide addition. The PB2 cap-binding site and PA endonuclease are located at one end of the main body of the polymerase, as depicted by the red and purple balls in this model. The capped RNA-cleavage and initiation steps are identical in the absence and presence of TRIM25. In contrast, the onset of RNA chain elongation is blocked in the presence of TRIM25, as discussed in the text. In the lower left panel, a small arrow indicates that NP has to be displaced in order for the RNA chain to enter the polymerase and be copied. These models contain aspects of the polymerase models shown in figure 3 of (Velthuis and Fodor, 2016).
TRIM25 binds influenza vRNPs at various locations in an RNA-dependent manner
To determine whether TRIM25 binding of influenza vRNPs is RNA-dependent, one mixture of human FLAG-TRIM25 (see methods) and vRNPs was first treated with RNase A to remove viral RNA from the vRNPs. Glycerol gradient analysis of this mixture was compared to that of a duplicate TRIM25-vRNP mixture that was not treated with RNase A. Because viral RNA is on the outside surface of influenza vRNPs (Ye et al., 2006), it is susceptible to RNase digestion (Duesberg, 1969; Ye et al., 2006). After RNase digestion, the resulting nucleoprotein complexes have lower molecular weights than the vRNPs that were not RNAse-treated (Figure 5A), but nonetheless these nucleoprotein complexes do not fall apart, as previously shown by others (Duesberg, 1969; Ye et al., 2006). TRIM25 did not bind to these RNA-free oligomeric nucleoprotein complexes, demonstrating that TRIM25 binding of vRNPs is dependent on the presence of viral RNA in the vRNPs. Variable amounts of free TRIM25 were observed at the top of the gradients. We also confirmed RNase dependence of TRIM25 binding to vRNPs in the context of minigenome conditions. 293T RIG-I knockout cells were transfected as described for Figure 3 and nuclear extracts were used to immunoprecipitate FLAG-tagged TRIM25 (Figure 5B). NP immunoprecipitated with both human and gibbon TRIM25, whereas NP was not detected when nuclear extracts were treated with RNase (Figure 5B). It is not surprising that TRIM25 binding of influenza vRNPs is RNA-dependent because previous results have established that TRIM25 has RNA-binding activity (Choudhury et al., 2014; Kwon et al., 2013; Manokaran et al., 2015). In fact, TRIM25 binds both influenza virion RNA (vRNA) and its complementary RNA (cRNA), and we did not detect a difference in the binding of influenza vRNA or cRNA between gibbon and human TRIM25 proteins (Figure S7B).
To identify the site(s) on the vRNPs at which human and gibbon TRIM25 bind, His-human TRIM25, His-gibbon TRIM25, or a His-SUMO control, was mixed with vRNPs, adsorbed onto electron microscopy (EM) grids, and subsequently incubated with Ni-NTA-nanogold particles that bind to His tags. The resulting complexes were visualized by EM (Figure 5C). The vRNPs were seen to be in their native double-helical conformation. Both human and gibbon TRIM25 molecules (detected by nanogold particles) bound to the vRNPs, although there was a small amount of nonspecific binding to the His-SUMO control. TRIM25 binding was observed at various different locations on the vRNPs, and a preferential TRIM25 binding site on the vRNPs was not observed. Gibbon TRIM25 bound significantly more vRNPs than did human TRIM25 (Figure 5C and 5D). In addition, a substantial number of individual vRNPs contained multiple gibbon TRIM25 molecules, whereas only a few individual vRNPs contained multiple human TRIM25 molecules. These results show that gibbon TRIM25 has a higher affinity than human TRIM25 for binding influenza vRNPs. In contrast, as stated above, gibbon and human TRIM25 have similar affinities for binding influenza vRNA (Figure S7B), indicating that TRIM25 recognizes features of vRNPs in addition to the viral RNAs on the surface, most likely NP structures/sequences.
TRIM25 inhibits viral mRNA synthesis after the production of capped RNA primers
To determine whether TRIM25 binding to vRNPs leads to inhibition of the transcription activity of the vRNP-associated viral polymerase, we carried out capped RNA-primed viral mRNA synthesis with purified viral RNPs. Globin mRNA was used as the source of capped RNA primers. As shown in Figure 6A, increasing amounts of purified human TRIM25 or gibbon TRIM25 inhibited capped RNA-primed synthesis of viral mRNAs. Gibbon TRIM25 was approximately four-fold more effective than human TRIM25 in inhibiting viral mRNA synthesis, reflecting the difference in the binding of the two TRIM25 molecules to vRNPs. TRIM25 inhibition leads to a decrease in the amount of the full-length viral mRNAs synthesized without an increase in short RNA products. This pattern of inhibition is diagnostic of inhibition of the initiation of viral mRNA synthesis rather than inhibition resulting from premature termination of RNA chain elongation.
Figure 6. TRIM25 inhibits the onset of mRNA chain elongation.

(A) Capped RNA-primed viral mRNA synthesis using vRNPs and globin mRNA as a primer was carried out in vitro. The indicated amounts of human or gibbon TRIM25 (T25) were added to the reaction mixture. RNA products were resolved by electrophoresis on 5% SDS-PAGE. Transcripts were quantified in Image J by measuring band intensities of each lane. The relative amounts of transcripts are shown in a graph below each sample, normalized to the sample containing no TRIM25. (B) 32P-cap-labeled, 42 base-long RNA was incubated with 0.5 ¼g of vRNP in the absence of NTPs (lanes 1-3) or the presence of the four NTPs (lanes 5-7), and where indicated, 3 ¼g of human or gibbon TRIM25. Labeled RNAs were resolved by electrophoresis on 10% SDS-PAGE. The amount of 14 nt RNA primer was quantified in Image J for lanes 1-3 and normalized to the sample containing no TRIM25. Data for panels A and B are representative of two experimental replicates.
Accordingly, we focused on the effect of TRIM25 on the initiation of viral mRNA synthesis. The first step in capped RNA-primed viral mRNA synthesis is the cleavage of a capped RNA by the polymerase-associated endonuclease to generate capped RNA fragments of 12-14 nucleotides in length that serve as primers (Plotch et al., 1981). This reaction requires the participation of the PA endonuclease domain and the PB2 cap-binding domain of the viral polymerase (Dias et al., 2009; Guilligay et al., 2008; Yuan et al., 2009). If TRIM25 interacts directly with the tripartite polymerase of the vRNPs, it is likely that this endonuclease activity would be inhibited. To assay endonuclease activity, a 42 nucleotide-long capped RNA containing 32P only in its cap structure was incubated with the vRNPs in the absence of ribonucleotide triphosphates (NTPs) (Figure 6B, lanes 1-3). Neither gibbon (Gi) TRIM25 nor human (Hu) TRIM25 inhibited endonuclease activity, as seen by the equal accumulation of the 14 nucleotide RNA primer generated in all three lanes. In the presence of the four NTPs and the absence of TRIM25, the generated capped primers were used to produce full-length viral mRNAs, which migrate together in a diffuse band near the top of the 10% gel used in this analysis (Figure 6B, lane 7). Both human and gibbon TRIM25 inhibited the synthesis of full-length viral mRNAs from these capped primers, and gibbon TRIM25 was a more potent inhibitor (Figure 6B, lanes 5-7, light exposure; lanes 8-10, dark exposure). The 10% gel enabled us to discover whether any RNA transcripts that are smaller than the smallest full-length viral mRNA were produced. We did not detect any such short transcripts even with very dark exposures of the gels, confirming the results of Figure 6A that TRIM25 does not cause premature termination of elongating RNA chains. Hence it is likely that the minimal amount of full-length mRNAs produced in the presence of TRIM25 result from vRNPs that lack bound TRIM25. In addition, we found that the 14-nucleotide-long capped primers decreased to the same extent in the absence or presence of TRIM25. As discussed below, these results are compatible with the mechanism of viral mRNA synthesis elucidated by recent structural studies (Pflug et al., 2014; Reich et al., 2014; Velthuis and Fodor, 2016).
Discussion
Here we identify a previously unknown TRIM25 anti-influenza virus activity that is inhibited by the NS1 protein of influenza A viruses. Specifically, using both overexpression of human TRIM25 and CRISPR-Cas9 deletion of TRIM25 from human cells, we demonstrate that human TRIM25 targets the RNA synthesis of influenza virus. We show that the NS1 protein blocks TRIM25 from inhibiting influenza viral RNA synthesis, and that this TRIM25 inhibitory function is independent of its ligase activity.
Minigenome assays enabled us to show that TRIM25 targets influenza vRNPs, thereby inhibiting viral mRNA synthesis catalyzed by the tripartite viral polymerase associated with these vRNPs. Because inhibition of viral RNA synthesis by TRIM25 occurred in the absence of functional RIG-I in both infected cells and minigenome assays, we were able to reconstitute this inhibition in vitro with purified components, definitively divorcing this restriction activity from activation of the host interferon response. Viral RNA replication is also catalyzed by viral polymerase molecules that are in the form of RNPs containing either vRNA or cRNA (Velthuis and Fodor, 2016; York et al., 2013). We therefore propose that a similar inhibition of viral RNA replication would result from TRIM25 binding to all influenza virus-specific RNPs. Particularly because we also showed that endogenous TRIM25 binds to vRNPs in the nucleus of virus-infected cells, we conclude that TRIM25 binding to influenza virus-specific RNPs is responsible for the inhibition of viral RNA synthesis in infected cells. Several other TRIM proteins have been shown to restrict influenza A virus, but the exact mechanisms have not been determined in these cases (Di Pietro et al., 2013; Fu et al., 2015; Liu et al., 2014).
TRIM25 blocks influenza viral RNA synthesis and virus replication directly without the need for cellular signaling through the interferon induction pathway. Our use of a TRIM25 protein of a nonhuman primate (gibbon) facilitated the elucidation of the TRIM25 mechanism of inhibition of influenza A virus RNA synthesis. Gibbon TRIM25 is a more potent inhibitor of influenza A virus RNA synthesis than human TRIM25, presumably because human influenza A viruses have not been exposed to gibbon TRIM25. The high anti-influenza virus activity of gibbon TRIM25 and the other nonhuman primate TRIM25 proteins that were tested suggests that nonhuman primate species also have immune systems directed against influenza-like viruses. It is not known whether the anti-influenza activity of TRIM25 plays a role in regulating cross-species transmission of influenza viruses, an interesting issue for future studies.
Our results provide a model for the mechanism of TRIM25 inhibition of viral mRNA synthesis by the vRNP-associated polymerase that is consistent with recent structural studies of the influenza viral polymerase (Pflug et al., 2014; Reich et al., 2014; Velthuis and Fodor, 2016) (Figure 7). We demonstrate that TRIM25 binding to vRNPs requires the presence of the constituent viral RNAs of vRNPs, which are accessible on the outside surface of the vRNPs (Arranz et al., 2012; Ye et al., 2006; Zheng et al., 2013). A requirement for RNA exposed on the outer surface of vRNPs would explain at least in part the lack of TRIM25 inhibition of Sendai virus: its viral RNA is fully sequestered in the interior of its vRNP (Ruigrok et al., 2011). Because gibbon TRIM25 has a higher affinity than human TRIM25 for influenza vRNPs, but not for its constituent vRNAs, it is likely that TRIM25 recognizes features of vRNPs in addition to the viral RNAs on the outside surface. We interpret these results to mean that TRIM25 binds to both viral RNA and NP proteins within the context of the vRNP, thereby clamping the two together and blocking the movement of viral RNA along the vRNP surface.
This interpretation provides an explanation for the effects of TRIM25 on vRNP-catalyzed mRNA synthesis that we observed, and underlies the model of TRIM25-mediated inhibition of viral mRNA synthesis depicted in Figure 7. We showed that TRIM25 binding to vRNPs does not inhibit the cap-dependent endonuclease of the viral polymerase that cleaves capped RNAs to generate short capped RNA primers. This reaction requires the activities of a cap-binding domain in the PB2 protein and an endonuclease domain in the PA protein, and requires that these two domains are positioned at a specific distance from each other (Pflug et al., 2014; Reich et al., 2014). Because this positioning requires multiple interactions between all three polymerase subunits, we conclude that all three polymerase subunits are not perturbed by TRIM25 binding. In addition, because the catalytic site in the PB1 protein is stabilized by multiple interactions with the PA and PB2 subunits (Pflug et al., 2014; Reich et al., 2014; Wright et al., 2013), it is unlikely that TRIM25 could negatively impact the PB1 catalytic site without disturbing PA and PB2 functions. Consequently, TRIM25 most likely acts after these initial steps in the initiation of viral mRNA synthesis. Because we did not detect any prematurely terminated RNA chains, it is reasonable to propose that TRIM25 inhibits the onset of mRNA chain elongation. To elongate viral mRNA chains the viral polymerase needs to pull in the viral RNA template and at the same time displace the NP protein that is associated with the viral RNA (Velthuis and Fodor, 2016) (onset of elongation step in Figure 7A). However, one or more TRIM25 molecules that are bound to a vRNP would act as clamps on the vRNP-associated viral RNA, thereby blocking the movement of the viral RNA into the polymerase (onset of elongation step in Figure 7B). By this mechanism TRIM25 does not directly inhibit polymerase activity, but rather deprives the polymerase access to the viral RNA template. This model is also consistent with the EM images that show TRIM25 binding at various places along the length of vRNPs, with no preferential binding site. No matter where TRIM25 binds it would block the movement of the viral RNA into the polymerase. We propose that this TRIM25 clamp would also deprive the polymerase of access to vRNA and cRNA templates during viral RNA replication.
In addition, we find that the short capped primers, which are not elongated in the presence of TRIM25 and the four NTPs, are degraded. This is compatible with the current model of the mechanism of viral mRNA synthesis (Pflug et al., 2014; Reich et al., 2014; Velthuis and Fodor, 2016). Previous results indicated that the PB2 cap-binding site is responsible for positioning the capped RNA primer at the catalytic site for nucleotide addition (Pflug et al., 2014; Reich et al., 2014; Velthuis and Fodor, 2016), where the primer begins its binding to the 3’ end of the vRNA template (the initiation step in Figure 7A and 7B). Once this positioning is completed, the PB2 cap-binding site dissociates from the cap of the capped RNA primer (Braam et al., 1983; Velthuis and Fodor, 2016), so that this PB2 site can then participate in the generation of more capped RNA primers. As a result of the dissociation of the PB2 cap-binding site, elongating capped viral mRNAs are free to exit the polymerase (onset of elongation step in Figure 7A). This dissociation would also allow capped RNA primers that are not elongated in the presence of TRIM25 to exit the polymerase (onset of elongation step in Figure 7B). The released small capped RNA primers lacking 3’ poly A would then be susceptible to degradation by nucleases.
The human genome encodes approximately 100 TRIM E3 ubiquitin ligases (Han et al., 2011). Although many TRIMs have been shown to possess antiviral activities (Rajsbaum et al., 2014), detailed mechanisms are known for only a small number of TRIMs. The best characterized example is TRIM5α, which restricts HIV-1 by forming a hexagonal net over the surface of the cytoplasmic viral capsid (Grütter and Luban, 2012; Li et al., 2016). Here we provide another detailed mechanism of TRIM restriction, which again involves binding to a unique viral structure. The unifying features of the TRIM family remain to be determined, however, sensing and inhibition of macromolecular viral complexes such as influenza vRNPs or retroviral capsids may emerge as a common characteristic of TRIM restriction.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Sara Sawyer (ssawyer@colorado.edu). MTAs from either the University of Colorado Boulder or the University of Texas at Austin will be required to acquire any viruses or viral vectors used in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293T cells (human embryonic kidney, ATCC #CRL-3216), MDCK cells (canine kidney, ATCC #CCL-34) and CRFK cells (feline kidney, ATCC #CCL-94) were cultured in Dulbecco’s modified Eagle media (Sigma; #D6429). A549 cells (human lung carcinoma, ATCC #CCL-185) were cultured in F12K media (Corning; #10-025-CV). All media was supplemented with 10% fetal bovine serum (Sigma; #F2442), 2 mM L-glutamine (Invitrogen; #25030-081), and 1% antibiotics (Corning; #30-002)(complete media) at 37°C and 5% CO 2. Since all cell lines were obtained from renowned international repositories they were not authenticated or tested for mycoplasma.
Viruses
Influenza H3N2 influenza A Ud and pH1N1 A/California/04/09 (California) virus stocks were grown in 10-day-old fertilized chicken eggs, and virus titers were determined using plaque assays in MDCK cells. Influenza B virus stocks (B/Florida/4/2006 Yamagata lineage) were obtained through BEI Resources (NIAID, NIH, NR-41795) and virus titers were determined using plaque assays in MDCK cells. Sendai virus stocks (formerly parainfluenza virus 1) were obtained through BEI resources (NIAID, NIH, NR-3227) and titered using a hemagluttination assay. Viruses for single-cycle infection assays were packaged in 293T cells by co-transfection of plasmids encoding viral proteins and VSV-G, along with a transfer vector, as follows: HIV-1 (pMDLg/pRRE (Addgene, #12251), pRSV-Rev (Addgene, #12253), pMD2.G (Addgene, #12259), pRRLSIN.cPPT.PGK-GFP.WPRE(Addgene, #12252)), FIV (pFP93 (Barraza and Poeschla, 2008), pCMV-VSV-G (Addgene, #8454), pGIN-SIN:GFP (Barraza and Poeschla, 2008)), NB-MLV (pCS2-mGP (Yamashita and Emerman, 2004), pCMV-VSV-G (Addgene, #8454), pLXCG (Yamashita and Emerman, 2004). After 48 hours, supernatant containing viruses was harvested, filtered, and frozen. Viruses were titered on CRFK cells by measuring percent GFP-positive cells along a volume gradient of virus supernatant.
METHOD DETAILS
TRIM25, TRIM5α, and SUMO expression constructs
TRIM25 genes (human, GenBank #NM005082; orangutan, GenBank #KX000923; agile gibbon, GenBank #KX000924; talapoin, GenBank #KX000925) were amplified from cDNA libraries constructed from total mRNA from: human 293T cells (ATCC, #CRL-3216), orangutan B-lymphocytes (Coriell, #PR01052), agile gibbon fibroblasts (Coriell, #PR00773), or talapoin fibroblasts (Coriell, #PR00716). DNA copies of mRNA was generated with the Superscript III First-Strand Synthesis System (Thermo, #18080051) using oligo(dT) primers. Primate TRIM25 and human TRIM5α were amplified with the primers listed in Table S1 using PCR Supermix High Fidelity (Thermo, #10790020). Constructs were TA-cloned into pCR4 (Invitrogen, #K4575-01). An N-terminal FLAG tag was added by PCR and these tagged constructs were TA-cloned into the Gateway entry plasmid pCR8 (Invitrogen, #K2500-20). An LR Clonase II reaction (Invitrogen, #11791-100) was used to transfer these constructs into a Gateway-converted pLPCX retroviral vector (Clontech, # 631511). To generate the C13/16A TRIM25 RING mutants, site-directed mutagenesis was carried out using pLPCX-TRIM25 plasmid as template and primers that contained the appropriate mutations (Table S1). The SUMO gene (S. cerevisiae Smtp3) with an N-terminal 6xHis tag was cloned into a pETDuet-1 (EMD Millipore, #71146) vector for protein expression in E. coli.
Immunoblotting and antibodies
293T, A549, or CRFK cells were lysed in 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1% NP-40, 2.5 mM MgCl2, 5 mM EDTA, 10% glycerol, and supplemented with Complete protease inhibitor (Roche, #11836170001). After quantifying protein concentration using a Bradford assay (Thermo, #23200), 20 μg of whole cell extract was resolved using a 10% SDS-polyacrylamide gel run at 150V. Proteins were transferred to a 0.45 μm nitrocellulose membrane (GE Healthcare, #10600002) using a wet transfer apparatus run at 100V for 1 hour at 4°C. The membrane was blocked with TBST (0.1%) containing 3% nonfat dry milk for 30 minutes at room temperature. Blocked membranes were incubated with primary antibody diluted in TBST (0.1%) for 1 hour at room temperature, washed three times with TBST (0.1%), incubated with secondary antibody diluted in TBST (0.1%) containing 3% nonfat dry milk for 40 minutes at room temperature, washed three times with TBST (0.1%), and then developed using the ECL Plus detection reagent (GE Healthcare, #RPN2132). Influenza A PB2 polymerase subunit was detected using a 1:1000 dilution of mouse anti-PB2 antibody, which was kindly provided by Juan Ortín (Ochoa et al., 1995). Influenza A NS1 was detected using a 1:2000 dilution of rabbit anti-NS1 antibody generated against the H3N2 Ud NS1 protein that was expressed in bacteria (Kuo et al., 2010). The goat antibody against the major structural proteins (HA, NP, M1) of Udorn virus was kindly provided by Dr. Robert A. Lamb (Chen et al., 2007) and used at a 1:10000 dilution. Commercial primary antibodies used: mouse anti-FLAG (SydLabs, #PA000274-P-210, 1:1000 dilution), mouse anti-Penta-His (Qiagen, #34698, 1:1000 dilution), mouse anti-TRIM25 (BD Biosciences, #610570, 1:1000 dilution), mouse anti-RIG-I (EMD Millipore, #MABF297, 1:1000 dilution), total IRF3 was detected with rabbit anti-IRF3 (Santa Cruz, #sc-9082, 1:1000 dilution), phosphorylated IRF3 was detected with rabbit anti-p-IRF3 (Abcam, #76493, 1:1000 dilution), mouse anti-Mre11 (GeneTex, #70212, 1:1000 dilution), mouse anti-NP (BEI Resources, #NR-4282, 1:1000 dilution), mouse anti-β-actin (Santa Cruz, #sc-47778, 1:1000 dilution). Commercial secondary antibodies used: donkey anti-mouse horseradish peroxidase-conjugated (Thermo Scientific, #32430, 1:10000 dilution), donkey anti-rabbit horseradish peroxidase-conjugated (Thermo Scientific, #32460, 1:10000 dilution), donkey anti-goat horseradish peroxidase-conjugated (Santa Cruz, #sc-2020, 1:10000).
Generation of stable cell lines
To generate retroviral vectors for transduction of TRIM25 genes into CRFK or A549 cells, 293T cells were seeded at a concentration of 1×106 cells/well in a 6-well dish. Cells were transfected with 2 μg pLPCX construct (empty or encoding a TRIM protein), 1 μg pCS2-mGP encoding MLV gag-pol, and 0.2 μg pC-VSV-G at a final 1:3 ratio of μg DNA to μl TransIT-293 transfection reagent (Mirius, #2705). Supernatants containing the retrovirus were collected after 48 hours, passed through a 0.2 μm filter, and used to infect CRFK or A549 cells in the presence of polybrene. After 24 hours medium containing 8 μg/ml puromycin was added to select for transduced cells. Cells resistant to puromycin were passaged and expanded for at least two weeks before testing for expression of FLAG-TRIM25 or FLAG-TRIM5α using immunoblots probed with anti-FLAG antibody.
Retrovirus infections
For infection assays, CRFK stable cells lines were plated at a concentration of 7.5×104 cells/well in a 24-well plate and infected with HIV-1, FIV, or NB-MLV such that 30-50% of the control cell line was infected. Two days post-infection, cells were fixed in 2% paraformaldehyde for 15 minutes, washed three times with 2 mL FACS buffer (DPBS supplemented with 2% FBS and 1 mM EDTA), resuspended in 500 μl FACS buffer, and analyzed by flow cytometry for expression of GFP using the BD Bioscience Fortessa cell analyzer. All experiments were carried out in triplicate.
Assaying influenza titers, proteins, and RNAs
Influenza H3N2 influenza A Ud and pH1N1 A/California/04/09 (California) virus stocks were grown in 10-day-old fertilized chicken eggs, and virus titers were determined using plaque assays in MDCK cells. CRFK cells or human A549 cells in triplicate were infected with the indicated pfu/cell of Udorn or California virus, followed by incubation at 37 °C in serum-free complete medium supplemented with 1.0 ¼g/ml N-acetylated trypsin. To measure virus replication, aliquots of the medium were taken at the indicated times after infection, and virus titers were determined by plaque assays in MDCK cells. To measure viral protein production, CRFK cells or A549 cells were infected with the indicated pfu/cell of Udorn or California virus. Cells collected at the indicated times after infection were extracted using lysis buffer (150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 1% NP-40, 2.5 mM MgCl2, 5 mM EDTA, 10% glycerol) supplemented with Complete protease inhibitor. After quantifying protein concentration using a Bradford assay (Thermo, #23200), 20 μg of cell extracts were analyzed by immunoblots probed with an antibody against detergent-treated Udorn virus provided by Robert A. Lamb (Chen et al., 2007). This antibody detects the NP and M1 proteins of influenza A viruses, and surprisingly detects not only the Udorn virus HA, but also the HA of California virus. Where indicated, a retrovirus encoding either the PR8 wild-type or R38A/K41A mutant NS1 protein was incubated for 48 hours with the CRFK cells expressing human TRIM25 prior to Ud infection. These two retroviruses were generated in the same way as described above for the retroviruses expressing TRIM proteins. To measure viral mRNA and vRNA production, infected CRFK or A549 cells were harvested at the indicated time after infection using the RNeasy Mini Kit (Qiagen, #74104). RNA (500 ng) was then used to generate cDNA using either oligo(dT) or virus-specific primers (Table S1) with Superscript III First-Strand Synthesis System (Invitrogen, #18080-051), and the cDNA was used as a template in qPCR with Power SYBR Green PCR Master Mix (Applied Biosystems, #4367659) and virus- or host-specific primers (Table S1). Cycling for qPCR was performed using Life Technologies’ Vii-A7 (CRFK cell experiments) or Bio-Rad’s CFX96 Touch (A549 cell experiments) real-time PCR detection systems. All samples were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels.
Generation of RIG-I/TRIM25 knockout cells
293T or A549 cells were seeded into a 6-well dish to achieve 70% confluency the following day. Twenty four hours later, cells were transfected with pLentiCRISPRv2 (Addgene, #52961) containing a target sequence complimentary to the first exon of either RIG-I or TRIM25 (Figures S4 and S5C, D; see Table S1 for primers containing guide sequence). Once fully confluent, cells were re-seeded into a 10 cm dish with complete DMEM media containing 1 μg/ml puromycin to select for cells expressing Cas9, and 72 hours later, cells were diluted and seeded into a 96 well dish at 1 cell/well in complete DMEM media. Wells that contained a single colony were expanded until enough cells were available for genomic DNA extraction and sequencing of RIG-I or TRIM25 loci using primers listed in Table S1. For each candidate cell line, RIG-I and TRIM25 PCR products were cloned and ten independent clones were sequenced to verify homozygous knockout (Figures S4 and S5C, D).
Cellular fractionation and immunoprecipitation
A549 cells were seeded into a 10 cm dish and mock treated or infected with Udorn at MOI = 0.002 for 24 hours. Cells were approximately 90% confluent at the time of fractionation. 293T cells were seeded into 6-well dishes and transfected with minigenome components as described below. Fractionation into cytoplasmic and nuclear fractions was carried out as described previously (Zhao et al., 2016). Cells were trypsinized, washed with cold PBS, and pelleted at 400xg for 8 min. For each 10 cm plate of cells, the cell pellet was resuspended in 500 μl Buffer C (10 mM Tris-HCl pH 7.4, 10 mM KCl, 0.2% NP-40, 1 mM EDTA, 1 mM dithiothreitol, Complete protease supplement) and gently pipetted ten times. An additional 500 μl Buffer C was then added before incubating the sample for 10 min on ice. Samples were centrifuged at 1000xg for 10 min and the resulting supernatant collected as the cytoplasmic extract used for immunoblotting. Nuclear pellets were then washed twice with 1 mL Buffer C, resuspended in 250 μl of Buffer N (50 mM Tris-HCl pH 7.4, 200 mM NaCl, 0.5% NP-40, 0.2 mM EDTA, 1 mM dithiothreitol, Complete protease supplement, 1 unit Turbo DNase (Thermo, #AM2238), and rotated at 4°C for 30 minutes. After clarifying nuclear extracts by centrifugation at 1000xg for 10 min, 30 μl was used for immunoblotting, and the remainder was used for immunoprecipitation with TRIM25 or FLAG antibody. For samples requiring RNase treatment, RNase A (50 ng/μl, Thermo Scientific, #EN0531) and RNase I (1 U/ml, Thermo Scientific, #EN0601) were added to the cleared nuclear extracts. To carry out immunoprecipitation, nuclear extracts were pre-cleared with 20 μl protein G Dynabeads (Invitrogen, #1003D) at 4°C for 30 min. At the same time, 1.25 μg of either mouse anti-TRIM25 or normal mouse IgG control antibody (Sigma, #12-371) was bound to 20 μl protein G Dynabeads at 4°C for 30 min. Pre-cleared lysate was collected, added to the bead and antibody mixture, and then rotated at 4°C for 2 hours. Beads were then washed 4 times for 5 minutes using wash buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM MgCl2, 50 μM ZnSO4) and eluted in either 30 μl 1X Laemmeli buffer (Figure 4C) or 30 μl wash buffer supplemented with 250 ng/ml 3xFLAG peptide (Sigma, #F4799)(Figures 5B and S3). β-actin was probed as a cytoplasmic marker and the DNA repair protein Mre11 was probed as a nuclear marker.
Minigenome assays
The dual luciferase minigenome assay was carried out as previously described (Marklund et al., 2012). Briefly, 293T cells in 6-well plates were transfected using TransIT-LT1 (Mirius Bio, #MIR2300) with the indicated amounts of TRIM25 plasmids or pLPCX empty vector, and 0.5 ¼g each of pcDNA3 plasmids expressing PB1, PB2 and NP, 0.4 μg PA and 0.2 ¼g of a pHH21 plasmid that expresses in the negative sense the firefly luciferase containing the 5′- and 3′- terminal regions of the Udorn NS vRNA. Where indicated, 2 μg of a pcDNA3 plasmid expressing the PR8 wild-type or R38A/K41A NS1 protein was also transfected. In addition, 0.2 ¼g of pcDNA3 plasmid expressing renilla luciferase was transfected to correct for differences in transfection efficiencies. The activities of the two luciferases were determined using the Dual-Glo Luciferase Assay System (Promega, #E2920) on a Mithras microplate luminometer. The ratio of firefly to renilla luciferase signal (relative luciferase units, RLU) was calculated and then normalized to the samples without TRIM25. The levels of expression of the indicated proteins were monitored by immunoblots. The 293T cells used in the minigenome experiments harbor a disrupted RIG-I that was generated by CRISPR-Cas9 as described in Figure S7.
Purification of His-TRIM25 and His-SUMO
The TRIM25 open reading frame was inserted into pFastBac-HTa (Invitrogen, #10712-024) vector downstream of a 6xHis tag using EcoRI and XhoI restriction sites. This plasmid was then transformed into DH10Bac cells (Invitrogen, #10359-016) to induce recombination of His-TRIM25 into a Bacmid. Bacmids were isolated and transfected into insect cells to generate baculovirus that harbors His-TRIM25. Sixty 15 cm plates of insect cells were then infected with TRIM25-harboring baculovirus, and cells were collected 48 hours later. Cells were lysed in 40 mL TRIM buffer (50 mM NaH2PO4 pH 8.0, 300 mM NaCl, 50 μM ZnSO4, 10% glycerol, 100 mM 2-Mercaptoethanol), dounced and sonicated, and centrifuged to pellet cell debris. Ten mL of 50% Ni-NTA slurry was added and rotated at 4°C for two hours. Beads were collected and washed (50 mM Tris-HCl, 150 mM NaCl, 20 mM imidazole, 0.5% NP-40) three times. Beads were eluted using wash buffer containing 250 mM imidazole. Eluates were subjected to fast performance liquid chromatography using a Superdex 200 3.2/300 size exclusion column (GE Healthcare, #29-0362-32). Peak fractions eluted at a volume consistent with TRIM25 tetramers (~285 kDa). However, a previous study that also observed apparent TRIM25 tetramers from a Superdex 200 column went on to show that TRIM25 from peak fractions actually exist as dimers in solution and that the apparent higher molecular weight of TRIM25 in gel filtration is likely due to its elongated shape (Li et al., 2014). His-SUMO was expressed in E. coli using the Rosetta 2 (DE3) strain. Cells were first grown to an OD600 of approximately 0.8 to 1.0 at 37°C and then induc ed with 1 mM IPTG for 6 hours at 37°C. Cells were harv ested and lysed using a sonicator (Branson Sonifer 250) for 15 min at 4°C. The lysis buffer co nsisted of 300 mM NaCl, 50 mM Tris pH 7.5, 5 mM 2-mercaptoethanol, and 5 mM imidazole. The lysate was clarified by centrifugation at 12,000xg at 4°C for 45 min. The supernatant was app lied to affinity purification using Ni-NTA resin. Bound protein was eluted using an elution buffer containing 500 mM NaCl, 50 mM Tris pH 7.5, 5mM 2-mercaptoethanol, and 300 mM imidazole.
Purification of viral RNPs
MDCK cells were infected with influenza A Udorn virus at a MOI=0.01. Media was collected at 48 hours post infection and clarified by centrifugation at 2600xg for 10 minutes at 4°. The clarified supernatant was then layered over 5 mL of 20% sucrose in buffer A (100 mM NaCl, 10 mM Tris-HCl pH 7.4, 2 mM EDTA) in an ultraclear centrifuge tube (Beckman, 25mm × 89mm). The virus was pelleted by ultracentrifugation at 28,000 rpm for 45 minutes at 4°C in a SW28 rotor. The virus pellet at the bottom was then resuspended in buffer A and loaded onto a linear sucrose gradient (15-60% w/w) with the same buffer and centrifuged at 30,900 rpm for 3 hours at 4 °C in a SW41 Ti rotor. The banded virus was co llected with a 20G needle, diluted with buffer A and pelleted again by ultracentrifugation at 30,900 rpm for 45 minutes at 4°C in a SW41 Ti rotor. The pelleted viruses were suspended in disruption buffer (5 mM MgCl2, 100 mM KCl, 1.5 mM dithiothreitol, 5% glycerol, 10 mM Tris-HCl pH 8.0, 1% Triton-X 100, 10 μg/mL lysolecithin) and incubated at 30°C for 15 minutes. To purify the vRNPs, a discontinuous glycerol gradient (1 mL 70%, 0.75 mL 50%, 0.375 mL 40%, 1.8 mL 33%, all v/v) was prepared in an ultraclear centrifuge tube (Beckman, 13mm × 51mm). The disrupted viruses were loaded on the glycerol gradient and centrifuged at 478,000 rpm for 3 hours and 45 minutes at 4°in a SW55 Ti rotor. Fractions were taken from the gradients and analyzed for the presence of nucleoprotein (NP, see Figure S6A). Fractions containing vRNPs as detected by NP immunoblots were pooled and used for downstream analyses.
Gradient analysis of TRIM25 binding to vRNPs
vRNPs and TRIM25 were mixed, and the mixtures were dialyzed in a buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10% glycerol for 12 hours at 4°C. TRIM25 was either baculovirus expressed and purified as described above (Figure 4A), or immunoprecipitated with anti-FLAG beads and eluted using FLAG peptide as described above using cell extracts containing plasmid-expressed human FLAG-TRIM25 (Figure 5A). Where indicated, the vRNP-TRIM25 mixture was treated with 10 ¼g/ml of RNase A for 10 minutes at 30°C prior to dialysis. The dialyzed mixtures were loaded onto a discontinuous glycerol gradient and ultracentrifuged as described above. Fractions of 400 ¼l were collected, and the proteins in each fraction were TCA precipitated. The precipitates were dissolved in 25 ¼l in SDS buffer and analyzed in immunoblotted for the presence of NP and TRIM25.
Transmission electron microscopy
Purified vRNP and TRIM25 samples were mixed at 1:1 NP-to-TRIM25 mass ratio and incubated for 1h at 4°C. The mixture was then added onto glow-discharged FCF 400-Cu grid (Electron Microscopy Sciences). After 1 min, extra liquid on the sample grid was removed by gently blotting the edge of the grid on filter paper. The grid was then placed upside-down on a droplet of 5 nm Ni-NTA-Nanogold (Nanoprobes) diluted 5-fold with 20 mM Tris-HCl pH 7.5, 150 mM NaCl and 5 mM imidazole, and incubated for 30 minutes at room temperature. Afterwards, the grid was washed twice with 5 mM imidazole and rinsed twice with water before staining with freshly prepared 0.75% uranyl formate solution for 90 seconds. After air-drying overnight, the grids were examined using JEOL 1230 High Contrast Transmission Electron Microscope at 80kV. For the His-SUMO control, 161 vRNPs were counted from 41 images; for Human TRIM25, 145 vRNPs from 37 images; for Gibbon TRIM25, 187 vRNPs from 30 images. For each image containing vRNPs, percentage of occupied vRNPs was calculated and the number of images was used to calculate standard error. For the calculation of nanogold particles per vRNP, data from all images was combined and the total number of vRNPs for eah sample was used to calculate standard error.
Capped RNA-primed transcription assays
Capped RNA-primed mRNA synthesis using globin mRNA as primer was carried out as described previously (Plotch et al., 1981). Briefly, 5 μl of 100 ng/μl vRNPs was incubated for 30 minutes at 30°C in a 50 μl reaction mixture containing 20 ng globin mRNA (Sigma, R1253-20UG), 50 mM Tris-HCl pH 7.4, 100 mM KCI, 10 mM NaCl, 1 mM DTT, 5 mM MgCl2, 1 mM ATP, 1 mM CTP, 1 mM GTP, and 0.5 μM [α-32P]UTP (3000 Ci/mmol). Where indicated, human or gibbon TRIM25 was added to the reaction mixture. RNA was cleaned up with Quick spin columns for radiolabeled RNA purification (Roche), and resolved on a 5% PAGE gel. To assay the cap-dependent endonuclease, the pGEM-9Zf(−) vector (Promega) was cleaved with Sal1 followed by transcription with SP6 RNA polymerase to yield a 42 base-long RNA. A 32P-labeled cap1 structure was added to the 5’ end of this RNA by incubation with the vaccinia capping enzyme and mRNA cap 2´-O-methyltransferase (both from New England Biolabs) in the presence of S-adenosylmethionine and (α-32P) GTP under the conditions described by the supplier of the enzymes. The cap-labeled 42 base-long RNA was purified by SDS gel electrophoresis. The assay for the cap-dependent endonuclease was carried out for 30 minutes at 30°C in a reaction mixture of 50 μl containing (32P)-cap-labeled 42 base-long RNA, 5 μl of vRNP, 50 mM Tris-HCl pH 7.4, 100 mM KCI, 10 mM NaCl, 1 mM DTT, 5 mM MgCl2. To assay for the incorporation of the capped primer into mRNA, ATP, GTP, CTP and UTP (each at 1 mM) was added to the reaction mixture. The capped RNA species were resolved by electrophoresis on 10% SDS PAGE gels.
Live imaging using confocal microscopy
C terminal GFP-tagged human or gibbon TRIM25 were generated by gateway cloning TRIM25 constructs that do not contain a stop codon (see Table S1 for primers) into the pcDNA6.2/C-emGFP-DEST destination vector (Thermo #V35520). These GFP-tagged constructs were used as templates for a subsequent gateway cloning reaction for placement into the pLPCX retroviral packaging plasmid. The pLPCX plasmids containing TRIM25-GFP constructs were used for packaging retrovirus in 293T cells, which was then used to generate stable cell lines in RIG-I/TRIM25 knockout A549 cells (see Materials and Methods for stable cell line generation). Single cells were grown up to establish clonal cell lines. For imaging, stable A549 cells were plated at a concentration of 15,000 cells/well in a black 96-well, #1.5 glass-bottom dish (MatTek #PBK96G-1.5-5-F) that was acid-etched to promote cell adherence. Cells were imaged on a Nikon TiE spinning disc confocal microscope using a 40X objective dry lens. Focal plane was adjusted to the point where nuclei were the largest. Images were taken at 1 minute intervals over 30 minutes (human TRIM25-GFP) or 60 minutes (gibbon TRIM25-GFP) and processed using ImageJ.
QUANTIFICATION AND STATISTICAL ANALYSIS
Immunoblot band quantitation
Band intensity quantification for immunoblots was performed using ImageJ v1.43u (NIH, http://rsb.info.nih.gov/ij)). Details for each analysis is provided in figure legends.
Flow cytometry
Flow cytometry data (Figure S1E) were collected using the BD Bioscience Fortessa cell analyzer. Data was then analyzed using FlowJo version 10 software (FlowJo, LLC).
Statistical Analysis
Averages, standard deviations, and standard errors were all calculated using Microsft Excel. Details regarding number of replicates and error bars can be found in figure legends. The GraphPad InStat v3.0 software (GraphPad Software, San Diego, California) was used for all statistical analyses. When two groups were compared, statistical significance was assessed by two-tailed unpaired Student’s t test. Single measurement comparisons in more than two groups were assessed by one-way ANOVA followed by a Tukey-Kramer post-hoc test for multiple comparisons. In accordance with current standard practice, only p-values < 0.05 were considered statistically significant.
DATA AVAILABILITY
All newly generated TRIM25 sequences from orangutan, agile gibbon, and talapoin have been deposited in GenBank (accession #KX000923-KX000925).
Supplementary Material
C-terminal GFP-tagged human TRIM25 (see Table S1 for primers) was cloned into the pcDNA6.2/C-emGFP-DEST destination vector (Thermo #V35520). This plasmid was used as a template for a subsequent gateway cloning reaction for placement into the pLPCX retroviral packaging plasmid. The pLPCX plasmid encoding TRIM25-GFP was used for packaging retrovirus in 293T cells, which was then used to generate stable cell lines in RIG-I/TRIM25 knockout A549 cells (see STAR Methods for stable cell line generation). Single cells were grown up to establish a clonal cell line. For imaging, stable A549 cells were plated at a concentration of 15,000 cells/well in a black 96- well, #1.5 glass-bottom dish (MatTek #PBK96G-1.5-5-F) that was acid-etched to promote cell adherence. Cells were imaged on a Nikon TiE spinning disc confocal microscope using a 40X objective dry lens. Focal plane was adjusted to the point where nuclei were the largest. Images were taken at 1 minute intervals over 30 minutes and processed using ImageJ.
C-terminal GFP-tagged gibbon TRIM25 (see Table S1 for primers) was cloned into the pcDNA6.2/C-emGFP-DEST destination vector (Thermo #V35520). This plasmid was used as a template for a subsequent gateway cloning reaction for placement into the pLPCX retroviral packaging plasmid. The pLPCX plasmid encoding TRIM25-GFP was used for packaging retrovirus in 293T cells, which was then used to generate stable cell lines in RIG-I/TRIM25 knockout A549 cells (see STAR Methods for stable cell line generation). Single cells were grown up to establish a clonal cell line. For imaging, stable A549 cells were plated at a concentration of 15,000 cells/well in a black 96-well, #1.5 glass-bottom dish (MatTek #PBK96G-1.5-5-F) that was acid-etched to promote cell adherence. Cells were imaged on a Nikon TiE spinning disc confocal microscope using a 40X objective dry lens. Focal plane was adjusted to the point where nuclei were the largest. Images were taken at 1 minute intervals over 60 minutes and processed using ImageJ.
Highlights.
TRIM25 binds to influenza ribonucleoproteins (vRNPs) in the nucleus
On vRNPs, TRIM25 acts as a clamp to inhibit viral RNA chain elongation
TRIM25 restriction of influenza RNA synthesis can be reconstituted in vitro
Influenza NS1 protein antagonizes this anti-influenza activity of TRIM25
Acknowledgments
We thank Jon Huibregtse, Maryska Kaczmarek, Tanya Paull, Paul Rowley, Alex Stabell, Cody Warren, and Julie Zhang. This work was supported by NIH grants (R01-GM-093086 to SLS and R01-AI-11772 to RMK) and the Welch Foundation (C-1565 to YJT). SLS is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease. NRM is supported by a graduate fellowship from the National Science Foundation, and a PDEP award from the Burroughs Wellcome Fund. The imaging work was performed at the BioFrontiers Institute Advanced Light Microscopy Core.
Footnotes
Competing Financial Interests
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arranz R, Coloma R, Chichón FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortín J, and Martín-Benito J (2012). The structure of native influenza virion ribonucleoproteins. Science 338, 1634–1637. [DOI] [PubMed] [Google Scholar]
- Barraza RA, and Poeschla EM (2008). Human gene therapy vectors derived from feline lentiviruses. Vet. Immunol. Immunopathol 123, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D, Venkatesh S, and Bieniasz PD (2012). Intrinsic cellular defenses against human immunodeficiency viruses. Immunity 37, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braam J, Ulmanen I, and Krug RM (1983). Molecular model of a eucaryotic transcription complex: functions and movements of influenza P proteins during capped RNA-primed transcription. Cell 34, 609–618. [DOI] [PubMed] [Google Scholar]
- Chen BJ, Leser GP, Morita E, and Lamb RA (2007). Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol. 81, 7111–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury NR, Nowak JS, Zuo J, Rappsilber J, Spoel SH, and Michlewski G (2014). Trim25 Is an RNA-Specific Activator of Lin28a/TuT4-Mediated Uridylation. Cell Reports 9, 1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, and Vicenzi E (2013). TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J Virol. 87, 4523–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias A, Bouvier D, Crépin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, and Ruigrok RWH (2009). The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458, 914–918. [DOI] [PubMed] [Google Scholar]
- Duesberg PH (1969). Distinct subunits of the ribonucleoprotein of influenza virus. Journal of Molecular Biology 42, 485–499. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol. 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Wang L, Ding H, Schwamborn JC, Li S, and Dorf ME (2015). TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 11, e1004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Kirchhofer A, Shin YC, Inn K-S, Liang C, Cui S, Myong S, Ha T, Hopfner K-P, and Jung JU (2008). Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proceedings of the National Academy of Sciences 105, 16743–16748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, Shin YC, Joo C-H, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, et al. (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. [DOI] [PubMed] [Google Scholar]
- Gack MU, Albrecht RA, Urano T, Inn K-S, Huang I-C, Carnero E, Farzan M, Inoue S, Jung JU, and García-Sastre A (2009). Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grütter MG, and Luban J (2012). TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Current Opinion in Virology 2, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilligay D, Tarendeau F, Resa-Infante P, Coloma R, Crépin T, Sehr P, Lewis J, Ruigrok RWH, Ortín J, Hart DJ, et al. (2008). The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol 15, 500–506. [DOI] [PubMed] [Google Scholar]
- Han K, Lou DI, and Sawyer SL (2011). Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet. 7, e1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz C, Stavnezer E, Krug RM, and Gurney T (1981). Influenza virus, an RNA virus, synthesizes its messenger RNA in the nucleus of infected cells. Cell 26, 391–400. [DOI] [PubMed] [Google Scholar]
- Huber VC, Kleimeyer LH, and McCullers JA (2008). Live, attenuated influenza virus (LAIV) vehicles are strong inducers of immunity toward influenza B virus. Vaccine 26, 5381–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inn K-S, Gack MU, Tokunaga F, Shi M, Wong L-Y, Iwai K, and Jung JU (2011). Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 41, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug RM, and Garcia-Sastre A (2013). The NS1 protein: a master regulator of host and viral functions. In Textbook of Influenza, 2nd edition, Webster RG, S Monto A, Braciale TJ, and Lamb RA, eds. John Wiley & Sons, Ltd., pp. 114–132. [Google Scholar]
- Kuo R-L, Li L-H, Lin S-J, Li Z-H, Chen G-W, Chang C-K, Wang Y-R, Tam E-H, Gong Y-N, Krug RM, et al. (2016). Role of N Terminus-Truncated NS1 Proteins of Influenza A Virus in Inhibiting IRF3 Activation. J Virol. 90, 4696–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo R-L, Zhao C, Malur M, and Krug RM (2010). Influenza A virus strains that circulate in humans differ in the ability of their NS1 proteins to block the activation of IRF3 and interferon-βtranscription. Virology 408, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SC, Yi H, Eichelbaum K, Föhr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, and Kim VN (2013). The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol 20, 1122–1130. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu H, Wu W, Zhuo W, Liu W, Zhang Y, Cheng M, Chen Y-G, Gao N, Yu H, et al. (2014). Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 24, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Chandrasekaran V, Carter SD, Woodward CL, Christensen DE, Dryden KA, Pornillos O, Yeager M, Ganser-Pornillos BK, Jensen GJ, et al. (2016). Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. eLife 5, e16269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li NL, Wang J, Shi P-Y, Wang T, Miller MA, and Li K (2014). Overlapping and distinct molecular determinants dictating the antiviral activities of TRIM56 against flaviviruses and coronavirus. J Virol. 88, 13821–13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, Tan HC, Sessions OM, Ward AM, Gubler DJ, et al. (2015). Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund JK, Ye Q, Dong J, Tao YJ, and Krug RM (2012). Sequence in the Influenza A Virus Nucleoprotein Required for Viral Polymerase Binding and RNA Synthesis. J Virol. 86, 7292–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa M, Bárcena J, la Luna, de S, Melero JA, Douglas AR, Nieto A, Ortin J, Skehel JJ, and Portela A (1995). Epitope mapping of cross-reactive monoclonal antibodies specific for the influenza A virus PA and PB2 polypeptides. Virus Research 37, 305–315. [DOI] [PubMed] [Google Scholar]
- Peisley A, Bin Wu, Xu H, Chen ZJ, and Hur S (2014). Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 508, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflug A, Guilligay D, Reich S, and Cusack S (2014). Structure of influenza A polymerase bound to the viral RNA promoter. Nature 516, 355–360. [DOI] [PubMed] [Google Scholar]
- Plotch SJ, Bouloy M, Ulmanen I, and Krug RM (1981). A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23, 847–858. [DOI] [PubMed] [Google Scholar]
- Rajsbaum R, Albrecht RA, Wang MK, Maharaj NP, Versteeg GA, Nistal-Villán E, García-Sastre A, and Gack MU (2012). Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 8, e1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajsbaum R, García-Sastre A, and Versteeg GA (2014). TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. Journal of Molecular Biology 426, 1265–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich S, Guilligay D, Pflug A, Malet H, Berger I, Crépin T, Hart D, Lunardi T, Nanao M, Ruigrok RWH, et al. (2014). Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 516, 361–366. [DOI] [PubMed] [Google Scholar]
- Resa-Infante P, Jorba N, Coloma R, and Ortín J (2014). The influenza virus RNA synthesis machine. RNA Biology 8, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, et al. (2001). The tripartite motif family identifies cell compartments. EMBO J 20, 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok R, Crépin T, and Kolakofsky D (2011). Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol 14, 504–510. [DOI] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Meth 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz J, Barlow PG, Hossain J, Kim E, Okada K, Kenniston T, Rea S, Donis RO, and Gambotto A (2010). A Candidate H1N1 Pandemic Influenza Vaccine Elicits Protective Immunity in Mice. PLoS One 5, e10492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, H M, Yu EY, An DS, Sabatini DM, Chen I, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, and Sodroski J (2004). The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853. [DOI] [PubMed] [Google Scholar]
- Velthuis Te, A. J and Fodor E (2016). Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol 14, 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PF, Neumann G, and Kawaoka Y (2013). Orthomyxoviruses. In Fields Virology, 6th edition, Knipe DM, et al. , eds. Lippincott Williams & Wilkins, pp. 1186–1243. [Google Scholar]
- Yamashita M, and Emerman M (2004). Capsid Is a Dominant Determinant of Retrovirus Infectivity in Nondividing Cells. J Virol. 78, 5670–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Krug RM, and Tao YJ (2006). The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444, 1078–1082. [DOI] [PubMed] [Google Scholar]
- York A, Hengrung N, Vreede FT, Huiskonen JT, and Fodor E (2013). Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proceedings of the National Academy of Sciences 110, 4238–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, et al. (2009). Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458, 909–913. [DOI] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, and Chen ZJ (2010). Reconstitution of the RIG-I Pathway Reveals a Signaling Role of Unanchored Polyubiquitin Chains in Innate Immunity. Cell 141, 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Elabd S, Hammer S, Solozobova V, Yan H, Bartel F, Inoue S, Henrich T, Wittbrodt J, Loosli F, et al. (2015). TRIM25 has a dual function in the p53/Mdm2 circuit. Oncogene 34, 5729–5738. [DOI] [PubMed] [Google Scholar]
- Zhao C, Sridharan H, Chen R, Baker DP, Wang S, and Krug RM (2016). Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat. Comms 7, 12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Olson J, Vakharia V, and Tao YJ (2013). The crystal structure and RNA-binding of an orthomyxovirus nucleoprotein. PLoS Pathog. 9, e1003624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C-terminal GFP-tagged human TRIM25 (see Table S1 for primers) was cloned into the pcDNA6.2/C-emGFP-DEST destination vector (Thermo #V35520). This plasmid was used as a template for a subsequent gateway cloning reaction for placement into the pLPCX retroviral packaging plasmid. The pLPCX plasmid encoding TRIM25-GFP was used for packaging retrovirus in 293T cells, which was then used to generate stable cell lines in RIG-I/TRIM25 knockout A549 cells (see STAR Methods for stable cell line generation). Single cells were grown up to establish a clonal cell line. For imaging, stable A549 cells were plated at a concentration of 15,000 cells/well in a black 96- well, #1.5 glass-bottom dish (MatTek #PBK96G-1.5-5-F) that was acid-etched to promote cell adherence. Cells were imaged on a Nikon TiE spinning disc confocal microscope using a 40X objective dry lens. Focal plane was adjusted to the point where nuclei were the largest. Images were taken at 1 minute intervals over 30 minutes and processed using ImageJ.
C-terminal GFP-tagged gibbon TRIM25 (see Table S1 for primers) was cloned into the pcDNA6.2/C-emGFP-DEST destination vector (Thermo #V35520). This plasmid was used as a template for a subsequent gateway cloning reaction for placement into the pLPCX retroviral packaging plasmid. The pLPCX plasmid encoding TRIM25-GFP was used for packaging retrovirus in 293T cells, which was then used to generate stable cell lines in RIG-I/TRIM25 knockout A549 cells (see STAR Methods for stable cell line generation). Single cells were grown up to establish a clonal cell line. For imaging, stable A549 cells were plated at a concentration of 15,000 cells/well in a black 96-well, #1.5 glass-bottom dish (MatTek #PBK96G-1.5-5-F) that was acid-etched to promote cell adherence. Cells were imaged on a Nikon TiE spinning disc confocal microscope using a 40X objective dry lens. Focal plane was adjusted to the point where nuclei were the largest. Images were taken at 1 minute intervals over 60 minutes and processed using ImageJ.
Data Availability Statement
All newly generated TRIM25 sequences from orangutan, agile gibbon, and talapoin have been deposited in GenBank (accession #KX000923-KX000925).