

Abstract
Mutations in the primate-specific proline-rich domain (PRD) of aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) are thought to cause Leber congenital amaurosis or dominant cone-rod dystrophy. The role of PRD and the mechanisms of PRD mutations are poorly uNderstood. Here, we have examined properties of hAIPL1 and effects of the PRD mutations on protein structure and function. Solution structures of hAIPL1, hAIPL11–316 with PRD truncation, and the P351∆12 and P376S mutants were examined by small angle X-ray scattering. Our analysis suggests that PRD assumes an extended conformation and does not interact with the FK506-binding and tetratricopeptide domains. The PRD truncation, but not PRD mutations, reduced the molecule’s radius of gyration and maximum dimension. We demonstrate that hAIPL1 is a monomeric protein, and its secondary structure and stability are not affected by the PRD mutations. PRD itself is an extended monomeric random coil. The PRD mutations caused little or no changes in hAIPL1 binding to known partners, phosphodiesterase-6A and HSP90. We also identified the γ-subunit of phosphodiesterase-6 as a novel partner of hAIPL1 and hypoth-esize that this interaction is altered by P351D12. Our results highlight the complexity of mechanisms of PRD mutations in disease and the possibility that certain mutations are benign variants.
Keywords: AIPL1, HSP90, phosphodiesterase-6, photoreceptor, retina, SAXS
Graphical Abstract
Mutations in the proline-rich domain (PRD) of human AIPL1 cause severe retinal diseases, yet the role of PRD and the mechanisms of PRD mutations are unknown. Here, we describe a SAXS-derived solution structure of AIPL1 and functional properties of disease-linked AIPL1-PRD mutants. This structure and functional analyses provide a framework for understanding the mechanisms of PRD in disease.
Mutations in aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) have been linked to one of the most severe forms of Leber congenital amaurosis (LCA), a severe, early-onset, inherited retinopathy (Kaplan et al. 1990; Sohocki et al. 2000a; Koenekoop 2004; den Hollander et al. 2008). AIPL1 is composed of two core domains, an N-terminal FK506-binding protein (FKBP)-like domain and a C-terminal tetratricopeptide (TPR)-domain with three tetratricopeptide repeats (Das et al. 1998; Sohocki et al. 2000a). Uniquely, AIPL1 proteins in primates contain a third proline-rich region (PRD) located C-terminally to the TPR domain (Fig. 1a). Mutations associated with LCA are found in all three domains of AIPL1 (Sohocki et al. 2000b; Dharmaraj et al. 2004; Stone 2007; Tan et al. 2012). The key role of AIPL1 in rods and cones is its function as a specialized chaperone for cGMP-specific phosphodiesterase-6 (PDE6), the effector enzyme in the phototransduction cascade (Liu et al. 2004; Ramamurthy et al. 2004). AIPL1-knockout in mice revealed markedly reduced stability and activity of PDE6, which was followed by rapid retina degeneration (Ramamurthy et al. 2004). This phenotype parallels that of PDE6 mutations causing retinal degeneration in humans and animal models due to elevation of intracel-lular cGMP that triggers photoreceptor cell death (Farber and Lolley 1974; Bowes et al. 1990; Pittler and Baehr 1991; McLaughlin et al. 1995; Dryja et al. 1999).
Fig. 1.

(a) Schematic representation of the domain structure of human aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1) protein. hAIPL1 consists of an FK506-binding proteins (FKBP)-like domain (blue), a TPR domain with three tetratricopeptide repeats (red), and a proline-rich domain PRD (yellow). Shown are the reported LCA4-linked AIPL1 mutations analyzed in this study. (b) Coomassie blue-stained gel showing purified hAIPL1 and mutants.
The mechanism of the chaperone/client relationships between AIPL1 and PDE6 is largely unknown. AIPL1 appears to be critical for the proper assembly of rod PDE6, which involves folding and prenylation of the catalytic PDE6A and PDE6B subunits, formation of the heterodimer PDE6AB, and its association with the regulatory PDE6γ-subunits (Pγ). In this process, AIPL1 interacts with farnesylated PDE6A (Ramamurthy et al. 2003; Kolandaivelu et al. 2009), utilizing direct binding of the farnesyl moiety to the FKBP domain (Majumder et al. 2013). The farnesyl/FKBP interaction is disrupted in the LCA-linked AIPL1 C89R mutant (Majumder et al. 2013). HSP90 is thought to be a probable partner of AIPL1 in the retina-specific chaperone complex for PDE6 (Hidalgo-de-Quintana et al. 2008; Kosmaoglou et al. 2008). Similarly to well-known interactions of TPR domain proteins with HSP90 (Taipale et al. 2010), the AIPL1 TPR domain binds to the C-terminal signature sequence MEVEED of HSP90 (Hidalgo-de-Quintana et al. 2008). Several LCA-associated mutations within the TPR domain of AIPL1 impacted its interaction with HSP90 (Hidalgo-de-Quintana et al. 2008). Furthermore, pharmacological inhibition of HSP90 reduced stability of PDE6 in mouse retina (Aguila et al. 2014). The mechanisms of PRD mutations in retina disease are entirely unclear. PRD was reported to contribute to generic chaperone function of human AIPL1 (hAIPL1) (Li et al. 2013). However, PRD is absent in the majority of vertebrate AIPL1 proteins, and therefore, it is not required for the folding/assembly of PDE6. Consequently, recessive PRD mutations either destabilize hAIPL1 or impede proper functions of its FKBP and TPR domains. Deletion of PRD modestly increased the affinity of hAIPL1 for HSP90, but the effects of PRD mutations have not been investigated (Li et al. 2013). In addition to recessive LCA, the PRD mutation P351∆12 with deletion of 12 bp at P351 was identified in a patient with dominant cone-rod dystrophy (Sohocki et al. 2000a,b). The dominant phenotype of P351∆12 was confirmed in a double-transgenic mouse model expressing both the mutant and WT hAIPL1 (Ku et al. 2015). Thus, the mechanism of P351∆12 may involve P351∆12 dimerization with WT hAIPL1. Alternatively, P351D12 interferes with PDE6 assembly independently of WT hAIPL1.
A recent analysis revealed a high degree of polymorphism in hAIPL1, which complicates establishing reliable disease causation of hAIPL1 variants. The study concluded that a significant fraction of hAIPL1 mutations found in LCA patients, including PRD mutations, are not likely to be pathological (Tan et al. 2012). Here, we have examined the solution structure of hAIPL1 and the effects of PRD deletion and mutations on the protein structure and function in order to ascertain the mechanism and/or the likelihood of disease causation.
Materials and methods
Cloning, expression, and purification of hAIPL1 and mutants
DNA sequences coding the full-length hAIPL1 and hAIPL11–316 lacking PRD were PCR-amplified from human retinal cDNA and cloned into the pET15b vector using NdeI/BamHI sites. hAIPL11–316 was chosen based on the model of mouse AIPL1 (Majumder et al. 2013), which shows that residues C-terminal to position 316 do not make intramolecular contacts with the FKBP and TPR domains. PRD of hAIPL1 (hAIPL1323–384) was PCR amplified from the hAIPL1 plasmid and cloned into the pET15b vector using NcoI/BamHI sites. The R302L and P376S mutations were introduced using standard QuikChange site-directed mutagenesis protocol. The P351 12 mutant was generated by 12 bp in-frame deletion (nt 1053–1064) in a two-step PCR procedure. In the first step, the hAIPL1 sequence C-terminal to the deletion was amplified with a forward primer containing sequence upstream and downstream of the deletion site and a reverse primer with a stop codon and BamHI site. This PCR product was used as a reverse primer in the second PCR amplification with a forward primer containing NdeI site. This resulting PCR product was then cloned into the pET15b vector using NdeI/BamHI sites. The His6-tagged hAIPL1 and mutant proteins were expressed in BL21-codon plus Escherichia coli cells by induction with 100 µM isopropyl1-thio-β-D-galactopyranoside at 16 °C overnight. hAIPL1-PRD (hAIPL1323–384) was expressed in BL21 (DE3) pLysS E. coli cells by induction with 100 µM isopropyl1-thio-β-D-galactopyranoside at 25°C for 5 h. The cell pellets were sonicated on ice (five 30-s pulses) in 50 mM Tris-HCl buffer (pH 8) containinure (Roche Applied Science, Indianapolis, IN, USA). The His6-tagged hAIPL1 and mutants proteins were purified over Ni-NTA resin (EMD Millipore: Billerica, MA, USA) using buffer A containing 250 mM imidazole for elution. Ni-NTA affinity-purified hAIPL1 and mutants were further purified by ion-exchange chromatography on a Mono Q5 column (Bio-Rad Laboratories, Hercules, CA, USA) and used in fluorescence binding assays and CD experiments. For analysis of the solution structure by small angle X-ray scattering (SAXS) and light scattering experiments, hAIPL1 and mutants were additionally purified by gel filtration chromatography on a HiLoad 16/600 Superdex 75 column (GE Healthcare, Pittsburgh, PA, USA) equilibrated against 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 50 mM L-arginine, 50 mM L-glutamic acid, 5% glycerol, and 5 mM DTT (buffer B).
Dynamic and static light scattering
Dynamic light scattering (DLS) was used to examine the degree of polydispersity of hAIPL1 samples. Static light scattering (SLS) was used to determine the molecular weight (MW) of hAIPL1 proteins. Purified hAIPL1 and mutants were concentrated up to 4 mg/mL and dialyzed against buffer B. Analyses of monodispersity and MW determination of dialyzed hAIPL1 and mutants samples were performed at 25°C using a DynaPro Nanostar instrument (Wyatt, Santa Barbara, CA, USA). The data were analyzed using the Dynamics 7.1.7 software (Santa Barbara, CA, USA).
Small angle X-ray scattering
SAXS data on hAIPL1, hAIPL11–316, and the P376S mutants were collected using the in-line FPLC purification system at the Advance Photon Source (18 ID-BioCAT, beamline) at the Argonne National Laboratories. Concentrated protein samples of hAIPL1 (15 mg/mL), P376S (15 mg/mL), hAIPL11–316 (10 mg/mL) were loaded on a Superdex 75 10/300 GL gel filtration column (GE Healthcare) equilibrated against buffer B. Data were collected on the fractions as they eluted off the Superdex 75 column. The fractions before the void volume were used for buffer substraction. The SAXS data on the P351∆12 mutant were collected at the Advanced Light Source (12.3.1 SIBYLS beamline) at the Lawrence Berkeley National Laboratories. Three different concentrations of the protein (1.9, 3.8, 5.8 mg/mL) were exposed to the synchrotron radiation for 0.5, 1, 2, 4 s each. Scattering from the buffer was subtracted, and the sample scattering data were analyzed by Primus/QT (33). AUTORG and DATGNOM were used to calculate the Rg (radius of gyration) and Dmax (maximum particle dimension) from a pairwise distribution function) in Primus/QT. The Kratky and P(r) plots in Fig. 3(d) and (e) for the P351∆12 mutant are scaled by factor of 0.1 for visualization purposes. This was done to bring them to a comparable scale with the other samples diluted during the in-line FPLC SAXS data collection.
Fig. 3.

(a–c) SAXS curves for hAIPL11–316, P351∆12, and P376S. (d) Scattering data hAIPL1, hAIPL11–316, P351∆12, and P376S replotted as Kratky plots indicate that the proteins are mostly globular with some unfolded region. (e) The pairwise distance distribution functions P(r) indicate that a maximum dimension of hAIPL11–316 (Dmax = 97 Ǻ) is similar to that of mAIPL1 (95 Ǻ) (Majumder et al. 2013) and smaller than hAIPL1 (139 Ǻ), P351∆12 (118 Ǻ), and P376S (129 Ǻ). The plots for P351∆12 in (d and e) are scaled by factor 0.1 to bring them to a comparable scale with the other samples diluted during the in-line FPLC SAXS data collection.
Modeling the solution structure of hAIPL1
Structure modeling of hAIPL1 was performed using the AllosMod-FoXS web server (Schneidman-Duhovny et al. 2010; Weinkam et al. 2012). The server generates hundreds of models starting with a user defined template, calculates small angle scattering profiles for each, and determines the best individual and ensemble fit to the experimental scattering data. The solution structure of mouse AIPL1 (mAIPL1) was used as a starting model for hAIPL1 (Majumder et al. 2013). The most probable conformations consistent with the starting model were sampled in 10 runs at MD temperature of 300 K generating 100 models in each run for a total of 1000 models. The models were ranked by Chi value to evaluate the best individual and minimum ensemble fit.
Preparation of the fluorescence peptide probes
The peptide corresponding to the C-terminus of mouse PDE6A (GAPASKSC) with the Cys residue S-farnesylated and carboxymethylated was custom made by AnaSpec, Inc. Conjugation of AMCA-X to the PDE6A peptide was performed for 1 h at 25°C using, respectively, 6 and 2 mM solutions of the reagents in 25% acetonitrile, 25% dimethylformamide, and 100 mM HEPES (pH 7.3). The peptide corresponding to the C-terminus of HSP90a (TSRMEVEED) was custom made by GenScript, Inc. Labeling of the HSP90 peptide with FITC was performed for 1 h at 25°C in 100 mM NaHCO3 (pH 8.0). The AMCA-labeled PDE6A peptide (AMCA-CtPDE6A-farnesyl) and the FITC-labeled HSP90 peptide (F-CtHSP90) were purified by reverse-phase HPLC. Pγ-subunits with a single Cys residue at position 35 or 68 were produced and labeled with 3-(bromo acetyl)-7-diethyl aminocoumarine (Pγ35BC, Pγ68BC) as previously described (Granovsky et al. 1998).
Fluorescence assays
Fluorescence resonance energy transfer assay of AMCA-CtPDE6A-farnesyl binding to hAIPL1 or mutants was performed on an F-2500 Fluorescence Spectrophotometer (Hitachi, Minato-ku, Tokyo, Japan) in 1 mL of 100 mM HEPES (pH 7.3) with excitation at 280 nm and emission at 440 nM. AMCA-CtPDE6A-farnesyl (100 nM) served as acceptor of AIPL1 Trp fluorescence. The assays of Pγ35BC or Pγ68BC (50 nM) binding to hAIPL1 and mAIPL1 proteins were performed with excitation at 445 nm and emission at 490 nm. Fluorescence polarization assay of F-CtHSP90 (300 nM) binding to hAIPL1 proteins was performed on a HORIBA Jobin Yvon Fluorescence Spectrophotometer with excitation at 490 nm and emission at 520 nm. Concentration of AMCA-CtPDE6A-farnesyl, F-CtHSP90, Pγ35BC (Pγ68BC), hAIPL1 or mutants, and mAIPL1 were determined using ɛ350 = 19 000, ɛ490 = 70 000, ɛ445 = 53 000, ɛ280 = 56 380/M cm, and ɛ280 = 57 870/M cm, respectively. PDE6AB used in assays with Pγ68BC and hAIPL1 was obtained by limited trypsinization of holoPDE6 as previously described (Artemyev et al. 1996). The data were fit with equation for binding with or without ligand depletion using Prism software (GraphPad Software Inc., San Diego, CA, USA) as previously described (Majumder et al. 2013). The Kd values are expressed as mean ± SE for at least three independent measurements.
PDE activity assays
PDE6 activity was measured using hypotonic extracts of mouse retina (Sinha et al. 2013) and [3H]cGMP as described (Muradov et al. 2006). Briefly, the hypotonic extracts of mouse retina (final concentration 0.02 µg/lL protein) containing holoPDE6 were incubated in 40 µL of 20 mM Tris–HCl (pH 7.5) buffer containing 120 mM NaCl, 2 mM MgSO4, 1 mM 2-mercaptoethanol, 0.1 U bacterial alkaline phosphatase, 10 µM [3H]cGMP (100 000 cpm) in the presence or absence of AIPL1 proteins for 15 min at 25°C. The reaction was stopped by the addition of AG1-X2 cation exchange resin (0.5 mL of 20% bed volume suspension). Samples were incubated for 6 min at 25°C with occasional mixing, and spun at 10 000 g for 3 min. A total of 0.25 mL of the supernatant was removed for counting in a scintillation counter.
Circular dichroism analysis
For CD analysis, hAIPL1 or mutants were dialyzed against 5 mM sodium phosphate buffer (pH 7.9) containing 0.1 mM DTT. A total of six scans of CD spectra were collected on 7 lM hAIPL1 or mutant protein samples in a 1-mm cuvette at 25°C over the range of 300–190 nm with an interval of 1 nm and scan speed of 100 nm/min using a Jasco J-815 CD spectrophotometer. Buffer substraction and spectra deconvolution were performed using Jasco Spectra II software and the SELCON program on the Dichroweb server (Sreerama and Woody 1993; Whitmore and Wallace 2004). Variable temperature CD measurements at 222 nm were conducted over the range of 20–80°C to determine the mid-points of thermal unfolding (Tm) of hAIPL1 and mutants.
Statistical analysis
The groups of measurements were compared with two-tailed unpaired t-test.
Results
Solution structure of hAIPL1
The structure of hAIPL1 in solution was analyzed by SAXS to determine relative orientations of the PRD and the FKBP/TPR-domains. Highly pure and monodisperse (polydispersity < 20%) preparations of hAIPL1 were obtained following the expression of the His6-tagged protein in E. coli and purification using Ni-affinity, ion-exchange, and gel filtration chromatographic steps (Fig. 1b). The SAXS data were collected in-line upon hAIPL1 elution from an FPLC column allowing for the scattering at a wide range of protein concentrations (from the protein peak to tail fractions). All SAXS scattering curves superimposed well reflecting no concentration dependence (data not shown). From the Guinier region (q • Rg < 1.3) of the scattering curve, the calculated Rg value was 39.8 Ǻ (Fig. 2). The Dmax value of 139 Ǻ was obtained from the pairwise distribution function (Fig. 3d).
Fig. 2.

Buffer-subtracted SAXS curve for hAIPL1. The linear portion of the Guinier region and the residuals after fit to the experimental data in the low q region such that q • Rg < 1.3 are shown in the inset.
In addition to hAIPL1, the SAXS data were collected on the mutant proteins with truncation (hAIPL11–316), the P351∆12, or P376S mutations of PRD (Fig. 3a–c). SAXS analysis indicated a smaller Rg (27.9 Ǻ) and Dmax (97 Ǻ) for hAIPL11–316 compared to hAIPL1 (Fig. 3d). The Rg and Dmax values of hAIPL11–316 were similar to the parameters of mouse AIPL1 derived with SAXS previously (Majumder et al. 2013). In contrast, the Rg values for P351∆12 (38.6 Ǻ) and P376S (38.5 Ǻ) were similar to the hAIPL1 values. The Dmax value for P351∆12 (118 Ǻ) appeared to be somewhat reduced compared to hAIPL1 (Fig. 3e).
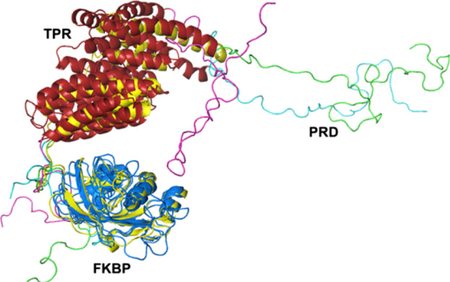
The quality of SAXS data at high q values for hAIPL1 allowed robust modeling of the solution structure. The modeling of hAIPL1 was performed using combined AllosMod-FoXS web server and the solution structure of mouse AIPL1 as a starting model (Majumder et al. 2013). The modeling yielded 1000 models with varying fits to the experimental SAXS data. The FKBP and TPR domains were relatively unchanged, but flexibility in the connecting loop between the two domains allowed sampling of a number of relative conformations. The PRD for which there was no template that could be used as a starting model, was sampled in a large number of conformations lacking any secondary structure. The top 20 models with low Chi values (1.8–2.1) had an extended conformation of the PRD that excludes potential interactions with the FKBP/TPR domains. The relative orientation of the FKBP and TPR domains in these models were also similar to that in the mouse AIPL1 solution structure (Fig. 4). In contrast, models with PRD tilting toward the FKBP/TPR domains were present in the model pool, but such models scored very poorly against the SAXS data with a Chi value of 5.0 or worse (Fig. 4). The best hAIPL1 model fit the SAXS data with the Chi value of 1.8 (Fig. 4). The best minimal ensemble of the conformations was represented by the best model (57%) and one of the top models with a Chi value of 2.0 (43%) (Fig. 4) (Pelikan et al. 2009). The Chi value for this ensemble is 1.5. The minimal ensemble indicates a moderate degree of freedom in orientation of the FKBP and TPR domains in hAIPL1.
Fig. 4.

(a) AllosMod-FoXS models of hAIPL1. The best fit model (Chi 1.8) combined with another good fit model (Chi 2.0) represents the best minimal ensemble of the conformations. The Chi 5.0 model is representative of models with PRD tilting toward the FK506-binding proteins (FKBP)/TPR domains. The FKBP domains of hAIPL1 models are superimposed with the mAIPL1 model (yellow) (Majumder et al. 2013). (b) The view is rotated −90° about the vertical axis from the view in (a). (c) Experimental SAXS data (black) and superimposed simulated scattering curves for the Chi 1.8 (green) and Chi 5.0 (magenta) models shown in (a).
The AIPL1 PRD is an extended random coil
The individual PRD domain hAIPL1-PRD (hAIPL1323–384) was expressed to examine its properties and secondary structure. The CD spectrum of hAIPL1-PRD with a minimum at 200 nm and a plateau approaching zero at 220–240 nm is typical of an unstructured random coil protein (Fig. 5a). The lack of defined secondary structure in hAIPL1-PRD is in agreement with the predictions of multiple secondary structure algorithms (www.expasy.org). The calculated MW of hAIPL1-PRD is ~ 7.5 kDa. However, hAIPL1-PRD eluted as a protein with an apparent MW of ~ 15 kDa upon gel filtration on a Superdex 75 column calibrated with globular proteins (data not shown). DLS and SLS measurements were conducted to examine the polydispersity and potential dimerization of hAIPL1-PRD. The DLS tests indicated a relatively low polydispersity (~ 20%) for hAIPL1-PRD. The hydrodynamic radius Rh of hAIPL1-PRD of 2.65 ± 0.05 is consistent with a linear polymer of 11.9 ± 0.9 kDa or a globular protein of 32.8 ± 1.3 kDa. SLS analysis, which is a more reliable technique to assess protein MW, yielded a MW of 10.6 ± 1.2 kDa. Altogether, the gel filtration, DLS, and SLS analyses suggest that individual hAIPL1-PRD is a monomeric protein that assumes predominantly an extended conformation. As the polydispersity of hAIPL1-PRD is low, the protein seems unlikely to sample simultaneously both compact and extended conformations.
Fig. 5.

(a) CD spectrum of hAIPL1-PRD indicates an unstructured random coil protein. The spectrum shows a minimum at 200 nm and a plateau approaching zero at 220–240 nm. (b) CD spectra of hAIPL1, hAIPL11–316, R302L, P351∆12, and P376S suggest similar secondary structures of the proteins.
The secondary structure and stability of hAIPL1 are not affected by the PRD mutations
To determine whether PRD or its mutations influence the protein secondary structure or thermal stability, hAIPL11–316, P351∆12, and P376S were examined with CD spectrometry. The LCA-linked mutation R302L located between the TPR and PRD domains was also included in the analysis. The average CD spectra for hAIPL1, and the mutant proteins at 25°C were nearly superimposable indicating that the hAIPL1 mutations or PRD deletion did not alter the secondary structure of the protein (Fig. 5b). The fractions of α-helix and β-strand determined using the Selcon 3 program from the Dichroweb server were similar for hAIPL1 and all the mutants (Table 1). The secondary structure composition of the hAIPL1 proteins was also similar to that of mouse AIPL1, which lacks PRD (Majumder et al. 2013). The mid-points of thermal unfolding (Tm) were determined as CD spectra changes at 222 nm over a range of temperatures. The Tm values for all hAIPL1 proteins (Table 1) and mouse AIPL1 (Tm = 47.4) were comparable. In addition, CD spectra were recorded for the equimolar mixture of hAIPL1 and P351∆12, as the mixture potentially mimics the protein presence in dominant cone-rod dystrophy. The secondary structure composition and the Tm value of the mixture were similar to those of hAIPL1 (data not shown).
Table 1.
Biochemical properties of hAIPL1 and the PRD mutants
| Protein | Helix (%) | Strand (%) | Tm (°C) |
Kd (µM) AMCA-CtPDE6A |
Kd (µM) F-CtHSP90 | Kd (µM) Pγ35BC |
|---|---|---|---|---|---|---|
| hAIPL1 | 43.5 | 10.9 | 45.0 | 0.11 ± 0.03* | 20.8 ± 3.4 | 0.99 ± 0.27 |
| R302L | 44.2 | 11.3 | 46.8 | 0.16 ± 0.02 | 27.2 ± 6.1 | 0.81 ± 0.07 |
| P351∆12 | 42.7 | 11.9 | 43.3 | 0.14 ± 0.04 | 32.8 ± 2.0 | 1.38 ± 0.33 |
| P376S | 42.5 | 11.5 | 46.2 | 0.20 ± 0.01 | 23.0 ± 2.2 | 0.95 ± 0.28 |
| hAIPL11–316 | 40.3 | 13.7 | 44.9 | 0.24 ± 0.05 | 20.8 ± 1.4 | 0.73 ± 0.31 |
Mean ± SE (n ≥ 3).
hAIPL1 and the PRD mutants are monomeric proteins
To test if the dominant phenotype of P351∆12 can be explained by the protein dimerization, we estimated MW of hAIPL1 and the PRD mutants using gel filtration and SLS. The calculated MW of the His-tagged hAIPL1 is 46.4 kDa. Elution profiles of hAIPL1, R302L, P351∆12, and P376S on a calibrated Superdex 200 column were consistent with monomeric AIPL1 proteins with MW of ~ 46 kDa (data not shown). According to SLS measurements, the MW values of the hAIPL1 proteins were as follows: hAIPL1, 47.1 ± 0.6; R302L, 51.9 ± 2.4; P351∆12, 52.8 ± 1.1; and P376S, 50.9 ± 0.6 kDa. Neither the gel filtration tests nor SLS measurements indicated the presence of dimers or higher oligomeric states of hAIPL1 and the mutants.
hAIPL1 and PRD mutants potently bind the farnesylated C-terminus of PDE6A
Interaction of hAIPL1, hAIPL11–316, R302L, P351∆12, and P376S with the prenylated C-termini of PDE6 was probed using the AMCA-labeled C-terminal PDE6A peptide GAPASKSC (AMCA-CtPDE6A-farnesyl), in which the Cys residue was S-farnesylated and carboxymethylated. The binding assay is based on fluorescence resonance energy transfer between Trp residues in hAIPL1 and AMCA. Using this assay, AMCA-CtPDE6A-farnesyl bound to hAIPL1 with Kd of 0.11 ± 0.03 µM (Fig. 6). Compared to hAIPL1, hAIPL11–316, R302L, and P376S displayed modest ~ 1.5–2 fold reductions in affinity to AMCA-CtPDE6A-farnesyl (Table 1). To determine if hAIPL1 can bind the geranylgeranyl moiety of PDE6B, we used N-acetyl-S-geranylgeranyl-L-cysteine (Enzo, Farmingdale, NY, USA) in competition studies with AMCA-CtPDE6A-farnesyl. Excess of the geranylgeranyl compound partially reversed AMCA-CtPDE6A-farnesyl binding to hAIPL1 (data not shown) suggesting that AIPL1 binds the geranylgeranyl moiety. However, the quantitative analysis was not possible due to low solubility of the geranylgeranyl compound in physiological solutions.
Fig. 6.

Binding of hAIPL1 and P351D12 to the C-terminus of PDE6A. (a) Emission spectra of hAIPL1 (100 nM) (dashed line) alone or mixed with AMCA-CtPDE6A-farnesyl (100 nM) (solid line; corrected for the probe alone) were recorded with excitation at 280 nm. (b). Increases in AMCA-CtPDE6A-farnesyl fluorescence due to fluorescence resonance energy transfer (FRET) (λex = 280 nm, λem = 440 nm) are plotted as a function of hAIPL1 or P351∆12 concentration and fit with an equation for binding with ligand depletion. Results from one of three similar experiments are shown.
HSP90 binding to the AIPL1 TPR domain is not significantly altered by the hAIPL1 PRD mutations
The C-terminal HSP90a peptide representing the key site of HSP90 interaction with TPR domains was labeled with FITC (F-CtHSP90) and utilized in the fluorescence polarization binding assay (Fig. 7). From the assay, the affinity of F-CtHSP90 for hAIPL1 (Kd = 20.8 ± 3.4 µM) is comparable to the reported affinity of HSP90 signature peptides for the aryl hydrocarbon receptor-interacting protein AIP (Morgan et al. 2012). Truncation of PRD had no effect on binding of F-CtHSP90 to hAIPL1 (Table 1). The PRD mutants bound to F-CtHSP90 similarly to hAIPL1 or slightly weaker (Table 1).
Fig. 7.

Binding of hAIPL1 and P351∆12 to the C-terminus of HSP9α. Fluorescence polarization (mP) of F-CtHSP90 is plotted as a function of hAIPL1 or P351∆12 concentration and fit with an equation for binding without ligand depletion. Results from one of three similar experiments are shown.
hAIPL1 interacts with the Pc-subunit of PDE6 and inhibits basal activity of holoPDE6
The inhibitory Pγ-subunit appears to be critical for the folding and/or assembly of holoPDE6 (Tsang et al. 1996). We have explored the potential interaction of hAIPL1 with Pγ using the fluorescently labeled protein Pγ35BC. A large fluorescence increase in Pγ35BC was observed in the presence of hAIPL1 that was reversible with the addition of excess unlabeled Pγ. This interaction between hAIPL1 and Pγ (Kd = 0.99 ± 0.27 µM) was independent of the presence of PRD, as hAIPL11–316 and mAIPL1 bound Pγ35BC with similar affinity (Fig. 8a, Table 1). Addition of hAIPL1-PRD had no effect on the Pγ35BC fluorescence, nor did it compete with hAIPL1 for Pγ35BC binding (data not shown). The R302L, P351∆12, and P376S mutants bound Pγ35BC comparably to hAIPL1 (Fig. 8a, Table 1).
Fig. 8.

(a) Binding of hAIPL1, hAIPL11–316, mAIPL1, and P351∆12 to Pγ35BC. Increases in Pγ35BC fluorescence due to binding of the hAIPL1 proteins (λex = 445 nm, λem = 490 nm) are plotted as a function of the proteins’ concentration and fit with an equation for binding without ligand depletion. Results from one of three similar experiments are shown. (b). Binding assays of hAIPL1 to Pγ68BC alone (50 nM) (●) or Pγ68BC pre-incubated with PDE6AB (50 nM) (■) were performed as in (a). The Kd values of 2.5 ± 0.8 µM (●) and 5.2 ± 1.7 µM (■) were not significantly different (p = 0.23). Results from one of three similar experiments are shown. (c) Basal activity of holoPDE6 in hypotonic extract of mouse retina was measured in the absence (100%) or presence of increasing concentrations of hAIPL1, P351∆12, and mAIPL1. The results of a representative experiment are shown. From three similar experiments, the Ki value of 0.15 ± 0.01 µM for mAIPL1 was somewhat lower compared to the Ki values for hAIPL1 (0.47 ± 0.07 µM, p = 0.01) and P351∆12 (0.45 ± 0.02 µM, p = 0.001).
To determine if hAIPL1 is capable of binding Pγ complexed with PDE6AB, we utilized Pγ68BC labeled with the reporter probe at Cys68. The fluorescence of Pγ68BC is not affected by its binding to PDE6AB (Granovsky et al. 1998). The affinity of hAIPL1 for Pγ68BC was slightly lower compared to Pγ35BC (Fig. 8b). hAIPL1 retained its ability to bind Pγ68BC after the latter formed a tight complex with PDE6AB (Fig. 8b). Considering this finding, we next examined the potential effects of hAIPL1, P351∆12, and mAIPL1 on basal activity of holoPDE. Interestingly, the AIPL1 proteins inhibited basal activity of holoPDE6 (Fig. 8c), while they had no effect on trypsin-activated PDE6AB (data not shown). mAIPL1 was a slightly more potent inhibitor of holoPDE6 compared to hAIPL1 or P351∆12 (Fig. 8c). Thus, AIPL1 and Pγ-PDE6AB-Pγ appear to form a complex in which basal PDE6 activity is suppressed.
Discussion
Structural information on AIPL1 is critical to understanding the mechanisms of the protein mutations leading to retina disease. Low resolution structures or models based on SAXS analyses are becoming a valuable tool in structural biology, particularly in the absence of atomic structures (Putnam et al. 2007; Jacques and Trewhella 2010; Petoukhov and Svergun 2013). No high resolution structures are available for AIPL1. However, NMR structures have been solved for the individual FKPB and TPR-domains of aryl hydrocarbon receptor-interacting protein, which shares significant sequence homology with AIPL1 (Morgan et al. 2012; Linnert et al. 2013). Previously, we have modeled the solution structure of mouse AIPL1 using the protein SAXS profile and the known structures of the FKPB and TPR domains (Majumder et al. 2013). Unlike non-primate AIPL1 proteins, hAIPL1 contains PRD of unknown function, in which recessive LCA-linked mutations (A336∆2, P376S) and a dominant cone-rod dystrophy-linked mutation (P351∆12) had been identified (Sohocki et al. 2000b; Dharmaraj et al. 2004; Stone 2007). We examine the solution structure of hAIPL1 with SAXS to gain insights into the potential function of PRD and the mechanisms of PRD mutations. A model of hAIPL1 in solution derived from the SAXS data indicated that PRD assumes an extended conformation and does not interact with the rest of the molecule. Consistent with the extended conformation of PRD in hAIPL1, the SAXS profile of the C-terminally truncated hAIPL11–316 yielded a markedly reduced Rg compared to the full-length protein. Protein sequences rich in Pro often form unstructured random coils unless an induced conformation is stabilized by intra- or inter-molecular interactions. Our CD analysis of the individual hAIPL1-PRD indicates that it is a random coil. Furthermore, results from gel filtration, DLS, and SLS experiments using isolated hAIPL1-PRD suggest that it is elongated similarly as PRD in the full-length hAIPL1. Thus, the conformation of hAIPL1 PRD is not fixed, but rather it is a flexible ensemble of extended conformations.
The apparent lack of direct communication between the PRD and FKBP/TPR-domains in the solution structure of AIPL1 raises the question of potential consequences of PRD mutations for the protein function. A 2-bp deletion in AIPL1-A336∆2 causes a frame shift that delays translation termination and leads to a 69-residue-long artificial adduct. This mutation results in expression of insoluble AIPL1 protein in transfected cells (Gallon et al. 2004). On the other hand, our CD analysis confirmed previous observations that P351∆12 and P376S are folded and stable proteins (Gallon et al. 2004). In contrast to previous observations (Gallon et al. 2004), the P376S (and P351∆12) mutation had no effect on the secondary structure composition of hAIPL1. Furthermore, the equivalent Rg values for hAIPL1, P351∆12 and P376S derived from the SAXS analyses suggest that the PRD mutations do not significantly alter the global conformation of hAIPL1. We tested if PRD or its mutations influence known interactions of hAIPL1 with the C-termini of PDE6A and HSP90 (Hidalgo-de-Quintana et al. 2008; Li et al. 2013; Majumder et al. 2013). Only minor variations in the affinity of mutant AIPL1 proteins for the PDE6A and HSP90 probes were detected.
We next considered the possibility that PRD mutations alter interactions of hAIPL1 with a previously unrecognized partner. The Pγ-subunit was examined as the candidate since it plays an essential role in assembly of PDE6. The PDE6AB heterodimer is reduced and non-functional in the Pγ-knock-out mice (Tsang et al. 1996). Furthermore, Pγ apparently binds PDE6AB near the prenylated C-termini (Barren et al. 2009; Zhang et al. 2015), and thus it might be well positioned for interaction with hAIPL1 during PDE6 maturation. Indeed, we found that Pγ interacts with hAIPL1. Moreover, hAIPL1 was able to bind Pγ associated in a tight complex Pγ-PDE6AB-Pγ and inhibit basal activity of holoPDE6. These findings represent the first indication that AIPL1 may interact with folded PDE6. Thus, they may provide insights into the mechanism of AIPL1 action during PDE6 folding and assembly. Yet, the hAIPL1/Pγ interaction was independent of PRD, and the PRD mutations did not appreciably affect the affinity of hAIPL1 for Pγ (Table 1). Overall, our analysis did not reveal functional deficiencies in the PRD mutants that may account for the loss of AIPL1 function in LCA. Such an outcome appears to be consistent with the solution structure of hAIPL1, which offers no structural rationale for a direct impact of PRD mutations on the FKBP and TPR-domains. Although functional defects of R302L and P376S may have been undetected, an alternative explanation is that these are benign variants rather than disease-causing mutations. This possibility is strongly supported by a recent study of AIPL1 variants in a large cohort of LCA patients, which in particular challenged pathogenicity of the R302L and P376S mutations (Tan et al. 2012).
The P351∆12 mutation linked to dominant cone-rod dystrophy cannot be a benign variant, as the mutant phenotype was confirmed in a mouse model (Ku et al. 2015). The phenotype was observed in the double-transgenic animals expressing P351∆12 and WT hAIPL1, but not in mice co-expressing P351∆12 and mouse AIPL1 (Ku et al. 2015). Dimerization of hAIPL1 and P351∆12 was suggested as a potential underlying mechanism of the dominant disorder (Ku et al. 2015). Our tests show that hAIPL1 and its PRD mutants are monomeric proteins with no propensity for dimerization. Hypothetically, P351∆12 may interfere with the function of WT AIPL1 even in the absence of protein dimerization. Two AIPL1 molecules may be forced into proximity by the HSP90 dimer and/or by the PDE6 subunits during assembly of the PDE6 catalytic dimer. Remarkably, the mouse model of cone-rod dystrophy revealed persistent decreases in the level of Pγ (Ku et al. 2015). In light of our identification of Pγ as a novel partner of hAIPL1, we speculate that this interaction might be altered by the P351∆12 mutation. Our results support an idea that PRD plays no essential role in normal functions of hAIPL1 including interactions with PDE6AB, Pγ, and HSP90. However, P351∆12 may affect the recruitment of Pγ to the PDE6-chaperone complex in the context of a structured cellular environment. Validation of this hypothesis will require reconstitution of the functional PDE6-chaperone complex outside living photoreceptor cells, which has been as yet unsuccessful. Our study suggests a new layer of complexity of the mechanisms of AIPL1 mutations in disease rooted in the intricate process of chaperoning of PDE6.
Acknowledgments and conflict of interest disclosure
This work was supported by the National Institutes of Health grant EY-10843 to N.O.A. A.M. is supported by a Pediatric Ophthalmology Career-Starter Research Grant from the Knights Templar Eye Foundation. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The SIBYLS beamline at the Advanced Light Source, Lawrence Berkeley National Laboratory is supported in part by the DOE program Integrated Diffraction Analysis Technologies (IDAT) under Contract Number DE-AC02-05CH11231 with the U.S. Department of Energy. For the SAXS data collection, we are grateful to the staff at 18-ID at the Advanced Photon Source, Argonne National Laboratory and at the SIBYLS beamline at the Advanced Light Source, Lawrence Berkeley National Laboratory. The authors declare no conflicts of interest.
All experiments were conducted in compliance with the ARRIVE guidelines.
Abbreviations used:
- AIPL1
aryl hydrocarbon receptor-interacting protein-like 1
- AMCA
6-((7-amino-4-methylcoumarin-3-acetyl)amino) hexanoic acid, succinimidyl ester
- BC
3-(bromo acetyl)-7-diethyl aminocoumarine
- CD
circular dichroism
- DLS
dynamic light scattering
- FKBP
FK506-binding proteins
- FRET
fluorescence resonance energy transfer
- LCA
leber congenital amaurosis
- PDE6AB
catalytic subunits of rod PDE6
- PDE6
photoreceptor phosphodiesterase-6
- PRD
proline-rich domain of human AIPL1
- Pγ
the inhibitory γ-subunit of PDE6
- SAXS
small angle X-ray scattering
- SLS
static light scattering
- TPR
tetratricopeptide repeat
References
- Aguila M, Bevilacqua D, McCulley C et al. (2014) Hsp90 inhibition protects against inherited retinal degeneration. Hum. Mol. Genet 23, 2164–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artemyev NO, Natochin M, Busman M, Schey KL and Hamm HE (1996) Mechanism of photoreceptor cGMP phosphodiesterase inhibition by its gamma-subunits. Proc. Natl Acad. Sci. USA 93, 5407–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barren B, Gakhar L, Muradov H, Boyd KK, Ramaswamy S and Artemyev NO (2009) Structural basis of phosphodiesterase 6 inhibition by the C-terminal region of the gamma-subunit. EMBO J 28, 3613–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML and Farber DB (1990) Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature 347, 677–680. [DOI] [PubMed] [Google Scholar]
- Das AK, Cohen PW and Barford D (1998) The structure of the tetratricopeptide repeats of protein phosphatase 5: implications for TPR-mediated protein-protein interactions. EMBO J 17, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmaraj S, Leroy BP, Sohocki MM et al. (2004) The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch. Ophthalmol 122, 1029–1037. [DOI] [PubMed] [Google Scholar]
- Dryja TP, Rucinski DE, Chen SH and Berson EL (1999) Frequency of mutations in the gene encoding the alpha subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci 40, 1859–1865. [PubMed] [Google Scholar]
- Farber DB and Lolley RN (1974) Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 186, 449–451. [DOI] [PubMed] [Google Scholar]
- Gallon VA, Wilkie SE, Deery EC, Newbold RJ, Sohocki MM, Bhattacharya SS, Hunt DM and Warren MJ (2004) Purification, characterisation and intracellular localisation of aryl hydrocarbon interacting protein-like 1 (AIPL1) and effects of mutations associated with inherited retinal dystrophies. Biochim. Biophys. Acta 1690, 141–149. [DOI] [PubMed] [Google Scholar]
- Granovsky AE, McEntaffer R and Artemyev NO (1998) Probing functional interfaces of rod PDE gamma-subunit using scanning fluorescent labeling. Cell Biochem. Biophys 28, 115–133. [DOI] [PubMed] [Google Scholar]
- Hidalgo-de-Quintana J, Evans RJ, Cheetham ME and van der Spuy J (2008) The Leber congenital amaurosis protein AIPL1 functions as part of a chaperone heterocomplex. Invest. Ophthalmol. Vis. Sci 49, 2878–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Roepman R, Koenekoop RK and Cremers FP (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res 27, 391–419. [DOI] [PubMed] [Google Scholar]
- Jacques DA and Trewhella J (2010) Small-angle scattering for structural biology–expanding the frontier while avoiding the pitfalls. Protein Sci 19, 642–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J, Bonneau D, Frezal J, Munnich A and Dufier JL (1990) Clinical and genetic heterogeneity in retinitis pigmentosa. Hum. Genet 85, 635–642. [DOI] [PubMed] [Google Scholar]
- Koenekoop RK (2004) An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv. Ophthalmol 49, 379–398. [DOI] [PubMed] [Google Scholar]
- Kolandaivelu S, Huang J, Hurley JB and Ramamurthy V (2009) AIPL1, a protein associated with childhood blindness, interacts with alpha-subunit of rod phosphodiesterase (PDE6) and is essential for its proper assembly. J. Biol. Chem 284, 30853–30861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmaoglou M, Schwarz N, Bett JS and Cheetham ME (2008) Molecular chaperones and photoreceptor function. Prog. Retin. Eye Res 27, 434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku CA, Chiodo VA, Boye SL, Hayes A, Goldberg AF, Hauswirth WW and Ramamurthy V (2015) Viral-mediated vision rescue of a novel AIPL1 cone-rod dystrophy model. Hum. Mol. Genet 24, 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zoldak G, Kriehuber T, Soroka J, Schmid FX, Richter K and Buchner J (2013) Unique proline-rich domain regulates the chaperone function of AIPL1. Biochemistry 52, 2089–2096. [DOI] [PubMed] [Google Scholar]
- Linnert M, Lin YJ, Manns A, Haupt K, Paschke AK, Fischer G, Weiwad M and Lucke C (2013) The FKBP-type domain of the human aryl hydrocarbon receptor-interacting protein reveals an unusual Hsp90 interaction. Biochemistry 52, 2097–2107. [DOI] [PubMed] [Google Scholar]
- Liu X, Bulgakov OV, Wen XH et al. (2004) AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc. Natl Acad. Sci. USA 101, 13903–13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder A, Gopalakrishna KN, Cheguru P, Gakhar L and Artemyev NO (2013) Interaction of aryl hydrocarbon receptor-interacting protein-like 1 with the farnesyl moiety. J. Biol. Chem 288, 21320–21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin ME, Ehrhart TL, Berson EL and Dryja TP (1995) Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc. Natl Acad. Sci. USA 92, 3249–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RM, Hernandez-Ramirez LC, Trivellin G, Zhou L, Roe SM, Korbonits M and Prodromou C (2012) Structure of the TPR domain of AIP: lack of client protein interaction with the C-terminal alpha-7 helix of the TPR domain of AIP is sufficient for pituitary adenoma predisposition. PLoS ONE 7, e53339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muradov H, Boyd KK and Artemyev NO (2006) Analysis of PDE6 function using chimeric PDE5/6 catalytic domains. Vision. Res 46, 860–868. [DOI] [PubMed] [Google Scholar]
- Pelikan M, Hura GL and Hammel M (2009) Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys 28, 174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petoukhov MV and Svergun DI (2013) Applications of small-angle X-ray scattering to biomacromolecular solutions. Int. J. Biochem. Cell Biol 45, 429–437. [DOI] [PubMed] [Google Scholar]
- Pittler SJ and Baehr W (1991) Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc. Natl Acad. Sci. USA 88, 8322–8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hammel M, Hura GL and Tainer JA (2007) X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys 40, 191–285. [DOI] [PubMed] [Google Scholar]
- Ramamurthy V, Roberts M, van den Akker F, Niemi G, Reh TA and Hurley JB (2003) AIPL1, a protein implicated in Leber’s congenital amaurosis, interacts with and aids in processing of farnesylated proteins. Proc. Natl Acad. Sci. USA 100, 12630–12635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy V, Niemi GA, Reh TA and Hurley JB (2004) Leber congenital amaurosis linked to AIPL1: a mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl Acad. Sci. USA 101, 13897–13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M and Sali A (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res 38, W540–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Majumder A, Belcastro M, Sokolov M and Artemyev NO (2013) Expression and subcellular distribution of UNC119a, a protein partner of transducin alpha subunit in rod photoreceptors. Cell. Signal 25, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohocki MM, Bowne SJ, Sullivan LS et al. (2000a) Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat. Genet 24, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohocki MM, Perrault I, Leroy BP et al. (2000b) Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol. Genet. Metab 70, 142–150. [DOI] [PubMed] [Google Scholar]
- Sreerama N and Woody RW (1993) A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem 209, 32–44. [DOI] [PubMed] [Google Scholar]
- Stone EM (2007) Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol 144, 791–811. [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF and Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol 11, 515–528. [DOI] [PubMed] [Google Scholar]
- Tan MH, Mackay DS, Cowing J et al. (2012) Leber congenital amaurosis associated with AIPL1: challenges in ascribing disease causation, clinical findings, and implications for gene therapy. PLoS ONE 7, e32330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH, Gouras P, Yamashita CK, Kjeldbye H, Fisher J, Farber DB and Goff SP (1996) Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science 272, 1026–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinkam P, Pons J and Sali A (2012) Structure-based model of allostery predicts coupling between distant sites. Proc. Natl Acad. Sci. USA 109, 4875–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore L and Wallace BA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32, W668–W673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, He F, Constantine R, Baker ML, Baehr W, Schmid MF, Wensel TG and Agosto MA (2015) Domain organization and conformational plasticity of the G protein effector, PDE6. J. Biol. Chem 290, 12833–12843. [DOI] [PMC free article] [PubMed] [Google Scholar]