

Abstract
Distinct populations of effector memory T cells use different homing receptors to traffic to skin and gut (1). Whether tissue selective T cells are needed for early rejection of a neoplasm growing in these tissues remains an open question (2). We chose to study an allogeneic tumor model because growth of such a fully mismatched tumor would signify a profound immune deficit. We implanted allogeneic tumor cells in the skin or gut of mice deficient in either α(1,3) fucosyltransferases IV and VII, enzymes critical for generating E- selectin ligands on skin homing T cells, or β7 integrin, a component of the α4β7 integrin ligand for the mucosal adressin MAdCAM. During the first 9 days after tumor implantation FucTVII−/− mice showed a profoundly impaired capacity to reject tumors growing in skin, but readily rejected tumors implanted in the gut. Rejection of tumors in skin was even more impaired in mice deficient in both FucTIV and FucTVII. This impairment was corrected by infusion of T cells from normal mice. By contrast, β7 integrin−/− mice showed profoundly impaired rejection of tumors in gut, but no defect in skin tumor rejection. These differences were unrelated to antigen recognition or effector function of T cells, since all strains of mice were capable of generating tumor specific CTL’s in vitro against the tumor cell line used in vivo. These results demonstrate that T cell homing defects in vivo impair immune surveillance of peripheral epithelial tissues in a specific and selective fashion.
Keywords: homing molecule, tumor immunity, skin, gut, immune drugs
Introduction
Cancers frequently arise in epithelial tissues that interface with the environment, including skin, lung, and GI tract. The emergence of dramatic clinical results using immune checkpoint inhibitor antibodies for multiple cancers has rekindled interest in tumor immunity (2, 3, 4); however, many unknowns still exist. For example, while many features of tumor immunity have been studied intensively, the role of tissue selective effector-memory T cell trafficking in the early rejection of tumors is incompletely understood (5, 6, 7, 8) . To begin to address this question, we used mice with genetically engineered deficiencies in key T cell skin- and gut-trafficking molecules and studied their impact on tumor immunity in these tissues. In order to set the highest possible threshold for immunosurveillance, we chose fully mismatched allogeneic tumors, mindful that such major antigenic differences would never occur in spontaneous tumors.
T cell homing to skin, in both humans and mice, depends on the interaction of dermal microvascular selectins with specific carbohydrate ligands for E- and P-selectin expressed on specialized effector and memory T cells [1]. These carbohydrate epitopes, including the Cutaneous Lymphocyte-associated Antigen (CLA) expressed on skin-homing effector and memory T cells in humans, are formed by the action of key glycosyltransferases to generate terminally sialylated, fucosylated lactosamines related to sialyl-LewisX (references/can find these in the Sackstein review, attached). Both α(1,3)-fucosyltransferase VII (FucTVII) and α(1,3)-fucosyltransferase IV (FucTIV) have been reported to mediate glycosylation steps critical for the generation of E-, and P-selectin ligands [9]. Mice deficient in FucTVII show diminished, although not absent, trafficking of T cells to skin; this defect is even more profound in mice lacking both FucTVII and FucTIV [9, 10].
T cell homing to gut lamina propria does not appear to depend on selectins, but rather on the integrin α4β7, which initiates the multi-step extravasation process by binding to Mucosal Addressin Cell Adhesion Molecule (MAdCAM) [7]. Naïve T cells express α4β7 at low levels, but α4β7 is much more highly expressed by effector and memory cells generated in gut-draining lymphoid tissue; these latter T cells selectively traffic to the laminia propria in the GI tract [8]. Mice deficient in α4β7 integrin have been shown to have altered immune cell populations in gut associated lymphoid tissue, and altered responses to gastrointestinal viruses and parasites [13,14,15,16,17].
To study the influence of these tissue selective T cell homing molecules on early tumor immunity, we chose a tumor cell line (J558L) that could grow successfully and progressively in both skin and gut of syngeneic mice, and was rapidly rejected in both sites by H-2 mismatched mice. We implanted tumor cells into skin or gut of allogeneic C57BL/6 (H-2d) mice deficient in organ-specific T cell trafficking molecules to assess the role of such T cells in early tumor immunity. Because J558L cells are H-2b and thus completely mismatched at all C57Bl/6 (H-2d) major histocompatability loci, any observed tumor growth represents a significant defect in surveillance.
Methods
Mice.
BALB/c and WT B6 mice were purchased from Charles River Laboratories Inc. (Wilmington, MA). FucTVII−/− B6, FucTIV−/− /VII−/− B6, and β7−/− B6 mice were kindly provided by John Lowe (Genentech, Inc.) and Norbert Wagner (University of Cologne), respectively. Strains were generated from C57BL/6, and inbred for well over 6 generations. Mice were housed under specific pathogen-free conditions at Harvard Institutes of Medicine’s animal facility. Experiments were approved by the Harvard Medical Area Standing Committee on Animals.
T cell cytotoxicity assay.
WT B6, FucTVII−/− B6, β7−/− B6 or syngeneic BALB/c mice splenocytes were co-cultured for 5 days with mitomycin C-treated BALB/c splenocytes to produce effector cell populations. CTL activities were assayed with CytoxiLux flow cytometric cytotoxicity assay kits (OncoImmunin, Gaithersburg, MD) [17]. Briefly, TFL2 fluorescent dye-labeled target J558L cells were cultured in triplicate with effector cells in 96-well plates at various effector/target (E/T) ratios for three hours at 37°C. Plates were centrifuged five minutes at 1400rpm. Cells were then incubated in fluorogenic caspase substrate (75 μl/well) 30 minutes at 37°C followed by two PBS washes. Samples were obtained via FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). Data were analyzed using Flowjo software (Tree Star, Inc., San Carlos, CA). Target cell killing percent (%target cells caspase staining) = [%(caspase+ TFL2+ cells) / (% total TFL2+)] x 100%.
Tumor transplantation into skin and cecum.
Murine J558L BALB/c plasmacytoma was grown in RPMI with 10% FCS, 2% L-glutamate, and penicillin/streptomycin. Injected cells were concentrated to 1×107 cells/mL RPMI (skin) or 3–4×107 cells/mL PBS (gut). For tumors in skin, mice were clipped 24–36 hours pre-transplant, anesthetized with Avertin, and then 106 live J558L cells were injected intradermally using a 30.5G needle. For tumors in gut, the lower abdomen was sterilely incised, and the cecum was externalized. Next,106 live J558L cells were injected into the cecal lamina propria, and the cecum was replaced intraperitoneally. Incisions were closed with 4–0 silk or nylon sutures as described previously [18]. Tumor sizes were greatest diameter of grossly visible tumor mass at skin and gut injection sites. For histopathologic examination, skin and gut samples were fixed in 10% formalin, paraffin embedded, and sections submitted for hematoxylin-eosin staining.
Tumor immunohistochemistry.
4 μm thick formalin-fixed, paraffin-embedded tissue sections were examined. Slides were pre-treated with 10 mM citrate, pH 6.0 (Zymed, South San Francisco, CA) for CD3 or 1.0 mM EDTA pH 8.0 (Zymed) for CD138 in a pressure cooker (Decloaking Chamber, BioCare Medical, Walnut Creek, CA). Pre-diluted polyclonal rabbit anti-CD3 antibody (DAKO, Carpintera, CA) or monoclonal anti-murine CD138 antibody (Pharmingen, San Diego, CA) diluted 1:250 was applied for 1 hour at room temperature. Rabbit anti-rat immunoglobulin antibody at 1:7500 dilution and DAKO Envision kits (DAKO) were used for detection.
CD3+ T cell immunohistochemistry.
6 μm-thick frozen tissue sections were fixed in −20°C acetone for 5 min, air dried, then incubated with 5μg/ml rat anti-mouse CD3 monoclonal antibody (mAb) (Antigenix America Huntington, NY) 30 min at 25°C. After extensive washing, sections were incubated with 1:200 biotinylated anti-rat IgG Ab (Vector, Burlingame, CA) for 30 min at 25°C, washed, then incubated with avidin-biotin conjugate (ABC; Vector) for 30 min at 25°C. Reaction product was detected by incubation with NovaRed (1 min), and sections were counterstained with hematoxylin.
Adoptive T cell transfer.
Primed T cells were obtained from skin-draining lymph nodes of WT B6 mice that had been injected with J558L cells 9 days previously. T cells were purified by negative selection using mouse CD3+ T cell enrichment columns (R & D systems, Minneapolis, MN). Next, 5 × 106 T cells were injected via tail vein into FucTVII−/− B6 mice 48 hours before J558L tumor cell transplantation. Similarly, J558L-naïve WT B6 CD4+ and CD8+ T cells were purified from unmanipulated WT B6 splenocytes using mouse CD4+ and CD8+ T cell enrichment columns (R & D systems, Minneapolis, MN). Next, 5 × 106 naive CD4+ and CD8+ T cells were injected via tail vein into FucTVII−/− B6 mice 48 hours prior to dermal injection of J558L tumor cells.
Statistical analysis.
Mean tumor size was compared between groups at pre-specified times using nondirectional two-sample t tests that account for unequal group variances. Two-sided p values < 0.05 were considered statistically significant.
Results
FucTVII−/− and α4β7−/− strains both generate J558L-reactive cytotoxic T cells.
To assess whether C57/BL6 (H-2b) mice lacking α4β7 integrin (β7−/− B6) or FucTVII (FucTVII−/− B6) could generate cytotoxic T cells to BALB/c targets in vitro, splenic T cells from each strain were activated in vitro via mixed lymphocyte reaction (MLR) against BALB/c splenocytes, and were then assayed for capacity to kill J558L (H-2d) cells. There were no significant differences in the magnitude or efficiency of T cell cytotoxicity among WT B6, β7−/− B6 and FucTVII−/− B6 (Figure S1). Neither transgenic strain showed defects in alloantigen responsiveness, nor generation of tumor-specific cytotoxic effector cells.
Allogeneic tumors implanted in gut grew progressively in mice deficient in β7 integrin but not deficient FucTVII.
To examine anti-tumor immune responses in gut, we injected J588L cells into cecal lamina propria of BALB/c, WT B6, FucTVII−/− B6 and β7−/− B6 mice. After 9 days, large tumors in gut were observed both grossly and histologically in both syngeneic BALB/c (positive control) and H-2 mismatched β7−/− B6 mice. By contrast, no tumor growth was detected grossly or histologically in FucTVII−/− B6 or WT B6 mice (Figure 1A and 2A). We assessed J588L immunohistochemistry using anti- CD138 (syndecan) antibodies. Large CD138-positive tumor cell aggregates were observed in cecal lamina propria in both the BALB/c and β7−/− B6 mice (Figure 2A). We next looked for T cell infiltration using anti-CD3 antibodies. Day 19 BALB/c mice still showed virtually no T cell infiltration (Figure 3H. Tumors in β7−/− B6 mice showed some CD3+ infiltration by day 9, and were much smaller or undetectable by day 14 (data not shown.) Therefore, during the first 9 days, gut tumor rejection was delayed by β7 integrin deficiency, but gut tumor immunity was preserved in FucTVII deficient mice.
Figure 1: (A) Early rejection of tumors in gut was seen in FucTVII−/− B6 mice but not in β7−/− B6 mice; (B) early rejection of tumors in skin was seen in β7−/− B6 mice but not in FucTVII−/− B6 mice , and CD3+ T cells from wild-type mice largely restored tumor rejection in skin of FucTVII−/− B6 mice.
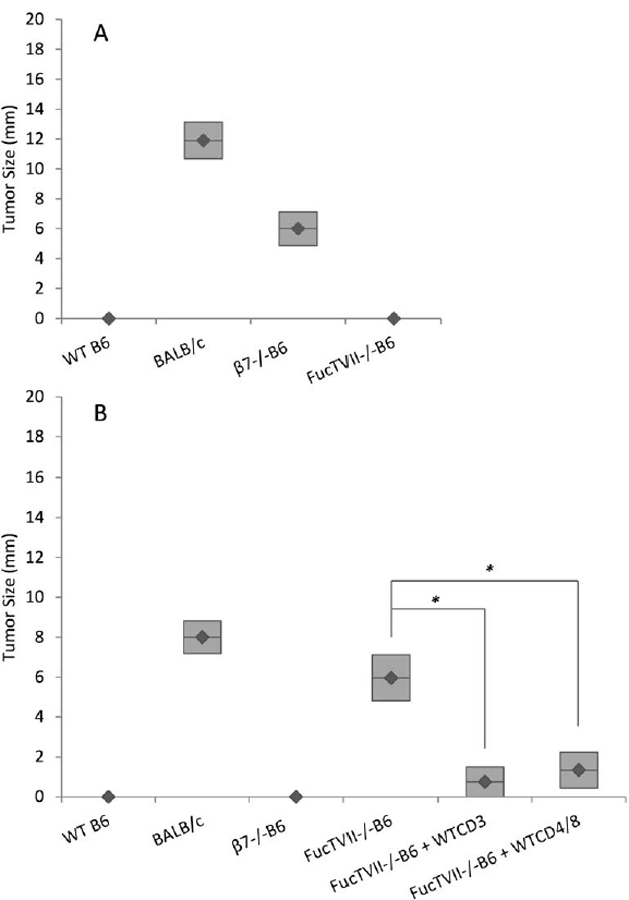
A. Tumors were measured 9 days after tumor cells were injected into gut. Large tumors were present in BALB/c (n=9) and β7−/− B6 mice (n=5), but not in WT B6 (n=3) and FucTVII−/− B6 (n=3) mice. B. Tumors were measured 9 days after tumor cells were injected into skin. Tumors were present in BALB/c (n=4) and FucTVII−/− B6 (n=8) mice, but not in WT B6 (n=7) and β7−/− B6 mice (n=5). Purified CD3+ cells from J558L tumor-primed WT B6 mice were injected into FucTVII−/− B6 mice (n=4) 2 days prior to tumor implantation. Purified CD4+ and CD8+ T cells from tumor-naïve WT B6 mice were injected into FucTVII−/− B6 mice (n=5) 2 days prior to tumor implantation. The bars represent the mean ± SE. Mean tumor size was significantly different for FucTVII−/−B6 and FucTVII−/−B6+WTCD3 (t(df = 9.99) = 3.78, p = 0.003) and for FucTVII−/−B6 and FucTVII−/−B6 + WTCD4/8 (t(df = 10.99) = 3.16, p = 0.009). All tests are 2-tailed t-tests. All tumor sizes in skin and gut are based on histologic examination.
Figure 2: (A) Tumors grew in gut of β7−/− B6 mice but not FucTVII−/− B6 mice; (B) tumors grew in skin of FucTVII−/− B6 mice but not β7−/− B6 mice.
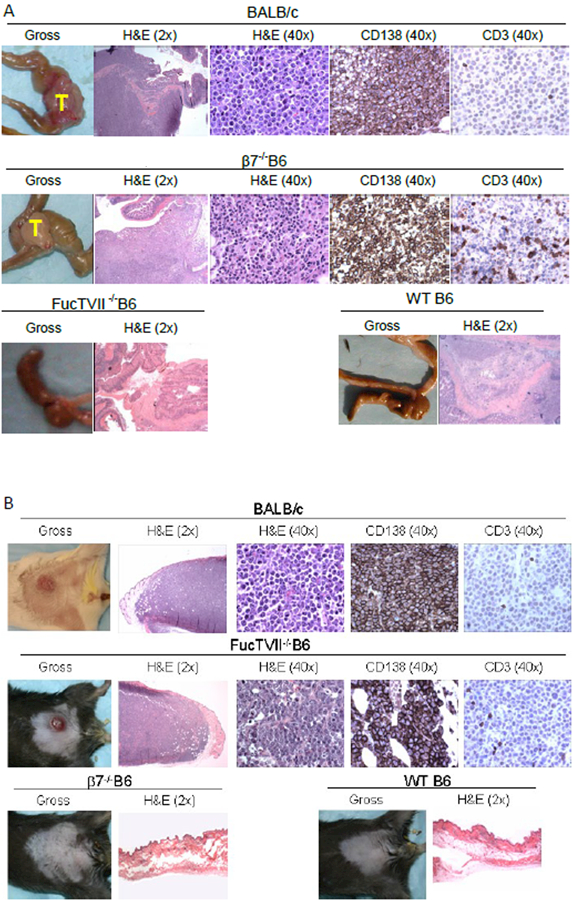
(a) Histology of J558L tumor implantation sites at 9 days after implantation showed large aggregates of CD138/Syndecan+ J558L tumor cells in cecal lamina propria in both BALB/c, and β7−/− B6 recipients, but not in WT B6 or FucTVII−/− B6 mice. Representative histopathology preparations are shown for each (magnifications in parentheses). (b) Tumors grew in skin of FucTVII−/− B6 mice but not β7−/− B6 mice. Histology of J558L tumor implantation sites at 9 days after implantation showed large dermal aggregates of CD138/Syndecan+ J558L tumor cells in both BALB/c and FucTVII−/− B6 recipients, but not in WT B6 or α4β7−/− B6 mice. Representative histopathology preparations are shown for each (magnifications in parentheses).
Figure 3: Infiltration of tumors in skin by CD3+ cells correlates with delay in allogeneic tumor rejection seen in FucTVII−/− and FucTIV−/−/VII−/− mice.
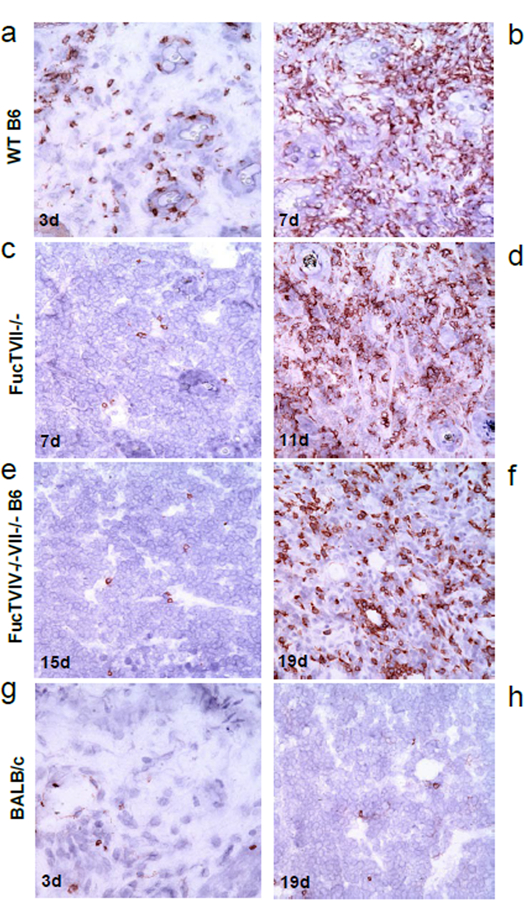
(a) Tumors from WT B6 mice showed mild-to-moderate CD3+ infiltrate as early as 3 days after tumor implantation. (b) Tumors from WT B6 mice showed robust infiltrate at 7 days. (c) Tumors from FucTVII−/− B6 mice showed minimal infiltrate at 7 days. (d) Tumors from FucTVII−/− B6 mice showed significant CD3+ infiltrate at 11 days, as tumors started to regress. (e) Tumors from double knockout FucTIV−/−/VII−/− B6 mice showed minimal infiltrate at 15 days. (f) Tumors from double knockout FucTIV−/−/VII−/−B6 mice showed significant CD3+ infiltrate at 19 days, as tumors started to shrink. (g) BALB/c showed minimal CD3+ infiltrate at 3 days. (h) BALB/c still showed minimal infiltrate at 19 days. Representative histopathology preparations are shown for each.
Allogeneic tumors grew progressively in skin of mice lacking FucTVII but not β7 integrin.
To assess the capacity of mice to reject the same tumor in skin, we injected J558L tumor cells intradermally into BALB/c, B6, β7−/− B6, and FucTVII−/− B6 mice, and measured tumors regularly (Figure 4). As expected, tumors grew progressively in skin of syngeneic BALB/c mice, and showed essentially no growth in allogeneic control B6 mice. In contrast, progressive tumor growth was seen in the skin of FucTVII−/− B6 mice for the first 9–12 days. A later time points, these tumors regressed. Figure 2B shows gross and histological appearance of tumors at day 9. At this time point, CD138+ tumors in skin of BALB/c mice and FucTVII−/− mice were readily identifiable and contained few CD3+ T cells. In contrast, on day 9 there was no gross or microscopic tumor in either WT B6 or in β7−/− B6 mice, consistent with successful early tumor rejection. Thus, FucTVII−/− mice showed profoundly impaired early rejection of allogeneic tumor cells in skin, but were able to rapidly reject the same tumor implanted in the gut mucosa. In contrast, β7−/− mice have impaired ability to reject allogeneic tumors implanted in gut, but have fully intact skin tumor immunity; that is, they can rapidly reject the same tumor in the skin that they did not reject in the gut.
Figure 4: J558L tumors showed early growth in skin of BALB/c and FucTVII−/− B6 mice but not WT B6 mice; early tumor growth was even greater in skin of double-knockout FucTIV−/−/VII−/− B6 mice than in single knockout FucTVII−/− B6 mice.
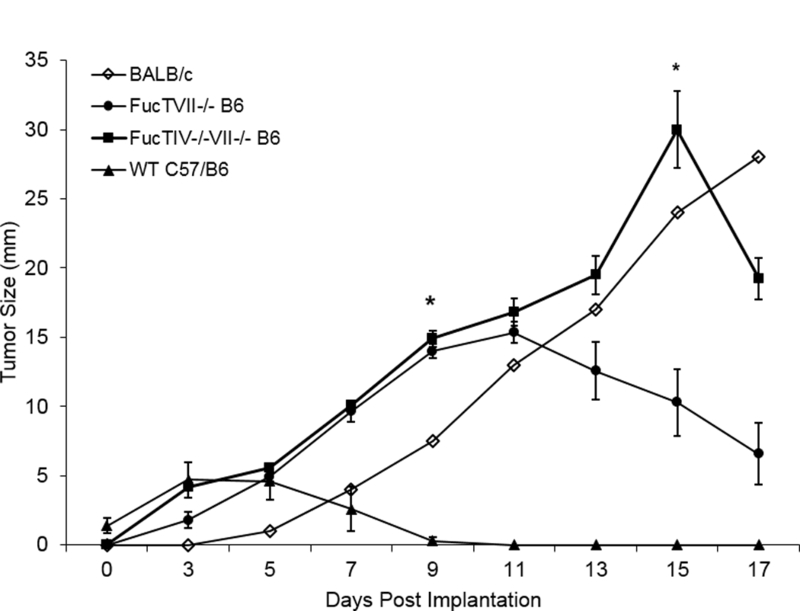
J558L cells were injected into abdominal skin and external dimensions of the resulting tumors were measured. Tumors grew progressively in the skin of syngeneic BALB/c mice (n=1) and allogeneic FucTVII−/− B6 mice (n=7) for the first 9–11 days. Tumors grew faster in double knockout FucTIV−/−/VII−/− B6 mice (n=11) than either BALB/c or single knockout FucTVII−/− B6 mice (n=5) (day 15, t = 5.31, p < 0.001). Tumors began to regress later in the double knockout FucTIV−/−/VII−/− B6 mice versus the single knockout FucTIV−/−/VII−/− B6 mice. In a separate experiment, J558L tumor cells injected into abdominal skin of allogeneic WT B6 mice (n=5) did not show significant growth. Of note, no tumor growth was seen in >15 WT B6 mice. Comparisons of mean tumor size at day 9 show tumor size was significantly larger in single knockout FucTVII−/− B6 mice (n=5) (t = 22.4, p < 0.001) and double knockout FucTIV−/−/VII−/− B6 mice (n=11) (t = 21.8, p < 0.001) than in WT B6 (n = 5). Comparisons of mean tumor size at day 15 show tumors were significantly larger in double knockout FucTIV−/−/VII−/− B6 mice (n=11) than single knockout FucTVII−/− B6 mice (n=5) (t = 22.4, p < 0.001). All tests are 2 tailed t-tests. The bars represent the mean ± SE. These tumor measurements were taken in vivo and include surrounding skin and associated inflammatory infiltrates, so tumor sizes may be overstated.
Accelerated tumor growth in skin of FucTVII−/− B6 mice was prevented by adoptive transfer of WT B6 T cells.
To assess further whether the role of FucTVII in rejecting tumors in skin depends on CD3+ T cells, we injected WT B6 T cells (FucTVII+/+) into FucTVII−/− B6 mice before tumor implantation. Two separate experiments were performed using primed or naïve WT B6 T cells, respectively. Primed CD3+ T cells were purified from skin-draining LN of WT B6 mice that had been injected with J558L cells 9 days prior and injected into FucTVII−/− B6 mice 2 days before tumor implantation. FucTVII−/− B6 mice that received primed WT B6 CD3+ T cells showed little or no tumor growth in skin at day 9, in contrast to the large tumors in skin of unmanipulated FucTVII−/− B6 mice (Figure 1B). To determine if this protective response depended on pre-exposure to antigen in skin, naïve CD4+ and CD8+ splenocytes were purified from unmanipulated WT B6 mice and injected into FucTVII−/− B6 mice 2 days before tumor implantation. As with primed T cells, FucTVII−/− B6 mice that received naïve WT B6 T cells showed little or no tumor growth in skin at day 9. Together these data suggest that tumor rejection of J588L cells in skin by B6 mice is dependent upon T cells that can express FucTVII, and upon the capacity of T cells to enter skin where they can recognize alloantigen and mediate anti-tumor effector functions.
Tumors grew more rapidly in skin of mice lacking both FucTIV and FucTVII.
Since skin leukocyte homing is diminished but not absent in mice lacking FucTVII, we performed additional experiments in mice that lack both FucTVII and FucTIV and therefore do not express any selectin ligands. We injected J558L cells intradermally into BALB/c, FucTVII−/− B6 and FucTIV−/− /VII−/− B6 mice. As before, tumors grew progressively in the skin of syngeneic BALB/c mice (Figure 4), and in skin of FucTVII−/− mice until day 9–12, after which tumors began to regress. In contrast, tumor growth in the skin of FucTIV−/− /FucTVII−/− B6 mice was very robust over the first 15 days, outstripping even that in syngeneic BALB/c mice (Figure 4). Tumors in FucTIV−/− /FucTVII−/− B6 mice subsequently shrank, however, and were ultimately rejected.
Delayed CD3 effector T cell infiltration of tumors in FucTVII−/− and FucTIV−/− /FucTVII−/− skin.
Despite early tumor growth in skin, both FucTVII−/− and FucTIV−/− /FucTVII−/− B6 mice ultimately rejected J558L tumors. We assessed whether this was associated with CD3+ T cell infiltration by performing immunohistochemistry of tumors from these mice at various time points (Figure 3). As expected, reduced or delayed T cell infiltration correlated with tumor rejection delays across the different mouse strains (Figure 3). WT B6 showed mild-to-moderate T cell infiltrates at day 3 (Figure 3a), and robust T cell infiltrates by day 7 (Figure 3b). Mice with deletion of FucTVII or FucTIV and FucTVII showed delays in both T cell infiltration and tumor regression. Tumors in skin of FucTVII−/− B6 mice showed little infiltrate at 7 days (Figure 3c), but significant infiltrate at 11 days as tumors started to regress (Figure 3d). FucTIV−/− /VII−/− B6 mice showed minimal infiltrate even at 15 days (Figure 3e); however, they showed significant CD3+ infiltrates at 19 days as tumors started to regress (Figure 3f). By comparison, BALB/c mice showed only trace infiltration of both early (day 3, Figure 3g) and late (day 19, Figure 3h) samples.
Discussion
Epithelial interfaces with the environment, including skin, gut, and lung, are frequent sites of injury, infection, and malignant tumor development. Distinct populations of memory effector T cells have been identified that traffic preferentially to skin and gut, respectively [1]. This trafficking is mediated or facilitated by expression of α4β7 on gut-homing memory effector T cells, and by E- and P-selectin ligands on skin-homing memory effector T cells [1]. In this study, we asked whether mice deficient in these skin or gut homing molecules would show a tissue-specific deficit in rejection of an immunogenic allogeneic tumor. If early tumor immunosurveillance is independent of tissue-specific T cell homing, mice deficient in homing molecules should have unimpaired early tumor immunity. On the other hand, if tumor immunosurveillance is related to tissue-specific T cell trafficking, then even highly immunogenic allogeneic tumors might be functionally ignored in a tissue-selective fashion when key trafficking molecules are absent. To investigate this hypothesis, H-2d tumors (J588L) were implanted in the skin and gut, respectively, of C57BL/6 (H-2d mismatched) (a) WT, (b) FucTVII−/−, and (c) β7 −/− mice as well as control (d) BALB/c (H-2d syngeneic) mice, and tumor growth was monitored over time. Although mindful that spontaneous tumors are never mismatched, we chose fully MHC-mismatched tumors to highlight the role of trafficking. In other words, survival of a fully mismatched tumor implies a very profound immunosurveillance defect.
The data demonstrate that unrestrained initial H-2d tumor growth occurred in the skin of FucTVII−/− B6 mice and the gut of α4β7−/− B6 mice (Figures 1 and 2). . However, FucTVII−/− mice readily rejected the same tumor in gut, and α4β7−/− mice readily rejected this tumor implanted in skin. These data, coupled with in vitro results confirming that both mouse strains produce functional cytotoxic T lymphocytes capable of recognizing J558L tumor antigens, suggest that the observed tumor immunity defect was the result of impaired tissue-specific homing of effector T cells, rather than impaired antigen recognition or response.
To test the hypothesis that FucTVII−/− mice have no other impediment to effective anti-tumor immunity than defective recruitment of T cells into skin, we adoptively transferred purified WT (FucTVII+/+) B6 T cells into FucTVII−/− B6 mice 48 hours before tumor implantation. Transferring either naïve or primed WT T cells prevented J588L tumor growth in the skin of FucTVII−/− mice (Figure 1). Thus, effector T cells expressing FucTVII, but not those deficient in FucTVII, were capable of recognizing and rejecting allogeneic tumors in skin of FucTVII deficient hosts. These results also demonstrated that FucTVII+ T cells were sufficient for tumor rejection without requiring other FucTVII+ bone marrow derived leukocytes, such as dendritic cells, macrophages, or neutrophils.
In a similar fashion we also show that the β7 integrin chain, which forms heterodimers with both α4 and αE integrin chains, is required for early rejection of allogeneic tumors in gut. Absence of β7 integrin allowed significant early growth of implanted H-2 mismatched tumors in gut, despite their profound immunogenicity. These experiments did not allow us to address the question of whether this effect was due to the absence of αΕβ7 versus α4β7, or both. However, absence of β7 integrin did not inhibit alloantigen recognition and response in vitro, or early rejection of H-2 identical tumors from skin, indicating that in a non-gut microenvironment β7−/− T cells traffic normally and exhibit potent antitumor activity.
It is of interest to note that mice deficient in FucTVII, which reduces but does not eliminate expression of T cell selectin ligands, showed delayed tumor suppression, but were ultimately able to recognize and suppress intradermal implanted tumor. To further elucidate the impact of T-cell surveillance defects on allogeneic tumor growth, we examined tumor growth in the skin of C57BL/6 mice lacking both fucosyltransferases IV and VII. These mice do not make any E- or P-selectin ligands and have a much more profound skin homing defect. We found that tumors grew more rapidly in FucTIV and VII double-knockout mice than in FucTVII single-knockouts, and that infiltrates were delayed longer in the double-knockouts (Figures 3 and 4). These in vivo data show that increasing the T cell homing defect correlates with increased tumor surveillance defect. Furthermore, although immunohistochemical stains showed profound inhibition of CD3+ T cell infiltration during early tumor growth in FucT deficient mice, development of significant T cell infiltrates at later time points was associated with tumor regression. It remains unclear by what mechanism T cells are ultimately able to enter skin based tumors in FucTIV and VII deficient mice, though we speculate that even a very few cells that enter by non-specialized means will eventually proliferate and mediate tumor suppression. It is important to recall that H-2 mismatched tumors are intrinsically more immunogenic than any other tumor, and that when anatomical barriers are disrupted by tumor growth, rejection is inevitable.
Immunosurveillance at epithelial interfaces with the environment can be thought of as occurring on several different levels [1]. Our results are consistent with a model of secondary immunosurveillance in which peripheral epithelial tissues such as skin and gut are patrolled by subsets of resident memory T cells expressing tissue-selective trafficking molecules that allow them to circulate or emigrate to tissues under normal, homeostatic conditions (i.e., in the absence of obvious inflammation) (19, 20, 21). These observations have important implications for studies that seek to induce immune responses in patients with widespread cancer. If our model is correct, then tumor vaccination delivered appropriately through skin or skin draining lymph nodes may help generate effector T cells that more efficiently enter skin )(22). Results from related studies in our laboratory support this prediction [23].
LIMITATIONS
One limitation of this study can also be viewed as a strength. Our hypothesis was that tissue specific trafficking molecules on skin and gut homing effector/memory T cells, respectively, were essential for the entry of tumor specific T cells into skin and gut and subsequent tumor rejection. We used a highly artificial tumor system (H-2 complete mismatch) to test our hypothesis in the most stringent possible way, as such tumors are highly immunogenic. We were surprised how large such tumors could grow in lamina propria and dermis in the absence of T cells that could specifically infiltrate skin or gut. Only when anatomical barriers were disrupted by large growing tumors could systemic T cells gain access to them, whereupon they were rapidly rejected.
Another limitation is that we used adoptive transfer pooled WT CD4 and CD8 cells to recover tumor immunity; this prevented analysis of relative contributions of each T cell subset.
Supplementary Material
{kind=link}
Acknowledgements
We are grateful to Carol Botteron for comments, and John D. Omobono and Christina Stephenson for help with preparation of the figures.
Financial support: This work was supported by NIH grants T32 AR07098, R01 AI040124, and R37 AI25082 (all to T.S.K), and was also supported in part by Frontiers: The Heartland Institute for Clinical and Translational Research CTSA KL2RR033177 (awarded to the University of Kansas Medical Center, J.W.). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Non-standard abbreviations:
- β7−/− B6
β7 integrin deficient C57/BL6 mice
- CLA
Cutaneous Lymphocyte-associated Antigen
- CTL
cytotoxic T lymphocyte
- FucTVII−/− B6
Fucosyltransferase VII deficient C57/BL6 mice
- MAdCAM
Mucosal Addressin Cell Adhesion Molecule
- PSGL-1
P-Selectin Glycoprotein Ligand-1
- TCR
T cell receptor
- WT B6
WT B6 wild-type C57/BL6 mice
Footnotes
Summary Sentence: Tumor immune surveillance of peripheral epithelial tissues depends on tissue selective T cell homing
References
- 1.Kupper TS, Fuhlbrigge RC (2004) Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol 4: 211–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shackstein R, Schatton T, & Barthel SR (2017). T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Laboratory Investigation, 97(6), 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, & Allison JP (2015). The future of immune checkpoint therapy. Science, 348(6230), 56–61. [DOI] [PubMed] [Google Scholar]
- 4.Adusumilli PS, Cha E, Cornfeld, Davis T, Diab A, Dubensky TW, … & Olwill SA (2017). New Cancer Immunology Agents in Development: a report from an associated program of the 31st Annual Meeting of the Society for Immunotherapy of Cancer, 2016. Journal for immunotherapy of cancer, 5(1), 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahoney KM, Rennert PD, & Freeman GJ (2015). Combination cancer immunotherapy and new immunomodulatory targets. Nature reviews Drug discovery, 14(8), 561. [DOI] [PubMed] [Google Scholar]
- 6.Dunn GP, Old LJ, Schreiber RD (2004) The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21: 137–148. [DOI] [PubMed] [Google Scholar]
- 7.Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140: 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zitvogel L, Tesniere A, Kroemer G (2006) Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol 6: 715–727. [DOI] [PubMed] [Google Scholar]
- 9.Smithson G, Rogers CE, Smith PL, Scheidegger EP, Petryniak B, et al. (2001) Fuc-TVII is required for T helper 1 and T cytotoxic 1 lymphocyte selectin ligand expression and recruitment in inflammation, and together with Fuc-TIV regulates naive T cell trafficking to lymph nodes. J Exp Med 194: 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erdmann I, Scheidegger EP, Koch FK, Heinzerling L, Odermatt B, et al. (2002) Fucosyltransferase VII-deficient mice with defective E-, P-, and L-selectin ligands show impaired CD4+ and CD8+ T cell migration into the skin, but normal extravasation into visceral organs. J Immunol 168: 2139–2146. [DOI] [PubMed] [Google Scholar]
- 11.Johansson-Lindbom B, Agace WW (2007) Generation of gut-homing T cells and their localization to the small intestinal mucosa. Immunol Rev 215: 226–242. [DOI] [PubMed] [Google Scholar]
- 12.Gorfu G, Rivera-Nieves J, Ley K (2009) Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr Mol Med 9: 836–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato S, Hokari R, Matsuzaki K, Iwai A, Kawaguchi A, et al. (2000) Amelioration of murine experimental colitis by inhibition of mucosal addressin cell adhesion molecule-1. J Pharmacol Exp Ther 295: 183–189. [PubMed] [Google Scholar]
- 14.Masopust D, Jiang J, Shen H, Lefrancois L (2001) Direct analysis of the dynamics of the intestinal mucosa CD8 T cell response to systemic virus infection. J Immunol 166: 2348–2356. [DOI] [PubMed] [Google Scholar]
- 15.Kuklin NA, Rott L, Feng N, Conner ME, Wagner N, et al. (2001) Protective intestinal anti-rotavirus B cell immunity is dependent on alpha 4 beta 7 integrin expression but does not require IgA antibody production. J Immunol 166: 1894–1902. [DOI] [PubMed] [Google Scholar]
- 16.Artis D, Humphreys NE, Potten CS, Wagner N, Muller W, et al. (2000) Beta7 integrin-deficient mice: delayed leukocyte recruitment and attenuated protective immunity in the small intestine during enteric helminth infection. Eur J Immunol 30: 1656–1664. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Chahroudi A, Silvestri G, Wernett ME, Kaiser WJ, et al. (2002) Visualization and quantification of T cell-mediated cytotoxicity using cell-permeable fluorogenic caspase substrates. Nat Med 8: 185–189. [DOI] [PubMed] [Google Scholar]
- 18.Yamori M, Yoshida M, Watanabe T, Shirai Y, Iizuka T, et al. (2004) Antigenic activation of Th1 cells in the gastric mucosa enhances dysregulated apoptosis and turnover of the epithelial cells. Biochem Biophys Res Commun 316: 1015–1021. [DOI] [PubMed] [Google Scholar]
- 19.Park CO, & Kupper TS (2015). The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nature medicine, 21(7), 668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, … & Clark RA (2015). Human skin is protected by four funcationally and phenotypically discrete populations of resident and recirculating memory T cells. Science translational medicine, 7(279), 279ra39–279ra39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park CO, Fu X, Jiang X, Pan Y, Teague JE, Collins N, … & Jung Y (2017). Staged development of long-lived T-cell receptor αβ TH17 resident memory T-cell population to Candida albicans after skin infection. Journal of Allgergy and Clinical Immunology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu L, Zhong Q, Tian T, Dubin K, Athale SK, & Kupper TS (2010). Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nature medicine, 16(2), 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RF, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 2012. February 29;483(7388):227–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vermeire S, Loftus EV Jr, Colombel JF, Feagan BG, Sandborn WJ, Sands BE, Danese S, D’Haens GR, Kaser A, Panaccione R, Rubin DT, Shafran I, McAuliffe M, Kaviya A, Sankoh S, Mody R, Abhyankar B, Smyth M. Long-term Efficacy of Vedolizumab for Crohn’s Disease. J Crohns Colitis 2017. April 1;11(4):412–424 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.