

Abstract
A simple method for the dimerization of phenylpropenoid derivatives is reported. It leverages electrochemical oxidation of p-unsaturated phenols to access the dimeric materials in a biomimetic fashion. The mild nature of the transformation provides excellent functional group tolerance, resulting in a unified approach for the synthesis of a range of natural products and related analogues with excellent regiocontrol. The operational simplicity of the method allows for greater efficiency in the synthesis of complex natural products. Interestingly, the quinone methide dimer intermediates are potent radical-trapping antioxidants; more so than the phenols from which they are derived – or transformed to – despite the fact that they do not possess a labile H-atom for transfer to the peroxyl radicals that propagate autoxidation.
Keywords: electrochemistry, antioxidants, phenylpropenoids, quinone methides, resveratrol
Graphical Abstract
Quinone methide dimers are prepared via mild anodic oxidation mediated by a cheap and readily available amine base with excellent yield and regiocontrol. This strategy provides rapid access to intermediates for the synthesis of phenylpropenoid oligomers in a catalytic fashion, providing a new tool for the total synthesis of these complex molecules.
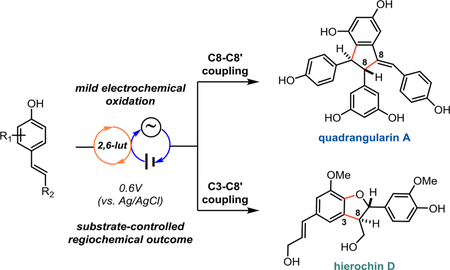
Polyphenolic natural products arising from stilbene- or styrene-derived monomers have drawn substantial interest due to a wide range of observed biological activities, including their widely-touted antioxidant properties.1–3 Their biosynthesis is hypothesized to proceed via the regio- and stereoselective coupling of phenoxyl radicals of form 2a–2c to generate products such as hierochin D (3) via C3–C8’ coupling and quadrangularin A (4) via C8–C8’ coupling (Figure 1A).3,4 Several of the synthetic efforts to recapitulate this strategy, including our own, have used enzymatic catalysis or single-electron oxidants, while others have targeted these molecules through de novo routes.5–7 Our previous approaches employed ferrocenium hexafluorophosphate as a mild stoichiometric oxidant to promote dimerization of protected resveratrol derivatives, which ultimately enabled the total synthesis of several dimeric and tetrameric resveratrol natural products.5c-d While these conditions were high-yielding and afforded excellent regioselectivity, we sought to develop a catalytic method that would translate more readily to other systems, especially those that may be prone to overoxidation. We previously investigated the use of photocatalysis to achieve this goal.5c Unfortunately, competing energy transfer pathways resulted in stilbene isomerization, while stoichiometric terminal oxidants led to decomposition of the products and/or starting materials. Therefore, an alternative method for catalytic phenol oxidation was desired.
Figure 1.

a)Phenylpropenoid oligomerization occurs via phenoxyl radical coupling.b) Our electrochemical approach to the oligomerization of phenylpropenoid scaffolds.
Several reports have demonstrated the utility of anodic oxidation in total synthesis,8–14 but efforts to apply this approach in the context of phenylpropenoid natural product total synthesis have been limited by low yields and lack of regiocontrol.15–18 Here, we present an electrochemical method for the regioselective dimerization of various hydroxystilbene derivatives (Figure 1B). The anodic oxidation reactions proceed at low potential and deliver the corresponding quinone methide dimers (QMDs) in good to excellent yields, providing rapid access to multiple natural product scaffolds in a selective fashion.
To determine whether the desired dimerization would be feasible under anodic oxidation conditions, we interrogated the oxidation potential of various 4-hydroxystilbene substrates using cyclic voltammetry (see page S38 of the Supporting Information). In all cases, direct oxidation of the substrate occurred above +0.8 V, however in the presence of 2,6-lutidine oxidation occurred below +0.6 V in all cases. With this information in hand (see page S43 of the Supporting Information for complete details), we developed conditions where anodic oxidation in the presence of 2,6-lutidine enables phenoxyl radical dimerization (Figure 2). The scope of this reaction was investigated by first varying the substituents of the resorcinol ring. A range of 3,5-di-tert-butyl-4-hydroxystilbene substrates were prepared from commercially available starting materials in 2–3 steps (see Supporting Information). Various protecting groups were tolerated on the resorcinol ring without diminishing the yield (5–7). When the tert-butyl substituents at C3 were exchanged with trialkylsilyl groups (8, 9), the yield diminished due to the decreased stability of the QMDs, which we have reported on previously.5d It was apparent that both electron-withdrawing and electron-donating groups were tolerated at each position of this aromatic ring as reactions proceeded with a wide-variety of electronically distinct substrates in excellent yields (10–20). The scope of this chemistry was extended to include dimerization of the ε-viniferin analogue (21/22), whose QMD product is the key intermediate in our recently reported syntheses of the natural products nepalensinol B and vateriaphenol C.5d As observed in the preparation of 5 and 8, the electrochemical yields of 23 and 24 were comparable to those obtained using a stoichiometric chemical oxidant, demonstrating the generality of this method for selective biomimetic synthesis of C8–C8’ resveratrol oligomers (Figure 3A). Regarding the observed diastereoselectivities in the synthesis of these QMD products, we presume that these dimers are in dynamic equilibrium with the corresponding phenoxyl radical monomers via C8–C8’ homolysis as we have previously characterized for QMDs 5, 8, 23, and 24.5d Therefore, the minimal diastereoselectivity observed for 5–20 is in agreement with prior methods for preparing these materials, as is the excellent diastereoselectivity observed for 23 and 24. The influence of additional stereocenters present in 23/24 presumably has a greater impact on the orientation in which the persistent radicals recombine, resulting in an increase in diastereoselectivity when moving from the dimer to the tetramer system.19 Importantly, the preparation of tert-butylated dimers (5-7, 10–20, 23) did not require chromatographic purification, which is highly advantageous for multi-step syntheses and provides an attractive alternative to stoichiometric dimerization methods.
Figure 2.

Substrate scope for the electrochemical dimerization of 4-hydroxystilbenes.
Figure 3.

a)Electrochemical dimerization delivers the key intermediate for the synthesis of resveratrol tetramers. 2,6-lut. = 20 mol%, Electrolyte = KPF6 (50 mM), Reference Electrode = Ag/AgCl. b)The removal of the C3-substituent results in a direct C3–C8’ dimerization, affording dihydrobenzofuran products. 2,6-lut. = 20 mol%, Electrolyte = KPF6 (50 mM), Reference Electrode = Ag/AgCl. c) Electrochemical synthesis of (±)-hierochin D. 2,6-lut. = 50 mol%, Electrolyte = KPF6 (50 mM), Reference Electrode = Ag/AgCl. d) Preparation of natural product analogue cores from QMDs.
The removal of one of the C3 blocking groups resulted in C3–C8’ dimerization, enabling access to the cores of the natural products δ-viniferin (27) and shegansu B (28) exclusively as the trans-diastereomers (Figure 3B), consistent with prior biomimetic dimerization studies of these types of hydroxystilbenes.3 As a result, the regioselectivity for this transformation is controlled exclusively by the presence or absence of a C3-blocking group. As described in Figure 1B, the analogous biosyntheses of the lignan class of natural products prompted us to investigate an extension of this electrochemical dimerization strategy. Subjection of coniferyl alcohol (29) to anodic oxidation under our optimized conditions afforded moderate conversion of the starting material; gratifyingly, a simple increase in the concentration of 2,6-lutidine from 20 mol% to 50 mol% afforded the neolignan natural product (±)-hierochin D in 53% yield (3, Figure 3C).
To explore the utility of the electrochemically-synthesized quinone methide dimers for the preparation of natural product-like indane and diquinane scaffolds, the QMDs described in Figure 2 were exposed to Lewis acids that promote intramolecular Friedel-Crafts cyclizations. While direct, double cyclization of 5 was successful in our previous synthesis of the resveratrol dimer pallidol,5c QMDs lacking the resorcinol moiety present in 5 did not react in this fashion. Instead, substrates with electron-donating groups at C12 (and their corresponding phenoxyl radicals) were susceptible to redox disproportionation conditions, returning half of the material as the stilbene precursor (reduction product) with loss of the remaining mass to oxidative decomposition (See Figure S1 on page S85 of the Supporting Information). The QMDs depicted in Figure 2 were, however, found to be suitable intermediates for the preparation of analogues of quadrangularin A (4). Base-mediated isomerization of one quinone methide followed by Lewis acid activation and Friedel-Crafts cyclization onto the remaining quinone methide forged a series of tert-butylated quadrangularin A analogues (30–33, Figure 3D).
With these derivatives in hand, we sought to extend our previous investigations of the radical-trapping antioxidant (RTA)20 activities of resveratrol derivatives, which had revealed that the quadrangularin A scaffold was more potent than either the pallidol scaffold or the parent resveratrol scaffold.5c Thus, the quadragularin A analogues were evaluated for their ability to inhibit co-autoxidations of PBD-BODIPY and 1-hexadecene in chlorobenzene at 37 °C (Figure 4).21 PBD-BODIPY enables quantitative determination of the reaction progress of the autoxidation by UV-vis spectrophotometry; its consumption by reaction with chain-carrying peroxyl radicals is associated with a loss of absorbance at 588 nm (Figure 4A). Kinetic analysis of the reaction progress data according to Eqs. 1 and 2 enables determination of the rate constant for the reaction of the RTA with peroxyl radicals (kinh) as well as the reaction stoichiometry (n) (Figure 4B). Representative data are shown in Figure 4C and kinetic parameters are tabulated in Figure 4D.
Figure 4.

a) PBD-BODIPY serves as the signal carrier in 1-hexadecene autoxidations. b) Determination of inhibition rate constants (kinh) and stoichiometries (n) for reactions of inhibitors with chain-carrying peroxyl radicals. c) Co-autoxidations of 1-hexadecene (2.9 M) and PBD-BODIPY (10 μM) initiated by AIBN (6 mM) in chlorobenzene at 37 °C (dashed black trac e) and inhibited by 5 μM substituted quadrangularin A analogues (dashed traces) and quinone methide dimers (solid traces). d) Inhibition rate constants and stoichiometries. e) Plausible mechanistic possibilities for the radical-trapping antioxidant activity of the quinone methide dimers; the DFT-optimized TS structure represents a model for the addition of (methyl)peroxyl radicals to the meso diastereomer of a model quinone methide dimer.
The quadrangularin A analogues 30–33 are all good RTAs. Their inhibition rate constants are roughly one order of magnitude greater than BHT (kinh = 2×104 M−1s−1), the quintessential hindered phenolic RTA.5c Substitution of the resorcinol ring has little impact on the reactivity of the quadrangularin A analogues; most kinh values are within experimental error of each other, and the maximum difference is less than a factor of 2. These results are consistent with our expectation that quadrangularin A and its tert-butylated derivative inhibit autoxidation via H-atom transfer from the (hindered) phenolic moiety. The number (n) of peroxyl radicals trapped per molecule is ca. 4 for all four compounds. Since compounds 30–33 contain two hindered phenols per molecule, the maximum expected stoichiometry is 4, corresponding to double that of BHT and other hindered phenols (e.g. the parent phenols, see Table S2).22
To our great surprise, the QMDs from which the quadrangularin A analogues were synthesized are very good RTAs.23 In fact, despite being devoid of phenolic moieties, the QMDs are >10-fold more reactive than the quadrangularin A derivatives (up to 36-fold more reactive for the 4-SMe derivative 32). Interestingly, monomeric quinone methides have been reported to be – at best – poor RTAs,24 suggesting that the QMD scaffold is special, perhaps due to the persistence of the quinone methide functionalities relative to those that have been previously studied.25
Mechanistic possibilities for the impressive reactivity of the QMDs are presented in Figure 4E. At first glance, mechanism 1 can be excluded solely on the basis of the magnitude of kinh determined from the inhibited autoxidations (kinh = 2–3×106 M−1s−1). H-atom transfer from carbon is well known to be a relatively slow reaction, even when highly thermodynamically favorable.26 Indeed, the CBS-QB3-predicted27 rate constants for HAT from a truncated form of the unsubstituted dimer to a model (methyl)peroxyl radical is a mere 6 M−1s−1 (see Supporting Information for the optimized structures, their energies, and the rate constants estimated therefrom). Mechanism 2 requires tautomerization of the QMD; however, upon exposing NMR samples of QMD to deuterated methanol (CD3OD), no deuterium incorporation was observed. Furthermore, if tautomerization were to occur, the phenolic tautomer would be expected to exhibit similar reactivity to the monomeric resveratrol derivative from which the QMD is derived (kinh ~105 M−1s−1). Mechanism 3 can be excluded solely on the basis of the magnitude of Keq for the QMD dissociation/recombination equilibrium (1.8 × 10−10 M),5d as it would necessitate the phenoxyl-peroxyl combination to proceed with a rate constant well in excess of the diffusion limit.
To the best of our knowledge, mechanism 4 has little precedent.23 Although the addition of peroxyl radicals to carbon-carbon double bonds is well-known, it generally features in the propagation of autoxidation of unsaturated substrates (e.g. styrene), not in its inhibition! Of course, the primary difference in the current context is that the resultant radical (a hindered phenoxyl) is unreactive to O2 and does not propagate the autoxidation. Moreover, not only does peroxyl radical addition produce a more stabilized phenoxyl radical, but the thermodynamics are greatly enhanced by aromatization of the quinone methide. Indeed, CBS-QB3 predicts ΔH = −31.5 kcal/mol for the addition of a model (methyl)peroxyl radical to a truncated form of the unsubstituted dimer along with a rate constant of 4 × 106 M−1s−1, in excellent agreement with experiment. Further exploration of the RTA activity of QMDs and how/why they differ from simple quinone methides are ongoing and will be reported in due course.
In summary, a general, regioselective method for the dimerization of 4-hydroxystilbenes and related cinnamyl alcohol derivatives has been realized utilizing electrochemical oxidation, providing an operationally simple strategy for the synthesis of complex phenylpropenoid scaffolds. The adoption of this method will result in a more streamlined approach for the synthesis of a wide range of naturally-occurring (and synthetic) phenylpropenoid oligomers, which will greatly enable studies of their potential biological activities. An example of such a (preliminary) study is included here, wherein we have found that the quinone methide dimers produced upon 4-hydroxystilbene oxidation are surprisingly potent RTAs.
Supplementary Material
Acknowledgements
The authors acknowledge the financial support for this research from the NIH NIGMS (R01-GM121656), the Camille Dreyfus Teacher-Scholar Award Program, and the University of Michigan. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 1256260 (for K. J. R.). The Ramón Areces Foundation (Becas para Estudios Postdoctorales) is acknowledged for a postdoctoral fellowship (for I. B.). This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada and the Canada Foundation for Innovation and through generous access to the computational resources of the Centre for Advanced Computing (cac.queensu.ca). JPC acknowledges the support of the Ontario Graduate Scholarships program. The authors thank Mr. Ethan Strobel for assistance with the preparation of key synthetic intermediates, as well as Dr. Mitch Keylor and Dr. Daryl Staveness for helpful suggestions in the preparation of this manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Vogt T, Mol. Plant 2010, 3, 2. [DOI] [PubMed] [Google Scholar]
- [2].Keylor MH, Matsuura BS, Stephenson CRJ, Chem. Rev 2015, 115, 8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Quideau S, Deffieux D, Douat-Casassus C, Pouységu L, Angew. Chem. Int. Ed 2011, 50, 586. [DOI] [PubMed] [Google Scholar]
- [4].For simplicity and consistency, we have chosen to denote the carbon involved in the dihydrobenzofuran formation as C3 throughout this report, though we recognize that for the mono-methoxylated species (3, 26, 28, and 29) this carbon is actually denoted as C5.
- [5].Examples of biomimetic approaches to resveratrol-derived natural products; Li W, Li H, Li Y, Hou Z, Angew. Chem. Int. Ed 2006, 45, 7609. Li W, Li H, Luo Y, Yang Y, Wang N, Synlett 2010, 8, 1247. Matsuura BS, Keylor MH, Li B, Lin Y, Allison S, Pratt DA, Stephenson CRJ, Angew. Chem. Int. Ed 2015, 54, 3754. Keylor MH, Matsuura BS, Griesser M, Chauvin J-PR, Harding RA, Kirillova MS, Zhu X, Fischer OJ, Pratt DA, Stephenson CRJ, Science 2016, 354, 1260. Li W, Dong T, Chen P, Liu X, Liu M, Han X, Tetrahedron 2017, 73, 3056. For additional examples, the reader is directed to references 2 and 3.
- [6].Examples of de novo approaches to resveratrol-derived natural products; Snyder SA, Zografos AL, Lin Y, Angew. Chem. Int. Ed 2007, 46, 8186. Nicolaou KC, Wu TR, Kang Q, Chen DY-K, Angew. Chem. Int. Ed 2009, 48, 3440. Jeffrey JL, Sarpong R, Org. Lett 2009, 11, 5450. Snyder SA, Wright NE, Pflueger JJ, Breazzano SP, Angew. Chem. Int. Ed 2011, 50, 8629. Snyder SA, Gollner A, Chiriac MI, Nature 2011, 474, 461. Snyder SA, Thomas SB, Mayer AC, Breazzano SP, Angew. Chem. Int. Ed 2012, 51, 4080. Jepsen TH, Thomas SB, Lin Y, Stathakis CI, de Miguel I, Snyder SA, Angew. Chem. Int. Ed 2014, 53, 6747. Klotter F, Studer A, Angew. Chem. Int. Ed 2014, 53, 2473. Soldi C, Lamb KN, Squitieri RA, Gonzalez-Lopez M, Di Maso MJ, Shaw JT, J. Am. Chem. Soc 2014, 136, 15142.. For additional examples, the reader is directed to references 2 and 3.
- [7].Reviews detailing both biomimetic and de novo approaches to the synthesis of lignan and neolignan oligomers; Ward RS, Chem. Soc. Rev 1982, 11 (2), 75. Sefkow M, Synthesis 2003, 2595. Pan J-Y, Chen S-L, Yang M-H, Wu J, Sinkkonen J, Zou K, Nat. Prod. Rep 2009, 26, 1251.
- [8].Moeller KD, Tetrahedron 2000, 56, 9527. [Google Scholar]
- [9].Sperry JB, Wright DL, Chem. Soc. Rev 2006, 35, 605. [DOI] [PubMed] [Google Scholar]
- [10].Frontana-Uribe BA, Little RD, Ibanez JG, Palma A, Vasquez-Medrano R, Green Chem. 2010, 12, 2099. [Google Scholar]
- [11].Tabakovic I, Gunic E, Juranic I, J. Org. Chem 1997, 35, 947. Kotani E, Takeuchi N, Tobinaga S, J. Chem. Soc., Chem. Commun 1973, 0, 550. For additional reading regarding anodic oxidation of aromatic rings, see Grimshaw J in Electrochemical Reactions and Mechanisms in Organic Chemistry, Elsevier Science B.V., Amsterdam, 2000, pp. 187–238.
- [12].Yan M, Kawamata Y, Baran PS, Chem. Rev 2017, 21, 13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Miller AK, Hughes CC, Kennedy-Smith JJ, Gradl SN, Trauner D, J. Am. Chem. Soc 2006, 128, 17057. [DOI] [PubMed] [Google Scholar]
- [14].Rosen BR, Werner EW, O’Brien AG, Baran PS, J. Am. Chem. Soc 2014, 136, 5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hong F-J, Low Y-Y, Chong K-W, Thomas NF, Kam T-S, J. Org. Chem 2014, 79, 4528. [DOI] [PubMed] [Google Scholar]
- [16].Chong K-W, Hong F-J, Thomas NF, Low Y-Y, Kam T-S, J. Org. Chem 2017, 82, 6172. [DOI] [PubMed] [Google Scholar]
- [17].Mori N, Furuta A, Watanabe H, Tetrahedron 2016, 72, 8393. [Google Scholar]
- [18].Yamamura S, Shizuri Y, Shigemori H, Okuno Y, Ohkubo M, Tetrahedron 1991, 47, 635. [Google Scholar]
- [19].Note that the assignment of 23/24 is based upon the cyclization outcome observed in reference 5d. For further discussion, see Romero KJ, Galliher MS, Pratt DA, Stephenson CRJ, Chem. Soc. Rev 2018, 47, 7851.
- [20].Ingold KU, Pratt DA, Chem. Rev 2014, 114, 9022. [DOI] [PubMed] [Google Scholar]
- [21].Haidasz EA, Van Kessel ATM, Pratt DA, J. Org. Chem 2016, 81, 737. [DOI] [PubMed] [Google Scholar]
- [22].Burton GW, Doba T, Gabe E, Hughes L, Lee FL, Prasad L, Ingold KU, J. Am. Chem. Soc 1985, 107, 7053. [Google Scholar]
- [23].The analyses of RTA activity for the QMDs were carried out using the mixture of diastereomers arising from the dimerization protocol.
- [24].Zolotova NV, Galieva FA, Tokareva MB, Denisov ET, Volod’kin AA, Kinetika i Kataliz (Engl. Transl.) 1979, 20, 48. [Google Scholar]
- [25].Volod’kin AA, Ershov VV, Russ. Chem. Rev 1988, 57, 336. [Google Scholar]
- [26].Zavitsas AA, Chatgilialoglu C, J. Am. Chem. Soc 1995, 117, 10645. [Google Scholar]
- [27].Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA, J. Chem. Phys 1999, 110, 2822. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.