

Conspectus
Antibiotics are the cornerstone of modern healthcare. The 20th century discovery of sulfonamides and β-lactam antibiotics altered human society immensely. Simple bacterial infections were no longer a leading cause of morbidity and mortality, and antibiotic prophylaxis greatly reduced the risk of infection from surgery. The current healthcare system requires effective antibiotics to function. However, antibiotic-resistant infections are becoming increasingly prevalent, threatening the emergence of a post-antibiotic era. To prevent this public health crisis, antibiotics with novel modes of action are needed. Currently available antibiotics target just a few cellular processes to exert their activity: DNA, RNA, protein, and cell wall biosynthesis. Bacterial central metabolism is underexploited offering a wealth of potential new targets that can be pursued toward expanding the armamentarium against microbial infections.
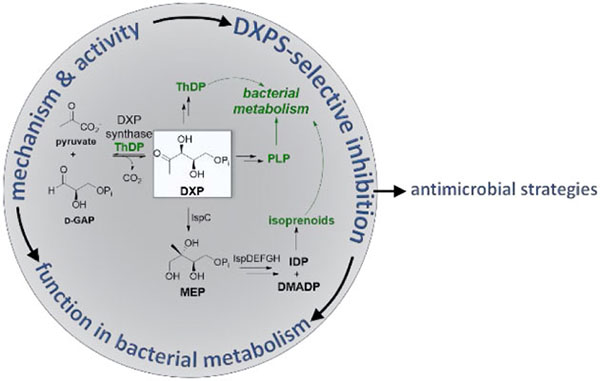
Discovered in 1997 as the first enzyme in the methylerythritol phosphate (MEP) pathway, 1-deoxy-d-xylulose 5-phosphate (DXP) synthase is a thiamin diphosphate (ThDP)-dependent enzyme that catalyzes the decarboxylative condensation of pyruvate and d-glyceraldehyde 3-phosphate (d-GAP) to form DXP. This five-carbon metabolite feeds into three separate, essential pathways for bacterial central metabolism: ThDP synthesis, pyridoxal phosphate (PLP) synthesis, and the MEP pathway for isoprenoid synthesis. While it has long been identified as a target for the development of antimicrobial agents, limited progress has been made towards developing selective inhibitors of the enzyme.
This Account highlights advances from our lab over the past decade to understand this important and unique enzyme. Unlike all other known ThDP-dependent enzymes, DXP synthase uses a random sequential mechanism which requires the formation of a ternary complex prior to decarboxylation of the lactyl-ThDP intermediate. Its large active site accommodates a variety of acceptor substrates lending itself to a number of alternative activities, such as the production of α-hydroxy ketones, hydroxamates, amides, acetolactate, and peracetate. Knowledge gained from mechanistic and substrate-specificity studies has guided the development of selective inhibitors with antibacterial activity and provides a biochemical foundation toward understanding DXP synthase function in bacterial cells. Although a promising drug target, the centrality of DXP synthase in bacterial metabolism imparts specific challenges to assessing antibacterial activity of DXP synthase inhibitors, and the susceptibility of most bacteria to current DXP synthase inhibitors is remarkably culture-medium-dependent. Despite these challenges, the study of DXP synthase is poised to reveal the role of DXP synthase in bacterial metabolic adaptability during infection ultimately providing a more complete picture of how inhibiting this crucial enzyme can be used to develop novel antibiotics.
Graphical Abstract
Discovery of DXP and DXP Synthase
The metabolite 1-deoxy-d-xylulose 5-phosphate (DXP) was discovered more than 15 years before the identification of DXP synthase (DXPS), the enzyme that catalyzes its formation from d-glyceraldehyde 3-phosphate (d-GAP) and pyruvate. In 1981, David and coworkers showed that the thiazole moiety of the essential cofactor thiamin diphosphate (ThDP) is derived from DXP in Escherichia coli.1 Eight years later, Hill et al. demonstrated DXP incorporation into the cofactor pyridoxal phosphate (PLP) in E. coli,2 indicating a critical role of DXP in bacterial primary metabolism (Scheme 1).
Scheme 1. DXP is used for the synthesis of ThDP, PLP, and isoprenoid precursors.

The blue atoms show where DXP is incorporated into ThDP, PLP, and IDP/DMADP.
From the discovery and characterization of the mevalonate pathway in the early 1960s until the early 1990s, it was assumed that this pathway3 was the sole source of the five carbon isoprenoid precursors, isopentenyl diphosphate (IDP) and dimethylallyl diphosphate (DMADP), in all living things. However, the non-mevalonate metabolite DXP was ultimately shown to be a precursor for isoprenoid biosynthesis in bacteria4,5, plants6 and apicomplexan parasites7 (Scheme 1). Isotopic labeling studies in bacteria suggested an orthogonal isoprenoid pathway originating from the glycolytic metabolites d-GAP and pyruvate.5,8 Equipped with this knowledge and the sequenced E. coli genome, multiple groups sought to identify a ThDP-dependent enzyme capable of catalyzing decarboxylative carboligation to produce DXP from d-GAP and pyruvate. These efforts uncovered DXPS, which shares sequence identity with ThDP-dependent enzymes transketolase (TK) and the pyruvate dehydrogenase E1 subunit (PDH), as is predicted by its requisite chemistry.9,10 The discovery of DXPS set the stage to elucidate the entire MEP pathway, with all seven enzymes identified in just four years. This pathway, beginning with the rate-limiting DXPS11,12, operates in most pathogenic bacteria, apicomplexan parasites, and the plastid of plant cells where it produces the essential isoprenoid precursors IDP and DMADP. As a precursor to ThDP, PLP, and isoprenoids, DXP is crucial for a great number of essential cellular processes, including the pentose phosphate pathway, the Krebs cycle, and amino acid and cell wall biosyntheses. Our studies of DXPS mechanism and function point to this unique enzyme as a key player in bacterial pathogen metabolism and promising target in the development of new antimicrobial strategies.13
DXP Synthase is Structurally and Mechanistically Unique Amongst ThDP Enzymes
DXPS catalyzes DXP formation via C2α-lactylthiamin diphosphate (LThDP) that is formed upon pyruvate binding and undergoes d-GAP-induced decarboxylation to produce the enamine. Subsequent carboligation with d-GAP leads to DXP (Scheme 2). Early studies conducted by Eubanks and Poulter14 provided the first evidence that DXPS could be mechanistically unique among ThDP-dependent enzymes. Their key finding from CO2 trapping experiments suggested that d-GAP binding accelerates CO2 release from the enzyme through formation of a ternary complex of enzyme, LThDP, and d-GAP (E-LThDP-GAP). This mechanism distinguishes DXPS from most ThDP-dependent enzymes that follow a classical ping-pong kinetic mechanism, in which CO2 release occurs in the absence of the second substrate. The ordered mechanism proposed by Eubanks and Poulter did not go unchallenged. In 2010, Matsue et al.15 proposed a ping-pong mechanism based on steady state kinetics alone. In the same year, single molecule force spectroscopy studies suggested enhanced binding of immobilized d-GAP to DXPS in the presence of pyruvate, which the authors suggested was supportive of the ordered mechanism proposed by Eubanks and Poulter.16 Our mechanistic studies have delved deeper to understand the d-GAP-dependent acceleration of CO2 release, toward establishing targetable features of this potential antimicrobial target (Scheme 2, Decarboxylation). We elucidated a random sequential preferred-order mechanism in which pyruvate and d-GAP each bind to free enzyme, with the E-pyruvate complex displaying higher affinity compared to E-GAP, en route to the E-LThDP-GAP ternary complex.17,18 Recent studies by Merkler19,20 further support a random sequential mechanism of DXPS from Plasmodium falciparum and Deinococcus radiodurans.
Scheme 2.

DXP synthase mechanism.
DXPS also differs structurally from other ThDP-dependent enzymes. It possesses a unique domain arrangement with the active site at the interface of two domains of the same monomer, in contrast to TK and PDH active sites which reside at the dimer interface.21 Further, the DXPS active site is approximately twice as large as TK or PDH active sites.22 These unique features permit DXPS to accept a broad range of acceptor substrates.
The unique random sequential mechanism coupled with the observation that d-GAP accelerates CO2 release raised an intriguing mechanistic question: Is d-GAP required for formation of the pre-decarboxylation intermediate, LThDP (Scheme 2), or does d-GAP induce LThDP decarboxylation? To answer this question, in collaboration with the Jordan lab at Rutgers University, we interrogated the early steps of the reaction coordinate using circular dichroism (CD)23 and NMR24.25 From these studies, two remarkable findings emerged: 1) LThDP is formed in a rate-limiting step and is stabilized on DXPS, and 2) LThDP undergoes rapid decarboxylation upon addition of d-GAP.25 These findings starkly contrast other ThDP-dependent enzymes that utilize classical ping-pong kinetics, as binding of the second substrate, d-GAP, occurs prior to the release of the first product, CO2. Thus, the reaction proceeds through a unique E-LThDP-GAP ternary complex, a targetable form of this bacterial ThDP-dependent enzyme.
The finding that d-GAP is a trigger of decarboxylation prompted investigation of the DXPS active site using two variants in which arginine is substituted with alanine (R420A and R478A, Figure 1), to study the two putative roles of d-GAP, as a trigger for LThDP decarboxylation and as an acceptor substrate in the subsequent carboligation. R420 and R478 form a cationic pocket near the active site entrance, providing a binding site for the phosphoryl group of d-GAP. Both R420A and R478A DXPS variants display greatly reduced affinity for d-GAP in DXP formation (i.e. increased Kmd-GAP), whereas both variants display affinity for pyruvate that is comparable to wild type. Despite the decreased affinity for d-GAP in DXP formation, d-GAP-induced LThDP decarboxylation (upstream of carboligation) occurs on these variants at rates comparable to wild type. Consistent with these findings, d-glyceraldehyde displays low affinity for carboligation18,26 on wild type DXPS, yet was also shown to induce LThDP decarboxylation at rates comparable to d-GAP-induced decarboxylation. These results distinguish the enzyme-GAP interactions that are important in the two roles of d-GAP on DXPS, indicating that the interaction of the d-GAP phosphoryl group with R420 and R478 is important during carboligation in the later steps of DXP formation, but is not required for its role to promote decarboxylation of LThDP on DXPS. The same study revealed the importance of Y392 (Figure 1) for d-GAP binding; however, Y392 is not crucial for d-GAP-induced decarboxylation or LThDP stabilization, as predicted by studies of the analogous residue on PDH.27 This highlights yet another distinct catalytic feature of DXPS. These findings, especially the requirement for R420 and R478 to anchor d-GAP for carboligation, give valuable insights for inhibitor design, detailed below.
Figure 1. DXP synthase active site.

The D. radiodurans DXPS crystal structure21 (PDB: 2O1X) highlighting residues of interest. Residue numbering without parentheses and with parentheses refers to the D. radiodurans and E. coli enzymes, respectively.
Conformational Flexibility of DXP Synthase
CD, fluorescence spectroscopy, and hydrogen – deuterium exchange mass spectrometry (HDX-MS) analyses have revealed conformational flexibility of DXPS in the absence of ligands, as well as conformational changes accompanying LThDP formation and decarboxylation, further distinguishing DXPS in ThDP-dependent enzymology. Hints of a conformational change upon d-GAP binding are evident by CD18,25 and fluorescence binding experiments.18–20 Stopped-flow CD analysis of d-GAP-induced LThDP decarboxylation on DXPS reveals a 20 – 30 msec delay between d-GAP addition and the exponential decrease in LThDP,18,25 possibly arising from a conformational change reorienting LThDP for decarboxylation. Additional evidence of d-GAP-induced conformational change comes from tryptophan fluorescence experiments performed by us on E. coli DXPS18, and by the Merkler lab where titration of D. radiodurans DXPS with d-GAP resulted in a Stokes shift of tryptophan fluorescence, indicating local environmental tryptophan changes upon d-GAP binding.19
In collaboration with the Jordan lab, we have gained deeper insights into DXPS conformational dynamics using HDX-MS.28 There are three mobile peptide regions near the active site that display interesting ligand-dependent HDX kinetics. The conformational diversity of DXPS in the absence of ligands is intriguing; whether this underlies alternative functions29,30 is unknown. Conformational changes upon substrate binding consistent with CD analyses are evident from HDX-MS. The pyruvate mimic methylacetylphosphonate (MAP) induces a closed conformation, while d-GAP or DXP induces an open conformation. Together these results suggest that as DXPS binds pyruvate and converts it to LThDP, the enzyme adopts a closed conformation to stabilize the LThDP intermediate. d-GAP binding then induces an open conformation leading to LThDP decarboxylation and following subsequent carboligation, product is released. Each conformation DXPS adopts along its reaction coordinate is potentially targetable.
Alternative Activities of DXP Synthase
Substrate-specificity and mechanistic studies have exposed remarkable flexibility of DXPS for alternative acceptor substrates and uncovered alternative activities that, collectively, provide critical insights for inhibitor design, and raise interesting questions about potential alternative functions of DXPS in bacterial cells. Early substrate-specificity studies were pursued to define active site binding determinants to guide inhibitor design. Our lab demonstrated that d-glyceraldehyde, lacking a phosphoryl group, is an alternative acceptor substrate, producing 1-deoxy d-xylulose (DX) in the absence of d-GAP.26 Straight-chain aliphatic aldehydes up to 6 carbons in length were also shown to be substrates.26 α-Branched aldehydes are not accepted, while β-branched aldehydes show modest turnover. Additionally, a second molecule of pyruvate can act as acceptor substrate to give acetolactate (Scheme 3).
Scheme 3. DXP synthase substrate and catalytic promiscuity.

Representative substituents are shown.
We expanded the substrate scope by testing aromatic aldehydes and nitroso aldehyde isosteres.22 Only electron-deficient aromatic aldehydes serve as acceptor substrates with 2-hydroxy-4,6-dinitrobenzaldehyde having the highest turnover efficiency (kcat/Km = 11 M−1s−1) of those tested. As more reactive isosteres of aldehydes, aromatic nitroso compounds proved to be better chemical tools to probe the DXPS active site through substrate-specificity analysis. Sterically demanding nitroso analogs display reasonable turnover efficiencies with Km values comparable to d-GAP, despite significant structural differences. Interestingly, nitroso alternative substrates are poor inhibitors of DXP formation (Scheme 3). In contrast to d-GAP, nitroso substrate binding is not impaired on R420A and R478A DXPS variants, suggesting that the nitroso acceptor substrates adopt distinct binding modes from d-GAP. Accordingly, active site volume calculations suggest DXPS has ample space to accommodate the nitroso substrates without interfering with d-GAP binding. In contrast, large acceptor substrates are not readily accommodated by PDH with its significantly smaller active site. Taken together, these results suggested a potential strategy to selectively target the large DXPS active site.
In addition to substrate promiscuity, DXPS also shows catalytic promiscuity. Our recent study to resolve discrepancies between the low, but significant, CO2 release rates observed by Eubanks and Poulter in the absence of d-GAP14 and the apparent stability of LThDP on DXPS observed by our lab in the absence of d-GAP25 revealed O2-induced decarboxylation of LThDP on DXPS, resulting in peracetate formation and solution deoxygenation.31 Biochemical, NMR, and EPR (electron paramagnetic resonance) experiments showed pyruvate is converted to acetate and bicarbonate via peracetate with concomitant O2 consumption. O2 incorporation into peracetate occurs via a ThDP-enamine radical and superoxide (Scheme 3). Importantly, conducting experiments anaerobically or permitting solution deoxygenation by DXPS-mediated oxidative pyruvate decarboxylation results in a stable E-LThDP complex determined by 14CO2 trapping and CD, in agreement with our previous reports.25 The oxygenase activity clearly impacts pre-steady state kinetic analyses to understand factors driving LThDP decarboxylation; however, the steady-state kinetics are comparable with or without O2, affirming a preference for d-GAP over O2 as acceptor substrate under these conditions. Taken together, the discovery of structurally distinct triggers of LThDP decarboxylation, combined with the conformational flexibility and observed catalytic and substrate promiscuity, could suggest DXPS can alter its function in response to cellular cues.
Selective Inhibition of DXP Synthase
Selectivity of DXPS inhibition over other ThDP-dependent enzymes is a primary challenge in targeting this enzyme. Ketoclomazone (1, Figure 2), a breakdown product of the herbicide clomazone (2), is an inhibitor of Chlamydomonas DXPS32 and has been used to probe isoprenoid biosynthesis in plants.33 In bacteria, 1 and 234 were shown to be weak inhibitors of DXPS from E. coli and Haemophilus influenza;15 selectivity of this inhibitor has not been studied in depth. The development of potent, selective DXPS inhibitors has been relatively slow given its role in isoprenoid biosynthesis was discovered nearly 20 years ago. Masini et al.35 suggested the polar nature of the enzyme active site could account for the low success with standard drug screening libraries. BASF screened a 100,000 compound library to identify MEP pathway inhibitors in Arabadopsis thaliana, and several inhibitors with micromolar potency emerged that could not be successfully optimized to enhance inhibition (3 and 4, Figure 2).36
Figure 2.

Known inhibitors of DXP synthase.
DXP Synthase Inhibitors Resembling Substrates and/or Cofactor.
The earliest mechanism-based DXPS inhibitor was fluoropyruvate (5), which found use as a mechanistic probe of DXPS.14 Fluoropyruvate acts by modifying the ThDP cofactor to produce a fluoro-LThDP intermediate that undergoes fluoride elimination to produce the dead-end acetyl-ThDP. Although useful as a mechanistic probe, fluoropyruvate indiscriminately inhibits ThDP-dependent pyruvate decarboxylases, and therefore, is not a promising antibiotic lead.
ThDP Mimics.
Deazathiamin diphosphate (6) potently inhibits D. radiodurans DXPS (Ki = 34 nM), whereas its non-phosphorylated analog 7 lacks potency, confirming the diphosphate group as a key binding element.37 Seeking more drug-like inhibitors, the Hirsch lab pursued ThDP mimics by a fragment modelling approach that afforded several micromolar inhibitors of D. radiodurans DXPS (e.g., 8, Figure 2).37 While mimicking the ThDP cofactor provides a heterocyclic drug-like scaffold to build from, DXPS enzymes bind ThDP with high affinity,37 and cross-toxicity with human ThDP-dependent enzymes is likely given the conserved ThDP binding sites. This challenge is exemplified by the work of Kozikowski, Crick, and coworkers38 to develop inhibitor 9, which is selective for M. tuberculosis DXPS in vitro, but toxic to human cells.
d-GAP Competitive Inhibitors.
Thus far, d-GAP competitive inhibitors are either non-selective or possess weak enzyme inhibitory activity. An oxime library screen by our lab identified 2,4,5-trihydroxybenzaldoximes (10) as low micromolar DXPS inhibitors39 that are competitive against d-GAP and uncompetitive against pyruvate, and thus appear to bind the E-LThDP complex. However, off-target activity in bacteria is likely due to the reactive catechol functionality. Using phage display, the Hirsch lab has discovered the first peptide-based d-GAP competitive inhibitor (11),40 albeit with weak enzyme inhibitory activity (Ki = 113 μM).
Alkylacetylphosphonate (alkylAP) Inhibitors.
Given the unique mechanism and large active site of DXPS, we have asserted that selective inhibition should be possible. Substrate-specificity studies in our lab22,26 provided a foundation to develop sterically demanding alkylacetylphosphonates (alkylAPs) as the first selective inhibitors of DXPS. MAP was previously characterized as a pyruvate mimic and inhibitor of ThDP-dependent pyruvate decarboxylases41; however, use of this small, indiscriminate inhibitor has been restricted to mechanistic studies. The ability of DXPS to process large aliphatic aldehydes and aromatic nitroso compounds as acceptor substrates22 suggested it might be possible to develop unnatural bisubstrate analogs bearing acetylphosphonate and hydrocarbon components to mimic pyruvate and unnatural acceptor substrates, respectively. As expected, synthetic alkylAPs were demonstrated to inhibit DXPS selectively22,42,43 over mammalian enzymes PDH and TK. Butylacetylphosphonate (BAP) exhibits reasonably high selectivity (KiPDH/KiDXPS of 60) and low micromolar inhibitory activity against recombinant DXPS from Yersina pestis, Mycobacterium tuberculosis, and Salmonella enterica.44 These early results supported the idea that DXPS enzymes, while distinct from other ThDP-dependent enzymes, are mechanistically similar across pathogens, making it a viable target in multiple important pathogens.
These first-generation alkylAPs demonstrate the feasibility of selectively inhibiting a bacterial ThDP-dependent enzyme based on its relaxed substrate specificity and large active site. Recently, we have advanced selective inhibitor development by creating bisubstrate inhibitor scaffolds with high potency and selectivity through targeting the distinct E-LThDP-GAP ternary complex. These inhibitors were accessed using copper catalyzed azide alkyne cycloaddition (CuAAC) chemistry to install polar functional groups mimicking d-GAP distal to the acetylphosphonate pyruvate mimic.45 This series contains several nanomolar DXPS inhibitors, the most potent of which (12, Figure 2) exhibits a Ki = 90 nM and slow, tight-binding behavior consistent with enzyme dynamics driving substrate-induced conformational changes on DXPS. Biochemical analysis with the R478A variant showing impaired binding to both d-GAP and 12 indicates inhibitor binding to the d-GAP binding site (Figure 3). Also competitive against pyruvate, 12 is highlighted as the first known bisubstrate inhibitor of DXPS targeting its unique mechanism. Remarkably, 12 exhibits 15,000-fold selectivity for DXPS over mammalian PDH E1 subunit, making it, by far, the most selective DXPS inhibitor to date. This inhibitor class offers valuable probes to study DXPS mechanism and guide selective inhibitor design.
Figure 3.

AlkylAPs targeting unique DXP synthase complexes.
Antimicrobial Activity of DXP Synthase Inhibitors
Evaluating antimicrobial activity of DXPS inhibitors, which target essential processes for metabolic adaptation during infection, requires non-conventional approaches, including assessment in a growth medium that mimics the infection microenvironment. Evaluating new antimicrobial agents in rich culture media is an industry standard set forth by the Clinical and Laboratory Standards Institute.46 However, growing evidence indicates that antibiotic activity in rich growth medium is poorly predictive of in vivo activity, and potentially interesting compounds can be overlooked as a result of restricting analysis to potency in rich medium alone.47 As for other inhibitors of central metabolism, assay growth medium profoundly impacts the activity of DXPS inhibitors. For example, BAP is >1,000-fold more potent against bacterial pathogens in defined, minimal medium than in rich medium,43,48 owing to differences in cellular entry and shifts in the relative requirements for isoprenoids, ThDP, and PLP in different growth environments.
While the centrality of DXPS in bacterial metabolism makes it a promising antimicrobial target, this complicates studies of this enzyme in its cellular context. We have examined DXPS inhibition in bacterial cells, uncovering particular challenges to the study of this enzyme in vivo. Prior to alkylAP inhibitor development, only a few DXPS inhibitors exhibited antibacterial activity (1, 2, and 9, Figure 2). Initially, the antibacterial activity of BAP was assessed according to CLSI guidelines46 in rich growth medium, demonstrating low millimolar growth inhibitory activity against E. coli, S. enterica serovar Typhimurium, Micrococcus sp., B. anthracis sterne and P. aeruginosa.44 Despite observing weak activity under these conventional growth conditions, we sought support for DXPS as the intracellular target of BAP through metabolite suppression and target overexpression experiments. We found that exogenous DX and thiamin, and DXPS overexpression all resulted in significant suppression of BAP growth inhibitory activity in E. coli.
The discovery that exogenous thiamin rescues growth of alkylAP-treated E. coli prompted our lab to explore more defined growth media. Cation-adjusted Mueller Hinton Broth (CAMHB) and Lysogeny Broth (LB) contain thiamin and other vitamins not readily available to bacterial pathogens in vivo.46,47 Remarkably, BAP activity against E. coli in defined M9-glucose minimal medium is 1000-fold more potent than in CAMHB (Figure 4).43 This trend holds across a series of alkylAPs, with BAP displaying the most potent activity. Growth-medium-dependent changes in BAP potency are also observed against S. enterica LT2, Pseudomonas fluorescens, Rhizobium radiobacter, Micrococcus sp., Klebisella pneumonae, Enterobacter cloacae, Bacillus thuringiensis HD 34, and Klebsiella oxytoca (Table 1).
Figure 4. BAP antibacterial activity strongly depends on medium.

a) BAP is 1000-fold more potent in M9 minimal medium compared to CAMBH. b) Nanomolar thiamin added to M9-glucose suppresses BAP activity. Adapted with permission from ref. 43.
Table 1. Antibacterial activity of BAP.
MIC values were reported by Sanders et al.43 unless otherwise indicated. Adapted with permission from ref. 43.
| BAP MIC (μM) | ||
|---|---|---|
| strain | CAMHB | M9 |
| Escherichia coli MG1655 | 2500 | 5 |
| Escherichia coli fsr-pET28a | 10753 | 43 |
| Escherichia coli clinical | >21505 | 11 |
| Salmonella enterica LT2 | 21505 | 11 |
| Pseudomonas fluorescens | >21505 | 43 |
| Rhizobium radiobacter | >21505 | 22 |
| Micrococcus sp. | 10753 | 86 |
| Klebsiella pneumonae | >21505 | 1376 |
| Enterobacter cloacae | >21505 | 688 |
| Bacillus thuringiensis HD 34 | >21505 | 86 |
| Klebsiella oxytoca | >21505 | 43 |
| Bacillus anthracis Sterne | 537644 | nd |
| Pseudomonas aeruginosa | 2150544 | nd |
Several factors underlie the medium-dependent activity of alkylAPs. Thiamin present in rich medium is partially responsible for diminished BAP activity, consistent with the requirement for ThDP in DXPS catalysis and for production of metabolites in all three metabolic pathways at this branch point (Scheme 1). In M9-glucose, nanomolar thiamin concentrations can rescue the growth of BAP-treated cells.43 Interestingly, while there is immediate metabolite suppression observed for other antimicrobial agents in the presence of downstream metabolites49, growth rescue of BAP-treated cells with thiamin alone is delayed.43 On the other hand, nutrient rich media containing a complex and undefined mixture of metabolites, amino acids, and vitamins that are likely used immediately allow bacterial cells to quickly overcome BAP-mediated growth inhibition by circumventing DXPS inhibition. Additionally, LC-MS/MS experiments revealed medium dependence of BAP accumulation in bacterial cells, with intracellular BAP levels significantly lower in E. coli grown in rich compared to minimal medium.48
Metabolite suppression and differential accumulation of BAP largely explain the medium-dependent activity of BAP. Although, it is also possible that changes in cell sensitivity to BAP reflect changes in target activity and/or demand of DXPS-dependent metabolite synthesis in different growth media. Evidence for such metabolic remodeling is provided by the medium-dependent relationship between the IspC inhibitor fosmidomycin and BAP. In rich medium, BAP and fosmidomycin strongly synergize, while in minimal medium these agents display an indifferent relationship,44,48 suggesting the relative metabolic requirements for isoprenoids, ThDP, and PLP are also impacted by the change in growth medium.
Challenges of Establishing Tools to Assess DXP Synthase Engagement In Vivo
Establishing alkylAPs as DXPS inhibitors in bacterial cells under nutrient limitation is challenging. Two common approaches to establish intracellular targets include target overexpression50 and exogenous supplementation of a downstream intermediate.49 However, overexpression of DXPS to high levels is toxic to cells43,51. In fact, significant suppression of BAP activity is only observed under conditions of basal DXPS expression from a leaky expression vector in rich culture conditions.44 The toxic effects of IPTG-induced DXPS overexpression in rich medium oppose the rescuing effects of increased target levels. Additionally, basal target overexpression is not possible in M9-glucose where alkylAPs are most potent, as the lac operon controlling enzyme expression is fully repressed by glucose. Downstream metabolite supplementation as a method to confirm the intracellular target is complicated by the position of DXP as a branch point metabolite. Supplementation with only downstream isoprenoids or PLP pathway intermediates does not rescue the effects of DXPS inhibition on cellular metabolism. Only thiamin supplementation undoes these inhibitory effects, due to its requirement in all three pathways and for DXPS catalysis itself. Further complicating supplementation experiments, all downstream products of DXPS are phosphorylated and thus have low permeability when supplied exogenously.
Conclusions and Perspective
Bacterial central metabolism is an underexplored target space for development of novel antimicrobial agents. Concerns about cross-toxicity of drugs targeting conserved metabolic processes, and the practice of culturing pathogens in non-relevant rich culture conditions, account for the lack of antibiotics targeting bacterial metabolism. However, there is renewed interest in this target space. It is now known that bacterial pathogens undergo metabolic remodeling in response to the rapidly changing, nutrient-limited host environment, resulting in infection-specific metabolic processes that are indispensable in vivo and therefore potential drug targets.47 DXPS is deeply interwoven into bacterial central metabolism making it a promising target for antibiotic development. Access to selective inhibitors of this enzyme will enable target validation and studies of enzyme function in relevant cellular contexts and could offer much needed novel antimicrobial agents.
We have progressed our understanding of the unique mechanism of DXPS over the past decade. Building from the early work of Eubanks and Poulter14, we elucidated the random sequential mechanism of DXPS revealing the remarkably stable LThDP intermediate, unique among ThDP-dependent enzymes. Broad substrate specificity and large active site volume further distinguish DXPS from other ThDP-dependent enzymes. Taken together with a newfound oxygenase activity, these features raise intriguing questions about other synthetic responsibilities of DXPS in the fluctuating in vivo environment during infection. Leveraging insights from mechanistic and substrate-specificity studies, we have advanced DXPS inhibitor development, with unnatural and natural alkylAP bisubstrate inhibitors exhibiting excellent potency and selectivity and emerging as tools for mechanistic and antimicrobial studies of DXPS. Notably, bisubstrate inhibitors targeting the unique mechanism of DXPS have provided 12 and analogs as the most potent, selective inhibitors of DXPS to date. Further, the discovery of the profound medium-dependence of in vivo alkylAPs has changed the way DXPS inhibitors are evaluated in our laboratory and also highlights the need for infection-site relevant growth conditions for the evaluation of antimicrobial agents, especially those targeting central metabolism.
These new insights into DXPS mechanism and function invoke more questions. How do acceptor substrates promote decarboxylation of the otherwise stable LThDP intermediate, and which molecular triggers of decarboxylation are important in vivo during infection? Given the promiscuity and large active-site volume of DXPS, what other activities does it possess and how might these alternative activities fit into bacterial metabolism in the context of pathogen metabolic adaption during infection? And, should these alternative activities be targeted? By what mechanisms do alkylAPs enter bacterial cells under nutrient limitation and how can inhibitor delivery be enhanced in fluctuating host growth environments? What is the molecular mechanism by which exogenous thiamin rescues growth of alkylAP-treated bacterial cells? And ultimately, can we exploit these unique attributes of DXPS mechanism and function to design improved antimicrobial agents that selectively target pathogenic bacteria without disturbing normal commensal bacteria in a human host?
Acknowledgements
This work was supported by funding from the National Institutes of Health (GM084998 for C.L.F.M. and D.B., T32 GM08018901 for D.B.).
Biography
David Bartee obtained his B.S. in Biochemistry and Molecular Biology from the University of Maryland, Baltimore County and earned his PhD in Chemical Biology at Johns Hopkins University in the lab of Caren Freel Meyers. His research focuses on developing novel antibiotic strategies targeting bacterial metabolism.
Caren L. Freel Meyers is an Associate Professor in the Department of Pharmacology and Molecular Sciences at The Johns Hopkins University School of Medicine. She earned her B.S. in Chemistry from Michigan Technological University, and her Ph.D. in Chemistry at the University of Rochester with Richard Borch. She held a teaching/research postdoctoral position at Purdue University with Professors Richard Borch and Mark Loudon and postdoctoral fellowship in chemical biology at Harvard Medical School with Christopher Walsh. Since 2005, her independent research program has focused on antimicrobial targets and drug delivery approaches toward developing new antimicrobial strategies.
References
- (1).David S; Estramareix B; Fischer JC; Therisod M 1-Deoxy-d-Threo-2-Pentulose: The Precursor of the Five-Carbon Chain of the Thiazole of Thiamine. J. Am. Chem. Soc 1981, 103, 7341–7342. [Google Scholar]
- (2).Hill RE; Sayer BG; Spenser ID Biosynthesis of Vitamin B6: Incorporation of d-1-Deoxyxylulose. J. Am. Chem. Soc 1989, 111, 1916–1917. [Google Scholar]
- (3).Buhaescu I; Izzedine H Mevalonate Pathway: A Review of Clinical and Therapeutical Implications. Clin. Biochem 2007, 40, 575–584. [DOI] [PubMed] [Google Scholar]
- (4).Broers STJ About the Early Steps of Biosynthesis of Isoprenoids in Escherichia Coli, PhD Dissertation no. 10978, Swiss Federal Institute of Technology, Zurich, 1994. [Google Scholar]
- (5).Rohmer M; Knani M; Simonin P; Sutter B; Sahm H Isoprenoid Biosynthesis in Bacteria: A Novel Pathway for the Early Steps Leading to Isopentenyl Diphosphate. Biochem. J 1993, 295 (Pt 2), 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schwarz MK Terpene Biosynthesis in Ginkgo Biloba: A Surprising Story, PhD Dissertation no. 10951, Swiss Federal Institute of Technology, Zurich, 1994. [Google Scholar]
- (7).Jomaa H; Wiesner J; Sanderbrand S; Altincicek B; Weidemeyer C; Hintz M; Türbachova I; Eberl M; Zeidler J; Lichtenthaler HK; Soldati D; Beck E Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. Science 1999, 285, 1573–1576. [DOI] [PubMed] [Google Scholar]
- (8).Rohmer M; Seemann M; Horbach S; Bringer-Meyer S; Sahm H Glyceraldehyde 3-Phosphate and Pyruvate as Precursors of Isoprenic Units in an Alternative Non-Mevalonate Pathway for Terpenoid Biosynthesis. J. Am. Chem. Soc 1996, 118, 2564–2566. [Google Scholar]
- (9).Lois LM; Campos N; Putra SR; Danielsen K; Rohmer M; Boronat A Cloning and Characterization of a Gene from Escherichia Coli Encoding a Transketolase-like Enzyme That Catalyzes the Synthesis of d-1-Deoxyxylulose 5-Phosphate, a Common Precursor for Isoprenoid, Thiamin, and Pyridoxol Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sprenger GA; Schörken U; Wiegert T; Grolle S; de Graaf AA; Taylor SV; Begley TP; Bringer-Meyer S; Sahm H Identification of a Thiamin-Dependent Synthase in Escherichia Coli Required for the Formation of the 1-Deoxy-d-Xylulose 5-Phosphate Precursor to Isoprenoids, Thiamin, and Pyridoxol. Proc. Natl. Acad. Sci 1997, 94, 12857–12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Miller B; Heuser T; Zimmer W Functional Involvement of a Deoxy-d-Xylulose 5-Phosphate Reductoisomerase Gene Harboring Locus of Synechococcus Leopoliensis in Isoprenoid Biosynthesis. FEBS Lett. 2000, 481, 221–226. [DOI] [PubMed] [Google Scholar]
- (12).Estévez JM; Cantero A; Reindl A; Reichler S; León P 1-Deoxy-d-Xylulose-5-Phosphate Synthase, a Limiting Enzyme for Plastidic Isoprenoid Biosynthesis in Plants. J. Biol. Chem 2001, 276, 22901–22909. [DOI] [PubMed] [Google Scholar]
- (13).Masini T; Hirsch AKH Development of Inhibitors of the 2 C -Methyl-d-Erythritol 4-Phosphate (MEP) Pathway Enzymes as Potential Anti-Infective Agents. J. Med. Chem 2014, 57, 9740–9763. [DOI] [PubMed] [Google Scholar]
- (14).Eubanks LM; Poulter CD Rhodobacter Capsulatus 1-Deoxy-d-Xylulose 5-Phosphate Synthase: Steady-State Kinetics and Substrate Binding. Biochemistry 2003, 42, 1140–1149. [DOI] [PubMed] [Google Scholar]
- (15).Matsue Y; Mizuno H; Tomita T; Asami T; Nishiyama M; Kuzuyama T The Herbicide Ketoclomazone Inhibits 1-Deoxy-d-Xylulose 5-Phosphate Synthase in the 2-C-Methyl-d-Erythritol 4-Phosphate Pathway and Shows Antibacterial Activity against Haemophilus Influenzae. J. Antibiot. (Tokyo) 2010, 63, 583–588. [DOI] [PubMed] [Google Scholar]
- (16).Sisquella X; de Pourcq K; Alguacil J; Robles J; Sanz F; Anselmetti D; Imperial S; Fernàndez-Busquets X A Single-Molecule Force Spectroscopy Nanosensor for the Identification of New Antibiotics and Antimalarials. FASEB J. 2010, 24, 4203–4217. [DOI] [PubMed] [Google Scholar]
- (17).Brammer LA; Smith JM; Wade H; Meyers CF 1-Deoxy-d-Xylulose 5-Phosphate Synthase Catalyzes a Novel Random Sequential Mechanism. J. Biol. Chem 2011, 286, 36522–36531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brammer Basta LA; Patel H; Kakalis L; Jordan F; Freel Meyers CL Defining Critical Residues for Substrate Binding to 1-Deoxy-d-Xylulose 5-Phosphate Synthase - Active Site Substitutions Stabilize the Predecarboxylation Intermediate C2α-Lactylthiamin Diphosphate. FEBS J. 2014, 281, 2820–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Battistini MR; Shoji C; Handa S; Breydo L; Merkler DJ Mechanistic Binding Insights for 1-Deoxy-d-Xylulose-5-Phosphate Synthase, the Enzyme Catalyzing the First Reaction of Isoprenoid Biosynthesis in the Malaria-Causing Protists, Plasmodium Falciparum and Plasmodium Vivax. Protein Expr. Purif 2016, 120, 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Handa S; Dempsey DR; Ramamoorthy D; Cook N; Guida WC; Spradling TJ; White JK; Woodcock HL; Merkler DJ; Merkler DJ Mechanistic Studies of 1-Deoxy-d-Xylulose-5-Phosphate Synthase from Deinococcus Radiodurans. Biochem. Mol. Biol. J 2018, 04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Xiang S; Usunow G; Lange G; Busch M; Tong L Crystal Structure of 1-Deoxy-d-Xylulose 5-Phosphate Synthase, a Crucial Enzyme for Isoprenoids Biosynthesis. J. Biol. Chem 2007, 282, 2676–2682. [DOI] [PubMed] [Google Scholar]
- (22).Morris F; Vierling R; Boucher L; Bosch J; Freel Meyers CL DXP Synthase-Catalyzed C-N Bond Formation: Nitroso Substrate Specificity Studies Guide Selective Inhibitor Design. ChemBioChem 2013, 14, 1309–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nemeria NS; Chakraborty S; Balakrishnan A; Jordan F Reaction Mechanisms of Thiamin Diphosphate Enzymes: Defining States of Ionization and Tautomerization of the Cofactor at Individual Steps. FEBS J. 2009, 276, 2432–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tittmann K; Golbik R; Uhlemann K; Khailova L; Schneider G; Patel M; Jordan F; Chipman DM; Duggleby RG; Hübner G NMR Analysis of Covalent Intermediates in Thiamin Diphosphate Enzymes. Biochemistry 2003, 42, 7885–7891. [DOI] [PubMed] [Google Scholar]
- (25).Patel H; Nemeria NS; Brammer LA; Freel Meyers CL; Jordan F Observation of Thiamin-Bound Intermediates and Microscopic Rate Constants for Their Interconversion on 1-Deoxy-d-Xylulose 5-Phosphate Synthase: 600-Fold Rate Acceleration of Pyruvate Decarboxylation by d-Glyceraldehyde-3-Phosphate. J. Am. Chem. Soc 2012, 134, 18374–18379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Brammer LA; Meyers CF Revealing Substrate Promiscuity of 1-Deoxy-d-Xylulose 5-Phosphate Synthase. Org. Lett 2009, 11, 4748–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Arjunan P; Sax M; Brunskill A; Chandrasekhar K; Nemeria N; Zhang S; Jordan F; Furey W A Thiamin-Bound, Pre-Decarboxylation Reaction Intermediate Analogue in the Pyruvate Dehydrogenase E1 Subunit Induces Large Scale Disorder-to-Order Transformations in the Enzyme and Reveals Novel Structural Features in the Covalently Bound Adduct. J. Biol. Chem 2006, 281, 15296–15303. [DOI] [PubMed] [Google Scholar]
- (28).Zhou J; Yang L; DeColli A; Freel Meyers C; Nemeria NS; Jordan F Conformational Dynamics of 1-Deoxy-d-Xylulose 5-Phosphate Synthase on Ligand Binding Revealed by H/D Exchange MS. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 9355–9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).James LC; Tawfik DS Conformational Diversity and Protein Evolution – a 60-Year-Old Hypothesis Revisited. Trends Biochem. Sci 2003, 28, 361–368. [DOI] [PubMed] [Google Scholar]
- (30).Copley SD Shining a Light on Enzyme Promiscuity. Curr. Opin. Struct. Biol 2017, 47, 167–175. [DOI] [PubMed] [Google Scholar]
- (31).DeColli AA; Nemeria NS; Majumdar A; Gerfen GJ; Jordan F; Freel Meyers CL Oxidative Decarboxylation of Pyruvate by 1-Deoxy-d-Xyulose 5-Phosphate Synthase, a Central Metabolic Enzyme in Bacteria. J. Biol. Chem 2018, 293, 10857–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mueller C; Schwender J; Zeidler J; Lichtenthaler HK Properties and Inhibition of the First Two Enzymes of the Non-Mevalonate Pathway of Isoprenoid Biosynthesis. Biochem. Soc. Trans 2000, 28, 792–793. [PubMed] [Google Scholar]
- (33).Ferhatoglu Y; Barrett M Studies of Clomazone Mode of Action. Pestic. Biochem. Physiol 2006, 85, 7–14. [Google Scholar]
- (34).Hayashi D; Kato N; Kuzuyama T; Sato Y; Ohkanda J Antimicrobial N-(2-Chlorobenzyl)-Substituted Hydroxamate Is an Inhibitor of 1-Deoxy-d-Xylulose 5-Phosphate Synthase. Chem. Commun 2013, 49, 5535–5537. [DOI] [PubMed] [Google Scholar]
- (35).Masini T; Kroezen BS; Hirsch AKH. Druggability of the Enzymes of the Non-Mevalonate-Pathway. Drug Discov. Today 2013, 18, 1256–1262. [DOI] [PubMed] [Google Scholar]
- (36).Witschel M; Röhl F; Niggeweg R; Newton T In Search of New Herbicidal Inhibitors of the Non-Mevalonate Pathway. Pest Manag. Sci 2013, 69, 559–563. [DOI] [PubMed] [Google Scholar]
- (37).Masini T; Pilger J; Kroezen BS; Illarionov B; Lottmann P; Fischer M; Griesinger C; Hirsch AKH De Novo Fragment-Based Design of Inhibitors of DXS Guided by Spin-Diffusion-Based NMR Spectroscopy. Chem. Sci 2014, 5, 3543–3551. [Google Scholar]
- (38).Mao J; Eoh H; He R; Wang Y; Wan B; Franzblau SG; Crick DC; Kozikowski AP Structure–activity Relationships of Compounds Targeting Mycobacterium Tuberculosis 1-Deoxy-d-Xylulose 5-Phosphate Synthase. Bioorg. Med. Chem. Lett 2008, 18, 5320–5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bartee D; Morris F; Al-khouja A; Freel Meyers CL Hydroxybenzaldoximes Are d-GAP-Competitive Inhibitors of E. Coli 1-Deoxy-d-Xylulose-5-Phosphate Synthase. ChemBioChem 2015, 16, 1771–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Marcozzi A; Masini T; Zhu D; Pesce D; Illarionov B; Fischer M; Herrmann A; Hirsch AKH Phage Display on the Anti-Infective Target 1-Deoxy-d-Xylulose-5-Phosphate Synthase Leads to an Acceptor–Substrate Competitive Peptidic Inhibitor. ChemBioChem 2018, 19, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).O’Brien TA; Kluger R; Pike DC; Gennis RB Phosphonate Analogues of Pyruvate. Probes of Substrate Binding to Pyruvate Oxidase and Other Thiamin Pyrophosphate-Dependent Decarboxylases. Biochim. Biophys. Acta - Enzymol 1980, 613, 10–17. [DOI] [PubMed] [Google Scholar]
- (42).Smith JM; Vierling RJ; Meyers CF Selective Inhibition of E. Coli1-Deoxy-d-Xylulose-5-Phosphate Synthase by Acetylphosphonates. Medchemcomm 2012, 3, 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sanders S; Vierling RJ; Bartee D; DeColli AA; Harrison MJ; Aklinski JL; Koppisch AT; Freel Meyers CL Challenges and Hallmarks of Establishing Alkylacetylphosphonates as Probes of Bacterial 1-Deoxy-d-Xylulose 5-Phosphate Synthase. ACS Infect. Dis 2017, 3, 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Smith JM; Warrington NV; Vierling RJ; Kuhn ML; Anderson WF; Koppisch AT; Freel Meyers CL Targeting DXP Synthase in Human Pathogens: Enzyme Inhibition and Antimicrobial Activity of Butylacetylphosphonate. J. Antibiot. (Tokyo). 2014, 67, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bartee D; Freel Meyers CL Targeting the Unique Mechanism of Bacterial 1-Deoxy-d-Xylulose 5-Phosphate (DXP) Synthase. Biochemistry 2018, 57, 4349–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).CLSI. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fifth Informational Supplement; CLSI document M100-S25; Wayne, PA: Clinical and Laboratory Standards Institute, 2015. [Google Scholar]
- (47).Murima P; McKinney JD; Pethe K Targeting Bacterial Central Metabolism for Drug Development. Chem. Biol 2014, 21, 1423–1432. [DOI] [PubMed] [Google Scholar]
- (48).Sanders S; Bartee D; Harrison MJ; Phillips PD; Koppisch AT; Freel Meyers CL Growth Medium-Dependent Antimicrobial Activity of Early Stage MEP Pathway Inhibitors. PLoS One 2018, 13, e0197638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zlitni S; Ferruccio LF; Brown ED Metabolic Suppression Identifies New Antibacterial Inhibitors under Nutrient Limitation. Nat. Chem. Biol 2013, 9, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Palmer AC; Kishony R Opposing Effects of Target Overexpression Reveal Drug Mechanisms. Nat. Commun 2014, 5, 4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Alper H; Fischer C; Nevoigt E; Stephanopoulos G Tuning Genetic Control through Promoter Engineering. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 12678–12683. [DOI] [PMC free article] [PubMed] [Google Scholar]