

Abstract
Structures of the four reaction states of the adenine riboswitch aptamer domain, including a transient intermediate state were solved by serial femtosecond crystallography. The structures not only demonstrate the use of X-ray free electron lasers for RNA crystallography but have also proven that transient states can be determined in real time by mix-and-inject crystallography. These results illustrate the structural basis for the ligand-induced conformational changes associated with the molecular “switch”.
Keywords: RNA, adenine riboswitch, serial femtosecond crystallography, X-ray free electron laser, time-resolved crystallography, diffusive mixing, phase transition, lattice conversion, reaction intermediate
Graphical Abstract
Using an X-ray free electron laser and a mix-and-inject serial crystallography approach, we have captured the ligand-induced structural conversion of the adenine riboswitch aptamer domain from apo, to intermediate, to final bound states. Like synchronized swimmers, millions of molecules undergo large conformational changes within the crystal lattice. These results provide the structural basis for the switching mechanism for regulating gene expression. Image: Joseph Meyer, Scientific Publications Graphics and Media, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research.
Introduction
Found in all three kingdoms of life, riboswitches are structural segments of certain mRNAs that function as cis-acting regulators of gene expression. They are usually located in the 5’-untranslated regions of mRNAs, and typically consist of two domains: an aptamer domain and a downstream expression platform (Fig. 1A). When the term “riboswitch” was coined about 15 years ago, it referred literally to the distinct conformational switching that takes place in an aptamer domain upon ligand binding, resulting in an altered pattern of gene expression [1, 2]. For most riboswitches, the core of the mechanism is a “switching sequence,” whose structure forms part of either the first helix (P1) of the aptamer domain, or the expression platform, depending on the ligand-triggered conformational changes [3]. How such structural changes are “transmitted” from the binding pocket to the “switching sequence,” and subsequently the expression platform, has been the focal point of extensive investigations [3]. A full understanding of the nature of binding and “switching” requires detailed structural information on unbound aptamers [3] as well as any transient intermediates [4].
Figure 1.
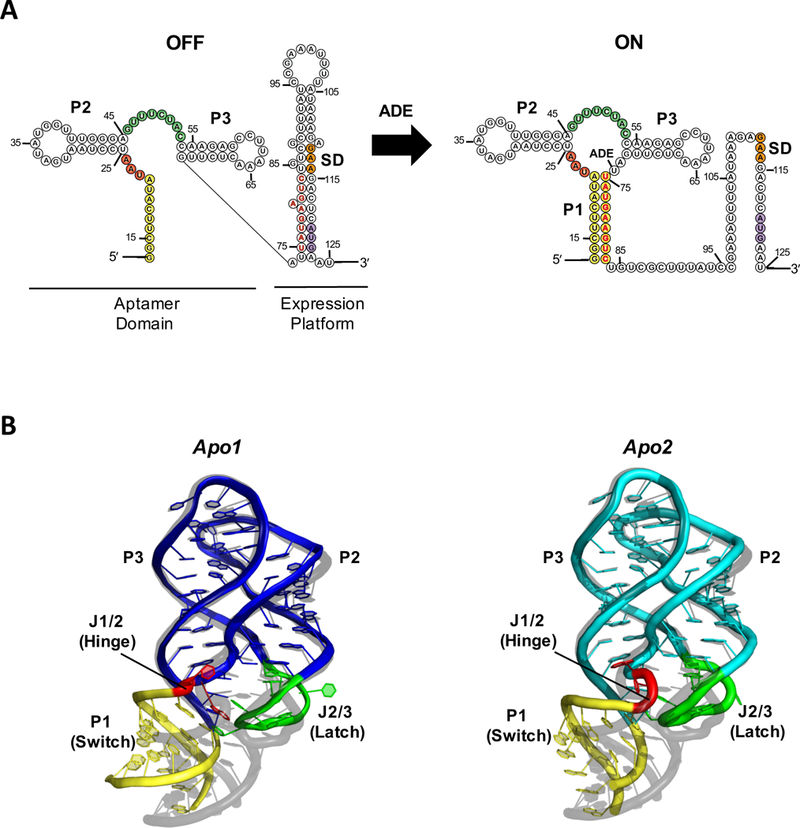
(A) Secondary structure of the V. vulnificus add adenine riboswitch and (B) Crystal structure of the ligand-free aptamer domain in apo1 (left, blue) and apo2 (right, cyan) states (PDB:5E54), aligned to the ligand-bound structure (grey, PDB:4TZX) for comparison. The P1 helix (yellow), J1/2 hinge (red), and J2/3 latch (green) regions, which exhibit the greatest structural differences among rA71 conformational states, are highlighted. The switching sequence is denoted in red bold letters, the Shine-Dalgarno in orange, and the start codon in purple. The sequence used for the apo-rA71 structure differs slightly from what is shown in (A) with a few stabilizing mutations in P1.
Challenges in RNA structure determination
A number of studies have suggested the presence of multiple conformational states of riboswitches, both in the presence and absence of ligand [5–8]. In particular, Zhang et al. have postulated that general equilibria exist between “dynamic incompetent” and “dynamic competent” apo states, and between the “dynamic competent” and “dynamic bound” states that precede the final “static bound” state [5]. However, such a hypothesis lacks direct structural evidence to support it. In order to prove its validity, structural information of a riboswitch in all of the described conformational states is necessary. To date, most determined riboswitch structures are ligand-bound and all currently known ligand-free structures of riboswitches either closely resemble their ligand-bound conformations, or exhibit only localized conformational changes [3, 9–14]. One exception is the glutamine riboswitch, where the structure of the apo state was determined in a complex with protein U1A with replacement of two hairpin loops [15]. Thus, the structure of a riboswitch aptamer domain in an apo state that differs significantly from its bound state has never been determined, and conformational changes induced by the absence or presence of ligand have not been directly visualized at the atomic level.
There is general lack of RNA structural information. Only 3% of the entries in the protein data bank are from RNA and RNA-containing complexes, of which fewer than 100 entries are unique and larger than 70 nucleotides. Limited RNA structural information is due in part to the intrinsic dynamics and conformational flexibility of RNA, as well as methodological limitations. With only four nucleotide building blocks and limited structural elements, RNA must be dynamic and conformationally flexible to achieve its diversity of biological functions. As a result, it is often difficult to obtain RNA crystals of sufficient size and order for single-crystal X-ray diffraction studies. Even when available, RNA crystals often exhibit high solvent content and elevated mosaicity, and their phosphate-rich contents make them highly susceptible to radiation damage [16]. Additionally, the ability to study RNA structure by nuclear magnetic resonance is limited by the large molecular weight of RNA and the chemical-shift similarities of nucleotide bases. New approaches and tools are therefore acutely needed to study RNA structure.
In 2009, the world’s first hard X-ray free-electron laser (XFEL) went into operation: the Linac Coherent Light Source (LCLS) at the SLAC National Accelerator Laboratory. An XFEL differs from conventional synchrotron X-ray sources in two significant ways: peak brilliance and femtosecond (fs) pulse duration. An XFEL pulse with the existing technology lasts between 10 and 100 fs and is over one billion times more brilliant than the current brightest synchrotron sources. One of the first biological applications of the XFEL was serial femtosecond crystallography (SFX) using micro and nanocrystals of proteins [17]. The ultra-short pulses enable “diffraction before destruction,” that is, a single diffraction pattern for each crystal can be recorded before the effects of radiation damage from the X-ray beam manifest. Unlike traditional crystallography, a new crystal must be delivered for each X-ray pulse, and then thousands of diffraction patterns are merged together[18]. The SFX method overcomes a fundamental problem in all biological imaging science, the ability to study the structures of macromolecules at room temperature with minimal effects from radiation damage. In addition, it eliminates a frequent bottleneck in crystallography of producing well-ordered crystals large enough for diffraction studies at a synchrotron.
The femtosecond time window and the brightness of the XFEL open unprecedented opportunities for structural biologists to study the time-evolution of biological processes at the atomic level. Fundamentally, all biological processes occur as a function of the spatial and temporal relationship of molecular events in the cellular environment. Since the XFEL is capable of taking instantaneous “snapshots” with femtosecond pulses, it will be possible to record “molecular movies” of chemical or biological processes, which occur on a slower picosecond to millisecond time scale. The first applications of time-resolved crystallography using an XFEL were optically triggered photoreactions [19–21]. However, this application is limited to photoactivated proteins, which comprise a very small population of all proteins. A broader application of the XFEL for time-resolved studies of ligand-activated biomacromolecules, which comprise the majority, is achieved by rapid mixing of ligand with the crystals prior to diffraction, termed mix-and-inject crystallography. Using this approach, a large number of biochemical systems, such as enzymatic, transport, drug-binding, and other ligand-triggered reactions, can be studied at the structural level in real time. The full course of reaction states—from initiation to activation, to transitional intermediates, to termination—can potentially be observed.
Perhaps one of the most profound impacts that the XFEL will have in the new era of biological science is to understand RNA structure and function. To date, SFX applications have been limited to proteins. The potential for SFX to address the myriad technical challenges associated with RNA structure determination has not been previously demonstrated. As we now know, functional RNA molecules are intrinsically dynamic and their functions are encoded not in primary sequences but in true four-dimensional descriptions of space and time. Therefore, functional insight requires not only snapshots of static RNA structures, but also the induced changes of those structures over time, i.e., movies that fully describe RNA functions in action. Here we discuss results demonstrating the capability of SFX to capture structural snapshots, in real time, of RNA in various conformational states.
Our initial indication of a multistage kinetic model for the adenine riboswitch aptamer domain (rA71), consisting of residues 13–83 (Fig. 1A) was obtained by stopped-flow fluorescence spectroscopy. The kinetics could be best explained by slow exchange between binding-incompetent (apo1) and binding-competent (apo2) forms of rA71, followed by adenine binding to form an intermediate complex, IB•ade. Adenine binding reorganizes the binding pocket, converting IB•ade to the final ligand-bound state, B•ade.
| (1) |
Verifying this four-state model required structure determination of the transient intermediate state, thus posing a significant challenge. To study the structural changes of rA71 upon ligand binding, we employed the mix-and-inject SFX strategy [4]. Such an experiment had been proposed previously in a proposal to LCLS in 2003 (http://www.slac.stanford.edu/pubs/slacreports/reports03/slac-r-611.pdf) and in the literature [22].
Structures of the apo states and comparison with the structure of the bound state
The structure of rA71 in the absence of ligand was determined to 2.3 Å resolution, with Rwork/Rfree of 0.213/0.256, an average B-factor of 77.2 Å2, and root mean square deviations of 0.007 Å and 1.62° for bond lengths and bond angles, respectively [4]. The asymmetric unit of apo-rA71 contains the two ligand-free conformers, apo1 and apo2, both of which differ significantly from the ligand-bound form (RCSB:4TZX [23]) in the three-way junction, primarily the hinge and latch regions, and in the P1 helix that contains the “switching sequence” (Fig. 1). The “hinge” is a pivot point for P1, whereas the “latch” locks the helix in place only when the ligand is bound. When compared to the bound form by superimposing the kissing-loop structures, the heavy-atom RMSDs are > 6 Å for the three-way junctions, and 6.5 Å and 10.3 Å for the P1 helices of apo2 and apo1, respectively. The kissing loop structures, on the other hand, differ only at A64, which is in an anti-configuration in both apo conformers and syn in the bound conformation.
Contrary to the previous conception [3], the ligand binding pockets in both apo conformers are highly ordered, but with different sets of base-stacking and hydrogen-bond interactions among key residues relative to the bound conformation. For example, U48, which is solvent-exposed in the ligand-bound form, flips in to form a wobble base pair either with ligand-recognition residue U74 (apo2), or U75 (apo1). U22, A23 (apo2 only), and U51 flip out of the binding pocket and are solvent exposed (Fig. 2A). The fact that the ligand is completely encapsulated by RNA in the bound structure implies that substantial rearrangements of the binding pocket are necessary for ligand entry or egress [6, 24]. The flexibility in the hinge (J1/2) and latch (J2/3) regions may facilitate such conformational fluctuations between the two apo states, as well as the changes upon ligand binding.
Figure 2.
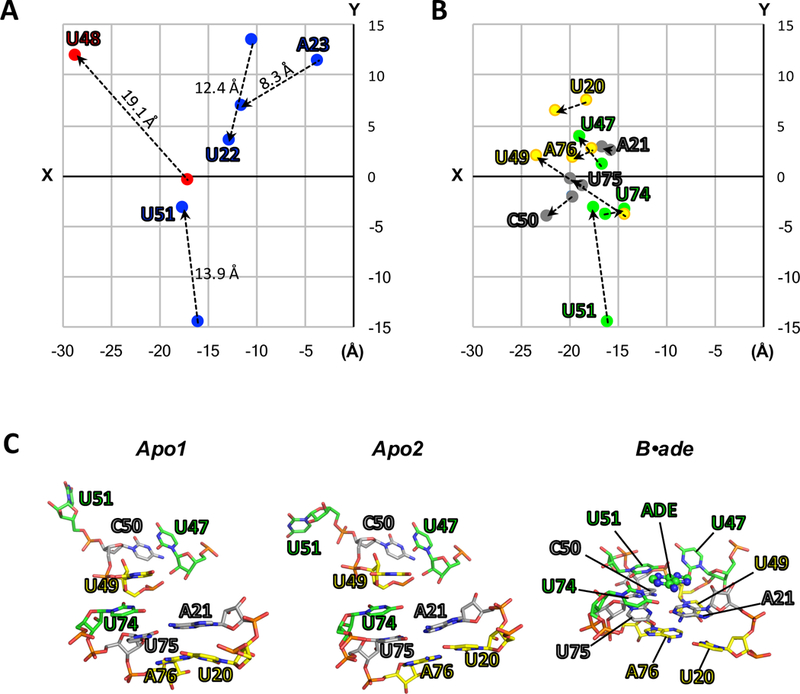
(A-B) Two-dimensional projections (XY-plane) of the structural coordinates (Å) of key residues for apo2 and B•ade. The structures of apo2 (PDB:5E54) and B•ade (PDB:4TZX) were aligned as in Fig. 1, and the XY-coordinates for O2 atoms (pyrimidines) or N6 atoms (purines) were taken directly from their respective PDB files and plotted. (A) “Swinging residues” that flip toward (blue) or away (red) from the ligand-binding pocket upon conversion to B•ade. The direction of movement is also indicated by dotted lines. Values reported above the dotted lines correspond to the distances in three dimensions. (B) Residues involved in the three ligand-facilitated base triples: U20-U49-A76 (yellow), A21-C50-U75 (grey), and U47-U51-U74-ade (green). (C) In the absence of ligand (apo1 and apo2), the base-triple interactions are broken. Ligand binding facilitates the formation of the base triples, as observed in the crystal structure of ligand-bound rA71 (PDB:4TZX). These interactions include ligand recognition by U74 and anchoring of P1 (residues U75 and A76) and the hinge (U20 and A21) via the latch (residues U47, U49, C50 and U51), ultimately leading to the stabilization of the P1 helix.
The hinge is a flexible region that facilitates the rotation of the P1 helix, and exhibits marked differences among all three structures, with RMSDs of ~7.8 Å (apo1 vs. B•ade) and ~5.9 Å (apo2 vs. B•ade). The hinge RMSD between the two apo conformers is also large (~7.2 Å) due to differences in residues U22 and A23. The angular differences in the helical axis of P1 relative to the ligand-bound form are ~15° (apo1) or ~22° (apo2). In addition, “kinking” at the hinge region causes a shift in P1 of ~2 Å (apo2) or ~8 Å (apo1) away from the latch, relative to the bound structure, and a compression of the major groove formed by helices P1 and P3, measuring ~10 Å in the two apo conformers, as compared to >16 Å for the ligand-bound conformer [4].
In the absence of ligand, the lack of key interactions in the binding pocket results in the partial disorder of P1, which includes the 3ʹ strand containing the “switching sequence.” Specifically, P1 is destabilized through the breaking of base triples U49-A76-U20, C50-U75-A21, and U47-U51-U74-Ade (Fig. 2B and2C), resulting in the loss of the coaxial stacking of helices P1 and P3, and the long-range interactions between P1 and the latch that lock P1 in place. The observed electron density for P1 was poorly defined in both apo conformations, particularly the 3′ strand containing the switching sequence, which could only be modeled up to residue 77 (apo1) or 80 (apo2) [4]. A number of studies have shown, for both the aptamer domain and full-length adenine riboswitch, that P1 is only marginally stable [25–29] and rapidly fluctuates between the “ON” and “OFF” states in the absence of a ligand, where the competing hairpin in the expression domain, which contains the “switching sequence” is formed at the expense of the P1 helix in the full-length riboswitch [8, 27–29].
The snapshot of the intermediate state
Our stopped-flow fluorescence kinetics data strongly indicated the presence of an intermediate bound (IB•ade) state between the binding-competent apo2 and final bound states [4]. This intermediate had a similar fluorescence intensity as the apo2, suggesting that U48, which is replaced by fluorescent 2-aminopurine, has a similar structural environment, specifically its stacking interactions. We were able to capture the structure of this intermediate state via our mix-and-inject experiment by employing a 10-second mixing delay. The crystal lattice was unchanged after 10 seconds of mixing, implying no major conformational changes had yet occurred. Nevertheless, the difference electron density between the apo and IB•ade structures revealed key local conformational changes in the binding pocket of apo2 only. In IB•ade, the adenine ligand displaces U48 and is base-stacked between U48 and U49, forming stacking interactions U49-ade-U48-U75, similar to the U49-U48-A21-U75 stacking in apo2 prior to binding. This explained the minor difference in U48–2AP fluorescence observed between the apo2 and IB•ade states. The U49-ade-U48-U75 stacking is similar to that in apo1, except that A21 is in place of the ligand. Thus, apo1 is in a “pseudo-intermediate” structural state, and must convert to apo2 in order to become binding competent. In this regard, apo1 may play a regulatory role, in which the rate of interconversion between the two apo states may vary in response to changes in the cellular environment, such as temperature or Mg2+ concentration, that are independent of ligand concentration.
The “switching”
Detailed structural knowledge of all four states in the aptamer-ligand interaction makes it possible to understand the structural basis for the “switching” action triggered by ligand binding. Like apo1 and apo2, the binding pocket of IB•ade is structured, but lacks the three base-triple interactions that lock the P1 helix in place. Ligand binding triggers the formation of the three base-triple interactions and alignment of helices P1 and P3 (Figs. 1B and2). This reconfiguration stabilizes the structure of P1, and thus the switching sequence, which is at the heart of the molecular switch.
After a mixing time of 10 minutes, our mix-and-inject results demonstrated that the switching action occurs in crystallo, resulting in a conversion of all molecules to the final ligand-bound state (Fig. 3). In addition to the large intramolecular changes associated with binding, half of the molecules rotate ~90°, resulting in a polymorphic phase transition in which the lattice was converted from monoclinic (P21) to orthorhombic (P21212). We presume that the diffraction quality (i.e., “survivability”) of crystals upon conversion depends on the spatiotemporal uniformity of the molecular changes throughout the crystal. Such uniform changes may occur cooperatively when the accumulation of IB•ade results in enough energy to overcome the opposing lattice constraints.
Figure 3.
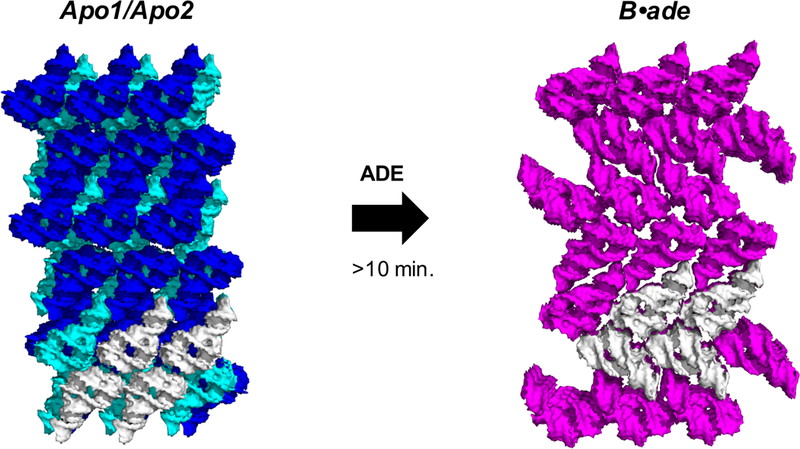
Large conformational changes induced after 10 minutes of ligand mixing result in a polymorphic phase transition and lattice conversion from a monoclinic (apo1 and apo2: blue and cyan respectively) to an orthorhombic space group (B•ade: magenta), which was accommodated in micro/nanocrystals of rA71. A subset of 4 related molecules (white) between the two structures illustrates that half of the molecules rotate ~90° upon conversion.
Final Remarks
This work was the first to demonstrate that real-time mix-and-inject crystallography is possible using an XFEL. Using this revolutionary structural biology method, we determined the structures of all four states of the adenine riboswitch aptamer domain, thereby illustrating the molecular basis for the switch. The mechanism begins with two interconverting apo conformers where ligand-binding pockets are distinctly different from the ligand-bound conformation. The binding of ligand results in a transitional intermediate and the subsequent formation of three base-triples and coaxial stacking of P1 with P3, which together constitute “switching” action and stabilize the P1 helix. As a result, the Shine-Dalgarno sequence in this translational riboswitch becomes accessible for translation initiation. The utility of mix-and-inject crystallography to structurally elucidate reaction mechanisms may have broad applications to study the dynamics of many biomacromolecules and their ligand/substrate interactions.
Acknowledgements
This work was supported in part by the NIH Intramural Research Programs of NCI and CIT and by the US Department of Energy, Office of Biological and Environmental Research, under Contract DE-AC02–06CH11357. Portions of this research were carried out at the Linac Coherent Light Source, a National User Facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The CXI instrument was funded by the LCLS Ultrafast Science Instruments (LUSI) project funded by the US Department of Energy, Office of Basic Energy Sciences. Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515.
Abbreviations
- rA71 71
nucleotide adenine riboswitch aptamer domain
- XFEL
X-ray free-electron laser
- SFX
Serial femtosecond crystallography
Reference
- 1.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL & Breaker RR (2002) Genetic control by a metabolite binding mRNA, Chem Biol. 9, 1043. [DOI] [PubMed] [Google Scholar]
- 2.Winkler WC & Breaker RR (2003) Genetic control by metabolite-binding riboswitches, Chembiochem. 4, 1024–32. [DOI] [PubMed] [Google Scholar]
- 3.Batey RT (2012) Structure and mechanism of purine-binding riboswitches, Q Rev Biophys. 45, 345–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stagno JR, Liu Y, Bhandari YR, Conrad CE, Panja S, Swain M, Fan L, Nelson G, Li C, Wendel DR, White TA, Coe JD, Wiedorn MO, Knoska J, Oberthuer D, Tuckey RA, Yu P, Dyba M, Tarasov SG, Weierstall U, Grant TD, Schwieters CD, Zhang J, Ferre-D’Amare AR, Fromme P, Draper DE, Liang M, Hunter MS, Boutet S, Tan K, Zuo X, Ji X, Barty A, Zatsepin NA, Chapman HN, Spence JC, Woodson SA & Wang YX (2016) Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography, Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Jones CP & Ferre-D’Amare AR (2014) Global analysis of riboswitches by small-angle X-ray scattering and calorimetry, Biochim Biophys Acta. 1839, 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilbert SD, Stoddard CD, Wise SJ & Batey RT (2006) Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain, Journal of molecular biology. 359, 754–68. [DOI] [PubMed] [Google Scholar]
- 7.Reining A, Nozinovic S, Schlepckow K, Buhr F, Furtig B & Schwalbe H (2013) Three-state mechanism couples ligand and temperature sensing in riboswitches, Nature. 499, 355–9. [DOI] [PubMed] [Google Scholar]
- 8.Rieder R, Lang K, Graber D & Micura R (2007) Ligand-induced folding of the adenosine deaminase A-riboswitch and implications on riboswitch translational control, Chembiochem. 8, 896–902. [DOI] [PubMed] [Google Scholar]
- 9.Garst AD, Heroux A, Rambo RP & Batey RT (2008) Crystal structure of the lysine riboswitch regulatory mRNA element, J Biol Chem. 283, 22347–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang L, Serganov A & Patel DJ (2010) Structural insights into ligand recognition by a sensing domain of the cooperative glycine riboswitch, Mol Cell. 40, 774–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serganov A, Huang L & Patel DJ (2008) Structural insights into amino acid binding and gene control by a lysine riboswitch, Nature. 455, 1263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenkins JL, Krucinska J, McCarty RM, Bandarian V & Wedekind JE (2011) Comparison of a preQ1 riboswitch aptamer in metabolite-bound and free states with implications for gene regulation, J Biol Chem. 286, 24626–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoddard CD, Montange RK, Hennelly SP, Rambo RP, Sanbonmatsu KY & Batey RT (2010) Free state conformational sampling of the SAM-I riboswitch aptamer domain, Structure. 18, 787–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vicens Q, Mondragon E & Batey RT (2011) Molecular sensing by the aptamer domain of the FMN riboswitch: a general model for ligand binding by conformational selection, Nucleic Acids Res. 39, 8586–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren A, Xue Y, Peselis A, Serganov A, Al-Hashimi HM & Patel DJ (2015) Structural and Dynamic Basis for Low-Affinity, High-Selectivity Binding of L-Glutamine by the Glutamine Riboswitch, Cell reports. 13, 1800–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golden BL (2007) Preparation and crystallization of RNA, Methods in molecular biology (Clifton, NJ). 363, 239–57. [DOI] [PubMed] [Google Scholar]
- 17.Boutet S, Lomb L, Williams GJ, Barends TR, Aquila A, Doak RB, Weierstall U, DePonte DP, Steinbrener J, Shoeman RL, Messerschmidt M, Barty A, White TA, Kassemeyer S, Kirian RA, Seibert MM, Montanez PA, Kenney C, Herbst R, Hart P, Pines J, Haller G, Gruner SM, Philipp HT, Tate MW, Hromalik M, Koerner LJ, van Bakel N, Morse J, Ghonsalves W, Arnlund D, Bogan MJ, Caleman C, Fromme R, Hampton CY, Hunter MS, Johansson LC, Katona G, Kupitz C, Liang M, Martin AV, Nass K, Redecke L, Stellato F, Timneanu N, Wang D, Zatsepin NA, Schafer D, Defever J, Neutze R, Fromme P, Spence JC, Chapman HN & Schlichting I (2012) High-resolution protein structure determination by serial femtosecond crystallography, Science. 337, 362–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlichting I (2015) Serial femtosecond crystallography: the first five years, IUCrJ. 2, 246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pande K, Hutchison CD, Groenhof G, Aquila A, Robinson JS, Tenboer J, Basu S, Boutet S, DePonte DP, Liang M, White TA, Zatsepin NA, Yefanov O, Morozov D, Oberthuer D, Gati C, Subramanian G, James D, Zhao Y, Koralek J, Brayshaw J, Kupitz C, Conrad C, Roy-Chowdhury S, Coe JD, Metz M, Xavier PL, Grant TD, Koglin JE, Ketawala G, Fromme R, Srajer V, Henning R, Spence JC, Ourmazd A, Schwander P, Weierstall U, Frank M, Fromme P, Barty A, Chapman HN, Moffat K, van Thor JJ & Schmidt M (2016) Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein, Science. 352, 725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tenboer J, Basu S, Zatsepin N, Pande K, Milathianaki D, Frank M, Hunter M, Boutet S, Williams GJ, Koglin JE, Oberthuer D, Heymann M, Kupitz C, Conrad C, Coe J, Roy-Chowdhury S, Weierstall U, James D, Wang D, Grant T, Barty A, Yefanov O, Scales J, Gati C, Seuring C, Srajer V, Henning R, Schwander P, Fromme R, Ourmazd A, Moffat K, Van Thor JJ, Spence JC, Fromme P, Chapman HN & Schmidt M (2014) Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein, Science. 346, 1242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barends TR, Foucar L, Ardevol A, Nass K, Aquila A, Botha S, Doak RB, Falahati K, Hartmann E, Hilpert M, Heinz M, Hoffmann MC, Kofinger J, Koglin JE, Kovacsova G, Liang M, Milathianaki D, Lemke HT, Reinstein J, Roome CM, Shoeman RL, Williams GJ, Burghardt I, Hummer G, Boutet S & Schlichting I (2015) Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation, Science. 350, 445–50. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt M (2013) Mix and Inject: Reaction Initiation by Diffusion for Time-Resolved Macromolecular Crystallography, Adv Cond Matter Phys. [Google Scholar]
- 23.Zhang J & Ferre-D’Amare AR (2014) Dramatic improvement of crystals of large RNAs by cation replacement and dehydration, Structure. 22, 1363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noeske J, Buck J, Furtig B, Nasiri HR, Schwalbe H & Wohnert J (2007) Interplay of ‘induced fit’ and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch, Nucleic acids research. 35, 572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nozinovic S, Reining A, Kim YB, Noeske J, Schlepckow K, Wohnert J & Schwalbe H (2014) The importance of helix P1 stability for structural pre-organization and ligand binding affinity of the adenine riboswitch aptamer domain, RNA Biol. 11, 655–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Palma F, Colizzi F & Bussi G (2013) Ligand-induced stabilization of the aptamer terminal helix in the add adenine riboswitch, Rna. 19, 1517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenleaf WJ, Frieda KL, Foster DA, Woodside MT & Block SM (2008) Direct observation of hierarchical folding in single riboswitch aptamers, Science. 319, 630–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neupane K, Yu H, Foster DA, Wang F & Woodside MT (2011) Single-molecule force spectroscopy of the add adenine riboswitch relates folding to regulatory mechanism, Nucleic Acids Res. 39, 7677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haller A, Altman RB, Souliere MF, Blanchard SC & Micura R (2013) Folding and ligand recognition of the TPP riboswitch aptamer at single-molecule resolution, Proceedings of the National Academy of Sciences of the United States of America. 110, 4188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]