

Summary
The 26S proteasome is the major proteolytic machine for breaking down cytosolic and nuclear proteins in eukaryotes. Due to the lack of a suitable assay, it is difficult to measure routinely and quantitatively, the breakdown of proteins by the 26S proteasome in vitro. In the present study, we developed an assay to monitor proteasome-mediated protein degradation. Using this assay, we discovered that epipolythiodioxopiperazines (ETPs) blocked the degradation of our model substrate in vitro. Further characterization revealed that ETPs inhibited proteasome function by targeting the essential proteasomal deubiquitinase Rpn11. ETPs also inhibited other JAMM proteases such as Csn5 and AMSH. An improved ETP with less non-specific effects, SOP11, stabilized a subset of proteasome substrates in cells, induced the unfolded protein response, and led to cell death. SOP11 represents a class of Rpn11 inhibitor and provides an alternative route to develop proteasome inhibitors.
GRAPHICAL ABSTRACT
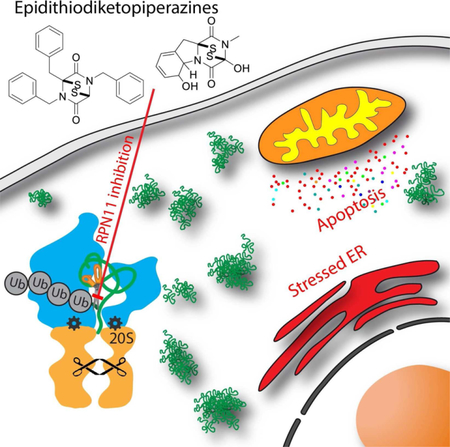
Introduction
The fungus Aspergillus fumigatus is one of the most common species to cause disease in immunocompromised individuals, such as AIDS patients and organ transplant recipients (Dolan et al., 2015). A. fumigatus infection causes aspergillosis and immunosuppression (Scharf et al., 2016). The secondary metabolites produced by Aspergillus are considered to be important virulence factors. Among them, gliotoxin, an epidithiodiketopiperazine (ETP), is the major and the most potent toxin (Scharf et al., 2016). Most of the biological activities of gliotoxin are derived from a pair of sulfur atoms that form an unusual, intramolecular disulfide bridge (Dolan et al., 2015; Scharf et al., 2016). Gliotoxin poisons animal cells by inactivating multiple key enzymes through conjugation of their thiol groups. Known targets of gliotoxin include nuclear factor κB (NF-κB), NADPH oxidase, and glutaredoxin (Pahl et al., 1996; Srinivasan et al., 2006; Tsunawaki et al., 2004). Glutathione (GSH) is important for gliotoxin uptake and mediates its cytotoxicity in animal cells, as it reduces gliotoxin to convert it into the toxic dithiol form (Dolan et al., 2015). Further studies have revealed a mechanism for its inhibition involving Zinc chelation: gliotoxin and other ETPs can inhibit HIF1α interaction with p300 by ejecting the Zn2+ from p300 through the formation of a Zn2+-ETP complex (Cook et al., 2009). This disrupts HIF1α activation and provides a plausible molecular basis for the anti-angiogenesis effects of ETPs.
The proteasome, the major cellular machine for protein degradation, is also reported as a target of gliotoxin (Kroll et al., 1999). Proteasomes are essential for various cellular processes including protein quality control, regulation of gene expression, and cell cycle progression. Structurally, the proteasome is composed of a 20S core particle (CP) and 19S regulatory particles (RP), which cap the ends of the CP. The RP recognizes polyubiquitinated substrates and processes them for insertion into the CP which contains the proteolytic active sites (Finley, 2009). There are three distinct catalytic peptidase activities identified in the CP: chymotrypsin-like, trypsin-like, and caspase-like (Heinemeyer et al., 1997). Drugs such as bortezomib and carfilzomib, which inhibit the active sites in the CP, are important therapeutic agents for the treatment of multiple myeloma (Dimopoulos et al., 2015). However, patients ultimately suffer relapse despite the clinical benefit conferred by these drugs. Therefore, novel drugs working through different mechanisms are needed. Recently, we and others discovered small molecule inhibitors targeting Rpn11, a JAMM protease that removes polyubiquitin chains from substrate proteins (Lauinger et al., 2017; Li et al., 2017; Perez et al., 2017). Inhibition of Rpn11 function results in proteasome malfunction and leads to cell death (Li et al., 2017). Previous research suggests that gliotoxin is a noncompetitive inhibitor of the chymotrypsin-like activity of the 20S proteasome (Kroll et al., 1999). However, the detailed mechanism of this inhibition remains unknown.
Herein, we developed an assay to measure the protein breakdown activity of the proteasome in purified systems and cell extracts and identified ETPs as a scaffold for inhibiting JAMM proteases. ETPs inhibit proteasome function by targeting the essential proteasomal deubiquitinase Rpn11. Identification of ETPs provides an alternative route to inhibit proteasome function and opens the door to the development of new Rpn11 inhibitors.
Results
Development of a proteasome substrate to monitor protein degradation in vitro
There is no quantitative method for the simple and rapid assessment of 26S proteasome protein degradation activity in vitro to date. The Suc-LLVY-amc substrate widely used for the evaluation of 20S proteasome activity does not accurately reflect protein breakdown because it only measures the chymotrypsin-like active site of the β5 subunit. Meanwhile, it has been shown that to block protein degradation, it is necessary to inhibit both the chymotryptic site, which is intrinsically the most sensitive to the commonly used 20S inhibitors, as well as either the tryptic or caspase site, which are about an order of magnitude less sensitive (Demo et al., 2007; Kisselev and Goldberg, 2005). In addition, Suc-LLVY-amc is not only cleaved by the 20S proteasome but also by other chymotrypsin-like proteases and by calpains (Giguere and Schnellmann, 2008).
To measure protein breakdown by the proteasome, we modified an existing method (Kim and Huibregtse, 2009) to generate a polyubiquitinated protein substrate, termed UbnGST– Wbp2 (WW domain-binding protein 2, n>30) using enzymatic approaches (Fig. 1A and Fig. S1A). Wbp2 was originally isolated from a mouse embryo library. It contains an N-terminal pleckstrin homology-glucosyltransferase (GRAM) domain and three C-terminal PPxY motifs, which interact with multiple WW domain-containing proteins (Chen and Sudol, 1995). A previous study showed that Wbp2 functions as a coactivator for estrogen receptor and is closely linked to the development of breast cancer (Chen et al., 2017). The original method described by Huibregtse and colleagues relies on Rsp5-mediated ubiquitination of a truncated Wbp2 (Kim and Huibregtse, 2009). We modified their method by inserting a C-terminal hexahistidine tag into the GST–Wbp2 construct and chemically labeling the purified protein with a cysteine-reactive fluorophore. Rsp5 assembles K63-linked ubiquitin chains and contains a WW-domain that recognizes the PPxY motifs in Wbp2. Swapping the HECT domain of Rsp5 with that from E6AP yields a chimeric enzyme that preferentially synthesizes K48-linked ubiquitin chains (Kim and Huibregtse, 2009). The ubiquitin linkages formed on our substrate treated with the wild-type and chimeric Rsp5 were quantified by mass spectrometry. 87% of the linkages formed by Rsp5 were via K63 (K63UbnGST-Wbp2), whereas 94% of those formed by the Rsp5–E6AP chimera were via K48 (K48UbnGST-Wbp2; Table S1).
Fig 1. Development of an assay to monitor protein degradation.
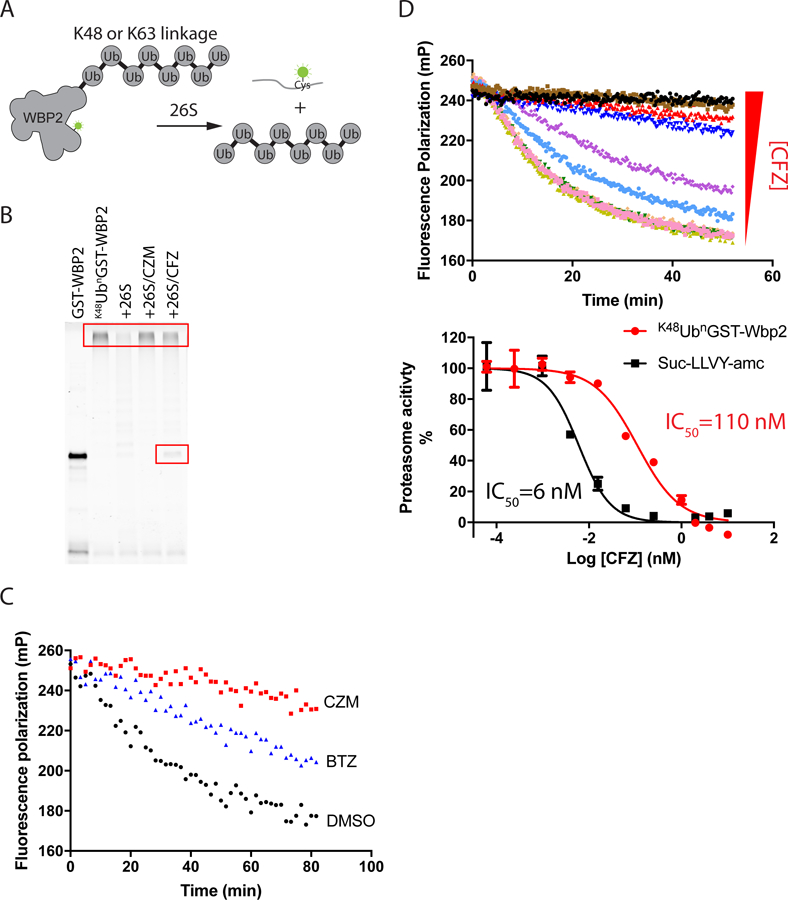
(A) Design of protein degradation assay. (B) 20 nM K48UbnGST–Wbp2 was incubated with 5 nM 26S proteasome at 37 ˚C for 2 hours in the absence and presence of different inhibitors (20 μM capzimin/CZM or 10 μM carfilzomib/CFZ). Reactions were fractionated by SDS-PAGE and analyzed using a Typhoon fluorescence scanner. Boxes mark the input high MW polyubiquitinated substrate and a deubiquitinated species that accumulated in reactions inhibited by CFZ (Verma et al., 2002). (C) Measurement of proteasome activity using the fluorescence polarization assay. K48UbnGST–Wbp2 (2.5 nM) was incubated with 26S proteasome (1 nM) at 37 °C in the absence or presence of different inhibitors (10 µM CZM or 1 µM bortezomib/BTZ). (D) Measurement of proteasome protein breakdown activity in cell lysate in response to increasing concentrations of carfilzomib. Shown are the reaction kinetics (upper panel) and dose-response (red curve in the bottom panel) of proteasome activity measured at 37 °C using K48UbnGST–Wbp2 as substrate and lysate from cells treated with different concentrations of carfilzomib. For comparison, a dose-response curve measured using Suc-LLVY-amc is plotted in black (bottom panel). Error bars represent s.d., n = 3 wells, from 1 representative of 3 independent experiments. See also Figure S1 and Table S1.
To test whether K63UbnGST-Wbp2 and K48UbnGST-Wbp2 were proteasome substrates, we incubated them with purified human 26S proteasomes and ATP, and analyzed the reactions by SDS-PAGE. The fluorescence-scanned gel showed that both proteins were degraded, consistent with the prior report (Kim and Huibregtse, 2009). The degradation was specific and was blocked by an Rpn11 inhibitor (capzimin, CZM), a 20S inhibitor (carfilzomib, CFZ) or a non-hydrolysable ATP analogue, AMP-PNP (Fig. 1B and Fig. S1B).
To simplify and accelerate data acquisition, we adapted the degradation assay to a format compatible with high throughput screening by monitoring the decrease in fluorescence polarization (FP) in a multi-well plate reader. The decreased fluorescence polarization indicated that the fluorescent UbnGST–Wbp2 substrate was cleaved into smaller species. Substrate degradation monitored in this fashion was specific in that it was inhibited by addition of 20S and Rpn11 inhibitors (Fig. 1C and Fig. S1C-D), or the slowly hydrolysable ATP analogue ATPγS (Fig. S1C), and was competed by a large excess of K48-linked Ub4 chains (Fig. S1E). Note that inhibition of the decrease in FP by the 20S inhibitor was not as complete as with the Rpn11 inhibitor (Fig. 1C), because there remained some decrease in FP due to substrate deubiquitination, which decreased its molecular weight (Fig. 1B).
To test whether the FP assay could be used to measure 26S proteasome activity directly in cell lysate, we lysed HEK293T cells treated with different concentrations of carfilzomib and performed the assay directly in the lysate by adding K48UbnGST-Wbp2 substrate and ATP. Ubiquitin aldehyde was included in the assay buffer to eliminate the interference of cysteine-based deubiquitinases (Hershko and Rose, 1987). The IC50 for inhibition of K48UbnGST-Wbp2 degradation by carfilzomib was 110 nM, which was ~20 fold higher than the IC50 obtained in the Suc-LLVY-amc assay (Fig. 1D). This is consistent with the observation that inhibition of protein breakdown requires blockade of at least two of the three active sites (Kisselev and Goldberg, 2005). This assay is also compatible with lysate prepared directly from tissue. In a recent study, we demonstrated that overexpression of the 11S proteasome subunit PA28α in the mouse retina does not alter ubiquitin-dependent protein degradation (Lobanova et al., 2018). This example illustrates how our assay can be used to monitor 26S proteasome activity in various disease states. It is worth noting that the K63UbnGST-Wbp2 substrate was not suitable for use in cell lysate, possibly due to the presence of a high level of K63 linkage-specific deubiquitinases (Cooper et al., 2009) (Fig. S1F).
Epidithiodiketopiperazines (ETPs) block protein degradation
Gliotoxin is the most well-known member of the family of epidithiodiketopiperazine compounds. Previous reports indicated that it functions as a 20S proteasome inhibitor targeting chymotrypsin-like activity, and the inhibitory effect depends on an intact disulfide bond in gliotoxin (Kroll et al., 1999). However, gliotoxin exists almost exclusively in the dithiol form after uptake into cells due to the reducing power of cellular glutathione (Bernardo et al., 2003). These contradictory observations motivated us to revisit the interaction between ETPs and the proteasome.
Taking advantage of our FP assay, we investigated the effects of gliotoxin and its core scaffold compound, SOP6, on the proteasome-mediated degradation of K48UbnGST–Wbp2 (Fig. 2A). Both SOP6 and gliotoxin slowed down the decline in fluorescence polarization, suggesting that ETP compounds inhibited proteasome function. To test this hypothesis, we treated HCT116 cells with different ETP compounds and immunoblotted for the accumulation of poly-ubiquitinated species. Gliotoxin and ETP analogues led to the accumulation of ubiquitinated species, which is a commonly used marker for proteasome inhibition (Fig. 2B). In addition, we tested ETP compounds in a UbG76V-GFP degradation assay (Chou and Deshaies, 2011). The results indicated that multiple ETP compounds blocked the degradation of pre-accumulated UbG76V-GFP, suggesting inhibitory effects on proteasome function (Table S2). Taken together, these results suggested that ETPs interfered with proteasome function in vitro and in cells. To identify the target of ETPs on the proteasome, we first examined their effects on the chymotrypsin-like activity of the 20S proteasome using the Suc-LLVY-amc fluorescence assay. Neither SOP6 nor gliotoxin showed inhibition of the chymotrypsin-like activity, indicating that reduced ETPs did not work as 20S proteasome inhibitors (Fig. 2C). Given that gliotoxin exists exclusively in the reduced form in cells (Bernardo et al., 2003), it is most likely that ETPs inhibit the proteasome through another mechanism.
Fig 2. ETPs inhibit protein degradation in vitro and in cells.
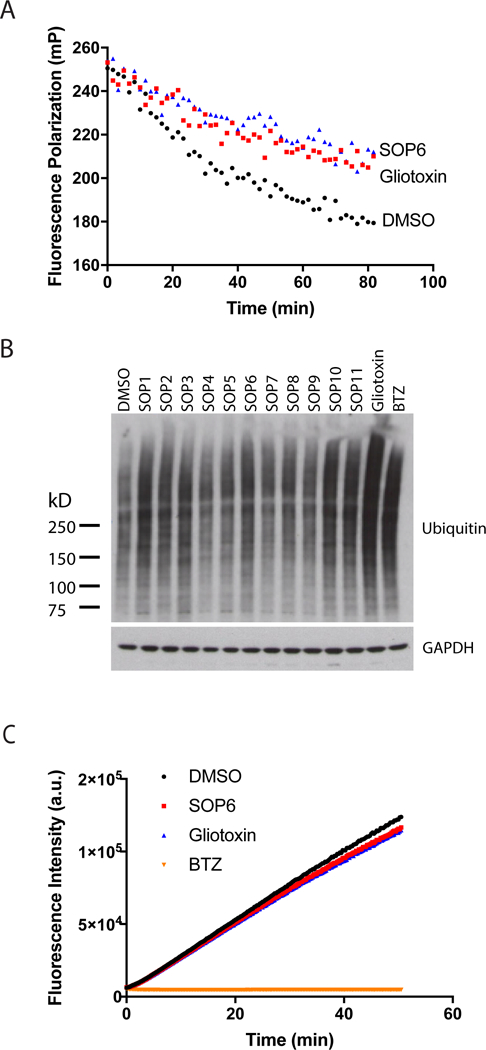
(A) SOP6 and gliotoxin block the processing of K48UbnGST–Wbp2 by the proteasome. K48UbnGST–Wbp2 was incubated with 26S proteasome at 37 °C in the absence or presence of SOP6 or gliotoxin (10 µM each). (B) ETP treatment caused accumulation of high-molecular weight ubiquitin conjugates. HCT116 cells were treated for 3 hours with 10 μM of the indicated ETPs, and the cell lysates were fractionated by SDS–PAGE and immunoblotted with antibodies against ubiquitin. (C) SOP6 and gliotoxin do not inhibit chymotrypsin-like activity of the proteasome. Suc-LLVY-amc (20 μM) was incubated with 15 nM purified human 26S proteasome in the absence or presence of SOP6, gliotoxin or BTZ (20 µM each).
ETPs inhibit Rpn11 and other JAMM proteases
Chetomin is an ETP that was initially identified as a toxic secondary metabolite from fungi (Geiger, 1949). Interestingly, chetomin was identified as the single positive hit in an HTS campaign for inhibitors that target the interaction between HIF1α and it coactivators p300 and CREB-binding protein (Kung et al., 2004). Subsequent research revealed that ETPs block the HIF1α-p300 interaction by extracting the zinc ion from the cysteine/histidine-rich domain 1 (CH1) of p300 (Cook et al., 2009).
The ubiquitin isopeptidase Rpn11 is the only essential proteasome subunit that is known to be zinc-dependent. Therefore, we surmised that ETPs might function as proteasome inhibitors by targeting Rpn11. To test this hypothesis, we evaluated the effect of ETPs on JAMM domain proteases and other metalloproteases (Fig. 3A, Table 1 and Table S2). The results suggested that ETPs specifically inhibited JAMM proteases, although the tested compounds did not show selectivity between different members of the JAMM family (Table 1 and Table S2). We further confirmed that ETPs not only inhibited Rpn11 function in the context of the intact proteasome (Fig. 3A) but also directly inhibited di-Ub cleavage mediated by purified Rpn11•Rpn8 heterodimer (Fig. 3B).
Fig 3. ETPs inhibit JAMM domain proteases.
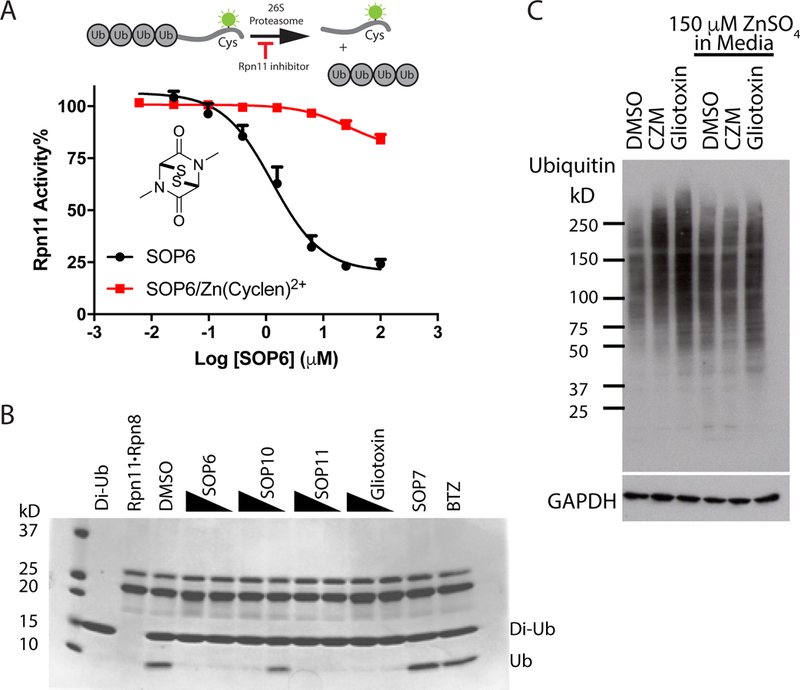
(A) ETPs inhibit Rpn11 activity by binding zinc. The IC50 for inhibition of Rpn11 activity by SOP6 was determined in the presence and absence of a Zn(cyclen)2+ coordination complex. The Ub4-peptide substrate was used for this assay as depicted. Error bars represent s.d., n = 4 wells, from 1 representative of 3 independent experiments. (B) Purified Rpn11•Rpn8 was incubated with K48-linked diubiquitin in the presence of 40 μM or 10 μM of the indicated compounds, and reactions were fractionated by SDS–PAGE and visualized with Coomassie blue. (C) Zn2+ counteracts gliotoxin induced accumulation of poly-ubiquitinated species. HCT116 cells were treated for 3 hours with 10 μM gliotoxin or CZM in the absence or presence of 150 μM ZnSO4 in the culture medium, and cell lysates were fractionated by SDS–PAGE and immunoblotted with antibodies against ubiquitin. See also Table S2.
Table 1.
Summary of IC50
| ID | Structure | IC50 (μM) | GI50 (μM) | ||||
|---|---|---|---|---|---|---|---|
| Rpn11 | Csn5 | AMSH | hCAII | MMP-2 | |||
| SOP6 | 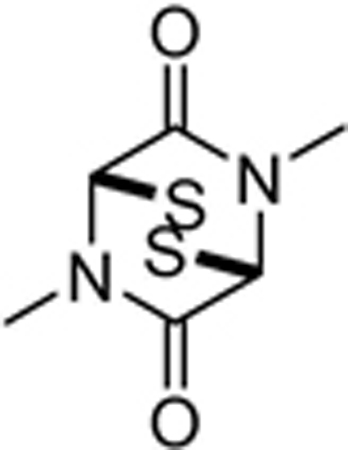 |
3.8±1.2 | 2.9±0.5 | 2.1±0.2 | >100 | >100 | 1.4±0.1 |
| SOP7 | 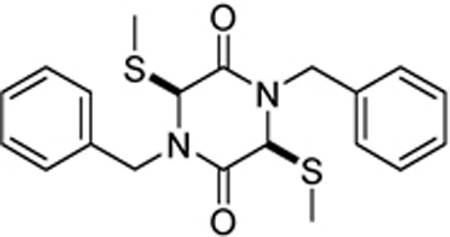 |
>100 | >100 | >100 | >100 | >100 | >100 |
| SOP10 | 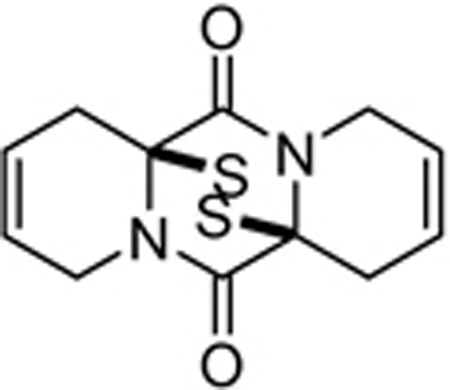 |
0.7±0.2 | 0.6±0.2 | 1.0±0.04 | >50 | >50 | 8.2±10 |
| SOP11 | 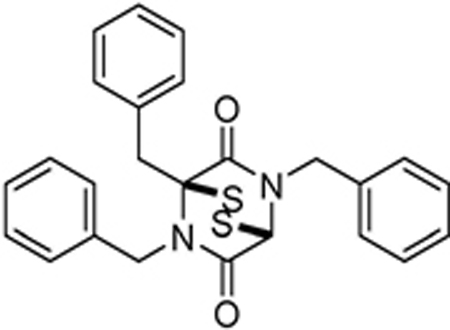 |
1.3±0.3 | 0.6±0.2 | 0.9±0.1 | >100 | >100 | 4.7±0.5 |
The dithiol/disulfide is required for the inhibitory effects of ETPs against HIF1α (Cook et al., 2009). Consistent with this, disrupting the disulfide bond in SOP7 completely abolished inhibitory activity towards Rpn11 (Table S2). We showed previously that compounds that inhibit Rpn11 via binding of zinc can be counteracted by the zinc coordination compound Zn(cyclen)2+, which titrates the inhibitor (Li et al., 2017; Perez et al., 2017). Zn(cyclen)2+ shifted the IC50 value of SOP6 from 1.2 μM to 28 μM, which implied that ETPs inhibited Rpn11 activity by chelating the catalytic Zn2+ ion (Fig. 3A). In addition, ZnSO4 added to cell culture medium blunted the effect of gliotoxin on Ub-conjugate accumulation (Fig. 3C). We conclude that ETPs inhibit proteasome function by targeting JAMM protease Rpn11 via binding to its catalytic zinc ion.
Cellular effects of ETPs
Gliotoxin, the most well-studied ETP, has multiple cellular effects such as perturbation of microfilament structure and induction of cell detachment (Jordan and Pedersen, 1986). However, cell detachment was not observed upon proteasome inhibition by either the Rpn11 inhibitor capzimin or the 20S proteasome inhibitor bortezomib (Fig. S2). Therefore, cell detachment is likely due to an effect of gliotoxin on a target other than Rpn11. To test whether this effect can be diminished through medicinal chemistry optimization, we monitored cell detachment induced by twelve different ETPs. Among them, SOP10 and SOP11 did not detach cells but retained inhibitory activity towards Rpn11 (Fig. 2B, Fig. 4A and Table 1). A previous study suggested that Rpn11 inhibition triggers a stress response that affects transcription (Lauinger et al., 2017). We therefore assessed the impact of SOP10 and SOP11 on transcription by monitoring highly unstable c-Myc mRNA in HCT116 cells. The level of c-Myc mRNA decreased upon treatment with these compounds, indicating negative effects of ETPs on transcription (Fig. 4B). Gliotoxin was reported to inhibit the activity of HOIP, a component of the linear ubiquitin chain assembly complex that mediates activation of IκB kinase (Sakamoto et al., 2015). We therefore tested the effect of ETPs on the degradation of IκB induced by TNF-α stimulation. Gliotoxin and chetomin strongly stabilized IκB and SOP6 and SOP8 slightly stabilized IκB, but other ETPs showed no effects on IκB degradation (Fig. S3).
Fig 4. Cellular effects of ETPs.
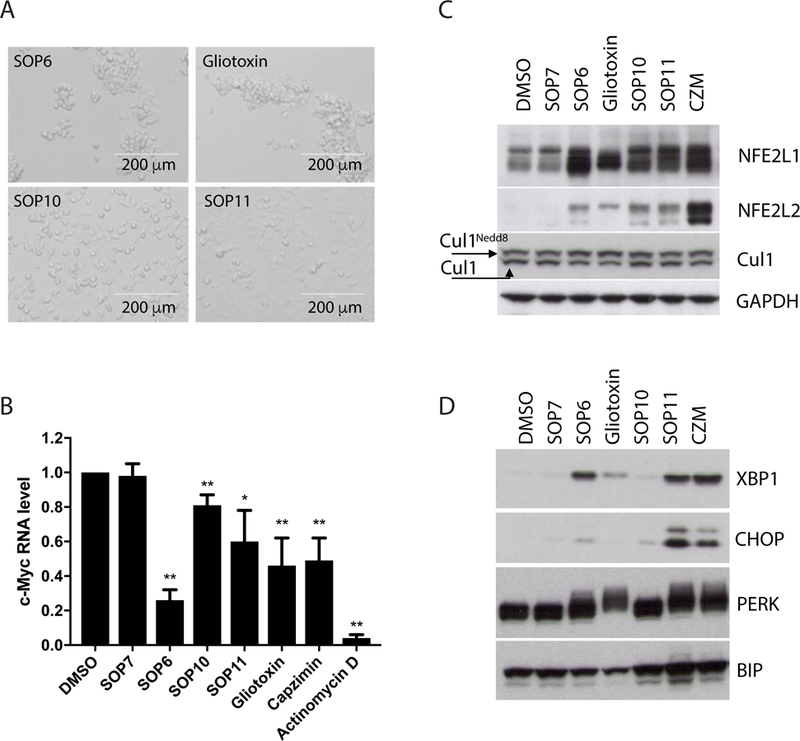
(A) SOP6 and gliotoxin induce cell detachment. Shown are microscopic images of HCT116 cells, taken 3 hours after treatment with 10 μM of the indicated ETPs. Note that when the cells detach, as in the top two panels, they appear more refractile and have a tendency to clump. (B) c-Myc mRNA expression, quantified by qRT-PCR, in HCT116 cells harvested 4 hours post treatment. 1 μM actinomycin D was included as positive control, and 10 μM other compounds were used. Data are normalized to GAPDH. Error bars mean ± s.d. from three independent experiments measured in triplicate each. * P<0.05, ** P<0.01 (C) HCT116 cells were treated for 6 hours with indicated compounds, and cell lysates were fractionated by SDS–PAGE and immunoblotted with antibodies against NFE2L1, NFE2L2, Cul1 and GAPDH (loading control for C and D). (D) Same as C, except that antibodies against XBP1, CHOP, PERK and Bip were used. See also Figure S2, Figure S3 and Figure S4.
To evaluate the impact of ETPs on proteasome function in cells in more detail, we evaluated the degradation of two endogenous proteasome substrates, NFE2L1 and NFE2L2. Nuclear-factor-erythroid-derived-2-related factor 1 (NFE2L1) is an unstable transcription factor that regulates the expression of genes that encode proteasome subunits. Upon inhibition of the proteasome, a processed form of NFE2L1 accumulates and induces transcription of proteasome subunit genes (Radhakrishnan et al., 2010; Steffen et al., 2010). Multiple ETPs including SOP10 and SOP11 mimicked capzimin and induced accumulation of processed NFE2L1 (Fig. 4C). Similarly, NFE2L2, the transcription factor that regulates antioxidant response, also accumulated after treatment with ETPs (Fig. 4C). We also examined other endogenous proteasome substrates including c-Myc, p53 and MDM2. ETPs showed little effect on those substrates (Fig. S4), which could be due to the negative impact of those compounds on transcription (Fig. 2B), weak potency in cells, or different substrate profiles from the conventional 20S proteasome inhibitors. Notably, the cell-detaching compounds SOP6 and gliotoxin caused accumulation of Nedd8-conjugated Cul1, whereas SOP10 and SOP11 did not (Fig. 4C). This suggests that SOP10 and SOP11 do not appreciably inhibit the JAMM enzyme Csn5 in cells.
Previous studies revealed that Rpn11 inhibition triggered an unfolded protein response (UPR) (Li et al., 2017). Surprisingly, only SOP11 provoked a pronounced UPR in cells, as determined by the accumulation of phosphorylated protein kinase R-like endoplasmic reticulum kinase (PERK), spliced X-box binding protein 1 (XBP1s), the transcription factor CCAAT/enhancer-binding protein homologous protein (CHOP) and Binding immunoglobulin protein (BiP) (Fig. 4D). Little UPR response was triggered by SOP10, which is in agreement with its weaker effects on the clearance of accumulated UbG76VGFP and high GI50 on cell proliferation (Table S2). Based on all of the results summarized above, we conclude that SOP11 is the most promising candidate for a selective Rpn11 inhibitor among the tested ETPs.
SOP11 blocks cancer cell proliferation
Inhibition of proteasome function results in cell death, which underlies the activity of the ‘omibs’ in chemotherapy of multiple myeloma. We and others previously showed that chemical inhibition of Rpn11 blocks cancer cell proliferation (Li et al., 2017; Song et al., 2017). We measured the effects of ETPs on the proliferation of HCT116 human colon cancer cells and calculated inhibition of cell growth (GI50) (Table S2). SOP6 was the most potent growth inhibitor. However, as exemplified by its effects on cell detachment, it may work through inhibiting multiple cellular targets in addition to Rpn11. The more selective compound SOP11 inhibited cell proliferation with a GI50 value of ~4.7 μM (Table 1). Of relevance to the potential for targeting Rpn11 in ‘omib’ refractory myeloma patients, we tested SOP11 using WT and bortezomib-resistant (BTZR) retinal pigment epithelial (RPE) cells (Wacker et al., 2012). SOP11 had the same GI50 against WT and BTZR RPE cells (~8 μM; Fig. 5A), indicating that ETP-based and ‘omib’ inhibitors worked through distinct mechanisms. In addition to inhibiting cell growth, ETPs induced cell apoptosis, as confirmed by immunoblotting for caspase-cleaved poly (ADP-ribose) polymerase (PARP1) (Fig. 5B). DNA damage is a common event during apoptosis, which is reflected by the increased phosphorylation of H2AX (γH2AX). ETPs strongly induced H2AX phosphorylation after 24 hours treatment, indicating the occurrence of the DNA damage response (Fig. 5B).
Fig 5. ETPs block cancer cell proliferation.
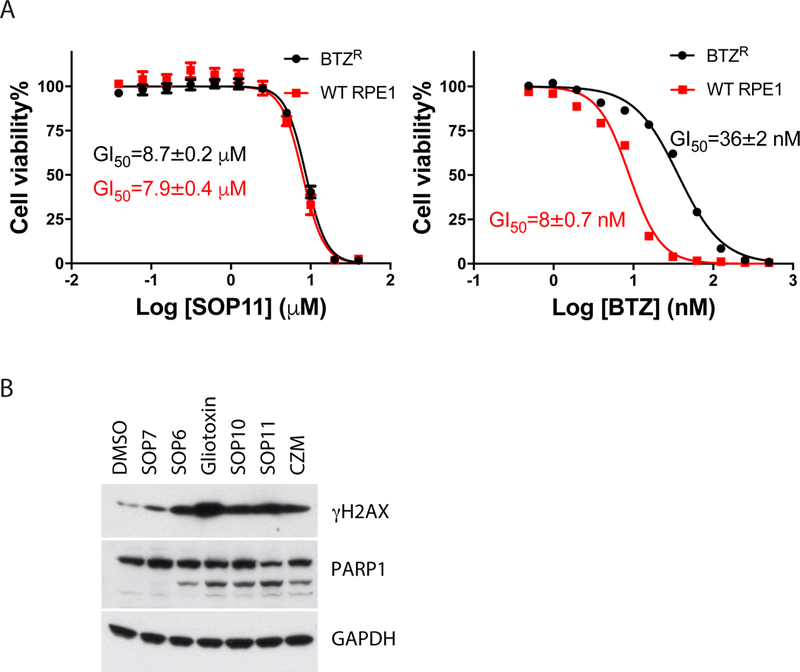
(A) Bortezomib-resistant cell line was not resistant to SOP11. WT or bortezomib-resistant (BTZR) RPE1 cells were treated with different concentrations of SOP11 or BTZ for 72 hours and then mixed with CellTiter-Glo reagent to estimate cell proliferation. Measured luminescence values were normalized to DMSO control, and data were fitted to a dose–response equation to determine the GI50 (50% cell growth inhibition). Error bars represent s.d., n = 4 wells, from 1 representative of 3 independent experiments. Error bars are too small to be visualized for some data points in this graph. (B) ETPs induce apoptosis. HCT116 cells were treated with 10 μM of the indicated ETPs or CZM for 24 hours. Western blot analyses of cell lysates were performed with antibodies against PARP1, γH2AX, and GAPDH, as indicated. See also Table S2.
Discussion
Gliotoxin is the most well-studied ETP. It targets multiple proteins and enzymes with important cellular functions. The proteasome, the central cellular machine for protein degradation, has been reported as a target of gliotoxin (Kroll et al., 1999). It inhibits the chymotrypsin-like activity of 20S proteasome at high concentrations under oxidative conditions. However, gliotoxin is rapidly reduced upon its uptake by the cell (Bernardo et al., 2003). We report here that multiple ETPs including gliotoxin inhibit the proteasome in vitro and in cells by targeting its essential deubiquitinase subunit Rpn11. Inhibition of Rpn11 function by gliotoxin and other ETPs led to proteasome malfunction and subsequent cell death. Our findings identify ETPs as an inhibitor scaffold for targeting Rpn11 and open the door to further development of more specific and potent Rpn11 inhibitors.
A key consideration in the characterization of inhibitors is their target specificity. Whereas gliotoxin is one of the most potent ETPs for inhibiting the proteasome, it is also relatively non-selective and induces morphological changes in cells that are not observed with the selective Rpn11 inhibitor capzimin (Jordan and Pedersen, 1986). To address this non-selectivity, we surveyed a panel of related molecules that share the core ETP scaffold and identified SOP11 as a candidate for future studies. Unlike gliotoxin, SOP11 did not cause the cell rounding phenotype. Like capzimin, SOP11 triggered an unfolded protein response and induced accumulation of polyubiquitin conjugates and the specific proteasome substrates NFE2L1 and NFE2L2. SOP11 also mimicked capzimin in that it did not inhibit zinc-dependent enzymes unrelated to Rpn11, such as human carbonic anhydrase and matrix metalloproteinase-2. However, SOP11 does inhibit other members of the JAMM family in vitro in addition to Rpn11, including Csn5 and AMSH. However, we did not observe effects on the Csn5 substrate Cul1 at concentrations of SOP11 that inhibited the proteasome. Thus, SOP11 is a promising starting point to develop Rpn11 inhibitors based on the ETP scaffold. However, considerable medicinal chemistry optimization will be required to generate ETPs that have sufficient potency and specificity to enable a rigorous test of the therapeutic potential of Rpn11 inhibition.
The proteasome is the central conduit through which all substrates of the ubiquitin-proteasome system (UPS) are degraded. Genomic mutations, aneuploidy, and other alterations in cancer cells can result in an imbalance between protein expression and the degradative capacity of the UPS (Cenci et al., 2008; Deshaies, 2014). This has the potential to render cancer cells more dependent on proteasome activity than normal cells. Inhibitors targeting the proteolytic center of the 20S proteasome, such as bortezomib, carfilzomib, and ixazomib, have been approved for the treatment of multiple myeloma and mantle cell lymphoma, thereby validating the hypothesis that at least some cancer cells have heightened dependence on proteasome activity. However, proteasome inhibitor therapy has not been successfully expanded to the treatment of solid tumors, potentially due to reduced sensitivity of solid tumor cells to proteasome inhibitors coupled with the pharmacological properties of the existing drugs (Deshaies, 2014). Our study provides an alternative to achieve proteasome inhibition, which might translate into a therapeutic approach in the future.
Changes in proteasome activity have been suggested to occur during aging and in neurodegenerative diseases (Saez and Vilchez, 2014). Proteasome dysfunction and decreased proteasome activity might contribute to aging-related diseases. For example, it has been proposed that toxic protein aggregates that accumulate in polyglutamine repeat diseases such as Huntington’s Disease may inhibit proteasome function (Diaz-Hernandez et al., 2006), although this has been controversial (Hipp et al., 2012; Ortega et al., 2007). One problem in addressing the role of proteasome activity in aging and disease has been a lack of suitable assays to directly measure the protein breakdown activity of the proteasome in cell lysates. Consequently, these studies rely on measuring the chymotryptic peptidase activity of the β5 subunit, which shows poor correlation with protein breakdown activity (Bence et al., 2001). The proteasome activity assay we developed in this study may help to shed light on these simple yet important issues.
STAR★Methods
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ubiquitin | Enzo Life Sciences | Cat# ADI-SPA-200-F |
| NFE2L1 | Cell Signaling | Cat# 8052S |
| NFE2L2 | Santa Cruz | Cat# sc-365949 |
| Cul1 | Bethyl | Cat# A303–373A |
| GAPDH antibody (FL-335) HRP | Santa Cruz | Cat# sc-25778 |
| GAPDH | Cell Signaling | Cat# 2118S |
| XBP1 | BioLegend | Cat# 647502 |
| CHOP | Cell Signaling | Cat# 2895P |
| PERK | Cell Signaling | Cat# 5683P |
| Bip | Cell Signaling | Cat# 3177P |
| γH2AX | Millipore | Cat# 05-636 |
| PARP1 | Cell Signaling | Cat# 5625S |
| IκBα | Abcam | Cat# ab32518 |
| c-Myc | Abcam | Cat# ab32072 |
| MDM2 | Thermo Fischer Scientific | Cat# PI700555 |
| p53 | Abcam | Cat# ab1101 |
| Chemical, Peptide and proteins | ||
| Alexa Fluor™ 488 C5 Maleimide | Thermo Fisher Scientific | Cat# A10254 |
| gliotoxin | Sigma-Aldrich | Cat# G9893 |
| TNF-α | Sigma-Aldrich | Cat# H8916 |
| Critical Commercial Assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7572 |
| Cell lines | ||
| HCT116 | ATCC | Cat# CCL-247™ |
| UbG76V GFP Hela cell line | T.F. Chou, Harbor-UCLA and LA BioMed | |
| RPE1 WT and BTZ-resistant cell lines | T.M. Kapoor, The Rockefeller University | |
| Software | ||
| Graphpad | Prism | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and request for resources and reagents should be directed and will be fulfilled by Raymond J. Deshaies (deshaies@caltech.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HCT116 cells and RPE1 cells were grown at 37 ˚C with 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum, penicillin-streptomycin, and 2 mM L-glutamine.
METHOD DETAILS
Plasmid construction and protein purification.
Purification of GST–Ube1 (E1), Rsp5 (E3 for generating K63-linked Ub chains) and Rsp5-E6AP (E3 for generating K48-linked Ub chains) was carried out as described previously (Kim and Huibregtse, 2009). UbcH5a (E2) was purchased from Boston Biochem Inc. The plasmid encoding Wbp2-C-K222 was a kind gift from Jon M. Huibregtse. A sequence encoding “LPETGHHHHHH” was inserted at the 3’ end of the Wbp2-C-K222 coding sequence through PCR reaction and the resulting construct was cloned into the pGEX-4T1 vector resulting in the addition of an N-terminal GST tag. To purify GST–Wbp2-C-K222-His6, Rosetta cells were transformed with the plasmid (RDB3386) and grown at 37 °C in Luria–Bertani broth containing 100 μg/mL ampicillin to an optical density of about 0.6 at 600 nm. Then, the cells were induced with IPTG (final concentration 0.5 mM) for three hours at 37 °C. After lysis of the cells by sonication, the fusion protein was purified from the soluble franction by GSTrap™ High Performance column (GE healthcare life sciences), followed by size-exclusion chromatography on a Superdex 200 prep grade column (GE healthcare life sciences).
Protein labeling.
GST–Wbp2-C-K222-His6 was covalently labeled with the fluorescent dye Alexa Fluor 488 C5 Maleimide (A10254, Thermo Fisher Scientific) on cysteine residues by incubation with a five-fold molar excess of the dye under the conditions recommended by the manufacturer. The reaction was quenched by the addition of 10 mM DTT. We then applied the reaction mixture to a Superdex 75HR column (GE Healthcare) to separate the free dye from the labeled protein. The concentration of the labeled protein and the degree of labeling were determined according to the manufacturer’s instructions.
In vitro ubiquitination and purification.
To ubiquitinate GST–Wbp2-C-K222-His6, two separate mixtures (E3 + substrate and GST–Ube1+UbcH5a+ubiquitin+ATP) were preincubated for two minutes and then combined to initiate the reaction. Ubiquitination was carried out at room temperature for 4 hours. The reaction mixture was purified by Ni-NTA affinity chromatography and desalting chromatography to remove E1, E2 and E3 and exchange the buffer.
Chemical Syntheses.
SOP1 to SOP9 was synthesized as previously reported (Sil and Hilton, 2013). Synthetic protocols for SOP10 and SOP11 were described below.
SOP-10
Ethyl-2-(allylamino)-2-(4-methoxybenzylthio)acetate (2).
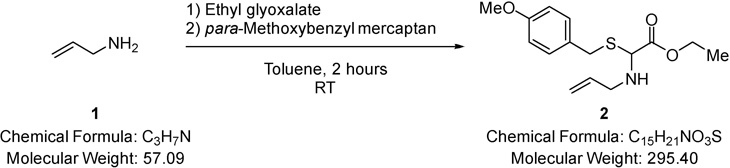
Allylamine (1.90 mL, 25.2 mmol) (1) was added to a solution of ethyl glyoxalate (5.00 mL of a 50% solution in toluene, 25.2 mmol) in toluene (30.0 mL) at room temperature (RT) and the solution stirred for 2 minutes, whereupon para-methoxybenzyl mercaptan (3.51 mL, 25.22 mmol) was added and the mixture stirred for 2 hours. Solvent was removed under reduced pressure and the residue purified via Biotage™ Horizon (3:1, petroleum spirit 40–60°C: ethyl acetate; snap 50 g) to give ethyl-2-(allylamino)-2-(4-methoxybenzylthio)acetate (6.00 g, 81%) (2) as a colourless oil; νmax/cm−1 3344 (N-H), 1732 (C=O); δ H (400 MHz; CDCl3) 1.27 (3H, t, J 7.1, CH2CH3), 1.89 (1H, br s, NH), 3.11 (1H, dd, J 13.9 & 6.4, NCH2), 3.28 (1H, dd, J 13.9 & 6.6, NCH2), 3.69 (2H, s, SCH2), 3.74 (3H, s, OCH3), 4.18 (2H, q, J 7.1, CH2CH3), 4.32 (1H, s, CHS), 5.06 – 5.10 (2H, m, CH=CH2), 5.72 –5.77 (1H, m, CH=CH2), 6.80 (2H, d, J 8.6, Ar-H), 7.23 (2H, d, J 8.6, Ar-H); δ C (101 MHz; CDCl3) 13.89 (CH2CH3), 32.85 (CH2), 47.59 (CH2), 54.99 (OCH3), 61.09 (CH2), 63.36 (CHS), 113.66 (Ar-C-H), 116.64 (CH=CH2), 129.72 (quaternary C), 129.78 (Ar-C-H), 135.07 (CH=CH2), 158.46 (quaternary C), 169.68 (CO); m/z 296 (100%, [M+H]+): Found [M+H]+ 296.1322, C15H22NO3S requires 296.1320.
2-(Allyl(2-ethoxy-1-(4-methoxybenzylthio)-2-oxoethyl)amino)-2-oxoethane-1,1-diyl diacetate (3).
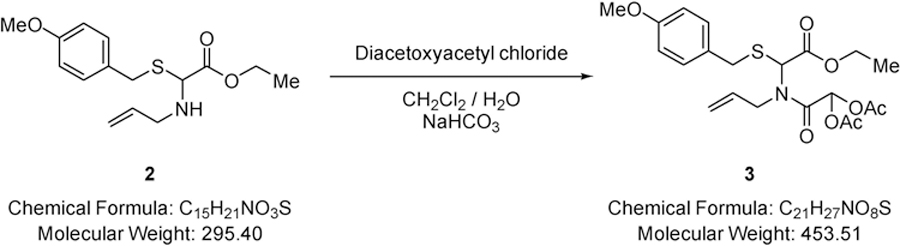
A solution of diacetoxyacetyl chloride (4.74 g, 24.4 mmol) in dichloromethane (15.0 mL) was added dropwise to a rapidly stirred biphasic mixture of ethyl-2-(allylamino)-2-(4-methoxybenzylthio)acetate (6.00 g, 20.3 mmol) (2) and sodium hydrogen carbonate (11.9 g, 142 mmol) in dichloromethane (200 mL) and water (100 mL) at RT and the resulting mixture stirred rapidly for 12 hours. The layers were separated and the aqueous phase extracted with dichloromethane (2 × 100 mL). The combined organic extracts were driedover MgSO4, filtered and solvent removed under reduced pressure to give 2-(allyl(2-ethoxy-1-((4-methoxybenzyl)thio)-2-oxoethyl)amino)-2-oxoethane-1,1-diyl diacetate (8.93 g, 97%) (3) as a colourless oil that did not require further purification; νmax/cm−1 1770 (C=O), 1743 (C=O), 1681 (C=O); δ H (400 MHz; CDCl3) 1.20 (3H, t, J 7.1, CH2CH3), 1.95 (3H, s, OC(O)CH3), 2.11 (3H, s, OC(O)CH3), 3.67 (2H, s, SCH2), 3.73 (3H, s, OCH3), 3.99 (1H, dd, J 17.7 & 5.4, NCH2), 4.09 (2H, d, J 7.1, CH2CH3), 4.25 (1H, dd, J 17.7 & 5.5, NCH2), 5.19 – 5.23 (2H, m, CH=CH2), 5.84 – 5.89 (1H, m, CH=CH2), 6.08 (1H, s, CHS), 6.80 (2H, d, J 8.6, Ar-H), 7.03 (1H, s, CH(OAc)2), 7.19 (2H, d, J 8.6, Ar-H); δ C (101 MHz; CDCl3) 13.70 (CH2CH3), 20.29 (C(O)CH3), 34.90 (CH2), 47.12 (CH2), 54.99 (OCH3), 60.77 (CHS), 62.08 (CH2), 83.86 (CH(OAc)2), 113.80 (Ar-C-H), 117.56 (CH=CH2), 128.48 (quaternary C), 130.02 (Ar-C-H), 133.22 (CH=CH2), 158.73 (quaternary C), 165.41 (CO), 167.14 (CO), 168.37 (CO), 168.69 (CO); m/z 454 (100%, [M+H]+): Found [M+H]+ 454.1539, C21H28NO8S requires 454.1536.
(3S,6S)-1,4-Diallyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (4).
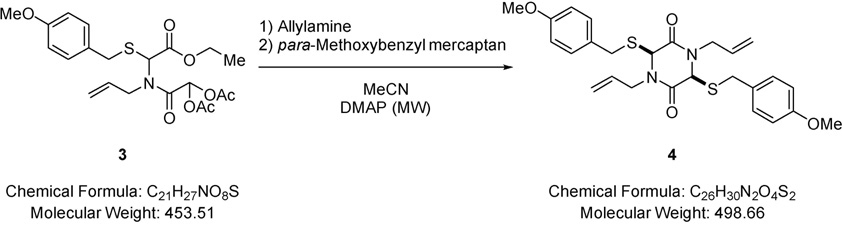
Allylamine (0.62 mL, 8.31 mmol) was added to a solution of 2-(allyl(2-ethoxy-1-((4-methoxybenzyl)thio)-2-oxoethyl)amino)-2-oxoethane-1,1-diyl diacetate (2.09 g, 4.62 mmol) (3) in acetonitrile (15.0 mL) followed by addition of para-methoxybenzyl mercaptan (1.29 mL, 9.23 mmol) and the resulting mixture stirred for 2 minutes. DMAP (0.28 g, 2.31 mmol) was added and the mixture heated in the microwave at 140 °C for 30 minutes, allowed to cool to room temperature and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (3:1, petroleum spirit 40–60 °C: ethyl acetate; snap 25 g) and further purified by recrystallisation (petroleum spirit 40–60 °C: ethyl acetate) to give (3S,6S)-1,4-diallyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (1.67 g, 72%) (4) as a colourless solid; m.p. 70–71 °C; νmax/cm−1 1675 (C=O), 1609 (C=C), 1465 (CH2), 832 (C-H); δ H (400 MHz, CDCl3) 3.82 – 3.91 (12H, m, NCH2 & OCH3 & SCH2), 3.98 – 4.02 (2H, m, NCH2), 4.45 (2H, s, CHS), 4.62 – 4.68 (2H, m, CH=CH2), 4.99 – 5.05 (2H, m, CH=CH2), 5.48 – 5.54 (2H, m, CH=CH2), 6.88 – 6.90 (4H, m, Ar-H), 7.36 – 7.38 (4H, m, Ar-H); δ C (101 MHz, CDCl3) 36.74 (CH2), 45.85 (CH2), 55.56 (OCH3), 58.20 (CHS), 114.28 (CH=CH2), 119.78 (CH2), 129.04 (quaternary C), 130.91 (Ar-C-H), 130.95 (Ar-C-H), 159.36 (quaternary C), 165.09 (CO); m/z 499 (100%, [M+H]+); Found [M+H]+ 499.1723, C26H31N2O4S2 requires 499.1725.
(3S,6S)-1,3,4,6-Tetraallyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (5).
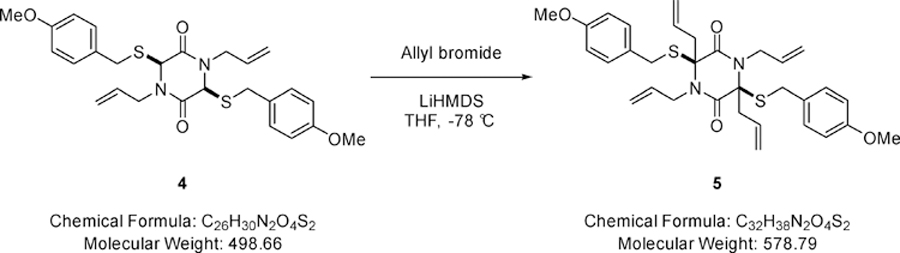
LiHMDS (4.14 mL of a 1M solution in tetrahydrofuran, 4.14 mmol) was added dropwise to a solution of (3S,6S)-1,4-diallyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (0.83 g, 1.65 mmol) (4) and allyl bromide (0.43 mL, 4.96 mmol) in tetrahydrofuran (20.0 mL) at −78 °C and the resulting mixture stirred at this temperature for 1 hour and 1.5 hours at 0 °C. Saturated aqueous sodium hydrogen carbonate (10.0 mL) was added and solvent removed under reduced pressure. The residue was partitioned between water (10.0 mL) and dichloromethane (20.0 mL). The aqueous phase was extracted with dichloromethane (2 × 10.0 mL) and the combined extracts were dried (MgSO4), filtered and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (4:1, petroleum spirit 40–60 °C: ethyl acetate; snap 10 g) to give (3S,6S)-1,3,4,6-tetraallyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (0.53 g, 55%) (5) as a pale yellow oil; νmax/cm−1 1656 (C=O), 1610 (C=C), 1001 (C-O-C), 835 (C-H); δ H (400 MHz, CDCl3) 2.69 (2H, d, J 14.2, CH2CH=CH2), 3.21 (2H, d, J 14.1, CH2CH=CH2), 3.79 (6H, s, OCH3), 3.94 – 3.99 (2H, m, SCH2), 4.05 – 4.09 (2H, m, SCH2), 4.23 – 4.27 (4H, m, NCH2), 5.13 – 5.22 (2H, m, CH=CH2), 5.25 (4H, dd, J 10.2 & 1.2, CH=CH2), 5.40 (2H, dd, J 10.3 & 1.4, CH=CH2), 5.52 – 5.63 (2H, m, CH=CH2), 6.02 – 6.07 (2H, m, CH=CH2), 6.81 – 6.85 (4H, m, Ar-H), 7.20 – 7.24 (4H, m, Ar-H); δ C (101 MHz, CDCl3) 35.66 (CH2), 41.66 (CH2), 47.69 (CH2), 55.42 (OCH3), 73.15 (quaternary C), 114.20 (CH=CH2), 118.63 (CH2), 121.15 (CH2), 128.06 (quaternary C), 130.53 (CH=CH2), 131.28 (Ar-C-H), 133.62 (Ar-C-H), 159.05 (quaternary C), 164.94 (CO); m/z 601 (100%, [M+Na]+); Found [M+Na]+ 601.2176, C32H38N2O4S2Na requires 601.2170.
(±)-(1S,4S)-1,2,4,5-Tetraallyl-7-thia-2,5-diazabicyclo[2.2.1]heptane-3,6-dione 7-sulfide (6).
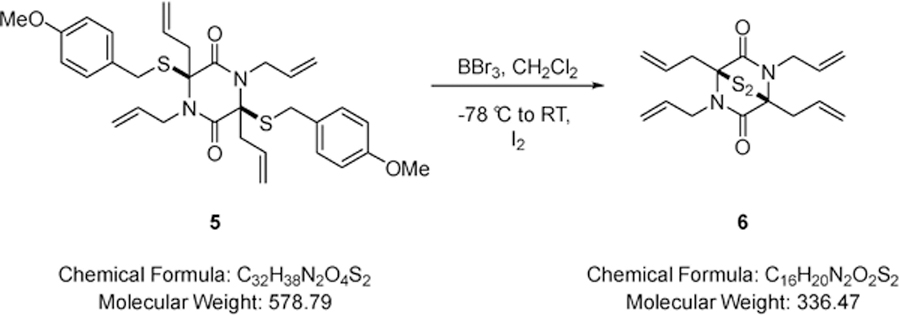
Boron tribromide (0.14 mL, 1.50 mmol) was added dropwise to a solution of 1,3,4,6-tetraallyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (0.44 g, 0.75 mmol) (5) in dichloromethane (20.0 mL) at −78 °C. The resulting mixture was stirred for 15 minutes whereupon Rochelle’s salt (20.0 mL of a saturated aqueous solution) was added and the biphasic mixture stirred for 15 minutes until the yellow colour had dissipated. The resulting mixture was stirred at room temperature for 10 minutes, whereupon iodine was added portionwise until the colour due to iodine just persisted and stirring was maintained for 2 minutes. Sodium thiosulfate was added and the mixture stirred for 10 minutes, diluted with dichloromethane (10.0 mL) and water (20.0 mL). The layers were separated and the aqueous phase extracted with dichloromethane (2 × 20.0 mL). The combined organic extracts were dried over MgSO4, filtered and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (4:1, petroleum spirit 40–60 °C: ethyl acetate; snap 10 g) and further purified by recrystallisation (petroleum spirit 40–60 °C: ethyl acetate) to give (±)-(1S,4S)-1,2,4,5-tetraallyl-7-thia-2,5-diazabicyclo[2.2.1]heptane-3,6-dione 7-sulfide (0.10 g, 41%) (6) as a colourless solid; m.p. 180–182 °C; νmax/cm−1 1639 (C=O, C=C), 1462 (CH2), 749 (CH2); δ H (400 MHz, CDCl3) 3.20 –3.27 (2H, m, CH2CH=CH), 3.28 – 3.36 (2H, m, CH2CH=CH), 4.04 (2H, dd, J 16.4 & 6.8, NCH2), 4.62 (2H, dd, J 16.2 & 4.8, NCH2), 5.21 – 5.41 (8H, m, CH=CH2), 5.89 – 5.94 (2H, m, CH=CH2), 6.12 – 6.17 (2H, m, CH=CH2); δ C (101 MHz, CDCl3) 36.44 (CH2), 45.46 (CH2), 74.50 (quaternary C), 118.31 (CH=CH2), 120.60 (CH=CH2), 131.74 (CH=CH2), 132.01 (CH=CH2), 165.02 (CO); m/z 337 (100%, [M+H]+); Found [M+H]+ 337.1456, C16H21N2O=S2 requires 337.1448.
(±)-(6aS,12aS)-7,10-Dihydro-6a,12a-epithiodipyrido[1,2-a:1’,2’-d] pyrazine-6,12(1H,4H)-dione 13-sulfide (SOP-10).
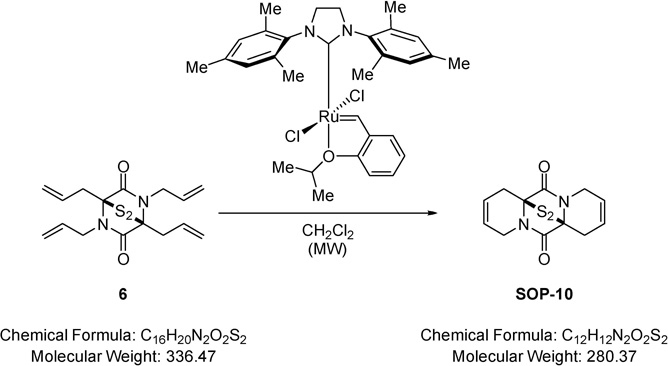
Method A:
A solution of (±)-(1S,4S)-1,2,4,5-tetraallyl-7-thia-2,5-diazabicyclo[2.2.1]heptane-3,6-dione 7-sulfide (0.04 g, 0.12 mmol) (6) in dichloromethane (2.00 mL) was degassed and taken under nitrogen atmosphere. To this (1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(o-isopropoxyphenylmethylene)ruthenium (Grubbs-Hoveyda second-generation catalyst) (0.01 g, 0.01 mmol) was added and the resulting mixture heated at 80 °C for 30 minutes in the microwave, cooled to RT and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (4:1, petroleum spirit 40–60 °C: ethyl acetate; pipette column) and further purified by recrystallisation (petroleum spirit 40–60 °C: ethyl acetate) to give (6aS,12aS)-7,10-dihydro-6a,12a-epithiodipyrido[1,2-a:1’,2’-d]pyrazine-6,12(1H,4H)-dione 13-sulfide (0.01 g, 39%) (SOP-10) as a colourless solid; m.p. 135–137 °C; νmax/cm−1 1683 (C=O, C=C), 1351 (C-H), 1321 (C-H); δ H (400 MHz, CDCl3) 2.60 – 2.72 (2H, m, CH2CH=CH), 3.29 – 3.40 (2H, m, CH2CH=CH), 3.87 – 4.08 (4H, m, NCH2), 5.86 – 5.97 (4H, m, CH=CH); δ C (101 MHz, CDCl3) 28.60 (CH2), 42.30 (CH2), 70.29 (quaternary C), 121.14 (CH=CH), 122.32 (CH=CH), 165.15 (CO); m/z 303 (100%, [M+Na]+); Found [M+Na]+ 303.0253, C12H12N2O2S2Na requires 303.0238.
Method B:
A solution of (±)-(1S,4S)-1,2,4,5-tetraallyl-7-thia-2,5-diazabicyclo[2.2.1]heptane-3,6-dione 7-sulfide (0.04 g, 0.13 mmol) (6) in dichloromethane (2.00 mL) was degassed and taken under nitrogen atmosphere. To this (1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro (phenylmethylene)(tricyclohexylphosphine)ruthenium (Grubbs second-generation catalyst) (0.01 g, 0.01 mmol) was added and the resulting mixture heated at 80 °C for 30 minutes in the microwave, cooled to RT and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (4:1, petroleum spirit 40–60 °C: ethyl acetate; snap 5 g) and further purified by recrystallisation (petroleum spirit 40–60 °C: ethyl acetate) to give (6aS,12aS)-7,10-dihydro-6a,12a-epithiodipyrido[1,2-a:1’,2’-d]pyrazine-6,12(1H,4H)-dione 13-sulfide (0.01 g, 7%) (SOP-10) as a colourless solid; identical spectroscopic data to that obtained previously for (SOP-10).
SOP-11
Ethyl-2-(benzylamino)-2-((4-methoxybenzyl)thio)acetate (8).
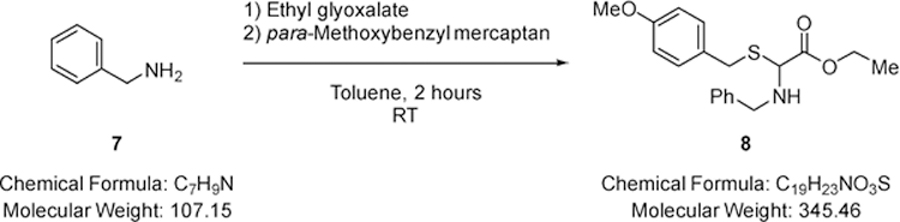
Benzylamine (2.76 mL, 25.2 mmol) (7) was added to a solution of ethyl glyoxalate (5.00 mL of a 50% solution in toluene, 25.2 mmol) in toluene (20.0 mL) at room temperature (RT) and stirred for 2 minutes, whereupon para-methoxybenzyl mercaptan (3.54 mL, 25.2 mmol) was added and the mixture stirred for 2 hours. Solvent was removed under reduced pressure and the residue purified via Biotage™ Horizon (3:1, petroleum spirit 40–60 °C: ethyl acetate; snap 50 g) to give ethyl-2-(benzylamino)-2-((4-methoxybenzyl)thio)acetate (7.56 g, 87%) (8) as a colourless oil; νmax/cm −1 3347 (N-H), 1732 (C=O); δ H (400 MHz; CDCl3) 1.31 (3H, t, J 7.2, CO2CH2CH3), 1.88 (1H, br s, NH), 3.72 (1H, d, J 13.1, SCH2), 3.75 –3.77 (2H, m, NHCH2), 3.79 (3H, s, OCH3), 3.87 (1H, d, J 13.1, SCH2), 4.22 (2H, q, J 7.2, CO2CH2CH3), 4.35 (1H, s, CH), 6.84 (2H, d, J 8.7, Ar-H), 7.23 – 7.29 (7H, m, Ar-H); δ C (101 MHz; CDCl3) 14.15 (CO2CH2CH3), 33.23 (SCH2), 49.22 (NHCH2), 55.28 (OCH3), 61.41 (CO2CH2CH3), 63.71 (CH), 114.00 (Ar-C-H), 127.19 (Ar-C-H), 128.41 (Ar-C-H), 128.48 (Ar-C-H), 130.07 (Ar-C-H), 138.79 (quaternary C), 158.76 (quaternary C), 169.97 (CO2CH2CH3); m/z 346 (100%, [M+H]+): Found [M+H]+ 346.1477, C19H24NO3S requires 346.1482.
2-(Benzyl(2-ethoxy-1-(4-methoxybenzylthio)-2-oxoethyl)amino)-2-oxoethane-1,1-diyl diacetate (9).
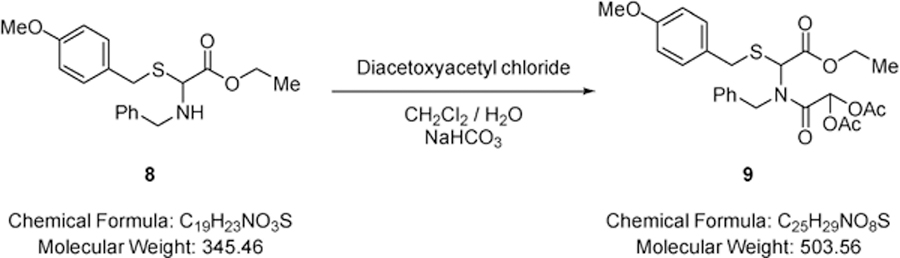
A solution of diacetoxyacetyl chloride (5.11 g, 26.3 mmol) in dichloromethane (20.0 mL) was added dropwise to a rapidly stirred biphasic mixture of ethyl-2-(benzylamino)-2-((4-methoxybenzyl)thio)acetate (7.56 g, 21.9 mmol) (8) and sodium hydrogen carbonate (12.9 g, 153 mmol) in dichloromethane (150 mL) and water (100 mL) at RT and the resulting mixture stirred rapidly for 12 hours. The layers were separated and the aqueous phase extracted with dichloromethane (2 × 100 mL). The combined extracts were dried over MgSO4, filtered and solvent removed under reduced pressure to give 2-(benzyl(2-ethoxy-1-((4-methoxybenzyl)thio)-2-oxoethyl)amino)-2-oxoethane-1,1-diyl diacetate (10.5 g, 95%) (9) as a colourless oil that did not require further purification; νmax/cm−1 1770 (C=O), 1739 (C=O), 1685 (C=O); δ H (400 MHz; CDCl3) 1.15 (3H, t, J 7.1, CH2CH3), 1.95 (3H, s, OC(O)CH3), 1.98 (3H, s, OC(O)CH3), 3.72 (2H, s, SCH2), 3.75 (3H, s, OCH3), 3.98 – 4.02 (2H, m, CH2CH3), 4.59 (1H, d, J 17.4, NCH2), 4.86 (1H, d, J 17.4, NCH2), 5.85 (1H, s, CHS), 6.79 (2H, d, J 8.4, Ar-), 6.92 (1H, s, CH(OAc)2), 7.16 – 7.27 (7H, m, Ar-H); δ C (101 MHz; CDCl3) 13.76 (CH2CH3), 20.20 (C(O)CH3), 20.25 (C(O)CH3), 35.49 (CH2), 48.68 (CH2), 55.18 (OCH3), 61.65 (CHS), 62.21 (CH2CH3), 83.99 (CH(OAc)2), 113.97 (Ar-C-H), 126.79 (Ar-C-H), 127.66 (Ar-C-H), 128.48 (Ar-C-H), 128.65 (quaternary C), 130.21 (Ar-C-H), 135.72 (quaternary C), 158.87 (quaternary C), 165.76 (CO), 167.16 (CO), 168.34 (CO), 168.62 (CO); m/z 504 (100%, [M+H]+): Found [M+H]+ 504.1688, C25H30NO8S requires 504.1692.
1,4-Dibenzyl-3-methoxy-6-((4-methoxybenzyl)thio)piperazine-2,5-dione (10).
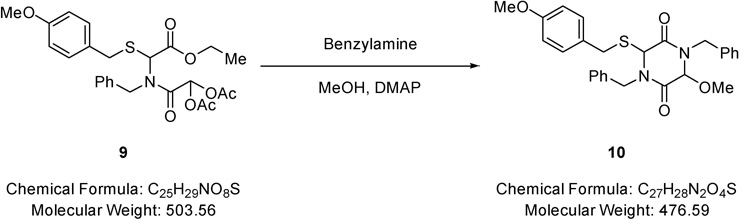
Benzylamine (0.21 mL, 1.93 mmol) was added to a solution of 2-(benzyl(2-ethoxy-1-((4-methoxybenzyl)thio)-2-oxoethyl)amino)-2- oxoethane-1,1-diyl diacetate (0.65 g, 1.29 mmol) (9) in methanol (15.0 mL) and the resulting mixture stirred for 2 minutes. DMAP (0.08 g, 0.64 mmol) was added and the resulting mixture stirred for 5 hours at RT and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (3:1, petroleum spirit 40–60 °C: ethyl acetate; snap 25 g) and further purified by recrystallisation (petroleum spirit 40–60 °C: ethyl acetate) to give 1,4-dibenzyl-3-methoxy-6-((4-methoxybenzyl)thio)piperazine-2,5-dione (0.49 g, 79%) (10) as a colourless solid; m.p. 85–88 °C; νmax/cm−1 3030 (C-H), 3005 (C-H), 1671 (C=O), 1357 (CH3), 1002 (C-O-C), 832 (C-H); δ H (400 MHz, CDCl3) 3.53 (3H, s, OCH3), 3.62 (1H, d, J 14.5, NCH2), 3.87 – 3.90 (4H, m, SCH2 & OCH3), 4.09 (1H, d, J 13.8, SCH2), 4.25 (1H, d, J 14.9, NCH2), 4.66 (1H, s, CHO), 4.45 (1H, s, CHS), 5.18 (2H, d, J 14.7, NCH2), 6.91 – 6.95 (2H, m, Ar-H), 7.16 – 7.29 (6H, m, Ar-H), 7.36 (6H, dd, J 16.4 & 9.2, Ar-H); δ C (101 MHz, CDCl3) 36.29 (CH2), 45.93 (CH2), 48.07 (CH2), 55.39 (OCH3), 57.66 (OCH3), 85.50 (CHS), 114.29 (CHO), 128.03 (Ar-C-H), 128.19 (Ar-C-H), 128.53 (Ar-C-H), 128.66 (Ar-C-H), 128.75 (Ar-C-H), 129.01 (Ar-C-H), 130.88 (Ar-C-H), 134.98 (quaternary C), 135.30 (quaternary C), 159.27 (quaternary C), 162.73 (CO), 166.29 (CO); m/z 477 (100%, [M+H]+); Found [M+H]+ 477.1841, C 27H29N2O4S requires 477.1848.
3,4-Tribenzyl-6-methoxy-3-((4-methoxybenzyl)thio)piperazine-2,5-dione (11).
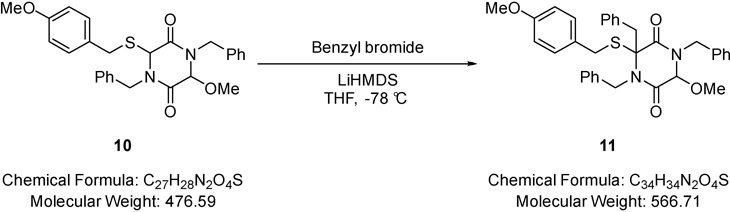
LiHMDS (0.50 mL of a 1M solution in tetrahydrofuran, 0.50 mmol) was added dropwise to a solution of 1,4-dibenzyl-3-methoxy-6-((4-methoxybenzyl)thio)piperazine-2,5-dione (0.20 g, 0.41 mmol) (10) and benzyl bromide (0.05 mL, 0.41 mmol) in tetrahydrofuran (2.00 mL) at −78 °C and the resulting mixture stirred at this temperature for 1.5 hours and 1 hour at 0 °C. Saturated aqueous sodium hydrogen carbonate (10.0 mL) was added and solvent removed under reduced pressure. The residue was partitioned between water (10.0 mL) and dichloromethane (20.0 mL). The aqueous phase was extracted with dichloromethane (2 × 10.0 mL) and the combined extracts were dried (MgSO4), filtered and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (3:1, petroleum spirit 40–60 °C: ethyl acetate; snap 10 g) to give 1,3,4-tribenzyl-6-methoxy-3-((4-methoxybenzyl)thio)piperazine-2,5-dione (0.14 g, 59%) (11) as a pale yellow oil; νmax/cm−1 3004 (C-H), 1670 (C=O), 1453 (CH2), 1357 (CH3), 1000 (C-O-C); δ H (400 MHz, CDCl3) 3.18 (1H, d, J 10.9, CH2), 3.32 (1H, d, J 14.2, CH2), 3.38 (1H, d, J 8.8, SCH2), 3.44 (3H, s, OCH3), 3.73 (1H, d, J 9.1, SCH2), 3.78 (3H, s, OCH3), 4.05 (1H, d, J 14.2, NCH2), 4.13 (1H, s, C HO), 4.65 (1H, d, J 14.7, NCH2), 4.75 (1H, d, J 14.7, NCH2), 5.30 (1H, d, J 14.7, NCH2), 7.01 (6H, dd, J 11.5 & 4.5, Ar-H), 7.14 – 7.17 (3H, m, Ar-H), 7.29 – 7.33 (8H, m, Ar-H), 7.54 – 7.57 (2H, m, Ar-H); δ (101 MHz, CDCl3) 34.61 (CH2), 43.51 (CH2), 47.21 (CH2), 47.91 (CH2), 55.41 (OCH3), 82.93 (OCH3), 113.98 (CHO), 127.63 (Ar-C-H), 128.13 (Ar-C-H), 128.36 (Ar-C-H), 128.60 (Ar-C-H), 128.82 (Ar-C-H), 129.03 (Ar-C-H), 129.39 (Ar-C-H), 129.56 (Ar-C-H), 130.19 (Ar-C-H), 130.48 (Ar-C-H), 130.65 (Ar-C-H), 133.87 (quaternary C), 134.70 (quaternary C), 135.44 (quaternary C), 137.89 (quaternary C), 159.02 (quaternary C), 164.23 (quaternary C), 165.06 (CO), 166.07 (CO); m/z 589 (100%, [M+Na]+); Found [M+Na]+ 589.2107, C34H34N2O4SNa requires 589.2137.
(3S,6S)-1,3,4-Tribenzyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (12).
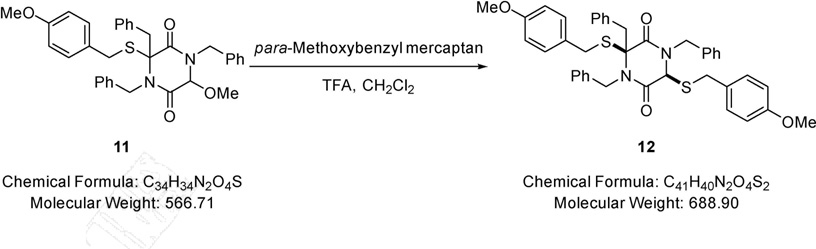
TFA (1.00 mL, 13.0 mmol) was added to a solution of 1,3,4-tribenzyl-6-methoxy-3-((4-methoxybenzyl)thio)piperazine-2,5-dione (0.18 g, 0.32 mmol) (11) in dichloromethane (5.00 mL) at RT followed by the addition of para-methoxybenzyl mercaptan (0.13 mL, 0.95 mmol) and the resulting mixture stirred at this temperature for 18 hours. After, the solution was diluted in dichloromethane (10.0 mL) and washed with saturated aqueous sodium hydrogen carbonate (10.0 mL). The aqueous phase was extracted with dichloromethane (2 × 10.0 mL) and the combined extracts were dried (MgSO4), filtered and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (4:1, petroleum spirit 40–60 °C: ethyl acetate; snap 10 g) to give (3S,6S)-1,3,4-tribenzyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (0.20 g, 91%) (12) as a pale yellow oil; νmax/cm−1 3032 (C-H), 3005 (C-H), 1666 (C=O), 1356 (CH3), 1001 (C-O-C), 833 (C-H), 730 (C-H); δ H (400 MHz, CDCl3) 3.77 – 3.78 (2H, m, CH2), 3.79 (3H, s, OCH3), 3.84 (3H, s, OCH3), 3.87 – 3.89 (4H, m, SCH2 & NCH2), 3.97 – 4.06 (2H, m, SCH2), 4.27 (1H, s, CHS), 4.62 (1H, d, J 15.1, NCH2), 5.28 (1H, d, J 15.1, NCH2), 6.51 – 6.54 (3H, m, Ar-H), 6.75 – 6.81 (4H, m, Ar-H), 6.84 – 6.88 (4H, m, Ar-H), 7.00 – 7.05 (4H, m, Ar-H), 7.13 – 7.20 (4H, m, Ar-H), 7.27 – 7.31 (2H, m, Ar-H), 7.59 – 7.63 (2H, m, Ar-H); δ C (101 MHz, CDCl3) 35.31 (CH2), 36.93 (CH2), 43.44 (CH2), 46.98 (CH2), 47.70 (CH2), 55.38 (OCH3), 58.20 (OCH3), 75.78 (quaternary C), 113.96 (CHS), 127.84 (Ar-C-H), 128.50 (Ar-C-H), 128.58 (Ar-C-H), 128.62 (Ar-C-H), 128.81 (Ar-C-H), 128.90 (Ar-C-H), 129.17 (Ar-C-H), 129.86 (Ar-C-H), 130.35 (Ar-C-H), 130.65 (Ar-C-H), 130.74 (Ar-C-H), 130.81 (Ar-C-H), 130.96 (Ar-C-H), 133.96 (quaternary C), 134.27 (quaternary C), 134.41 (quaternary C), 135.74 (quaternary C), 137.79 (quaternary C), 158.96 (quaternary C), 159.17 (quaternary C), 163.98 (CO), 165.99 (CO); m/z 711 (100%, [M+Na]+); Found [M+Na]+ 711.2355, C41H40N2O4S2Na requires 711.2327.
(±)-(1S,4S)-1,2,5-Tribenzyl-7-thia-2,5-diazabicyclo[2.2.1]heptane-3,6-dione 7-sulfide (SOP-11).
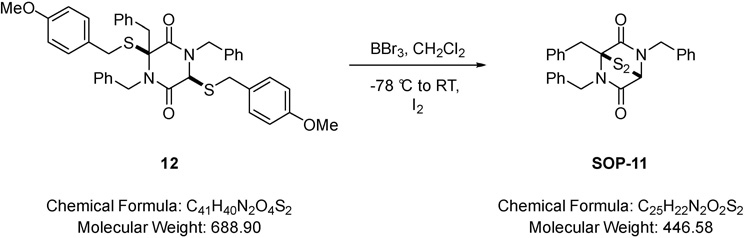
Boron tribromide (0.70 mL of a 1M solution in dichloromethane, 0.70 mmol) was added dropwise to a solution of (3S,6S)-1,3,4-tribenzyl-3,6-bis((4-methoxybenzyl)thio)piperazine-2,5-dione (0.12 g, 0.17 mmol) (12) in dichloromethane (2.00 mL) at −78 °C. The resulting mixture was stirred for 15 minutes whereupon aqueous sodium hydrogen carbonate (10.0 mL of a saturated aqueous solution) and methanol (1.00 mL) were added and the biphasic mixture stirred for 15 minutes until the yellow colour had dissipated. The resulting mixture was stirred at room temperature for 10 minutes, whereupon iodine was added portionwise until the colour due to iodine just persisted and stirring was maintained for 2 minutes. Sodium thiosulfate was added and the mixture stirred for 10 minutes, diluted with dichloromethane (10.0 mL) and water (20.0 mL). The layers were separated and the aqueous phase extracted with dichloromethane (2 × 20.0 mL). The combined organic extracts were dried over MgSO4, filtered and solvent removed under reduced pressure. The residue was purified via Biotage™ Horizon (3:1, petroleum spirit 40–60 °C: ethyl acetate; snap 10 g), followed by HPLC separation and further purified by recrystallisation (petroleum spirit 40–60 °C: ethyl acetate) to give (±)-(1S,4S)-1,2,5-tribenzyl-7-thia-2,5-diazabicyclo[2.2.1]heptane-3,6-dione 7-sulfide (0.04 g, 50%) (SOP-11) as a colourless solid; m.p. 122–124 °C; νmax/cm−1 1641 (C=O), 1438 (CH2), 749 (C-H); δ H (400 MHz; CDCl3) 3.62 – 3.68 (1H, m, CH2), 3.73 – 3.79 (1H, m, CH2), 4.23 (1H, d, J 15.2, NCH2), 4.34 (1H, d, J 15.0, NCH2), 4.86 (1H, d, J 14.9, NCH2), 5.12 (1H, d, J 15.0, NC H2), 5.26 (1H, s, CHS), 7.12 – 7.24 (15H, m, Ar-H); δ C (101 MHz; CDCl3) 36.39 (CH2), 46.11 (CH2), 48.78 (CH2), 64.13 (CHS), 76.53 (quaternary C), 127.38 (Ar-C-H), 127.98 (Ar-C-H), 128.17 (Ar-C-H), 128.68 (Ar-C-H), 128.74 (Ar-C-H), 128.80 (Ar-C-H), 128.98 (Ar-C-H), 129.40 (Ar-C-H), 130.42 (Ar-C-H), 133.96 (quaternary C), 134.54 (quaternary C), 136.00 (quaternary C), 164.83 (CO), 165.31 (CO); m/z 469 (100%, [M+Na]+): Found [M+Na]+ 469.1097, C25H22N2O2S2Na requires 469.1101.
SDS-PAGE analysis of proteasome activity.
20 nM UbnGST–Wbp2 and 4 nM purified human 26S proteasome were incubated at 37 ˚C for 2 hours in the presence of different compounds plus 1 mM ATP. The reactions were stopped with 2× SDS sample buffer and analyzed by SDS–PAGE. The gel was scanned using Typhoon™ FLA 9500 biomolecular imager (GE Healthcare life sciences) with filter for fluorophore Alexa 488.
Fluorescence polarization assay.
The fluorescence polarization assay was performed in a low-volume 384-well solid black plate to which the components were added in the following sequence: 1) 5 µl compound in 3% (v/v) DMSO or 3% DMSO control, 2) 5 µl of 26S proteasome (Enzo life science), 3) 5 µl of 5 nM UbnGST–Wbp2 to initiate the reaction. Fluorescence polarization was measured using a plate reader with excitation at 480 nm and emission at 520 nm (PHERAstar, BMG Labtech).
Evaluation of ubiquitin linkage.
Substrate samples were fractionated on an SDS-polyacrylamide gel and digested in-gel with trypsin. Di-Ubs of all 7 linkages (Boston Biotech) were used to quantify the standard heavy Ub linkages. The standard heavy Ub linkages were subsequently used as internal standards to quantify the linkages in substrates ubiquitinated by Rsp5-WT or Rsp5-E6AP. Mass spectrometry was carried out on a Thermo Orbitrap Velos.
Rpn11•Rpn8 di-Ub cleavage assay.
Di-ubiquitin cleavage assay was performed in 40 mM HEPES, pH7.5, 100 mM NaCl, 100 mM KCl and 5% (v/v) glycerol. Rpn11•Rpn8 dimer (5 μM) was incubated with di-UbK48 and different concentrations of inhibitors at 30 °C. The reactions were stopped with 2× SDS sample buffer after 2 hours and analyzed by SDS– PAGE.
hCAII assays.
hCAII was expressed and purified as previously reported (Martin et al., 2013). Human carbonic anhydrase II (hCAII) assays were carried out in clear Costar 96-well plates. Each well contained a volume of 100 µL including buffer (50 mM HEPES buffer, pH 8.0), protein (10 nM hCAII), inhibitor (100 µM), and substrate p-nitrophenyl acetate (500 µM). The protein and inhibitor were incubated in solution at 30 oC for 10 min prior to the addition of the p-nitrophenyl acetate to initiate the reaction. The change in absorbance was monitored at 405 nm for 15 min. The negative control wells, containing no inhibitor, were arbitrarily set as 100% activity. Readings from background wells, which did not contain protein, were subtracted from the active assay wells to account for background hydrolysis activity caused by the buffer. The assays were performed in triplicate. The data were normalized to values measured for uninhibited enzyme. Assay data are reported as the mean±standard deviation.
MMP-2 assays.
MMP-2 and OmniMMP fluorogenic substrate were purchased from Enzo Life Sciences. The assays were carried out in black NUNC 96-well plates. Each well contained a volume of 100 µL including buffer (50 mM HEPES, 10 mM CaCl2, 0.05% (v/v) Brij-35, pH 7.5), human recombinant MMP (1.16 U MMP-2, Enzo Life Sciences), inhibitor (100 µM), fluorogenic OmniMMP substrate (4 mM Mca-Pro-Leu -Gly-Leu-Dpa-Ala-Arg-NH2·AcOH, Enzo Life Sciences). The mixture of protein and inhibitor was pre-incubated in solution at 37 oC for 30 min, followed by the addition of the substrate to initiate the reaction. The change in fluorescence was monitored for 30 min by BioTek synergy HT fluorescence plate readers with excitation and emission wavelengths at 320 and 400 nm, respectively. The control wells, containing no inhibitor, were arbitrarily set as 100% activity. MMP activity was defined as the ratio of fluorescence increase in the inhibitor wells relative to the negative control wells, expressed as a percentage. The assays were performed in triplicate. The data were normalized to values measured for uninhibited enzyme. Assay data are reported as the mean±standard deviation.
UbG76V-GFP degradation assay.
A stable HeLa cell line with the ubiquitin fusion degradation reporter UbG76V-GFP were seeded on 384-well plates (5000 cells/well) and grown for 18 hours. Cells were treated with modified DMEM (without phenol red, folic acid, riboflavin, and vitamin B12) containing MG132 (4 µM) for 1 hour and washed twice with pre-warmed PBS. Modified DMEM containing FBS (2.5%), cycloheximide (CHX) (50 µM), and DMSO or a test compound (0.1, 0.19, 0.39, 0.78, 1.56, 3.13, 6.25, 12.5, 25 and 50 µM) was added to each well. Plates were imaged on the ImageXpress Micro microscope (Molecular Devices) after 60 to 240 min.
IκBα degradation assay.
Hela cells were pre-treated with DMSO or compounds as indicated for 10 minutes and then treated with 25 ng/mL of recombinant human TNF-α (Promega) for 30 minutes. Cells were lysed with RIPA buffer with protease inhibitor cocktail, fractionated by SDS–PAGE and immunoblotted with antibodies against IκBα or GAPDH.
Quantitative real-time PCR of HCT116 cell RNA.
Cells were seeded in 6-well plates at 5 ×105 cells/well in DMEM medium for 16 hours before drug treatment. Compounds were added at 10 μM each except for actinomycin D, which was used at 1 μM as a positive control. RNA was isolated using RNeasy kit (Qiagen). cDNA was synthesized with QuantiTect Reverse Transcription Kit (Qiagen). Quantitative real-time PCR was performed using Roche Lightcycler 480 and SYBR green I master kit. Sequences of primers used for qRT-PCR were as follows: c-Myc: Fwd, 5’-caccagcagcgactctga; Rev, 5’-gatccagactctgaccttttgc; GAPDH: Fwd, 5’-acccactcctccacctttgac; Rev, 5’-ctgttgctgtagccaaattcgt.
QUANTIFICATION AND STATISTICAL ANALYSIS
Graphpad Prism 7 was used for statistical analysis of the data from proteasome activity assay, Rpn11 activity assay, cell viability assay and quantitative real-time PCR. For each analysis, total n and S.D. are presented in the figure legend. Data were fitted to a dose-response equation (three parameters) to determine the IC50 or GI50. Quantitative real-time PCR data were analyzed by Student’s t test (two-tailed). Differences were statistically significant when P<0.05.
Supplementary Material
Highlights.
An HTS assay is developed to measure protein breakdown activity of 26S proteasome
It allows measurement of 26S proteasome activity in cell or tissue lysate
ETPs inhibit JAMM proteases including the proteasomal deubiquitinase Rpn11
ETPs block proliferation of cancer cells including those resistant to bortezomib
The deubiquitinase Rpn11 is a component of 26S proteasome, is required for proteasome activity, and is a new target for proteasome inhibition in cancer. We develop an assay that enables measurement of the proteasome’s protein breakdown activity in cell lysate and identify ETPs as a new class of Rpn11 inhibitors.
Significance.
We develop an assay to measure the protein breakdown activity of the proteasome, and use this assay to demonstrate that gliotoxin and other epidithiodiketopiperazines (ETPs) inhibit proteasome activity by targeting the essential deubiquitinase Rpn11. These molecules quench protease activity by chelating the active site Zn2+ ion in Rpn11. An improved ETP, SOP11, stabilizes proteasome substrates, triggers the unfolded protein response and blocks proliferation of cancer cells. Importantly, it does not cause cells to round up, an off-target effect observed with gliotoxin and other ETPs. In addition to characterizing proteasome inhibitors like ETPs, our assay can be used to monitor the protein degradation activity of the proteasome in cell lysates, which may facilitate studies on modulation of proteasome activity in response to disease, aging, and metabolic state.
Acknowledgements
We thank J.M. Huibregtse (The University of Texas at Austin) for the vector expressing GST–Wbp2-C-K222, Rsp5 and Rsp5-E6AP, A. Martin (University of California, Berkeley) for the pETDuet-1 vector expressing the Rpn11–Rpn8 dimer, T.M. Kapoor (The Rockefeller University) for providing the RPE1 WT and BTZ-resistant cell lines, H. Park for testing the cell lines for mycoplasma contamination, A. Bowers (University of North Carolina) for the discussion on gliotoxin and D. Sherman for critical reading of our manuscript. This work was supported by grants from the Caltech Gates Grubstake Fund and Amgen to R.J.D. and from the NIH (CA164803) to R.J.D. and S.M.C.. S. Hilton and B. Santos was supported by the funding from FCT (grant No. SFRH/BD/65630/2009).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
R.J.D. is a founder, shareholder and member of the Scientific Advisory Board of Cleave Biosciences, which is engaged in discovery and development of drugs that target enzymes involved in ubiquitin-dependent protein degradation. R.J.D is currently Senior Vice President of discovery research at Amgen and a Visiting Associate at the California Institute of Technology. S.M.C. is a co-founder and has an equity interest, and receives income as member of the Scientific Advisory Board for Cleave Biosciences and is a co-founder, has an equity interest, and a member of the Scientific Advisory Board for Forge Therapeutics. Both companies may potentially benefit from the research results of certain projects in the laboratory of S.M.C. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Data and Software Availability
All data are available upon request to the Lead contact.
References
- Bence NF, Sampat RM, and Kopito RR (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Bernardo PH, Brasch N, Chai CL, and Waring P (2003). A novel redox mechanism for the glutathione-dependent reversible uptake of a fungal toxin in cells. J Biol Chem 278, 46549–46555. [DOI] [PubMed] [Google Scholar]
- Cenci S, Pengo N, and Sitia R (2008). Proteotoxic stress and cell lifespan control. Mol Cells 26, 323–328. [PubMed] [Google Scholar]
- Chen HI, and Sudol M (1995). The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci U S A 92, 7819–7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang H, Huang YF, Li ML, Cheng JH, Hu P, Lu CH, Zhang Y, Liu N, Tzeng CM, et al. (2017). WW domain-binding protein 2: an adaptor protein closely linked to the development of breast cancer. Mol Cancer 16, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TF, and Deshaies RJ (2011). Quantitative cell-based protein degradation assays to identify and classify drugs that target the ubiquitin-proteasome system. J Biol Chem 286, 16546–16554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KM, Hilton ST, Mecinovic J, Motherwell WB, Figg WD, and Schofield CJ (2009). Epidithiodiketopiperazines block the interaction between hypoxia-inducible factor-1alpha (HIF-1alpha) and p300 by a zinc ejection mechanism. J Biol Chem 284, 26831–26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, and Cohen RE (2009). K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J 28, 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, et al. (2007). Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 67, 6383–6391. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ (2014). Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol 12, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Valera AG, Moran MA, Gomez-Ramos P, Alvarez-Castelao B, Castano JG, Hernandez F, and Lucas JJ (2006). Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J Neurochem 98, 1585–1596. [DOI] [PubMed] [Google Scholar]
- Dimopoulos MA, Richardson PG, Moreau P, and Anderson KC (2015). Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol 12, 42–54. [DOI] [PubMed] [Google Scholar]
- Dolan SK, O’Keeffe G, Jones GW, and Doyle S (2015). Resistance is not futile: gliotoxin biosynthesis, functionality and utility. Trends Microbiol 23, 419–428. [DOI] [PubMed] [Google Scholar]
- Finley D (2009). Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78, 477–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger WB (1949). Chetomin an antibiotic substance from Chaetomium cochliodes; composition and functional groups. Arch Biochem 21, 125–131. [PubMed] [Google Scholar]
- Giguere CJ, and Schnellmann RG (2008). Limitations of SLLVY-AMC in calpain and proteasome measurements. Biochem Biophys Res Commun 371, 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemeyer W, Fischer M, Krimmer T, Stachon U, and Wolf DH (1997). The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processing. J Biol Chem 272, 25200–25209. [DOI] [PubMed] [Google Scholar]
- Hershko A, and Rose IA (1987). Ubiquitin-aldehyde: a general inhibitor of ubiquitin-recycling processes. Proc Natl Acad Sci U S A 84, 1829–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Patel CN, Bersuker K, Riley BE, Kaiser SE, Shaler TA, Brandeis M, and Kopito RR (2012). Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J Cell Biol 196, 573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan TW, and Pedersen JS (1986). Sporidesmin and gliotoxin induce cell detachment and perturb microfilament structure in cultured liver cells. J Cell Sci 85, 33–46. [DOI] [PubMed] [Google Scholar]
- Kim HC, and Huibregtse JM (2009). Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol Cell Biol 29, 3307–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev AF, and Goldberg AL (2005). Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol 398, 364–378. [DOI] [PubMed] [Google Scholar]
- Kroll M, Arenzana-Seisdedos F, Bachelerie F, Thomas D, Friguet B, and Conconi M (1999). The secondary fungal metabolite gliotoxin targets proteolytic activities of the proteasome. Chem Biol 6, 689–698. [DOI] [PubMed] [Google Scholar]
- Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, et al. (2004). Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 6, 33–43. [DOI] [PubMed] [Google Scholar]
- Lauinger L, Li J, Shostak A, Cemel IA, Ha N, Zhang Y, Merkl PE, Obermeyer S, Stankovic-Valentin N, Schafmeier T, et al. (2017). Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases. Nat Chem Biol 13, 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yakushi T, Parlati F, Mackinnon AL, Perez C, Ma Y, Carter KP, Colayco S, Magnuson G, Brown B, et al. (2017). Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat Chem Biol 13, 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Li J, Travis AM, Hao Y, Klingeborn M, Skiba NP, Deshaies RJ, and Arshavsky VY (2018). Increased proteasomal activity supports photoreceptor survival in inherited retinal degeneration. Nat Commun 9, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Hann ZS, and Cohen SM (2013). Metalloprotein-inhibitor binding: human carbonic anhydrase II as a model for probing metal-ligand interactions in a metalloprotein active site. Inorg Chem 52, 12207–12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega Z, Diaz-Hernandez M, and Lucas JJ (2007). Is the ubiquitin-proteasome system impaired in Huntington’s disease? Cell Mol Life Sci 64, 2245–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL, Krauss B, Schulze-Osthoff K, Decker T, Traenckner EB, Vogt M, Myers C, Parks T, Warring P, Muhlbacher A, et al. (1996). The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-kappaB. J Exp Med 183, 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C, Li J, Parlati F, Rouffet M, Ma Y, Mackinnon AL, Chou TF, Deshaies RJ, and Cohen SM (2017). Discovery of an Inhibitor of the Proteasome Subunit Rpn11. J Med Chem 60, 1343–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, and Deshaies RJ (2010). Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell 38, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez I, and Vilchez D (2014). The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr Genomics 15, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto H, Egashira S, Saito N, Kirisako T, Miller S, Sasaki Y, Matsumoto T, Shimonishi M, Komatsu T, Terai T, et al. (2015). Gliotoxin suppresses NF-kappaB activation by selectively inhibiting linear ubiquitin chain assembly complex (LUBAC). ACS Chem Biol 10, 675–681. [DOI] [PubMed] [Google Scholar]
- Scharf DH, Brakhage AA, and Mukherjee PK (2016). Gliotoxin--bane or boon? Environ Microbiol 18, 1096–1109. [DOI] [PubMed] [Google Scholar]
- Sil BC, and Hilton ST (2013). A Mild and Convenient Base-Catalysed Approach to Disubstituted Epidithiodiketopiperazines. Synlett 24, 2563–2566. [Google Scholar]
- Song Y, Li S, Ray A, Das DS, Qi J, Samur MK, Tai YT, Munshi N, Carrasco RD, Chauhan D, et al. (2017). Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene [DOI] [PMC free article] [PubMed]
- Srinivasan U, Bala A, Jao SC, Starke DW, Jordan TW, and Mieyal JJ (2006). Selective inactivation of glutaredoxin by sporidesmin and other epidithiopiperazinediones. Biochemistry 45, 8978–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen J, Seeger M, Koch A, and Kruger E (2010). Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell 40, 147–158. [DOI] [PubMed] [Google Scholar]
- Tsunawaki S, Yoshida LS, Nishida S, Kobayashi T, and Shimoyama T (2004). Fungal metabolite gliotoxin inhibits assembly of the human respiratory burst NADPH oxidase. Infect Immun 72, 3373–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Aravind L, Oania R, McDonald WH, Yates JR 3rd, Koonin EV, and Deshaies RJ (2002). Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615. [DOI] [PubMed] [Google Scholar]
- Wacker SA, Houghtaling BR, Elemento O, and Kapoor TM (2012). Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol 8, 235–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.